NÖRO-ONKOLOJİ
Yazanlar: Erdem Tüzün,
Figen Hanağası, Pulat Akın Sabancı, Gülşen Akman Demir, Jale Yazıcı
Son güncelleştirme tarihi: 10.12.2019
Sinir
sisteminin tümörlerle çeşitli ilişkilerini inceleyen bir bilim dalı olan nöroonkolojinin
kapsamı içinde primer sinir sistemi tümörlerinin yanı sıra sistemik kanserlerin
nörolojik etkileri (metastaz, lokal yayılım, radyoterapi veya kemoterapi yan
etkileri, tümörün hematolojik-metabolik uzak etkileri ve paraneoplastik
sendromlar) yer alır. Bu ikinci gruptaki tümörler primer tümörlere oranla daha
sık görülürler.
Primer
beyin tümörleri merkezi sinir sistemi (MSS) hücrelerinden köken alan heterojen
bir grubu tanımlamak için kullanılır. Primer veya sekonder (metastatik) beyin
tümörlerinde tümörün davranış özellikleri birbirinden farklılık gösterse de,
sıklıkla karşılaşılan klinik tablolardan biri kafa içi basınç artışı
sendromudur (KİBAS). Ayrıca tümörün lokalizasyonuna göre değişen çeşitli
nörolojik belirti ve bulgular ve/veya epileptik nöbetler görülebilir. Bu
bölümde tümörlerin yarattığı klinik tabloların üzerinde durulmayacak, tümöre
bağlı ortaya çıkan metabolik sorunların sinir sistemi üzerindeki etkileri
kısaca sıralanacaktır. Dileyen okur bu kitapta yer alan ve bu konuların
anlatıldığı diğer bölümlere başvurabilir (Bakınız: Kafa İçi Basıncı Değişiklikleri, Epilepsi, Sinir Sisteminin Nütrisyonel
Hastalıkları).
Epidemiyoloji
Beyin
tümörleri tüm kanserlerin yaklaşık %2’sini oluşturur. Ancak çocukluk yaş
grubunda kanserlerin lösemilerden sonra en sık nedeni MSS tümörleridir. Beyin
tümörlerinin insidansı 100.000’de 21 olarak bildirilmiştir. Kranial
metastazlar, primer beyin tümörlerinden daha sık görülürler. Metastazlar,
kanserli hastaların %10-15’inde görülür ve en sık metastaz görülen bölge
beyindir. Günümüzde kanserli hastaların yaşam süresi uzadığı için beyin
metastazlarının görülme sıklığı da artmıştır. Ayrıca son yıllarda özellikle
endüstrileşmenin fazla olduğu ülkelerde beyin tümörlerinin insidansının arttığı
bildirilmektedir. Kansere bağlı ölümler arasında primer beyin tümörleri görece
küçük bir bölümü oluşturur (yılda yaklaşık 100.000’de 3000). Buna karşılık
diğer kanserlerin SSS’yi tutması ile ilişkili ölüm çok daha sık görülür.
Amerika
Birleşik Devletleri’nde ortalama 300 milyonluk bir nüfusta tüm yaş gruplarında
her yıl yaklaşık 27.000 yeni primer beyin tümörü tanısı konmaktadır. ABD’de
2019 yılında 87.000 (26.000 malign, 61.000 benign) beyin tümörü teşhisi
konulacağı öngörülmektedir Türkiye’de ise her yıl 15.000 civarında vakaya beyin
tümörü tanısı konduğu tahmin edilmektedir. Türkiye’de primer beyin tümörleri
insidansının 10-22/ 100.000 olduğu tahmin edilmektedir. 2018 verilerine göre 82
milyon nüfusa sahip ülkemizde yıllık ortalama 13.000 yeni primer beyin tümörü
hastası olduğu düşünülmektedir.
Beyin
tümörleri tüm kanserler içinde yukarıda verildiği gibi küçük oranda görülmesine
rağmen morbidite ve mortalite oranları diğer kanserlerle karşılaştırılınca
oldukça yüksektir. Amerika Birleşik Devletleri’nde primer malign beyin
tümörleri ve SSS tümörlerinde yıllık ölüm oranlarının 100.000’de 4,25 olduğu
bildirilmiştir. Tüm yaş ve ırklar dahil edildiğinde malign beyin tümörlerinde 5
yıllık sağkalım oranı %33’dür. Primer beyin tümörlerinde ileri yaş, iyi sağlık
durumu ve düşük patolojik evre iyi prognostik faktörlerdir. Buna karşılık tanı
sırasında kognitif bozukluğun bulunması, Karnofski Performans Skorunun düşük
olması, tümörün konuşma, motor, bellek, görme gibi kritik işlevlerden sorumlu
beyin bölgelerinde (“eloquent” alan) olması ve ameliyatta tümörün total
çıkarılamaması kötü prognostik faktörlerdir.
MSS
tümörlerini primer ve sekonder olarak ayırırsak, sekonder tümörlerin (yani
metastatik tümörler) primere göre çok daha sık olduğu görülür. Bunlar içinde
karsinomlar ön plandadır ve en sık da akciğer kanserinin metastazı görülür.
Diğer sekonder tümörler ise hematolojik maligniteler ve melanositik
tümörlerdir. Hematolojik malignitelerden olan lenfomalar çoğunlukla metastatik
olsa da primer MSS lenfoması olarak da görülebilir. Benzer şekilde melanositik
tümörler primer de olabilir.
Merkezi
sinir sisteminin primer tümörleri (metastaz dışındakiler) başlıca glial ve
glial olmayan tümörler olarak gruplanabilir. Glial tümörler içinde erişkinlerde
en sık glioblastom, çocukluk çağında pilositik astrositom görülür. Son yıllarda
çocukluk çağında H3K27M mutasyonu ile tanımlanan orta hat gliomu ayrı bir
antite olarak belirlenmiştir.
Erişkinde
astrositer tümörler %65 (Grade IV GBM dahil), oligodendrogliomlar %18.3,
epandimomlar da %5.9 ile glial tümörleri oluştururlar. Çocukluk çağında
pilositik astrositom ve epandimomlar % 50’lik görülme sıklığı ile glial
tümörleri oluşturur. Erişkinde çok daha nadir, çocuklarda daha sık görülen bir
tümör grubu da embriyonal tümörlerdir, yerleşim infratentoryal ise
medulloblaston adını alırlar. Yeni moleküler bilgiler ışığında
medulloblastomlar ikiye ayrılır; iyi prognoza sahip WNT-grubu ve daha düşük
sağkalıma sahip alt gruplar.
Meningiomlar
parankim dışı tümörlerdir. Ekstra-aksiyel tümör olarak da adlandırılırlar.
Primer MSS tümörlerinin yaklaşık % 30’unu oluştururlar.
Diğer
tümörler; germ hücreli tümörler, kraniofaringiom, hemanjiyoblastom, nöronal
tümörler, mikst glio-nöronal tümörler, histiositozlar olarak sayılabilir.
Ayrıca hipofiz adenomları da sık görülen tümörlerdir. Primer intrakranial
tümörlerin % 20’sini oluştururlar.
Beyin ve
MSS tümörlerinin oluşumundaki risk faktörleri arasında genetik ve çevresel
nedenler sayılabilir. Şimdiye kadar bilinen genetik nedenler içinde
nörofibromatozis (pontoserebellar köşe tümörleri), von Hippel-Lindau sendromu
(serebellar hemanjioblastomlar), Li-Fraumeni sendromu, Turcot sendromu ve Golin
sendromu sayılabilir. Olası çevresel nedenler arasında radyasyona maruziyet,
infeksiyonlar, polivinil klorür, N-nitröz bileşenleri ve polisiklik karbonlara
maruziyet sayılabilir. MSS tümörleri çoğunlukla sporadik olmakla birlikte bazen
herediter olarak yukarıda sayılan çeşitli sendromlarda görülürler.
Sınıflama
Dünya
Sağlık Örgütü (DSÖ) beyin tümörlerinin ilk sınıflamasını 1979 yılında yapmış ve
1993, 2000, 2007 yıllarında gelişmelere bağlı olarak güncellemelerle
yenilemiştir. Son olarak 2016 yılında DSÖ tarafından beşinci güncel sınıflama
yayınlanmıştır. Eski sınıflamalar, beyin tümörü hücrelerinin mikroskobik
özeliklerine göre histolojik farklılık temelinde yapılmıştır. Buna karşılık
DSÖ’nün 2016 yılında yaptığı son sınıflamada histopatolojik özelliklerin yanı
sıra, son 20 yıldır tümörogenezisin etyolojisi alanında kaydedilen gelişmelere
bağlı olarak moleküler tıp temelli bir ayırıma da gidilmiştir. Merkezi sinir
sistemi tümörleri 2007 ve 2016 sınıflaması arasındaki en önemli fark 2007’nin
histogeneze ve hücre kökenine dayalı olması, 2016 sınıflamasının ise moleküler
değişiklikleri ön plana çıkarıp histopatoloji ile bütünleştirmesidir. Bu sınıflama
ile beyin tümörlerinin prognozu hakkında da bilgi verilmektedir. Sınıflamada
katmanlı tanı formatına geçilmiştir. Tanı katmanları aşağıda sıralanmaktadır:
Katman 1:
Entegre (bütüncül) tanı
Katman 2:
Histolojik tanı
Katman 3:
DSÖ evrelemesi (grad)
Katman 4:
Moleküler bilgi
Tabakalı
tanı kavramında katman 1 son ve bütüncül tanı içindir. Eğer diğer katmanların
tamamı hakkında bilgi varsa bütüncül tanı konulabilir. Katman 2 histolojik
sınıflamayı kapsar (örneğin, astrositom, oligodendrogliom). Katman 3 evrelemedir
(örneğin, DSÖ evre II, III). Evreleme atipi, damarlanma, mitoz özellikleri ile
karakterize büyüme potansiyelini içerir. Son olarak katman 4, tümörün moleküler
özelliklerini içerir (örneğin, IDH-mutasyonu, 1p/19q ko-delesyon).
Nöropatologlar bu katmanlı tanı kavramında katman 2 ve 3’ten sorumludur. Ancak
bazı tümörler için katman 4 bilgisi yoksa bütüncül tanı, yani dolayısıyla tam
tanı, konulamaz. Bu durum tanıda “NOS (not otherwise specified)” /
“tanımlanamadı” şeklinde belirtilmektedir. Tümörleri moleküler özelliklerine
göre sınıflamak klinisyene doğru ve güvenilir bilgi verir, erişkin ve bazı
pediyatrik tümörleri birbirinden ayırmayı sağlar ve prognoz hakkında fikir
ortaya koyar.
Örnek
verilecek olursa bütünleşmiş tanısı oligodendrogliom, DSÖ grad II izositrat
dehidrogenaz (IDH) mutasyonu, 1p/19q ko-delesyonu olarak tanı konulmuş bir
beyin tümörü vakasında histolojik tanının oligodendrogliom fenotipi
olduğu, evrelemesinin II olduğu ve moleküler testler sonucunda IDH mutasyonu ve
1p/19q ko-delesyonu saptandığı anlaşılır. Diffüz glial bir tümörde IDH
mutasyonu ve 1p/19q ko-delesyonu varsa iyi prognoz göstergesidir.
En son
sınıflamaya göre primer beyin tümörlerinin listesi Tablo 1’de yer
almaktadır. Bu son sınıflamadaki en önemli değişiklikler diffüz gliomlar,
medullablastomlar ve embriyonal tümörlerde olmuştur.
Tablo1. Dünya Sağlık Örgütü-Merkezi Santral Sinir Sistemi Tümörlerinin
Sınıflaması
|
Diffüz Astrositik ve Oligodendroglial Tümörler |
Koroid Pleksus Tümörleri |
|
Diffüz astrositom, IDH*-mutant |
Koroid pleksus papillomu |
|
Gemisitositik astrositom, IDH mutant |
Atipik koroid pleksus papillomu |
|
Diffüz astrositom, IDH-wild (doğal)
tipi
|
Koroid pleksus karsinomu |
|
Diffüz astrositom, sınıflandırılmamış |
|
|
|
Nöronal ve Mikst Nöronal Glial Tümörler |
|
Anaplastik astrositom, IDH-mutant |
Disembryoplastik nöroepitelyal tümör |
|
Anaplastik astrositom IDH-wild (doğal) tipi |
Gangliositom |
|
Anaplastik astrositom, sınıflandırılmamış |
Gangliogliom |
|
|
Anaplastik gangliogliom |
|
Glioblastom, IDH-wild (doğal) tipi |
Displastik serebellar gangliositom (Lhermitte Duclos hastalığı) |
|
Dev hücreli glioblastom |
|
|
Gliosarkom |
Desmoplastik infantil astrositom ve gangliogliom |
|
Epiteloid glioblastom |
Papiller glionöronal tümör |
|
Glioblastom, IDH-mutant |
Rozet formlu glionöronal tümör |
|
Glioblastom, sınıflandırılmamış |
Diffüz leptomeningeal glionöronal tümör |
|
|
Santral nörositom |
|
Diffüz orta hat gliomu, H3 K27M-mutant |
Ekstraventriküler nörositom |
|
|
Serebellar liponörositom |
|
Oligodendrogliom, IDH-mutant ve 1p/19q-ko-delesyonlu |
Paragangliom |
|
Oligodendrogliom, sınıflandırılmamış |
|
|
|
Pineal Bölge Tümörleri |
|
Anaplastik oligodendrogliom, IDH-mutant ve 1p/19q-ko-delesyonlu |
Pineositom |
|
Anaplastik oligodendrogliom, sınıflandırılmamış |
Pineal parenkimal tümör (orta diferansiye) |
|
|
Pineoblastom |
|
Oligoastrositom, sınıflandırılmamış |
Pineal bölgenin papiller tümörü |
|
Anaplastik oligoastrositom, sınıflandırılmamış |
|
|
|
Embriyonel Tümörler |
|
Diğer Astrositik Tümörler
|
Medullablastom, genetik olarak tanımlanmış |
|
Pilositik astrositom |
Medullablastom, WNT-aktive |
|
Pilomiksoid astrositom |
Medullablastom, SHH-aktive ve TP53-mutant |
|
Subepandimal dev hücreli astrositom |
Medullablastom, SHH-aktive ve TP53-wild tipi |
|
Pleomorfik ksantoastrositom |
Medullablastom, non-WNT/non-SHH |
|
Anaplastik pleomorfik ksantoastrositom |
Medullablastom, grup 3 |
|
|
Medullablastom, grup 4 |
|
Epandimal Tümörler |
Medullablastom, histolojik olarak tanımlanmış |
|
Subepandimom |
Medullablastom, klasik |
|
Miksopapiller epandimom |
Medullablastom, desmoplastik/nodüler |
|
Epandimom |
Medullablastom, yaygın nodülariteli |
|
Papiller epandimom |
Medullablastom, büyük hücreli, anaplastik |
|
Berrak hücreli epandimom |
Medullablastom, NOS |
|
Tanisitik epandimom |
|
|
Epandimom, RELA füzyon – pozitif |
Embriyonel tümör (çok katlı rozet,C19MC-değişmiş) |
|
Anaplastik epandimom |
Embriyonel tümör (çok katlı rozet, sınıflandırılmamış) |
|
|
Medulloepitelyom |
|
Diğer Gliomlar |
SSS Nöroblastomu |
|
3. Ventrikülün kordoid gliomu |
SSS Ganglionöroblastomu |
|
Anjiosentrik gliom |
SSS embriyonel tümörü, sınıflandırılmamış |
|
Astroblastom |
Atipik teratoid/rabdoid tümör |
|
|
SSS’nin rabdoid özellikli embriyonel tümörü |
|
|
|
|
|
Melanositik Tümörler |
|
|
Meningeal melanositoz |
|
|
Meningeal melanositom |
|
|
Meningeal melanom |
|
|
Meningealmelanomatoz |
|
|
|
|
Kranial ve Paraspinal Sinir Tümörleri |
Lenfomalar |
|
Schwannom |
SSS’nin diffüz büyük B hücreli lenfoması |
|
Hücresel schwannom |
İmmun yetersizliğe bağlı SSS lenfomaları |
|
Pleksiform schwannom |
AIDS’e bağlı diffüz büyük B hücreli lenfoma |
|
Melanositik schwannom |
EBV-pozitif diffüz büyük B hücreli lenfoma |
|
Nörofibrom |
Lenfomatoid granülomatozis |
|
Atipik nörofibrom |
İntravasküler büyük B hücreli lenfoma |
|
Pleksiform nörofibrom |
Düşük gradlı SSS’nin büyük B hücreli lenfoması |
|
Perinörinom |
SSS’nin T hücreli ve NK/T hücreli lenfoması |
|
Hibrid sinir kılıfı tümörü |
Anaplastik büyük hücreli lenfoma, ALK pozitif |
|
Malign periferik sinir kılıfı tümörü (MPSKT) |
Anaplastik büyük hücreli lenfoma, ALK negatif |
|
Epiteloid MPSKT |
Dura’nın MALT lenfoması |
|
Perinöral diferansiyasyonlu MPSKT |
|
|
Psammamatöz meningiom |
Histiositik Tümörler |
|
Anjiomatöz meningiom |
Langerhans hücreli histiositoz |
|
Mikrokistik meningiom |
Erdheim – Chester hastalığı |
|
Sekretuar meningiom |
Rosai- Dorfman hastalığı |
|
Lenfoplazmasitten zengin meningiom |
Juvenil ksantrogranülom |
|
Melaplastik meningiom |
Histiositik sarkom |
|
Kordoid meningiom |
|
|
Berrak hücreli meningiom |
Germ Hücreli Tümörler |
|
Atipik meningiom |
Germinom |
|
Papiller meningiom |
Embriyonel karsinomu |
|
Rabdoid meningiom |
York sac tümörü |
|
Anaplastik (malign) meningiom |
Koryokarsinom |
|
|
Teratom |
|
Mezenkimal, non-meningotelyal tümörler |
Matür teratom |
|
Soliter fibröz tümör/hemanjioperisitoma**(WHO 2016 sınıflamasına
göre) |
İmmatür teratom |
|
Grad1 |
Malign transformasyonlu teratom |
|
Grad 2 |
Mikst germ hücreli tümör |
|
Grad 3 |
|
|
Hemanjioblastom |
Sella Bölgesi Tümörleri |
|
Hemanjiom |
Kranyofaringioma |
|
Epiteloid hemanjioendotelyom |
Adamantinomatöz kranyofarengioma |
|
Anjiosarkom |
Papiller kranyofarengioma |
|
Kaposi sarkomu |
Sella bölgesinin granüler hücreli tümörü |
|
Ewing sarkomu/PNET |
Pituisitom |
|
Lipom |
İğsi hücreli onkositom |
|
Anjiolipom |
|
|
Hipernom |
Metastatik tümörler |
|
Liposarkom |
|
|
Desmiod tip fibromatoz |
|
|
Miyofibroblastom |
|
|
İnflamatuar miyofiblastik tümör |
|
|
Benign fibröz histiositom |
|
|
Fibrosarkom |
|
|
İndiferansiye pleomorfik sarkom/malign fibröz histiositom |
|
|
Leiomiyom |
|
|
Leimiyosarkom |
|
|
Rabdomiyom |
|
|
Rabdomiyosarkom |
|
|
Kondrom |
|
|
Kondrosarkom |
|
|
Osteom |
|
|
Osteokondrom |
|
|
Osteosarkom |
|
|
|
|
*(IDH: İzositrat
dehidrogenaz)
Patogenez
Primer beyin
tümörlerinin % 5’den azı genetik yatkınlık sonucu oluşur. Hastaların çoğunda
tanımlanabilir risk faktörleri saptanmaz. İyonize radyasyon primer beyin
tümörlerinde tartışmasız risk faktörüdür. Diğer çevresel toksinlerin beyin
tümörlerine yol açtığı ile ilgili kesin bir bilgi yoktur. Büyük ölçekli
çalışmalar yapılmasına rağmen cep telefonu kullanımı da dahil radyofrekanslı
elektromanyetik alanların beyin tümörlerini arttırdığına dair kesin bir veri
bilinmemektedir. Ancak elde edilen bilgilerin toplamı uzun dönem cep telefonu
kullanımının gliom riskinde çok hafif bir artış yapabileceği yönündedir. DSÖ’ye
bağlı Uluslararası Kanser Araştırma Ajansı radyofrekans-elektromanyetik
alanların insanlar için olası karsinojenik olabileceğini bildirmiştir. Ancak bu
alan halen tartışmalıdır.
Primer
beyin tümörlerinin patogenezi tartışılırken başlıca iki ana hipotez üzerinde
durulur. Bunlardan 1800’lü yıllardan bu yana öne sürülen ilki; beyin
tümörlerinin embriyonel kalıntılardan ve kök hücrelerin diferansiyasyon sırasında
blastik değişim göstermesinden kaynaklandığını savunan hipotezdir. Bugün daha
fazla kabul gören diğer hipotez ise olgunlaşmış erişkin hücrelerin ardışık
mutasyonlar sonucunda kontrolsüz proliferasyonu ve dediferansiyasyonu
hipotezidir. Son yıllarda bu ikinci hipotezi doğrulayan pek çok onkogen ve
tümör baskılayıcı gen tanımlanmıştır. Tümör baskılayıcı genlerden en önemlisi
17. kromozomdaki P53 genidir. P53 geni hücre siklusunun düzenlenmesinde, DNA
hasarına cevapta ve programlanmış hücre ölümünde (apoptoz) önemli rol oynar.
Normal işlev gösteren P53 geninin glioblastom hücre serilerini baskıladığı,
mutant P53 geninin ise transformasyon gösteren astrositlerin ölümsüzleşmesinde
rol oynadığı gösterilmiştir. P53 geninin çalışmasını düzenleyen başka genler de
mevcuttur: Bunlardan 12. kromozomda bulunan MDM2 geni P53 geninin işlevini
baskılar. Yine, 6. kromozomda yer alan sikline bağımlı kinaz (CDK:cyclin
dependent kinase) genleri tarafından kodlanan P21 proteini hücre siklusunu
durdurur. Bunlar dışında hücre siklusu üzerine etkili proteinleri kodlayan
birçok başka gen de tanımlanmıştır. Neoplazi oluşumunda hücre siklusu üzerinde
etkili genlerin dışında, büyüme faktörlerinin ve reseptörlerinin de rolü
vardır. Bunlar arasında epidermal büyüme faktörü (EGF) ve reseptörü (EGFR),
trombosit kaynaklı büyüme faktörü (PDGF) ve reseptörü (PDGFR), fibroblast
büyüme faktörü (FGF) ve damar endotel büyüme faktörü (VEGF) sayılabilir. Hücre
gelişiminde rol oynayan bu büyüme faktörlerinin anormal derecede artması hücre
içi sinyal iletimi ile mitozu arttırmaktadır. Tümörlerde genellikle hem büyüme
faktörlerini hem de reseptörlerini kodlayan genlerde artış olduğu
gözlenmektedir. Bunların dışında henüz geni ve kodladığı proteini belirlenmemiş
bazı kromozom bozuklukları da tanımlanmıştır. Bütün bu sayılanlardan başka
apoptozun neoplazi gelişimindeki rolü de ortaya konmuştur. Apoptozda rol
oynayan başlıca iki yol vardır: Birincisi hücre yüzeyinde bulunan FAS molekülü
ve buna karşı gelen FAS ligandı (FASL), ikincisi de hücre içinde bulunan BAX ve
BCL-2 proteinleri. FAS ve BAX apoptozu indüklerken BCL-2 apoptozu önler. Daha
önce sözü edilen P 53 proteini de FAS ve BAX aktivasyonu yaparken, BCL-2’yi
baskılar. Apoptoz yeteneğinin kaybı hem neoplazi gelişimini indükler, hem de
tedaviye direnç sağlar. Bu nedenle pro-apoptotik proteinlerin arttırılması
yakın gelecekte birincil tedavi hedeflerinden biri haline gelebilir.
Gliomlar
Gliomlar
erişkinlerde primer beyin tümörlerinin %75’ini oluşturur. Glial
(astrositler, oligodendrositler, mikroglia hücreleri) veya prekürsör
hücrelerden kaynaklanan nöroektodermal kökenli tümörlerdir. Bu tümörlerin
2016’daki yeni sınıflamasında önemli değişikliklere gidilmiştir. Bu
değişiklikler ile tanı kriterleri, tanı testine yaklaşım, evreleme, prognoz ve
tedavi planı hakkında önemli farklılıklar olmuştur. Moleküler ve histolojik
parametrelerin bu sınıflanma ile yenilenmesi genomik, kanser ve MSS
immünolojisi alanları sayesinde olmuş ve nöroonkoloji alanında yeni bir çığır
açmıştır. DSÖ’nün 2016’dan önceki sınıflamasında tüm astrositik tümörler
birlikte sınıflandırılmıştır. Şimdi kullanılan sınıflamada; sınırları belirli
gliomlar DSÖ evre I ve diffüz infiltre gliomlar ise DSÖ evre II-IV arasında
büyüme paternlerine ve IDH mutasyonunun varlığı veya yokluğuna göre sınıflandırılmıştır.
Düşük gradlı diffüz astrositomlar: DSÖ sınıflamasına göre evre 2 düzeyindeki astrositik tümörler ve
oligodendriogliomlar bu gruba girer. Yüksek diferansiasyon vardır, nükleer
atipi vardır, yavaş büyür ancak çevre dokulara yaygın infiltrasyon gösterirler.
Genellikle 30-40 yaş arası genç erişkinlerde sıktır. Yerleşim sıklığı
açısından, supratentoriyal bölge birinci, beyinsapı ise ikinci sırayı alır.
Nöroradyolojik olarak genellikle sınırları belirsiz, solid, pek kontrast
tutmayan bir kitle olarak görülür. BT’de hipodens, MRG’de T1 ağırlıklı
kesitlerde hipointens, T2 ağırlıklı kesitlerde hiperintens görünür (Şekil 1).
Tedavi, tümör yerleşimi riskli değilse, tümörün cerrahi olarak çıkarılması
şeklindedir; mümkün olduğunca geniş total rezeksiyon hedeflenir. Bu durum
“maksimum güvenli cerrahi” (maximal safe resection) olarak tanımlanmaktadır.
Kısaca hastada morbidite oluşturmadan kitlenin çıkarılabilecek en geniş
bölümünün çıkarılması hedeflenmelidir. Kitlenin %78’inden fazlasının alınması
durumunda cerrahinin prognoza olumlu etkisi olduğu gösterilmiştir. Düşük gradlı
diffüz gliomlarda IDH1/2 mutasyonu ve 1p/19q ko-delesyonu durumuna göre tanısal
ve prognostik faktörler belirlenir. IDH mutant ve 1p/19q ko-delesyonu olan
tümörler en iyi prognoza sahiptirler. Bunu IDH mutant ve 1p/19q intakt
tümörler, en son olarak IDH doğal (wild) tip tümörler izler. Özellikle 40 yaşın
üstünde, nörolojik semptomlara sahip, büyük tümörü olanlar (5cm²’den büyük)
veya subtotal rezeksiyon yapılanlar yüksek riskli düşük gradlı diffüz
gliomlardır. Özellikle IDH doğal (wild) tipe sahip olan gliomlar bu grupta en
yüksek riskli tümörler olarak görülürler. IDH mutant yüksek riske sahip düşük
gradlı gliomlara cerrahi sonrası radyoterapi standart tedavidir. Ayrıca
kemoterapi de verilir. Cerrahi sonrasında ortalama sağkalım süresi 6-8 yıldır.
Bazen zaman içinde daha habis formlara dönüşüm görülebilir.


Şekil 1. Düşük gradlı glial tümör hastasına ait MRG kesitleri. Aksiyal
kesitte sağ oksipital lobda falks komşuluğunda T2 kesitlerde hiperintens, T1 kontrastlı
kesitte kontrast tutmayan lezyon.
Oligodendrogliom: Başlıca
oligodendroglial hücrelere benzeyen hücrelerce oluşturulan diffüz infiltratif
bir tümördür. Nükleer atipi, hücresel çeşitlilik ve mitotik aktivite arttığında
anaplastik oligodendrogliom adını alır; bu durumda bazen vasküler proliferasyon
ve nekroz da gösterebilir. Bunların dışında hem astrositik hem oligodendrositik
hücrelerden kaynaklanan oligoastrositomlar da mevcuttur. Oligodendrogliomlar
genellikle 3.- 4. onyılda görülürse de bazen çocuklarda da görülebilir.
Anaplastik oligodendrogliom ise 4.-5. onyılda daha sıktır. Oligodendrogliomlar
sıklıkla frontal ve temporal bölgede derin ak maddede yerleşir. Nöroradyolojik
olarak kitle etkisi olan, BT’de hipo veya izodens görünen, kalsifikasyon
gösterebilen, MRG’de T1 ağırlıklı kesitlerde hipointens, T2 ağırlıklı
kesitlerde hiperintens görünen ve sınırları belirgin olan lezyon görülür. BT’de
kalsifikasyon göstermesi kitlenin oligodendrogliom olabileceğini hakkında ciddi
şüphe uyandırmalıdır (Şekil 2). Hafif kontrast tutabilir, etrafı
ödemlidir. Bazen tümör içine kanama veya kist bulunabilir. Özellikle anaplastik
formda nekrotik kist, kanama, kalsifikasyon ve kontrast tutulumu daha
belirgindir. Çok nadiren oligodendrogliomlar BOS yoluyla yayılabilir. Tedavide
genellikle iyi diferansiye tümörlerde cerrahi yeterlidir; anaplastik olanlarda
cerrahinin ardından radyoterapi ve bazen kemoterapi uygulanır.
Oligodendrogliomda rezeksiyon sonrası ortalama sağkalım yaklaşık 4,5 yıl iken,
anaplastik formda biraz daha kısadır. Özellikle kromozom IDH-mutant ve
1p/19q-alelik kaybı bulunan tümörlerin kemosensitif olmaları dolayısıyla
tümörün bu yönden incelenmesi mümkün olursa prognostik önem taşıyabilir.



Şekil 2. Sol frontal yerleşimli oligodendrogliom hastasına ait preop MRG.
T2 aksiyal kesitte sınırları düzensiz sol lateral ventrikül frontal boynuzuna
baskı yapmış hiperintens kitle görüntüsü. T1 kontrastlı kesitte kontrast
tutmayan hipointensite. Aynı hastanın kranial BT tetkikinde tümör bölgesinde
kalsifikasyona ait hiperdensite görülmekte.
Anaplastik astrositom: “Malign”
astrositom veya yüksek gradlı astrositom adı da verilir. Diffüz yüksek gradlı
tümörler grubunda DSÖ grad III olarak yer
alır. Fokal veya yaygın anaplazinin yanı sıra, yoğun proliferasyon ve çevre
dokulara yaygın infiltrasyon gösterir. Başlangıç yaşı düşük gradlı
astrositomlara oranla daha geçtir (ortalama 41 yaş) ve daha hızlı ilerler.
Hemisferik tutulum yapabilir, yani tümör birden çok lobu aynı zamanda invaze
etmiş olabilir. Nöroradyolojik olarak sınırları belirsiz, solid bir kitle
olarak görülür. BT’de hipodens, MRG’de T1 ağırlıklı kesitlerde hipointens, T2
ağırlıklı kesitlerde hiperintens görünür. Düşük gradlı astrositomların tersine
kısmen kontrast tutulumu vardır ve ödem etkisi daha belirgindir. (Şekil 3) Esas
olarak cerrahi tedavi yapılması ve daha sonra kemoterapi ve radyoterapi
önerilir. Tedavi yüz güldürücü olmamakla birlikte, 40 yaş altı genç hastalarda
1,5 yıllık sağkalım oranı % 60 civarındadır.


Şekil 3. Sol temporal yerleşimli anaplastik astrositom. Kontrastlı T1
kesitlerde düzensiz sınırlı yüksek kontrast tutulumu.
Glioblastom: En “malign”
ve en sık astrositik tümördür. Çok az diferansiasyon gösterir, yoğun mitoz,
damar proliferasyonu ve nekroz içerir, çevre dokulara yoğun infiltrasyon
vardır. Erişkinlerde en sık görülen beyin tümörüdür. Genellikle 45-70 yaş arası
erişkinlerde sıktır (ortalama 53 yaş), ancak daha gençlerde de görülebilir ve
hızlı ilerler. Genellikle supratentoriyal yerleşim gösterir; frontotemporal
veya parietal yerleşim sıktır; bazal ganglia bölgesine ve korpus kallozumdan
karşı hemisfere geçiş sıktır; bu şekilde her iki hemisferi de tutan gliomlara
kelebek gliomu adı da verilir. Makroskopik veya mikroskopik olarak multifokal
olabilir. Nöroradyolojik olarak düzensiz sınırlı, ortasında nekrotik kavite
bulunan, çevresinde halkasal düzensiz kontrast tutulumu gösteren bir kitle
olarak görülür, etrafı ödemlidir (Şekil 4). Bazen BOS’ta protein
artışına ve lenfositik pleositoza yol açabilir. Nadiren de meningial
gliomatozis görülebilir. MSS dışına yayılım çok nadirdir. Prognozu kötüdür;
ortalama beklenen yaşam süresi 15.4 aydır. Cerrahide amaç hastada ek morbidite
oluşturmadan çıkarılabilecek en geniş tümör hacminin çıkarılmasıdır (Maximal
safe resection). Tümörün yerleşim yerine göre eloquent / “değerli”
alanlardaki bazı tümörler total olarak çıkarılamayabilirler. Ek olarak
radyoterapi ve kemoterapi yapılır.
Özellikle
tümörü büyük oranda çıkarılabilmiş, genç ve genel durumu iyi hastalarda oral
temozolamid ile kemoterapi, sağkalım şansını arttırabilir. Son yıllarda
O-metilguanin-DNA-metil transferaz (MGMT) geni “promoter” bölgesinin metillenmiş
olmasının temozolamid ve radyoterapiye cevabı önemli oranda arttırdığına dair
veriler elde edilmiştir. Radyoaktif/kemoterapötik implantlar ve stereotaktik
radyocerrahi de denenmektedir. Bunların dışında son yıllarda giderek artan
sıklıkta hedefli biyolojik tedaviler kullanılmaktadır. Yüksek gradlı glial
tümörler anjiojenik yapıları çok güçlü tümörlerdir. Bu nedenle son yıllarda
yapılan çalışmalarda özellikle tümörün yeni damar yapma potansiyelini
hedefleyen yaklaşımlar ön plana çıkmıştır. Tümörün yeni damar yapmasından
sorumlu temel moleküller VEGF (Vascular endothelial growth factor) ve VEGF
reseptörleridir. Günümüzde VEGF‘yi bağlayarak etkinliğini azaltan humanize
monoklonal antikor bevacizumab ve VEGFR 1 ve 2’yi bloke eden reseptör tirozin
kinaz inhibitörleri (sorafenib ve sunatinib) mevcuttur. Bu tedavilerde hedef
tümör hücresi değil tümörün mikro çevresidir. Bu ajanların kemoterapi
ilaçlarının dokuya penetrasyonlarını arttırdığı da gösterilmiştir ve
bevacizumab, glioblastoma multiforme tedavisinde Amerikan Gıda ve İlaç
Dairesi’nin (FDA) onayını almıştır. Yüksek gradlı glial tümörlerde hedeflenen
diğer genetik yollar arasında EGFR (epidermal growth factor), mTOR, p53, PI3K
sayılabilir.


Şekil 4. Sağ frontal yerleşimli GBM tümörlü hastaya ait MRG görüntüleri.
T2 kesitte 1.5 cm’lik orta hat şifti görülmekte. T1 kontrastlı kesitte bu
derecede kitle etkisine sebep olan tümörün aslında 2.5 cm çapında olduğu ve T2
de görünen hiperintansitenin çoğunun ödem olduğu anlaşılmakta. Diğer önemli
detay olarak da GBM’lerde tümörün çevresinin düzensiz kontrast tuttuğu,
merkezinin ise hipointens olduğu bu kesitte görülmekte.
Pilositik astrositom: Düşük evreli
astrositomlar arasındadır, ancak çok net sınırlarının olması nedeniyle farklı
kategoride ele alınır. Genellikle kistik komponenti vardır. Çoğunlukla ilk
yirmi yılda olmak üzere çocuklarda ve genç erişkinlerde sıktır. SSS’nin pek çok
bölgesinde görülebilir. En sık görüldüğü lokalizasyonlar serebellum, optik
sinirler (optik sinir gliomu), optik kiazma-hipotalamus (kiazmatik/ hipotalamik
gliom), talamus-bazal ganglia, beyinsapı, serebral hemisferler, daha nadiren de
medulla spinalistir. Nöroradyolojik olarak sınırları belirgin ve genellikle
kontrast tutan bir kitle ve çevresinde kist olarak görülür. Kistik özellik
göstermesi tanıyı kolaylaştırır (Şekil 5). Yavaş büyür, bazen stabilize
olabilir, hatta gerileyebilir. Bilinen en iyi huylu primer beyin tümörüdür.
Malign transformasyon göstermez. Çok nadiren BOS yoluyla yayılabilir. Prognozu
iyidir. Nadiren ölümcüldür. Total rezeksiyon yapıldığında 10 yıllık sağkalım
%100’e yakındır. Buna karşın subtotal rezeksiyonlarda oranlar %75 civarındadır.
Genetik yatkınlık söz konusu olabilir. Tip 1 nörofibromatozlu hastalarda en sık
görülen SSS tümörüdür.
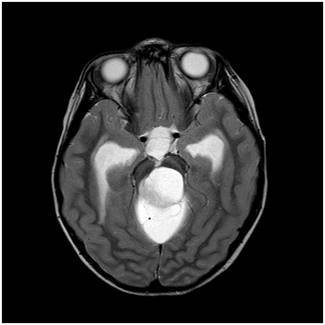

Şekil 5. Arka
çukur yerleşimli pilositik astrom hastasına ait MRG görüntüleri. Büyükçe bir
kist ve kenarında daha küçük solid tümör izlenmekte.
Epandimom: Ventrikülleri
kaplayan epandimal tabakadan kaynaklanan nöroektodermal kaynaklı tümörlerdir.
DSÖ 2016 sınıflamasında epandimal tümörler 3 evre ve farklı antiteye
bölünmüştür. Evre 1’deki epandimomlar diğer evredekilerden histopatolojik
özelliklerine göre ayrılırlar. Buna karşın grad II ve grad III’ün birbirinden
ayırt edilmesi prognostik açıdan büyük önem taşır. 2017’de uluslararası bir
uzlaşma grubu epigenetik ve genetik özellikleri, MSS lokalizasyonu ve yaş
grubuna göre 9 farklı epandimom grubunu moleküler özelliklerini de katarak tanımlamıştır.
Nükleer atipi, hücresel çeşitlilik ve artmış mitotik aktivite gösteren, bazen
vasküler proliferasyon ve nekroz da içerebilen tümörlere anaplastik epandimom
adı verilir. Subepandimom ve sadece kauda-konus bölgesinde yerleşen
miksopapiller epandimom genellikle düşük gradlı tümörlerdir. Ek olarak, RELA
füzyon pozitif-epandimom yeni bir antite olarak eklenmiştir. C11orf95 ve RELA
gen füzyonu mutasyonları supratentoriyal epandimomaların %7’den fazlasında
bulunur ve kötü prognoza sahiptir. RELA füzyon aktivitesi NF-kB yolağında
kontrol edilemeyen tümör aktivitesi ile sonuçlanır. Epandimomlar 1 ay-81 yaş
arasında bütün yaş gruplarında görülebilir. Epandimomlar ventriküllerin
herhangi bir bölgesinde yer almakla birlikte en sık 4. ventrikülden kaynaklanır.
Bu dağılım yaşa göre değişir. İnfratentoriyal yerleşim çocuklarda daha sıkken,
erişkinlerde spinal ve infratentoriyal yerleşim eşit orandadır. Nöroradyolojik
görünümü; sınırları düzgün bir kitle şeklindedir. Kistik komponenti olabilir,
değişen oranlarda kontrast tutabilir (Şekil 6). Hidrosefali sık görülür.
Bazen komşu normal nöral dokuyu infiltre eder. BOS yoluyla, tohumlama (seeding)
tipi metastaz yapması sıktır. Son iki özellik anaplastik formda sık görülür.
Bazen uzak metastaz dahi görülebilir. Tedavi olarak genellikle tümör
rezeksiyonuna ek olarak BOS yoluyla yayılıma karşı radyoterapi uygulanmaktadır.
Ancak radyoterapi uygulanacak alanın genişliği tartışmalıdır. Anaplastik formda
kemoterapi de eklenebilir. Çocuklarda prognoz kötü olmakla birlikte spinal
epandimomlarda prognoz oldukça iyidir.

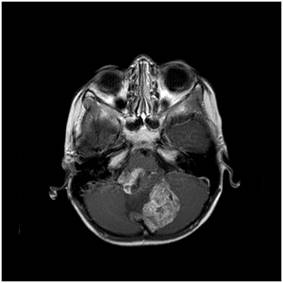
Şekil 6. Arka çukur epandimomuna ait MRG görüntüleri. T1 kontrastlı
kesitlerde düzensiz kontrast tutulumu görülmekte. Köken aldıkları doku gereği
subaraknoidal sisternaları dolduran bu tümör, T1 kontrastlı kesitte de
görüldüğü gibi her iki Foramen Lushka’dan geçerek beyin sapı sisternlarını
kaplamış.
Medulloblastom: Çocuklarda en
sık görülen primer beyin tümörüdür. Serebellumda yerleşen infiltratif ve malign
bir ilkel nöroektodermal tümördür. İlkel nöroektodermal tümörlerin ortak bir
öncü hücreden kaynaklandığı düşünüldüğünden farklı MSS hücre gruplarına doğru
farklılaşma gösterirler. En sık 7 yaş civarı olmak üzere genellikle çocukluk
çağında ortaya çıkar ve hastaların üçte ikisi erkektir. Çoğunlukla vermiste
yerleşim gösterir ve 4. ventriküle uzanır. BOS dolaşım yolunda tıkanıklığa
sebep olması nedeniyle çoğu hastada tümör hidrosefaliye yol açmaktadır ve bu
vakalarda başvuru hidrosefali kliniği ile olmaktadır. Nöroradyolojik olarak
yoğun ve homojen kontrast tutulumu gösteren solid kitle şeklinde görülür (Şekil
7). Leptomeningial yayılım sıktır ve bu da meninkslerde nodüler veya yaygın
kontrast tutulumuna yol açar. Yeni sınıflamada alt gruplar tanımlanmıştır ve
prognozları değişkenlik gösterir. Tedavide genellikle cerrahinin ardından
radyoterapi uygulanır. Bu tedavi ile 10 yıllık sağkalım oranı yaklaşık %45
civarındadır, ancak 3 yaşından küçük çocuklarda radyoterapiden kaçınıldığından
bu olgularda kemoterapi uygulanır. Yaşın küçük olması, cerrahi rezeksiyonun tam
olmaması ve BOS yoluyla yayılım bulunması kötü prognoz işaretleridir.



Şekil 7. Üç yaşında arka çukur medulloblastom tümörü olan hastaya ait MRG
görüntüleri. Aksiyal T2 ve T1 kontrastlı kesitlerde içerisinde küçük kistik
bölümlerin de olduğu heterojen tümör görünümü. Sagital kesitte tümörün BOS
geçişine engel olarak sebep olduğu hidrosefali de görülmekte. Lateral ve üçüncü
ventrikül dilate ve korpus kallozum yaylanmış görünümdedir.
Meningiom
Meningiomlar
araknoidal (meningotelyal) “cap” hücrelerinden kaynaklanan çoğunlukla iyi huylu
tümörlerdir. Nadiren atipik veya anaplastik meningiom görülebilir. En sık 6. ve
7. onyıllarda görülür. Hastaların üçte ikisi kadındır. Ailevi olgular vardır.
Özellikle tip 2 nörofibromatozda meningiomlar gelişir. Meningiomlar araknoidal
kılıfın her bölgesinden kaynaklanabilir. En sık falks yerleşimlidir. Sık
görüldüğü diğer bölgeler, konveksite, olfaktör oluk, sfenoid kanat, parasellar
bölge, optik sinirler, petroz çıkıntı, tentoryum ve arka çukurdur. Spinal
kanalda en sık dorsal bölgede yer alır. Meningiomlar çok yavaş büyür ve
bulunduğu bölgeye göre değişmekle birlikte, çok büyümedikçe belirti
vermeyebilir. Nöroradyolojik olarak kalsifikasyonlar gösteren ve homojen
kontrast tutan izodens bir kitle olarak görülür; genellikle dura ile birleştiği
yerde bir “dural kuyruk” bulunur (Şekil 8). Etrafı ödemlidir. Tümörün
cerrahi olarak çıkarılması kalıcı tedavi sağlar. DSÖ 2016 sınıflamasında beyin
parenkimini invaze eden meningiomlar invazif meningiom olarak
sınıflandırılmıştır. Cerrahi girişimin mümkün olmadığı (örneğin, kavernöz
sinüs) 3 cm’den küçük boyutta meningioma sahip hastalarda stereotaktik
radyocerrahi ve malign meningiomlarda radyoterapi yapılabilir.

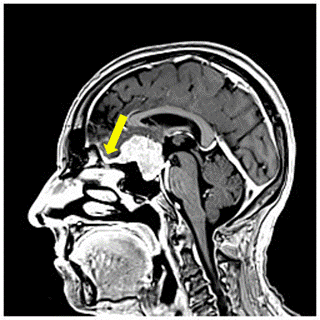
Şekil 8. Solda, kontrastlı koronal MRG’de sağ parietal yerleşimli falks
meningiomu. Sağda, kontrastlı sagital MRG kesitinde tüberkulum sella meningiomu
izlenmekte. Her iki tümöre ait dura kuyruğu (dural tail) sarı ok ile
işaretlenmiştir.
Nöroma
Bu grupta
yer alan Schwannom ve nörofibrom iyi huylu periferik sinir tümörleridir. Çok
daha nadir olarak malign formları görülebilir. Bu iki tümör tipi de ailevi MSS
tümörü sendromlarında sık görülen tümörlerdir. Birincisi periferik siniri
çevreleyen iyi diferansiye Schwann hücrelerinden oluşur; ikincisinde ise
Schwann hücrelerinin yanı sıra, perinöral-benzeri hücreler, fibroblastlar ve
diğer hücreler de bulunur. Schwannomlar 4.-6. onyıllar arasında sık olmakla
beraber her yaş grubunda görülebilir; nörofibrom da her yaş grubunda
görülebilir. Schwannom genellikle baş/boyun bölgesindeki ve ekstremitelerin
ekstansör taraflarındaki sinirlerden kaynaklanır. Spinal köklerde ve kranial
sinirlerde de sıktır. Özellikle VIII. kranial sinirde Schwannom sık görülür
(vestibüler nörinom/akustik nörinom gibi); serebellopontin köşe tümörü
olarak da isimlendirilen bu durumda serebello-pontin köşe sendromu bulguları
ortaya çıkar (Şekil 9) (Bakınız: Kranial sinirler: Kısa anatomi/fizyoloji, muayene ve
bozuklukları, Kranial nöropatiler). Nörofibrom ise kranial sinirlerde görülmez. Genellikle bir cilt
nodülü şeklindedir, bazen cilt ve cilt altını yaygın olarak tutar ve bütün
ekstremitenin büyük görünmesine yol açar (elephantiasis neuromatosa). Kranial
sinirlere ait Schwannom MRG’de düzgün sınırlı, bazen kistik komponentli,
kemikte erozyon yapmış bir kitle olarak görülür; homojen kontrast tutan bu tip
tümörlerin tedavisi cerrahidir.


Şekil 9. Sağ pontoserebellar köşe tümörü. Akustik nörinom (vestibuler
schwannom). Tümörün akustik kanala giren bölümü kontrastlı aksiyal MRG’de
(sağdaki resim) sarı ok ile işaretlenmiştir. Tümör sebebiyle beyin sapı bası altında
görülmektedir.
Kranyofaringiom
Çocuklarda
ve gençlerde görülen iyi huylu bir epitelyal tümördür. Rathke kesesi
kalıntılarından geliştiği düşünülür. Hipofiz sapına komşu yerleşir. Genellikle
suprasellar yerleşimlidir; bazen sella içine de uzanır. Optik kiazma basısına
bağlı görme yakınmaları ve endokrin bozukluklarla kendini gösterir. Heterojen
görünümde yer yer kistik ve yer yer kalsifikasyonlu bir tümördür (Şekil 10).
Solid kısımları kontrast tutar. BT’de kalsifikasyonlar hiperdens görünür.
Kolesterol içeren bir tümör olduğundan MRG’de kontrastsız T1 ağırlıklı
kesitlerde yer yer hiperintens görünebilir. Tedavi cerrahidir; ancak her zaman
tümörün tamamı çıkarılamayabilir. Hipofiz sapı komşuluğu nedeniyle tümörü total
çıkarmaya zorlamak diabetes insipidus gibi endokrin bozukluklara sebep
olabilir. Bu durumda nüksler sıktır. Radyoterapi yapılabilir, ancak yararı
tartışmalıdır. Endokrin bozukluğun düzeltilmesi önemlidir.


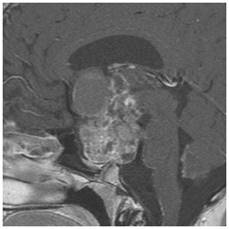
Şekil 10. Suprasellar yerleşimli kranyofaringioma hastasına ait MRG
görüntüleri. Heterojen görünümde, dağınık şekilde kontrast tutan, yer yer
kistik yer yer solid tümör. Sagital kesitlerde tümörün kistik kısmının 3.
ventrikül içerisine kadar girdiği görülmekte.
Primer Merkezi Sinir Sistemi
Lenfoması
MSS’de lenfoma
sıklıkla metastatik olarak görülür. Ancak daha seyrek olarak ekstra-nöral bir
odak olmaksızın sinir sisteminde lenfoma gelişebilir. Önceki yıllarda bu
tümörlerin retiküloendotelyal sistemden kaynaklanan histiositik sarkom olduğu
düşünüldüğünden retikulum hücreli sarkom veya mikrogliom gibi isimler
verilmiştir. Ancak daha sonra immünhistokimyasal yöntemlerin gelişmesi ile bu
tümörün aslında malign bir non-Hodgkin lenfoma tipi olduğu anlaşılmıştır.
Son yıllar içinde
primer serebral lenfoma sıklığında belirgin bir artış dikkati çekmektedir.
Amerika Birleşik Devletleri’nde görülen bütün primer MSS tümörlerinin yaklaşık
%3’ünü primer MSS lenfoması oluşturur; bütün non-Hodgkin lenfomaların ise yine
%2-3’ünü oluşturur. MSS lenfoması insidansında görülen artışın önemli bir
nedeni AIDS’li veya transplantasyon nedeniyle immünsüpresif tedavi gören ve
diğer nedenlerle immün yetersizliği olan hastalarda sık ortaya çıkmasıdır.
İmmün yetersizliği olan hastalarda serebral lenfomanın etyolojisinde
Ebstein-Barr virüsünün rol oynadığı düşünülmektedir. AIDS’e bağlı lenfoma
sıklığı son yıllarda kullanılan etkin antiretroviral tedaviler sayesinde düşüşe
geçmiştir.
Primer
serebral lenfomaların yaklaşık % 90’ı B-lenfosit kökenlidir (diffüz büyük B
hücreli lenfoma). T-lenfosit kökenli lenfomalar çok daha nadirdir. İmmün
yetersizliği olmayan hastalarda 6.-7. onyıllarda ortaya çıkarken, konjenital
immün yetersizliği olan çocuklarda 10 yaş civarında, transplantasyon hastaları
ve AIDS’lilerde ise 3. ve 4. onyılda sıktır. Genelde erkeklerde daha sık
görülür; AIDS’lilerde ise lenfomalı hastaların % 90’ı erkektir.
Klinik
Bulgular: Primer MSS lenfoması tutulan bölgeye göre
klinik bulgu verir. Genellikle periventriküler bölgede en sık frontal lobda,
giderek azalan sıklıkta temporal, parietal ve oksipital loblarda, arka çukurda,
bazal ganglia ve korpus kallozumda, nadiren de omurilikte yerleşir.
Leptomeningial yayılım % 30-40 olguda görülür. T-lenfosit lenfoması arka
çukuru, özellikle serebellumu daha fazla tutar; leptomeningial yayılım da daha
sıktır.
Genelde
fokal nörolojik bulgular ön plana çıkar, yarıya yakın hastada davranışsal ve
kognitif bozukluklar ortaya çıkarken %30’unda KİBAS bulguları gelişir. Tümör
genellikle derin ak maddeyi tuttuğu için epileptik nöbet görülmesi nadir bir
durumdur. Göz tutulumu primer olabileceği gibi MSS tutulumuna sekonder olarak
da gelişebilmekte, asemptomatik olabileceği gibi tutulan bölgeye göre bulanık
görme ya da görme alanı defekti de olabilmektedir.
Tanı: Dikkat edilmesi gereken bir konu da patolojik, klinik,
laboratuvar ve ileri tanı yöntemleri ile sistemik lenfomanın MSS tutulumu ile
primer MSS lenfomasının ayrılmasıdır. Uluslararası MSS Lenfoması Çalışma
Grubunun yayınladığı kılavuza göre bu hastaların değerlendirilmesinde patolojik
inceleme ve immünfenotiplemenin yanı sıra, ayrıntılı sistemik ve nörolojik
muayene, HIV serolojisi, kontrastlı kranial MRG, göz muayenesi, serum LDH
düzeyi, toraks-abdomen-pelvis BT incelemesi, kontrendikasyon yoksa BOS
sitolojisi ve “flow” sitometri testleri, kemik iliği aspirasyon biyopsisi, göz
tutulumu şüphesinde vitrektomi/koryoretinal biyopsi ve erkek hastalarda testis
muayenesi ile ultrason incelemesi yapılması önerilmektedir. Sistemik hastalığın
yayılımının ayrımında tüm vücut PET/BT incelemesi ve BOS’ta immünglobulin
düzeyleri, hafif zincirlerin ve polimeraz zincir reaksiyon analizinin yapılması
klinisyenin tercihine bırakılmıştır.
BT’de hipodens veya
izodens, MRG’de hiperintens veya izointens görünen tek veya multipl lezyonlar
görülür. Genellikle solid olur, nadiren kistiktir. Sıklıkla yoğun kontrast
tutulumu gösterir (Şekil 11). İmmün yetersizliği olan hastalarda
halkasal kontrast tutulumu görülebilir. Diffüzyon ağırlıklı MRG incelemesinde
hücresellikte artış nedeniyle kısıtlanma görülebilir. Daha çok supratentoriyal
yerleşir, periventriküler yerleşim sıktır. Beyin hemisferlerini, korpus
kallozumu, bazal gangliayı, talamusu, serebellumu, meninksleri ve nadiren de
medulla spinalis ve kafa çiftlerini tutabilir. Bazal ganglia yapılarında
kontrast tutan ayna görüntüsünün patognomonik olduğu söylenir. Tümör ile normal
doku arasındaki sınır keskin değildir, etrafındaki ödem de diğer tümörlerde
görülenden daha azdır. Nadir görülen medulla spinalis tutulumu prognoz
açısından en kötü formdur. Bazen leptomeningial kontrast tutulumu gözlenebilir.
Metastatik lenfomalar ise sıklıkla leptomeningial tutulum yapar, parenkim
lezyonu nadirdir.


Şekil 11. Merkezi sinir sistemi lenfoması hastasına ait kranial MRG
kesitleri. Periventriküler yerleşim, orta derecede kontrast tutulumu, çevresel
ödem görülmekte.
Primer serebral
lenfomalı olguların %35-60’ında BOS’ta pleositoz görülür. Ancak sitolojik
inceleme ile hastaların sadece % 5-30’una tanı konabilir. Oysa metastatik
lenfomalar sıklıkla leptomeningial tutulum yaptığından BOS sitolojisi % 70-95
oranında tanı koydurucu olabilir.
Stereotaktik biyopsi
tanı için günümüzde en çok tercih edilen yöntemdir. Ancak biyopsi
planlanıyorsa, kortikosteroide duyarlı bir tümör olduğu için, biyopsinin
tanısal değerini azaltacağından, işlem öncesinde kortikosteroid verilmemesi
gerektiği unutulmamalıdır. Histopatoloji %90’ın üzerindeki vakada diffüz
büyük B hücreli lenfomadır.
Tedavi: Cerrahi girişimin tedavide yeri
yoktur. Kortikosteroidler tümör hücrelerinin tamamen kaybolmasına yol açabilir.
Bu sebeple hayalet tümör (ghost tumor) diye de adlandırılır. Kortikosteroid
tedaviye radyolojik yanıtın olması, SSS’nin demiyelinizan, otoimmün ya da
inflamatuar hastalıklarla ayırıcı tanısında yeterli değildir.
Tedavi,
indüksiyon ve konsolidasyon fazı olmak üzere 2 aşamalıdır. İndüksiyon tedavisinde
kemoterapi yapılır ve radyolojik cevaba bakılır. Bu evrede tam radyolojik yanıt
olsa da nüks olasılığı yüksektir. Konsolidasyon tedavisi ise radyoterapi,
kemoterapi ya da yüksek doz kemoterapi ve otolog kemik iliği naklini
içermektedir.
Yeni tanı
konulmuş hastalarda ilk basamakta kan-beyin bariyerini geçebilmesi için yüksek
doz metotreksat ve sitozin arabinozid tedavisinin düşünülmesi gerekir.
Kullanılan diğer ajanlar arasında temozolamid, rituksimab, prokarbazin,
vinkristin, karmustin, etoposid, tiotepa ve siklofosfamid sayılabilir. Bu
ajanlar metotreksat ve sitozin arabinozidden farklı olarak yüksek dozlar
kullanılmadan da kan-beyin bariyerini geçebilmektedir. Sadece yoğun kemoterapi
verilerek tedaviye cevap alınabilir. Ancak, bugüne dek herhangi bir tedavinin
diğerine net olarak üstünlüğü gösterilmemiştir, immün yetmezliği olan
hastalarda bağışıklık sistemini güçlendirici yaklaşımlarda bulunulmalıdır.
İntratekal kemoterapinin belirgin bir yararı gösterilememiştir.
Primer MSS lenfomasının
radyosensitif bir tümör olması ve diffüz infiltratif karakteri nedeniyle
tedavide tüm beyin radyoterapisinin yeri vardır. Spinal ışınlamanın yararı
gösterilememiş ve morbiditeyi arttırdığı saptanmıştır. Yüksek doz kemoterapiye
rağmen hastalık ilerliyorsa standart dozda (40-45 Gy) tüm beyin radyoterapisi
önerilmektedir. Son yıllarda yapılan çalışmalarda yüksek doz kemoterapi sonrası
yapılan kontrollerde progresyon yoksa terapötik etkide azalma olmaksızın tüm
beyin radyoterapisi dozu azaltılarak (20-30 Gy) verilebilir. Böylelikle yüksek
doz radyoterapinin yan etkilerinden kaçınılmış olur.
Yüksek
doz metotreksat tedavisi sonrasında beyin radyoterapisine bakılmaksızın otolog
kemik iliği nakli yapılmasının genç hastalarda sağ kalıma fayda sağladığı
gösterilmiştir.
Prognoz: Tedaviye cevap genellikle
iyidir, ancak sıklıkla tekrarlar görülür. Tedavi sonrası ortalama sağkalım bir
yılı biraz aşar.
Hastanın
yaşı ve genel klinik durumu birçok çalışmada ana prognostik faktörlerdendir ve
5 yılı aşan sağkalımlar bildirilmiştir. İyi prognoz işaretleri immün
yetersizlik bulunmaması, tek intrakranial lezyon, meningial ve/veya ventriküler
yayılımın olmaması, yaşın 60’ın altında olması ve hastanın genel durumunun iyi
olmasıdır. Ayrıca iki kür kemoterapi sonrası radyolojik yanıtın tam olması da
iyi prognoz belirteçleri arasında sayılabilir. IELSG’nin (International
Extranodal Lymphoma Study Group) kötü prognoz belirteçleri arasında, 60 yaş
üstü hasta, ECOG (Eastern Cooperative Oncology Group) performans skorunun >1
olması, serum LDH yüksekliği, yüksek BOS proteini ve derin beyin yapılarının
tutulması yer alır.
Diğer Tümörler
Yukarıda
anlatılanlar dışında pek çok farklı MSS tümörü vardır. Bunlar arasında ilkel
nöroektodermal tümörlerden nöroblastom, retinoblastom, vasküler kökenli
hemangioblastom, pineal dokudan kaynaklanan pineoblastom ve pineositoma,
nöronal kökenli nöroblastom, ganglionöroma, ganglionöroblastoma, ilkel notokord
kalıntısından gelişen kordoma, kolloid kistler, kolesteatoma, karotis
bifurkasyonunda yerleşen paraganglioma ve glomus jugulare tümörü ile görece sık
görülen ve ortaya çıkardığı görme bozuklukları ve nöro-endokrin bozukluklarla
tanınan ancak nöro-onkoloji kapsamı dışında ele alınması gereken hipofiz
tümörleri sayılabilir. Okur bu konuların ayrıntıları için nöroşirurji
kitaplarına başvurabilir.
Ailesel MSS Tümörleri
Eskiden
beri bazı ailelerde tümör kümelenmeleri olduğu bilinmektedir. Son yıllarda
moleküler genetik çalışmalar ile bunların altında yatan genetik patoloji de
ortaya konmuş, bu sayede de genel anlamda kanser oluşum mekanizmaları daha iyi
anlaşılmıştır. Tümü otozomal dominant geçiş gösteren bu sendromlar Tablo 2’de
sıralanmıştır.
Nörofibromatosis
Tip I: von Recklinghausen hastalığı olarak da
adlandırılır. Tanı için şu kriterlerden en az ikisi bulunmalıdır:
1) Çapı
puberte öncesinde > 5mm, puberte sonrasında > 15 mm olan 6 veya daha
fazla “café au lait” sütlü kahverenginde cilt lekesi; 2) İki veya daha fazla
nörofibrom; 3) Aksiller veya inguinal çillenme; 4) Optik sinir gliomu; 5)
Belirgin bir kemik lezyonu; 6) Birinci derecede bir akrabada tip I
nörofibromatozis bulunması.
Nörofibromatosis Tip II: Genetik
olarak tip I nörofibromatozisten ayrı bir tablodur ve von Recklinghausen
hastalığı olarak adlandırılmaz. Tanı kriterleri:
1) Bilateral akustik
nörinom (Schwannom) (Şekil 12) veya 2) Birinci derecede bir
akrabada tip II nörofibromatosis bulunması, ilave olarak, ya a)
Unilateral akustik nörinom, ya da b) aşağıdakilerden ikisi: meningiom,
schwannom, gliom, nörofibrom, arka subkapsüler lens opasitesi, serebral
kalsifikasyon; veya 3) aşağıdakilerden ikisi: a) Unilateral akustik
nörinom; b) Multipl meningiom; c) Schwannom, gliom, nörofibrom,
arka subkapsüler lens opasitesi, serebral kalsifikasyon.
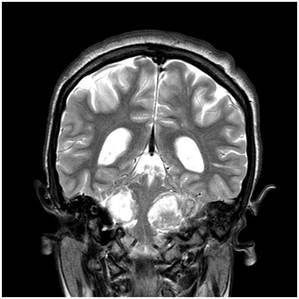

Şekil 12. NF Tip II’li hastaya ait kranial MRG görüntüleri. Bilateral
akustik nörinom mevcut. Kitlelere bağlı beyin sapı baskı altında.
Tablo 2. Ailesel MSS Tümörleri
|
Sendrom |
Gen |
Kromozom |
Sinir
sistemi |
Cilt |
Diğer |
|
Nörofibromatozis
I |
NF1 |
17q11 |
Nörofibromlar, MPNST, optik
gliom, astrositom |
“Café au lait”, aksiller
çiller |
İris hamartomu, kemik
lezyonları, lösemi, feokromasitoma |
|
Nörofibromatozis
II |
NF2 |
22q12 |
Bilateral akustik nörinom,
periferik nörinomlar (Schwannom), meningiomlar, spinal epandimomlar,
astrositomlar, vs. |
(-) |
Arka lens opasitesi,
retinal hamartom |
|
von
Hippel Lindau |
VHL |
3p25 |
Hemangioblastom |
(-) |
Renal ca., retinal
hemangioblastom, feokromasitoma, organ kistleri |
|
Tuberoz
Skleroz |
TSC1 TSC2 |
9q34 16p13 |
Subepandimal dev hücreli
astrositom, |
Anjiofibromlar “peau chagrin”,
subungual fibrom |
Kardiyak rabdomiyom, adenomatöz GİS akciğer-
böbrek kistleri, vs. |
|
Li-Fraumeni |
TP53 |
17p13 |
Astrositomlar, PNET |
(-) |
Meme ca., lösemi, kemik ve
yumuşak doku sarkomları, adrenal korteks ca. |
|
Cowden |
PTEN (MMAC1) |
10q23 |
Serebellumda displastik
gangliositom |
Trişilemmom, fibromlar |
Kolonda hamartomatöz
polipler, tiroid tm, meme ca. |
|
Turcot |
APC MLH1
hPSM2 |
5q21 3p21 7p22 |
Medulloblastom
glioblastoma |
(-) “café au lait” |
Kolorektal polipler
kolorektal polipler |
|
Nevoid
bazal ca. Sendromu |
PTCH |
9q31 |
Medulloblastom |
Multipl bazal ca. |
Çene kistleri, over
fibromları, iskelet anomalileri |
MPNST: Malign periferik sinir
kılıf tümörü, PNET: İlkel nöroektodermal tümör
Sistemik
Kanserin Nörolojik Etkileri
Sistemik
kanserin nörolojik komplikasyonları nöroloji ile uğraşan hekimin günlük
pratiğinde giderek artan sıklıkta ve önemli bir yer tutmaktadır. Nüfusun yaş
ortalamasının artması, endüstriyel toksinlere daha fazla maruz kalma gibi
nedenlerle kanser insidansı giderek artmaktadır. Ayrıca, yeni tedavi
yöntemlerinin gelişmesi ile kanserli hastaların sağkalım süresi uzamakta,
infeksiyon ve metabolik sorunlarla başa çıkmada geliştirilen yeni yöntemler de
sağkalım üzerinde olumlu etki göstermektedir. Bunlara bağlı olarak zaman içinde
görülebilecek nörolojik problemler de artmaktadır. Ayrıca kanserli hastalarda
görülebilen nörolojik komplikasyonlar genellikle acilen müdahale edilmesi
gereken ciddi, ancak tedavi ile kısmen de olsa rahatlatılması mümkün olan
sorunlardır. Bu nedenlerle nöroloji pratiğinde önemli bir yeri vardır. Kanserli
bir hastada belirti veren sinir sistemi bölgesine göre aranması gereken
komplikasyon türleri Tablo 3’de verilmektedir.
Tablo 3. Kanserli hastada belirti veren bölgeye göre aranması gereken
nörolojik sorun.
|
Bölge |
Nörolojik sorun |
|
Beyin |
|
|
Omurilik ve kauda ekuina |
|
|
Kranial ve periferik sinir |
|
|
Nöromüsküler bileşke |
|
|
Kas |
|
Tümör Yayılımına Bağlı
(Metastatik) Etkiler
Nörolojik
yakınmaları olan kanserli hastaların yaklaşık yarısında bu yakınmalar doğrudan
tümör invazyonuna bağlıdır. Kanserli hastaların yaklaşık %25’inde beyin
parenkimine, dura veya meninkslere metastaz görülür; %5’inde ise spinal
metastaz vardır. Yılda 100.000 kişilik nüfus başına MSS metastazı ile giden
hastalık oranı 4 ile 11 arasında değişir. Beyin metastazı sıklığı yaşla
birlikte artar. Bunların dışında, doğrudan MSS’yi tutmamakla birlikte kemik
metastazları da özellikle medulla spinalis basıları yaratarak önemli nörolojik
sorunlara yol açabilmektedir. Özetle, kanserli olduğu bilinen bir hasta
nörolojik tablo geliştirdiği takdirde öncelikle metastaz olasılığı düşünülmelidir.
Tümörün
sinir sistemine ulaşması çeşitli yollardan olur. Beyin parenkim metastazlarında
yayılım türü hematojendir. Bu yollardan biri olarak Batson venöz pleksusundan
da söz etmek gerekir: Batson venöz pleksusu olarak bilinen, vertebral venöz
pleksus pelvisten kranial venöz sinüslere uzanan ve kapakçık içermeyen bir
sistemdir. Bu sistem aracılığı ile iki yöne doğru da hematojen metastazlar
gerçekleşebilir. Leptomeningial metastaz ise perinöral lenfatikler kanalıyla ya
da beyin metastazlarından BOS’a dökülen hücreler sonucu oluşur.
Bunların dışında tümörün kendi kitlesinin veya lenf nodu ya da uzak
metastazlarının çevre dokuyu infiltre etmesi söz konusu olabilir.
Beyin Metastazı
Beyin
metastazının erişkinde en sık nedenleri akciğer kanseri, melanom, meme kanseri,
renal kanser, kolorektal kanser ve lenfoma gibi tümörler olmakla birlikte,
olguların %10’unda metastazın kaynağı belirlenemez. Çocuklarda ise en sık beyin
metastazı nedenleri sarkomlar, nöroblastomlar ve germ hücreli tümörlerdir.
Diğer yandan, özellikle sık beyin metastazı yapan kanserler akciğer, meme,
böbrek, kolorektal kanserler ve melanomdur. Melanomların % 75’i, testis
kanserlerinin % 57’si, akciğer kanserlerinin % 35’i beyin metastazı yapar.
Beyin dokusuna hemen hiç metastaz yapmayan bazı kanserler de vardır. Bunlar
arasında prostat, özofagus, orofarinks ve melanom dışı cilt kanserleri yer
alır. Buna karşın, prostat, meme kanseri, multipl miyelom, Hodgkin hastalığı ve
non-Hodgkin lenfoma ile birlikte dural metastaz görülür. Beyin parenkim metastazlarının
%80’i hemisferlerde % 20’si arka çukurda yer alır; ancak pelvik organların ve
kolon kanserinin metastazları arka çukuru tercih eder.
Beyin
metastazları diğer tümörler gibi başlıca KİBAS veya epileptik nöbetlerle
karşımıza çıkarlar. Ayrıca bulundukları bölgeye özgü klinik belirtiler de
verebilirler. Birden fazla sayıda (multipl) olduklarında tanı güçlüğü
yaratırlar. Bazen de davranış değişikliği, uyanıklık kusuru ve silik nörolojik
bulgularla genel bir ensefalopati tablosuna yol açabilirler.
Nöroradyoloji:
BT’de metastazlar izodens veya hipodens düzgün sınırlı lezyonlar
olarak görülür. Çoğunlukla kan akımının yavaşladığı, gri ve beyaz cevher
sınırında, subkortikal bölgede yerleşirler. Yoğun homojen veya çevresel
kontrast tutulumu vardır, etrafı ödemlidir. BT’de kitle görülmemesi metastaz
olasılığını ortadan kaldırmaz, kuşku varsa MRG yapılmalıdır. Günümüzde birçok
merkezde metastaz değerlendirmesi için ilk sırada yapılan inceleme
MRG’dir. Metastazlar MRG’de T1 ağırlıklı kesitlerde hipointens, T2 ağırlıklı
kesitlerde hiperintens görünür. Belirgin kontrast tutulumu gösterir (Şekil
13). Metastatik lezyon sayısı da primer tümör tipi hakkında bilgi
verebilir. Tek lezyon varsa, böbrek, meme, tiroid, akciğer adenokarsinomu akla
gelmelidir. Küçük hücreli akciğer kanseri ve melanom çok sayıda metastaz yapar.
Bazen kontrastsız BT’de lezyonlarda kanama gözlenebilir. En sık kanayan
metastazlar melanom, koryokarsinom, akciğer, tiroid, adrenal ve sürrenal
kanserlerine aittir.


Şekil 13. Akciğer kanseri nedeniyle takipli hastada görme sorunu olması
üzerine çekilen kranial MRG kesitleri. Sol oksipital bölgede, kontrast tutan
subkortikal yerleşimli kranial metastaz görülmekte.
Ayırıcı
Tanı: Metastatik hastalığın ayırıcı
tanısında primer beyin tümörleri, infeksiyöz lezyonlar, aşağıda sözü edilecek
olan paraneoplastik sendromlar, bazı durumlarda demiyelinizan hastalık akla
gelmelidir.
Tedavi: Akut iyilik hali sağlanması için antiödem etkisi nedeniyle
kortikosteroidler kullanılabilir. Standart olarak günde 16 mg deksametazon ile
başlanabilir, etki sağlanmazsa 48 saatte bir doz arttırılarak 100 mg/gün dozuna
kadar çıkılabilir. Ancak çok uzun kullanımda steroidlerin bilinen sakıncaları
gündeme gelecektir. Kontrastlı MRG ile gösterilmiş tek bir lezyon varsa, primer
tümör de kontrol altına alınmışsa, metastazın rezeksiyonu düşünülmelidir.
Cerrahi girişimin ardından radyosensitif tümörlerde postoperatif radyoterapi
yapılır. Bu grup hastalara tüm beyin radyoterapisi yapılırken son zamanlarda
kognitif fonksiyonların da korunması amaçlanarak kaviter radyoterapi de
yapılmaya başlanmıştır. Yaygın metastazları olan ve cerrahi girişim yapılamayan
hastalara tüm beyin palyatif radyoterapi uygulanırken 1-3 arasında metastazı
olan hastalara stereotaktik radyocerrahi (SRS) uygulanabilir. Kan beyin
bariyeri nedeniyle sistemik kemoterapinin bazı durumlar haricinde serebral
metastazlara pek etkili olmadığı düşünülmektedir. Radyoterapinin 48-72 saat
öncesinde 16 mg deksametazon veya eşdeğerinin verilmesi akut radyoterapi yan
etkilerini önlemek açısından yararlıdır. Tedaviye rağmen metastatik beyin
tümörlerinin prognozu kötüdür. Ancak lenfoma, bazı meme kanserleri,
koryokarsinom, testis kanseri gibi radyasyona duyarlı tümörlerde sağkalım daha
uzun olabilir.
Spinal Metastaz
Kanserli
hastalarda omuriliği ilgilendiren üç tip metastatik lezyon vardır. En sık
görülen epidural lezyonlardır. Bunların çoğunluğu omurlardaki kemik
metastazlarından kaynaklanır. Ayrıca paravertebral yapılardan ve epidural
boşlukta yer alan metastatik lezyonlardan da kaynaklanabilir. Bunun dışında tek
bir kitle halinde veya leptomeningial yayılım şeklinde intradural
ekstramedüller yerleşimli ve/veya intramedüller yerleşimli metastazlar da
görülebilir.
Epidural
Metastaz: Vertebralar, kemik metastazına yol açan kanserlerde % 70’e varan
oranlarda, en çok tutulan bölgelerdir. Çoğunluğu ağrı dışında asemptomatik
kalır. Bu ağrı, lokal ve şiddetlidir. Perküsyonla hassasiyet saptanır. Yaklaşık
üçte birlik bölümü epidural basıya yol açabilir. Nörolojik bulgu veren spinal
epidural metastazların yaklaşık % 25’i meme, % 15’i akciğer, % 10’u prostat
kanserine, % 10’u da lenforetiküler sistem tümörlerine aittir. Yaklaşık %
10-15’inde primer bölge belirlenemeyebilir. En sık dorsal vertebralarda
metastaz görülür.
Epidural
metastazlar başlıca üç mekanizma ile belirti verir. 1) Erken dönemde basıya
bağlı ak maddede ödem ve aksonal şişme görülür, bu dönemde basının ortadan
kaldırılması ile normal işlev kazanılabilir; 2) Özellikle ön ve arkadan basıda
spinal damarların sıkışması ile medulla spinalis infarktı gelişir; 3) Basıya
bağlı vazojenik ödem gelişir, bu durum steroidlere cevaplıdır.
Nörolojik
aciller kapsamında değerlendirilmesi gereken bu klinik tabloda, ağrı ortaya çıkan ilk bulgudur. Bölgesel veya radiküler
nitelikte olan ağrı, zaman içerisinde oldukça şiddetli bir nitelik kazanır.
Klinik bağlamda; hasta yatar durumdayken şiddetlenir, kişi otururken daha
rahattır. Ardından parestezi gibi duysal yakınmalar ve tutulan miyotoma özgü
hızlıca kas zaafı gelişir. Ayrıca etkilenen bölgeye göre seviye gösteren tam
veya kısmi medüller sendrom bulguları bulunabilir . Klasik medüller sendrom
bulguları dışında nadiren Lhermitte belirtisi, psödoklodikasyon, papilödem,
spinal miyokloniler ve çok ender olarak “moving toes” görülebilir. Epidural
metastazın ayırıcı tanısında epidural kanama, epidural abse, disk fıtıklaşması
ve diğer epidural kitleler akla gelmelidir.
İntradural
ve İntramedüller Metastaz: Dura içerisindeki
metastazlar metastatik spinal tümörlerin en azını, yaklaşık % 5 kadarını
oluşturur. İntradural ekstramedüller metastazlar bir sonraki leptomeningial
metastaz başlığı altında incelenecektir. Bu tür metastazlar nadiren intradural
bir kitle oluşturarak etki edebilirler. İntramedüller metastazlar ise daha çok
hematojen yolla olur. Bazen de leptomeningial tümör omuriliği infiltre eder.
Sonuçta oldukça nadir bir durumdur. İntramedüller metastazın en sık nedenleri;
akciğer tümörleri (yaklaşık % 50), meme kanseri ve Hodgkin, Hodgkin-dışı
lenfomalardır. Klinik tablosu, ağrı daha geri planda olmak üzere epidural bası
gibidir.

Şekil 14. Meme kanseri metastazlı hastaya
ait kontrastlı servikal MRG görüntüsü. C5 ve C6 seviyelerinde intradural
yerleşimli 2 adet ile serebellum superiorunda ve tentoryumun hemen üzerinde
görülen multipl metastazlar.
Nöroradyoloji: Gerek epidural bası, gerekse intramedüller metastaz kuşkusu olan
durumlarda en uygun inceleme yöntemi kontrastsız ve kontrastlı spinal MRG’dir.
Kemik metastazına bağlı epidural basılarda direkt radyografiler bile olguların
% 75’inden fazlasında sorumlu lezyonu gösterebilir. Bu durumda ilgili bölgenin
BT’si ve kemik sintigrafisi yararlı olabilir. Basının düzeyini belirlemek için
geçmişte myelografi ve intratekal kontrastlı BT gibi yöntemler kullanılırken,
günümüzde ayırıcı tanıda daha fazla bilgi vermesi ve intramedüller metastazları
göstermesi nedeni ile MRG ilk sırada tercih edilen yöntemdir. İntramedüller
metastazlar MRG’de solid, sınırları belirgin kitle şeklinde görülür. Genellikle
T1’de hipointens, T2’de hiperintenstir ve yoğun kontrast tutulumu gösterir (Şekil
14).. İntramedüller metastazların radyolojik ayırıcı tanısına primer
medüller tümörler, radyasyon myelopatisi, vasküler omurilik hastalığı,
paraneoplastik myelopati ve demiyelinizan myelopati girebilir.
Tedavi: Epidural metastaz basısı çok acil bir durumdur. Bugün yaygın
kabul gören tedavi şekli acil radyoterapi veya cerrahi dekompresyon sonrasında
radyoterapi yapılmasıdır. Ancak epidural bası saptanır saptanmaz diğer tedavi
seçenekleri gerçekleştirilene kadar beklemeden hemen yüksek doz steroid (100 mg
deksametazon veya 1000 mg metil prednizolon) yapılması gereklidir.
İntramedüller metastazların tedavisinde genellikle steroidler ve radyoterapi
yer alır. Metastazektomi çok nadir durumlarda düşünülebilir.
Leptomeningial
Metastaz
Meninkslerin
malign hücreler tarafından infiltre edilmesi leptomeningial metastaz olarak
adlandırılır. Bu hücreler lösemik hücrelerse bu duruma meningial lösemi (veya
lösemik menenjit), lenfoma hücreleri ise leptomeningial lenfomatoz (lenfomatoz
menenjit), kanser hücreleri ise meningial karsinomatoz (veya karsinomatoz
menenjit) adı verilir. Kanserli hastalarda leptomeningial metastaz sıklığı tam
olarak bilinmemekle beraber primer neoplazinin niteliğine göre değişkenlik
gösterir. Solid tümörlü hastalarda yaklaşık %4-15 arasında görülürken, primer
beyin tümörlerinde %1-2, hematolojik neoplazili hastalarda (lösemi, lenfoma) bu
oran %5-15 arasında olabilir. Ancak, bir otopsi çalışmasında nörolojik bulgu
veren kanserli olguların %19’unda saptanmış olması, muhtemelen hastaların bir
kısmında gözden kaçabildiğini düşündürmektedir. Leptomeningial metastaz
yapabilen diğer tümörler meme kanseri, küçük hücreli akciğer kanseri, melanom
ile non-Hodgkin lenfoma, akut lenfositik ve non-lenfositik lösemi gibi hematolojik
tümörlerdir. Leptomeningial metastaz genellikle geç dönemde, yaygın hastalıkla
birlikte ortaya çıkar. Çok daha seyrek olarak birkaç yılı bulan hastalıksız
dönemin ardından ortaya çıkabilir. Bazen de sistemik kanserin ilk belirtisini
oluşturduğundan klinik tanıyı koymak son derece önemlidir. Primeri bilinmeyen
karsinomatoz menenjit, olguların yaklaşık %5’ini oluşturmaktadır. Tümörlerde
sağkalımının artması ile orantılı olarak insidansı artar.
Leptomeningial
metastazlar genellikle multifokal belirti ve bulgular verir. BOS dolanımını
bozarak KİBAS’a yol açar; damarların parenkim içine girdiği Virchow-Robin
aralıklarını izleyerek parenkim invazyonu yapar ve epileptik nöbetlere veya
genel bir ensefalopati tablosuna yol açar; Virchow-Robin aralıklarına yayılmış
tümör hücreleri burada yer alan damarlardaki akımı bozarak iskemiye neden olur.
Bütün bunlara bağlı olarak hastalarda üç ana grupta belirtiler ortaya
çıkabilir. İlk grupta serebral hemisferik tutuluma bağlı başağrısı olabilir,
mental değişiklik görülebilir, bulantı-kusma olur, dengesizlik ve yürüme
güçlüğü olabilir, diabetes insipidus görülebilir. İkinci grup bulgu multipl
kranial sinir tutulumuna ait bulgulardır; üçüncü grupta da poliradiküler
tutuluma bağlı kauda ekuina sendromu, radiküler ağrı ve asimetrik refleks kaybı
sayılabilir. Seyrek olarak meningial iritasyon bulguları saptanabilir. Özetle;
merkezi sinir sistemine ilişkin farklı, birkaç anatomik lokalizasyon bulgusu
saptanan hastalarda bu tanı olasılığı kuvvetle düşünülmeli ve acilen tanı
koydurucu incelemeler yapılmalıdır. Leptomeningial metastaz yapmış solid
tümörlerin %30-80’inde eş zamanlı beyin metastazları da saptanmıştır. Önceden
de sözü edildiği gibi, sistemik kanseri bilinmeyen bir hasta meningial tutuluma
bağlı nörolojik tablo nedeniyle öncelikle nöroloğa başvuracaktır.
Tanı: Meningial tutulum tanısı BOS
incelemesinde malign hücre görülmesi ile konur. Ancak nörolojik bulguları olan
bir kanser hastasında BOS incelemesinden önce yer kaplayan bir kitleyi dışlamak
için kranial ve spinal MRG yapılmalıdır. MRG sırasında kontrast madde verilirse
meninkslerde dağınık lineer veya nodüler kontrast tutulumu gözlenebilir (Şekil
15). Yer kaplayıcı kitleyi dışlamanın yanı sıra, bazen lomber ponksiyon
sonrasında intratekal basıncın düşmesi sebebiyle meningial kontrast tutulumu
görülebileceğinden, BOS incelemesinin MRG sonrasında yapılması daha doğru
olacaktır. Gerekli BOS incelemeleri arasında basınç ölçümü, hücre sayımı,
protein ve şeker düzeyi ölçümü, sitolojik inceleme yer alır. İnfeksiyon kuşkusu
varsa buna yönelik incelemeler de yapılmalıdır. BOS’ta genellikle birkaç
hücreden birkaç yüz hücreye kadar değişebilen bir lenfositik pleositozun yanı
sıra malign hücreler de görülür. Protein düzeyi artmıştır, immünglobulin
düzeyleri de artabilir. İntratekal immünglobulin sentezini gösteren oligoklonal
bantlar saptanabilir. Buna karşın, sık görülmese de BOS şekerinin azalması
önemli bir bulgudur. Leptomeningial metastaz tanısında BOS incelemesinin en
önemli kısmı sitolojidir. Sitoloji laboratuvarına en az 4 ml BOS
gönderilmelidir. BOS’un bekletilmeden gönderilmesi çok önemlidir. Çok
önerilmemekle birlikte, laboratuvara hemen ulaşmayacaksa BOS alınır alınmaz
eşit miktarda saf alkol eklenerek fikse edilmesi denenebilir. İlk BOS
incelemesinde pozitif sitolojik sonuç elde etme şansı %50 civarındadır; bu oran
ikinci incelemede %80’i bulur. Eğer ilk alınan BOS’ta malign hücre
saptanamazsa, inceleme bir kere daha yinelenmelidir. Hala negatifse, ancak
klinik kuşku güçlüyse üçüncü kez de alınabilir. Mümkünse rutin sitolojik
incelemenin yanı sıra immünhistokimyasal çalışmalar ve yüzey belirteci
çalışmaları da yapılmalıdır. Bunların dışında BOS’ta tümör belirteçleri
(karsinoembriyojenik antijen, alfa-fetoprotein, beta-koryonik gonadotropin)
kandaki konsantrasyonlardan çok daha yüksek konsantrasyonda bulunursa özellikle
primer tümör hakkında ipucu vermesi bakımından yararlı olabilir. Bazı
durumlarda, özellikle kauda ekuina sendromu söz konusu ise, BOS alınırken
intratekal kontrast madde verilerek miyelografi yapılabilir. Kök kılıfları
içinde dolum defektleri görülmesi tanıyı kuvvetle destekler. Ayrıca radyoizotop
BOS akım çalışmaları da BOS dolanımının bozulduğu bölgeleri göstermek açısından
yararlıdır. Klinik olarak ayırıcı tanıda en önemli grup MSS infeksiyonlarıdır.
İnfeksiyonda en önemli ayırıcı özellik ateş ve ense sertliği gibi bulguların ön
planda olması, fokal nörolojik bulguların pek görülmemesidir.


Şekil
15.
Leptomeningeal metastazlı hastaya ait kranial MRG görüntüleri. Sağdaki
kontrastlı aksiyal kesitte sulkuslardaki yaygın kontrast madde tutulumu
görülmekte. Ayrıca beyin sapı çevresindeki subaraknoid sisternalar net şekilde
seçilemiyor.
Tedavi: Leptomeningial metastaz tedavisinde radyoterapi ile birlikte
kemoterapi uygulanır. Radyoterapinin semptomatik bölgelere sınırlı tutulması
toksisite açısından daha olumlu olabilir. Kemoterapinin ne yolla verileceği
halen tartışmalı ise de, intratekal uygulama sıkça tercih edilir. Ancak ne
şekilde tedavi edilirse edilsin, solid tümöre bağlı leptomeningial metastazlı
hastalarda prognoz kötüdür. Hastalar tedavi edilmediğinde birkaç haftalık
sağkalım söz konusudur. Tedavi, yerleşmiş nörolojik defisitleri düzeltmese bile
hastalık progresyonunu yavaşlatabilir veya durdurabilir; medyan sağkalımın
tedavi ile birkaç aya çıkması mümkündür. Ayrıca BOS dolanım bozukluğu varsa
ventriküloperitoneal şant takılabilir. Solid tümöre bağlı leptomeningial
metastazı olan bütün hastalar yoğun tedavi almak zorunda değildir. Yaşam
beklentisi 3 aydan kısa olan, genel durum bozukluğu bulunan (Karnofsky
performans skalası <%60) ve MSS tümör yükü çok fazla olan hastalarda sadece
destek tedavisi yapmak yeterli olacaktır. Hematolojik neoplazilerde ise prognoz
çok daha iyidir. Lösemi hastalarında %75 oranında stabilizasyon ve düzelme
görülür. Lenfomada da prognoz solid tümörlere oranla çok daha iyidir. Bu
nedenle bu hastalar ısrarlı bir şekilde tedavi edilmelidir.
Neoplastik Pleksopati
Kanserlerin
periferik sinir sistemine metastaz yapması nadir bir durumdur. Yaklaşık %1 kanserli
hastada pleksus tutulumu görülür. Brakiyal pleksopati, meme ve akciğer
kanserlerinin brakiyal pleksusu infiltre etmesiyle oluşur. Jinekolojik
kanserler, prostat kanseri, sarkomlar, lenfoma ya da kolorektal kanserler
lumbosakral pleksopati yapabilir. Baş boyun kanserleri, lenfoma, akciğer
kanseri ya da meme kanserleri servikal C1-C4 köklerinin ventral ramusunu
tutarak klinik bulgu verebilirler. Burada dikkat edilmesi gereken durum ise
tutulan bölge, daha önceden verilen radyoterapi alanına giriyorsa mevcut
durumun kanser tutulumu mu, yoksa radyoterapiye sekonder mi olduğunun ayırt
edilmesi gerekliliğidir. Neoplastik pleksopatide öncelikle şiddetli ağrı
başlar, sonrasında zaaf ve duyu kusuru gelişir. Radyasyona bağlı pleksopatide
ise tablo ağrısız olarak ilerler ve EMG’de tipik olarak miyokimik
deşarjlar saptanır. Neoplastik tutulumun tanısında MRG ve PET-BT incelemeleri
de fayda sağlamaktadır (Bakınız: Pleksuslar ve
Hastalıkları).
Nörolojik
bulgu gösteren kanserli hastaların yarısında nörolojik tabloya neden olan
herhangi bir metastaz veya tümör yayılımı saptanmaz. Bu durumda çok farklı
nedenler nörolojik tablodan sorumlu olabilir. Bu nedenler aşağıda
sıralanmıştır.
İnfeksiyöz, Metabolik,
Vasküler Etkiler
İnfeksiyonlar: Kanserli hastalarda hem tümöre bağlı gelişen immünsüpresyon, hem
de tedaviye bağlı kemik iliği baskılanması sonucu infeksiyonlar sık görülür.
Bunlar arasında MSS infeksiyonları da yer alır (Bu konunun ayrıntıları için
bakınız: Sinir Sistemi İnfeksiyonları). T-lenfosit ve
mononükleer fagosit sistemi bozuk olan hastalarda nötrofil işlevleri veya total
lökosit sayısı normal olsa bile; Listeria ve Nocardia gibi
bakteriyel infeksiyonlar, özellikle kriptokok gibi mantar, herpes zoster,
herpes simpleks ve sitomegalovirus infeksiyonları başta olmak üzere virus
infeksiyonları, toksoplazmoz gibi parazitozlar ve progresif multifokal
lökoensefalopati tablosu sık görülür. Granülositopenisi veya nötrofil işlev bozukluğu
olan hastalarda özellikle bakteriyel infeksiyonlara eğilim artar. Nötrofil
mutlak sayısı 1000/mm3’ün altında olan hastalarda enterik basiller (Psödomonas
grubu, E. coli, Klebsiella), streptokok, stafilokoklar ile
birlikte Listeria infeksiyonlarına ve kandida, aspergillus ve mukormikoz
gibi mantar infeksiyonlarına eğilim artar. B-lenfosit yetersizliği nadir
görülür. Bu durumda S. pneumoniae, H. influenzae ve N.
Meningitidis infeksiyonlarına eğilim artar.
Metabolik Etkiler: Kanserli hastalarda pek çok nedenle nütrisyonel ve metabolik
bozukluklar meydana gelebilir. Öncelikle kanserin tuttuğu organın işlevlerini
bozması ile o organ sistemine ait yetersizlikler olabilir; bunların arasında
hipoksi, karaciğer ve böbrek yetersizliklerine bağlı ensefalopatiler
sayılabilir. Var olan metabolik bozukluğa bağlı olarak metabolik
ensefalopatiler gelişebilir. Metabolik ensefalopati deyimi, bozulmuş
serebral metabolizma sonucu oluşan davranış değişikliklerini içerir. Akut ya da
subakut olarak gelişen bu tablonun klinik bulguları; konfüzyon, düşünce
kusurları, davranış bozuklukları, bilinç düzeyinde değişimler ve bazen de
anormal motor aktiviteler şeklinde özetlenebilir. Özetle; huzursuzluk, letarji,
emosyonel labilite, uyuklama ya da uykusuzluk gibi bulgulara tremor,
miyokloniler ve asteriksis gibi istemsiz hareketler eşlik ettiğinde bu tanı
olasılığı akla gelmelidir. Sıkça karşılaşılan bu klinik tablo altta yatan
nedene yönelik tedavi ile geriye döner. EEG’de yaygın yavaş dalga varlığı saptanır.
Ayrıca bu hastalarda sıvı-elektrolit dengesindeki bozukluklar, özellikle meme
kanserli hastalarda hiperkalsemi sıkça görülür. Kanserli hastalarda beslenme
bozuklukları ve kaşeksi de oldukça sıktır. Beslenme bozuklukları arasında en
sık görülenler tiamin eksikliğine bağlı Wernicke-Korsakoff sendromu, B12
vitamini eksikliği ve niasin eksikliğine bağlı pellegra sayılabilir. Bu
sonuncusu bazı kanserlerle görülen ve kanserin salgıladığı bazı peptidlere
bağlı diyare ataklarına yol açan karsinoid sendromda sıktır. Metabolik
bozukluklarla ilgili daha ayrıntılı bilgi ilgili bölümlerde bulunabilir
(Bakınız: Metabolik Ensefalopati ve Sinir Sisteminin Nütrisyonel
Hastalıkları).
Vasküler Etkiler: Sistemik arteroskleroz oranı genel popülasyona oranla kanserli
hastalarda daha düşük olmasına karşın, görülme sıklığı açısından serebrovasküler
komplikasyonlar metastatik komplikasyondan sonra ikinci sırada yer alırlar.
Kanserli hastalarda kanayıcı ve tıkayıcı serebrovasküler olaylar yaklaşık eşit
oranlarda görülür. Kanayıcı olaylar intraserebral kanamalar, kranial ve spinal
subdural ve epidural kanamalar ve nadiren subaraknoid kanama şeklinde karşımıza
çıkabilir. Kanayıcı olayların altında sıklıkla koagülasyon bozuklukları
bulunur. Kemik iliğinin tümör infiltrasyonu veya ilaçlarla baskılanmasına bağlı
trombositopeni, trombosit işlev bozuklukları, karaciğer yetersizliğine bağlı K
vitamini eksiklikleri kanserli hastalarda sık görülen kanama nedenleridir.
Trombositopenili hastalarda lomber ponksiyon yapılması gerekiyorsa öncesinde
trombosit süspansiyonu verilerek kanda trombosit sayısının 20,000/ mm3
düzeyinin üzerine çıkarılması gerekir, ayrıca ince iğne ile (20 veya 22 numara)
ve çok dikkatli bir şekilde ponksiyon yapılmalıdır. Aksi halde spinal subdural
veya epidural kanama gelişebilir. Bazen tümörün veya metastazlarının içine
kanama da görülebilir. En sık kanayan beyin metastazları melanom,
koryokarsinom, akciğer, renal ve sürrenal kanserlerine aittir. Kanserli
hastalarda görülebilen iskemik serebrovasküler olayların yaklaşık üçte birinin
altında klasik aterosklerotik damar darlıkları bulunur. Geri kalan çoğunluğunda
farklı nedenler söz konusudur. Bunlar arasında disemine intravasküler
koagülasyon (DIC), non-bakteriyel trombotik endokardit, septik emboli, tümör
embolisi, tümör basısı ile arter tıkanmasının yanı sıra meme, mide, akciğer
kanserleri ve lenfomada görülebilen trombotik mikroanjiyopati ile intravasküler
lenfoma sayılabilir. Kanserli hastalarda DIC tıkayıcı, trombotik vasküler
hastalıklar içerisinde ilk sırayı alır. Her türlü malignitede görülmekle
birlikte lökoz ve lenfomalarda daha sıktır. Genelde ölümle sonlanan bu klinik
tablo; akut gelişen ensefalopati, buna eklenen fokal ya da jeneralize
konvülziyonlar ile karakterizedir. Fokal nörolojik bulgu varsa hafiftir.
Serebral venöz oklüzyonlar ise her türlü sistemik kanserde görülmekle birlikte
lökoz ve lenfomalarda sıktır. Şiddetli başağrısını izleyen konvülziyonlarda bu
tanı akla gelmelidir. Yine bazı kanserlerin (jinekolojik, meme, akciğer
kanserleri ve hematolojik neoplaziler) salgıladığı pro-koagulan maddeler ile
bazı kanserlerde de (prostat, meme) dural metastazlar nedeni ile majör venöz
sinüslerde tıkanma görülebilir.
Tedavi Komplikasyonları
Kemoterapi Yan
Etkileri
Kanser tedavisinde kullanılan
kemoterapötiklere bağlı görülebilen nörolojik yan etkiler Tablo 4’de
sıralanmıştır. Sık görülen bazı özel durumlar burada biraz daha ayrıntılı
olarak ele alınacaktır.
Tablo 4. Nörotoksik etki gösteren kemoterapötikler
|
Akut ensefalopati
(delirium) Metotreksat (intravenöz
veya intratekal), kortikosteroidler, Sisplatin, Vinkristin, Asparaginaz,
Prokarbazin, 5-florourasil, Ara-C, Nitrozüreler, Siklosporin, İnterlökin-2,
İfosfamid/ mesna, İnterferonlar, Tamoksifen, VP-16, PALA, Bevasizumab,
Kapesitabin, Etoposid, Paklitaksel Kronik ensefalopati
(demansiyel sendrom) Metotreksat , BCNU, Ara-C,
Fludarabin Görme kaybı Tamoksifen, Galyum nitrat,
Nitrozüreler, Sisplatin Serebellar bozukluk/ ataksi 5-florourasil, Ara-C,
Prokarbazin, Vinkristin, Siklosporin A Aseptik menenjit Monoklonal antikorlar, OKT
3, Ara-C (intratekal), Metotreksat (intratekal) Başağrısı Tamoksifen, Temozolamid Epileptik nöbet Metotreksat, Etoposid,
Sisplatin, Vinkristin, Asparaginaz, Nitrojen mustard, Prokarbazin, BCNU,
Dakarbazin, Busulfan, Siklosporin, İfosfamid, Paklitaksel, Klorambusil Miyelopati (intratekal
ajanlar) Metotreksat, Ara-C, Tiotepa Periferik nöropati Vinka alkaloidleri,
Sisplatin, Prokarbazin, 5-azasitidin, Etoposid, 5-florourasil, Ara-C, Taksol,
Suramin, Mitotan, Bortezomab, Dosetaksel, Oksaliplatin, Teniposid, Talidomid |
Periferik
nöropati: Kemoterapiye bağlı periferik nöropati
genellikle distal-simetrik bir polinöropatidir. Nadiren fokal nöropatiler
görülebilir. Suramin demiyelinizan, duysal-motor nöropatiye yol açarken,
vinkristin aksonal duysal-motor nöropatiye yol açar. Her ikisi de tedavinin
kesilmesi ile bir miktar düzelebilir. Taksol ve sisplatin ise saf duysal
nöropatiye yol açar. Sisplatin ile sadece kalın lifler tutulurken, taksol ile
ince lifler de tutulur. Arka kök gangliyonundaki hasarın düzeyine bağlı değişmekle
birlikte tedavinin kesilmesi ile düzelebilir (Tablo 5).
Tablo 5. Periferik
nöropati ile ilişkili kemoterapi ajanları
|
İlaç |
Tip |
Klinik
Semptom |
Sonuç |
|
Botezomib |
Ağrılı,
duysal |
Ağrılı
distal parestezi |
Doz
azaltma/kesme ile geri dönüşümlü |
|
Sisplatin |
Kalın
lifler, duysal |
Uyuşukluk,
derin tendon reflekslerinde azalma, propriosepsiyon kaybı |
İlaç
kesilmesinde sonra da bir süre devam eder (coasting effect), geri dönüşümlü |
|
Oksaliplatin |
Akut
nöropati, kronik kalın lif duysal parestezi |
Orofarengial
allodini, soğuk hipersensitivitesi, infüzyon sonrası parestezi |
Akut:
Saatler içinde geri dönüşümlü Kronik:
İlaç kesimi sonrası aylar içinde geri dönüşümlü |
|
Vinca
alkaloidleri (Vinkristin, Vinblastin,Vindesin) |
Sensorimotor
ince lif, otonom, kranial sinirler |
Uyuşma,
ağrı, ekstansör kas zaafı, konstipasyon, baş dönmesi, oküler felç, fasyal
zaaf, vokal kord paralizisi |
İlaç
kesimi sonrası geri dönüşümlü |
|
Paklitaksel |
Sensorimotor
kalın ve ince lif |
Ağrılı
parestezi ve miyopati |
Geri
dönüşümlü |
|
Dosetaksel |
Sensorimotor
kalın ve ince lif |
Ağrılı
parestezi ve miyopati |
Geri
dönüşümlü |
|
Talidomid |
Sensorimotor |
Ağrılı
parestezi ve miyopati |
Geri
dönüşümlü |
|
Suramin |
Sensorimotor,
subakut demyelinizasyon, polinöropati |
Distal
ağrılı parestezi |
İlaç
kesimi sonrası geri dönüşümlü |
Metotreksat
toksisitesi: Bir folik asit antagonisti olan
metotreksat ile ilişkili akut veya kronik nörotoksisite oldukça iyi bilinir. En
sık görülen metotreksat toksisitesi tablosu aseptik menenjittir.
İntratekal metotreksat alan hastaların %10’unda görülür. Genellikle ilacın
verilmesinden 2-4 saat sonra başlar, 12-72 saat sürer. Daha nadir bir
komplikasyon ilacın intraventriküler uygulanmasından sonra görülebilen akut
ensefalopatidir. Bu durum bazen, özellikle kranial ışınlama sonrasında
yüksek doz intravenöz metotreksat uygulaması ile de görülebilir. Genellikle
24-48 saat içinde başlayan ve kendiliğinden düzelen konfüzyon, uykuya eğilim ve
nöbetlerle kendini gösterir. Yüksek doz intravenöz metotreksat sonrasında 5.-6.
günde görülen ve inme-benzeri tablolarla kendini gösteren subakut bir
ensefalopati görülebilir. Bu durum 1-2 gün içinde kendiliğinden düzelir.
Çok nadir olarak intratekal metotreksat tedavisinin bir komplikasyonu olarak transvers
miyelit görülür. Genellikle intratekal injeksiyondan sonra ilk 48 saat
içinde, nadiren ilk 2 hafta içinde ortaya çıkar. Düzelme değişkendir. Bazı
olgular tam düzelirken, bazılarında hiç düzelme görülmeyebilir. Metotreksat
tedavisinin en ciddi yan etkilerinden biri kronik lökoensefalopatidir.
Genellikle yineleyen yüksek doz intravenöz veya intratekal metotreksat
tedavisinden aylar veya yıllar sonra gelişir. Ani başlayan veya yavaş gelişen
mental gerileme ve davranış değişiklikleri ortaya çıkar. Seyir değişkendir;
bazen ılımlı bir demansiyel tablo olarak stabilize olurken, bazı durumlarda
ilerleyici bir gidişle spastik tetraparezi, nöbetler ve ölümle sonlanabilir.
Yaygın miyelin yıkımına bağlı olarak MRG’de yaygın ak madde hiperintensitesi
görünür. Metotreksatla birlikte kranial radyoterapi uygulanmış hastalarda
lökoensefalopati görülme riski ve şiddeti daha fazladır.
İmmün “checkpoint”
İnhibitörlerine Bağlı Nörolojik Komplikasyonlar
Son
yıllarda kanserde hedefli tedaviler olan monoklonal antikorların çeşitlenmesi ve
kullanımının yaygınlaşması ile birlikte, bazı nörolojik komplikasyonlar
görülmeye başlamıştır. Nörolojik komplikasyon ile ilişkisi bildirilen immün
“checkpoint” inhibitörü ajanlar arasında, CTLA-4 inhibitörü ipilimumab, PD-1
inhibtörü pembrolizumab ve nivolumab, PD-L1 inhibitörü atezolizumab, avelumab, durvalumab yer
almaktadır; bunlar bazen birbiri ile kombine edilerek de kullanılmaktadır ve
T-lenfosit yanıtlarını arttırmayı hedeflemektedir. Bunun sonucu olarak da pek
çok organa ait otoimmün hastalıklar ile birlikte nörolojik otoimmün hastalıklar
da görülebilmektedir. Bunların arasında inflamatuar miyopatiler, polimiyozit,
myasthenia gravis, otoimmün ensefalit, aseptik menenjit, transvers miyelit,
optik nörit, multipl skleroz, polinöropatiler, vaskülit gelişmesi sayılabilir.
Bu tür yan etkilerin gelişmesi, bu bölümün sonunda yer alan paraneoplastik
sendromları da hatırlatmaktadır.
Radyoterapi Yan
Etkileri
Radyoterapi
uygulanan SSS bölgesi, ortaya çıkacak yan etkiler açısından da belirleyicidir.
Sinir sisteminde radyasyon toksisitesi zamansal olarak genellikle üç ana başlık
altında incelenebilir: 1. Akut, 2. Erken-gecikmiş, 3. Geç-gecikmiş radyasyon
toksisitesi.
A. Radyasyon
Ensefalopatisi:
1. Akut
ensefalopati: KİBAS’a yol açmış büyük tümörlerde büyük bir fraksiyon
halinde (>300 cGy) radyoterapi uygulanmasından sonra ortaya çıkar. Öncesinde
steroid verilmemesi riski arttırır. Tedaviden hemen sonra veya birkaç saat
içinde nörolojik tabloda kötüleşme, başağrısı, bulantı-kusma, uyanıklık kusuru
ve ateş gelişir; genellikle kendine sınırlı bir tablo olsa da nadiren ölümcül
olabilir. Bu durumun önlenmesi için radyoterapi fraksiyonlarının 200 cGy’in
altında tutulması ve 24-72 saat öncesinden 16 mg/gün deksametazon başlanması
önerilmektedir.
2. Erken
gecikmiş ensefalopati: Gerek beyin tümörü nedeniyle gerekse profilaktik
olarak kranial radyoterapi alan hastalarda radyoterapiden sonraki 2 hafta-4 ay
içinde ortaya çıkar. Hastada başağrısı, uykuya eğilim başlar, eski nörolojik
bulgular tekrar belirir ve kötüleşir. Bu durumu klinik ve radyolojik olarak
primer tümörün nüksünden ayırt etmek çok zordur. BT ve MRG’de kontrast tutan,
etrafı ödemli bir bölge görülür. PET incelemesi ayrımı sağlayabilir. Bu
tablonun tümör nüksünden tek farkı, hem klinik hem de radyolojik tablonun
birkaç hafta içinde kendiliğinden düzelme göstermesidir. Steroidler bu
düzelmeyi hızlandırabilir. Bu nedenle böyle hastalar steroid tedavisi altında
sık görüntülemelerle izlenmelidir. Yapılmış histopatolojik incelemeler akson
kaybının geri planda olduğu demiyelinizasyonu gösterir.
3. Geç
gecikmiş ensefalopati: Radyasyon nekrozu genellikle beyin tümörü veya
baş-boyun tümörleri nedeniyle yapılan kranial radyoterapi tamamlandıktan
sonraki 1-2 yıl içinde ortaya çıkar. BT ve MRG’de beyin tümörlülerde eski tümör
bölgesinde, baş-boyun tümörlülerde de ışınlanan alandaki beyin parenkiminde
kontrast tutulumu ile çevresel ödem görülür. Bu durumun da klinik ve radyolojik
olarak primer tümörün nüksünden ayırt edilmesi çok zordur. Yukarıda söz
edildiği gibi, PET incelemesi veya MR spektroskopi ayrımı sağlayabilir.
Histolojik olarak radyasyon nekrozunun yukarıda sözü geçen kronik metotreksat
ensefalopatisinden ayırt edilmesi de güçtür. Kranial radyoterapi sonrasında
radyonekroz sıklığı %3-5 arasındadır. Olguların çok daha büyük bir kısmında ise
sadece kortikal ve subkortikal atrofi görülür. Radyoterapi ile birlikte
metotreksat da verilmişse lökoensefalopati gelişme riski artar.
B. Radyasyon
Miyelopatisi: (Ayrıca bakınız: Omurilik Hastalıkları)
1. Akut
miyelopati: Spinal radyoterapi sonrasında akut miyelopati görülüp
görülmeyeceği tartışmalı bir konudur. Tedavinin ilk günlerinde ortaya
çıkabileceği gibi haftalar içerisinde de oluşabilir. Genelde, boyun bölgesinin
ışınlanmasını izleyerek ortaya çıkan Lhermitte belirtisi ile tanınır.
Medulla spinalisdeki fokal demiyelizasyona bağlı olarak hasta başını fleksiyona
getirdiğinde gövde ve bacaklara, bazen de kollara yayılan, anlık bir
elektriklenme hissinden yakınır. Haftalar içerisinde spontan olarak düzelen bu
tablo için tedavi gerekli değildir.
2. Erken
gecikmiş miyelopati: Radyoterapiden sonraki 3-5 ay içinde ortaya çıkan
paresteziler ve Lhermitte bulgusu ile karakterizedir. Bu tablodan arka
kordonlarda ortaya çıkan demiyelinizasyonun sorumlu olduğu düşünülmektedir.
Genellikle de birkaç ay içinde kendiliğinden düzelir.
3. Geç
gecikmiş miyelopati: Geç radyasyon miyelopatisi başlıca üç şekilde görülür.
En sık görüleni 1-4 yıl içinde başlayıp hızlı veya yavaş bir progresyonla
paraparezi veya tetraparezi ile sonlanır. Klinik tablo; ağrısız ve öncelikle
medulla spinalisin daha çok radyasyon alan yarısı etkilenmiş olduğundan tam ya
da parsiyel bir Brown-Sequard sendromu olarak karşımıza çıkar. Benzer
bir klinik tabloya yol açabilecek epidural bası gibi olasılıkların dışlanması
için MRG yapılmalıdır. MRG basıyı dışlar; genellikle normaldir, ancak
omurilikte kontrast tutan hafif şiş bir bölge görülebilir. Radyoterapi bölgesi hakkında
ayrıntılı bilgi yoksa, ışınlanan bölgedeki vertebra korpuslarının yağlı doku
nedeniyle hiperintens görünmesi ışınlanan bölgenin sınırları hakkında ipucu
verir. Omurilik parenkim hasarı dışında pelvik ışınlamayı izleyerek alt
ekstremitelerde ortaya çıkan motor nöron sendromu ve intraspinal kanamalar da
spinal radyoterapinin geç komplikasyonları olarak karşımıza çıkabilir. Bu iki
durum da radyoterapiden uzun yıllar sonra görülebilir.
C. Radyasyona
Bağlı Kranial ve Periferik Nöropatiler
Radyasyona
bağlı kranial ve periferik nöropatiler nadiren de olsa görülebilir. Optik
nöropati radyoterapiden 6 ay-2 yıl sonra gelişen ağrısız tek veya iki yanlı
körlük ile ortaya çıkar. Papilödem görülebilir. MRG’de optik sinirde kontrast
tutulumu olabilir. Benzer şekilde diğer kranial sinirlerde de radyasyon hasarı
nadiren bildirilmiştir. Bunların dışında radyasyon pleksopatileri görülebilir.
Genellikle radyoterapiden bir yılı aşkın süre sonra geç-gecikmiş tablolar
ortaya çıkar. Ağrısız zaaf ve duyu kusuru olur. Radyasyon pleksopatisini olası
bir tümör infiltrasyonundan ayırmak önemlidir. Tümör infiltrasyonu genellikle
ağrılıdır. Ayrıca, iğne EMG’sinde tutulan kaslarda miyokimi varlığı radyasyon
pleksopatisi için oldukça tanı koydurucu sayılır (Bakınız: Pleksuslar
ve Hastalıkları
D. Radyasyonun
İndirekt Etkileri
Burada sözü
edilmesi gereken başlıca konular kranial ışınlamadan yıllar sonra ortaya çıkan ikincil
tümörler, büyük damar hastalığı ve hipotalamo-hipofizer yetmezlik
tablolarıdır. Çocukluk çağında kranial ışınlama yapılmış hastalarda ileride
meningiom, gliom, Schwannom gibi tümörlerin gelişme riski anlamlı olarak daha
yüksektir. Ortalama latans ise yaklaşık 17 yıldır. Benzer şekilde
radyoterapiden yıllar sonra damarsal komplikasyonlar gelişebilir. Radyoterapi
her boyda damar üzerinde olumsuz etki gösterir. Küçük damarlarda tıkanma
yukarıda söz edilen radyonekroza yol açar. Bunun dışında ışınlama penceresi
içinde kalan büyük serebral damarlarda da hızlanmış ateroskleroz görülür; ancak
ortaya çıkan darlıkların yerleşimi atipiktir. Bu durum radyoterapiden aylar
veya yıllar sonra belirti verebilir. Burada yaklaşım klasik aterosklerotik
hastalıktan farksızdır.
Paraneoplastik Nörolojik Hastalıklar
Genel özellikler
Paraneoplastik
nörolojik hastalıklar (PNH), periferik ve santral sinir sisteminin hemen hemen
tüm kısımlarını etkiler. Sıklıkla tutulan anatomik bölgeler arasında beyin,
omurilik, duysal ve otonom ganglionlar ve nöromüsküler bileşke bulunmaktadır. Bu
hastalıkların ortak yönü kanserin metastaz, vasküler komplikasyonlar, tedavi
yan etkileri ve fırsatçı infeksiyonlar gibi sık gözlenen mekanizmaları ile
değil genellikle otoimmün kökenli mekanizmalarla ortaya çıkmalarıdır.
Kanser hücreleri tarafından sinir sistemi dokusuna özgü antijenlerin (örneğin Hu,
N-metil D-aspartat reseptörü [NMDAR] gibi) anlatımı ve buna reaksiyon olarak
gelişen bağışıklık sistemi yanıtları PNH’nin temel patojenik mekanizması olarak
görülmektedir. Ayrıca timoma ve lenfoma gibi lenfoid doku içeren tümörlerin
bağışıklık sisteminin düzenleyici ve anti-inflamatuar mekanizmalarını bozması
da alternatif bir PNH gelişim mekanizması olabilir. Ancak, kanser eşliğinde
görmeye alışık olduğumuz pek çok nörolojik sendromun ve immünolojik yanıtın
kanseri olmayan hastalarda da ortaya çıktığı bilinmektedir. Bu durum PNH
fizyopatolojisi ile ilgili bilinmeyen pek çok özelliğin olduğunu
düşündürmektedir.
Patogenez
Paraneoplastik
sendromların neden ve nasıl ortaya çıktıkları tam olarak bilinmemekle beraber,
geçmiş yıllardan bu yana patogenezde bazı teoriler ileri sürülmektedir. Bugün
en yaygın kabul gören teori otoimmün teoridir. Otoimmün hastalığa ya fırsatçı
infeksiyona bağlı moleküler taklit, ya da tümörde bazı onko-nöral antijenlerin
ekspresyonunun yol açtığı düşünülmektedir. Ancak, paraneoplastik sendromun
sıklıkla erken dönemde kanser henüz sınırlı iken geliştiği, yani tümöre bağlı
immünsüpresyonun gelişmesinden önce ortaya çıktığı düşünülürse, fırsatçı
enfeksiyonların pek söz konusu olamayacağı görülmektedir. Bu durumda ikinci
olasılık daha akla yakın görünmektedir. Onko-nöral antijenler fetal yaşam
sonrası periferik dokularda kaybolan ve sinir sistemi içine sekestre olan
antijenlerdir. Sinir sisteminin bağışıklık sisteminden korunmuş yapısı içinde
yer alan bu ilkel antijenler, bazı tümörler tarafından da sergilenir ve tümöre
yönelen bağışıklık sistemi ögelerinin hedefi haline gelir. Bu antijenlerin
öncelikle CD4-hafıza T-lenfositlerini aktive ettiği düşünülmektedir. Aynı
zamanda, B-lenfositler aktive olur ve otoantikor üretimi başlar. Hücre içindeki
antijenlere (örn., Hu, Yo, CV2) karşı gelişen antikorlarla ilişkili PNH’ın
sitotoksik T-lenfosit temelli nöron hasarı sonucunda gerçekleştiği, hücre
membranı tarafından anlatımı yapılan antijenlere (NMDAR) karşı gelişmiş
antikorlarla ilişkili PNH’ın ise antikor temelli nöron işlev bozukluğu
sonucunda gerçekleştiği düşünülmektedir.
Tanı
PNH, sıklıkla çok iyi
tanımlanmış kendine özgü sendromlar şeklinde ortaya çıkar. PNH olgularında
sıklıkla saptanan çeşitli anti-nöronal antikorlar da tanıya yardımcı
olabilirler. Ancak PNH olgularının bir kısmında çeşitli sebeplerle antikor
saptanamaması veya henüz ilişkili antikorun tanımlanmamış olması mümkündür. Bu
sebeple klinik değerlendirme, PNH tanısının ilk basamağını oluşturur ve klinik
bulguların klasik PNH sendromları ile benzerliğinin saptanması önemlidir.
Görüntüleme yöntemleri tanıya yardımcı olmakla beraber nadiren spesifik
özellikler taşır. Örneğin T2 ağırlıklı kesitlerde mezial temporal lobda sinyal
artışı gözlenmesi otoimmün limbik ensefalit düşündürmekle beraber, bu bulgu
farklı infeksiyöz veya non paraneoplastik otoimmün ensefalitlerde de
görülebilir. BOS’da lenfositik pleositoz, IgG indeksinde artış ve
oligoklonal band (OKB) pozitifliği gibi çeşitli inflamasyon düşündüren bulgular
tanıya yardımcı olabilir. Ayrıca BOS incelemesi karsinomatöz menenjit ile
ayırıcı tanı için de gerekli bir incelemedir. EMG incelemeleri Lambert-Eaton
miyastenik sendromu (LEMS), nöromiyotoni ve duysal nöronopati gibi PNH
sendromlarında yol göstericidir. EEG’de saptanan anormal bulgular da MSS’nin
PNH’da tanıya yardım edebilir. EEG bulguları da diğer tanı yöntemleri gibi
spesifik bulgular vermez ama örneğin “extreme delta brush” adı verilen bulgu
bazı NMDAR ensefaliti olgularında görece sık saptanırken diğer antikorlarla
ilişkili PNH olgularında gözlenmez.
Antikor Testleri
Bazı PNH belli
antikorların bakılmasını akla getiren çok spesifik bulgularla başvurabilir.
Örneğin, sık tekrarlayan fasyobrakial distonik nöbetler “leucine rich, glioma
inactivated 1” (LGI1) antikoru ve over teratomu varlığı NMDAR antikoru tayinini
telkin ettirir. Ama PNH olgularının çoğunda bu gibi tipik bulgular olmadığından
geniş bir antikor paneli taraması yapılması gerekmektedir. Antikorlar olguların
bir kısmında serum örneklerinde saptanabilir. Ancak pek çok olguda antikorun
sadece BOS örneğinde pozitif saptanması da mümkündür. Ayrıca BOS örneği ile
yapılan antikor çalışmalarında yanlış pozitif ve yanlış negatif sonuçların son
derece düşük olduğu bildirilmiştir. Anti-Hu gibi çok klasik PNH antikorlarının
bile nörolojik bulgusu olmayan kanser hastalarında da saptanabildiği
unutulmamalıdır. Antikor düzeyleri tedavi sonrasında düşebilmekle beraber
antikor düzeyi ile tedavi kararı vermek veya tedavi etkinliğinin takibini
yapmak doğru değildir. Tam bir klinik iyileşme sonrasında antikor sonucu
pozitif olmaya devam edebileceği gibi, aktif klinik bulguları olan olgularda
immünsüpresyon sonrasında antikor testi negatif hale gelebilir.
Tümör Taraması
PNH şüphesinde hızlı
bir tümör taraması yapılması önemlidir. Tümörün gösterilmesi PNH tanısını
kolaylaştıracaktır. Ayrıca altta yatan tümör rezeksiyonu yapılmadan
immünsüpresif tedavilerin yararlılığı kısıtlı olacağından PNH hastasının
tedavisini de hızlandıracaktır. PNH olgularının büyük kısmında tümör nörolojik
bulguların başlangıcından çok sonra ortaya çıkmaktadır. İlk başvuruda tümör
saptanamayan olgularda tanı nörolojik bulgular ve antikor testleri ile
konmaktadır. Altta yatan tümöre ait bulguların sıklıkla ilk 1 yıl ve en geç 3-5
yıl içinde ortaya çıktığı bilinmektedir. Bu sebeple kuvvetli PNH şüphesi olan
olgularda tümör tarama testleri 5 yıl boyunca 3-6 ay arayla yapılmalıdır.
Kontrastlı tüm beden BT
incelemesi tarama amaçlı kullanılabilir. Tümör saptanma olasılığı yüksek
olgularda PET incelemesi de önerilmektedir. PET incelemesi küçük primer
tümörlerin ve metastazların gösterilmesini ve biyopsi için olası bölgelerin
belirlenmesini sağlar. Ailede kanser hikayesinin ve sigara kullanımı gibi
kanser risk faktörlerinin belirlenmesi ve CA125, CA15-3, prostat spesifik
antijen (PSA) ve karsinoembriyonik antijen (CEA) gibi kanser belirteçlerinin
serum düzeylerinin ölçülmesi de tanıya yardımcı olabilir. Antikor testlerinden
elde edilen sonuçlar da tümör araştırmasına yardımcı olabilir. Örneğin NMDAR
ensefalitinde over teratomuna yönelik kontrastlı pelvis MRG ve transvajinal
ultrason incelemesi, Yo antikoru pozitif olgularda meme kanserine ve
jinekolojik kanserlere yönelik kontrastlı pelvis MRG, mamografi ve pap smear
incelemeleri, Hu antikoru pozitif olgularda küçük hücreli akciğer kanserine
(KHAK) yönelik kontrastlı toraks BT incelemesi, Ma2 antikoru pozitif olgularda
testis kanserine yönelik ultrason incelemesi, “delta/notch-like epidermal
growth factor-related” (DNER) ve metabotropik glutamat reseptörü 1 (mGluR1)
antikoru pozitif olgularda Hodgkin lenfomasını saptamaya yönelik incelemeler
ilk sıraya alınabilir. Bazı PNH bulgularının tümörün saptanmasından ve hatta
tedavisinden uzun zaman sonra tümörün nüksü veya metastazı sırasında ortaya
çıkabileceği unutulmamalıdır. Ayrıca beyin metastazı bile olsa, hastanın klinik
tablosunu açıklamıyorsa paraneoplastik sendrom araştırılmalıdır.
Nöromüsküler Sistemi
Etkileyen PNH (Ayrıca Bakınız: Kas ve Nöromüsküler Kavşak
Hastalıkları, Periferik Sinirlerin Yaygın ve Çok Odaklı Hastalıkları)
Nöromüsküler bileşke,
kas, periferik sinir veya dorsal-otonomik ganglionların tutulumuyla seyreden
hastalıkları içeren bu grubun klasik sendromları saf duysal nöronopati,
otoimmün otonomik nöropati, LEMS, nöromiyotoni ve dermatomiyozittir.
Subakut Duysal
Nöronopati
Alt ekstremitelerden
başlayan parestezi-dizestezi ile ortaya çıkar ve günler/haftalar içinde
ilerler. Semptomlar genellikle asimetriktir, derin tendon refleksleri sıklıkla
kayıptır, derin duyu belirgin derecede bozulur ve kas gücü normaldir. Bazı
olgularda derin duyu kaybı baskın bulgudur. Bu yüzden duysal ataksi ve ellerde
atetoz benzeri hareketler sıklıkla gözlenir. Sinir sisteminde tutulan bölgeler
arka kök ve ganglion olduğundan BOS incelemesi sıklıkla patolojik sonuçlar
verir. Sıklıkla anti-Hu ve anti-CV2 antikorları ile ilişkilidir. EMG’de duysal
aksonal polinöropatiyle uyumlu olarak duysal aksiyon potansiyelleri
kaydedilemez, buna karşın motor iletiler normal sınırlardadır.
Otoimmün Otonomik
Nöropati
Yaygın otonomik
tutulumun nadir sebeplerindendir. Kuru göz, kuru ağız, anhidrozis, erektil
disfonksiyon ve aritmi bulgularına sık rastlanır. Sinir sisteminde hedef
otonomik ganglionlardır ve olguların yarısında nöronal nikotinik asetilkolin
reseptörünün α3β4 alt birimlerine karşı antikorlar (ganglionik
asetilkolin reseptör antikorları) saptanır. Bu antikor nörolojik bulgusu
olmayan akciğer kanseri olgularında da saptanabilir. Hastaların %10’unda timoma
bulunur ve bazı olgularda ek olarak myasthenia gravis de bulunabilir.
Lambert-Eaton Miyastenik Sendromu (LEMS )
Nöromüsküler
bileşkenin presinaptik bölgesinin otoimmün hastalığıdır ve yavaş ilerleyişli
güçsüzlük, çabuk yorulma ve çift görme bulguları ile seyreder. Sıklıkla ağız
kuruluğu, erkeklerde impotans gibi kolinerjik otonom bulgular eşlik edebilir.
Muayenede zayıf bulunan kas gücü, hastanın o kası çalıştırması sonucu fasilite
olur ve normale döner. Benzer şekilde hastalarda sıklıkla saptanan derin tendon
refleks kaybı eforla düzelebilir. Plazmaferez ve immünsüpresyona yanıtlı olabilir.
Semptomatik tedavide guanidin, 3,4-diaminopiridin ve antikolinesteraz yararlı
olabilir. Hastaların yaklaşık yarısında KHAK ve %90 kadarında presinaptik
voltaj kapılı kalsiyum kanallarına (VGKK) karşı antikor saptanır. VGKK
işlevlerinin bozulmasına bağlı olarak sinaptik boşluğa asetilkolin salınmasının
azalması sonucunda kas zaafı gelişir. VGKK antikoru pozitif olan olgularda LEMS
tablosuna serebellar dejenerasyon da eşlik edebilir ve bu olgularda KHAK ve Hu
antikoru bulunma olasılığı yüksektir. Ayrıca SOX isimli bir nöronal antijeni
tanıyan ve serebellumun Bergman glia hücrelerinin çekirdeklerine bağlandığı
için anti-glial nükleer antikor (AGNA) adı da verilen antikorların
paraneoplastik LEMS olgularının yarısında saptandığı bildirilmektedir. Ancak SOX
antikoru kanser olgularında yüksek oranda saptandığı için başka sebeplerle kas
zaafı gelişen kanser olgularında da bu antikorun pozitif bulunabileceği
unutulmamalıdır.
Nöromiyotoni
Isaacs sendromu veya
edinsel nöromiyotoni, periferik sinir aksonlarının artmış uyarılabilirliği
zemininde gelişen ağrılı kas kontraksiyonları ve kramplarla karakterize bir
hastalıktır. Hastaların bir kısmında timoma saptanması, myasthenia gravis ile
beraber görülebilmesi ve plazmafereze olumlu yanıt verebilmesi sebebiyle otoimmün
kökenli bir hastalık olduğu düşünülmüştür. Hastaların bir kısmında “contactin
associated protein-like 2” (CASPR2) antikoru saptanabilmekle beraber hastaların
çoğunda bilinen bir anti-nöronal antikor yoktur. İmmünoterapinin yanısıra
hastalar fenitoin ve karbamazepin gibi semptomatik tedavilerden faydalanabilir.
Morvan sendromunda nöromiyotoninin yanında ensefalit, otonomik bulgular ve
insomnia görülür. Nöromiyotoni gibi immünoterapiye yanıt verebilen bu olguların
büyük çoğunluğunda CASPR2 antikoru saptanır.
Dermatomiyozit
Haftalar-aylar içinde
ilerleyen güçsüzlük, kreatin kinaz yüksekliği, EMG incelemesinde miyopati
bulguları ve karakteristik deri bulguları ile ortaya çıkan bir hastalıktır.
Küçük bir grup olguda paraneoplastiktir ve yaşlılarda kanserle ilişkisi çok
daha sıktır. Meme, akciğer, over, mide, prostat ve kolonun adenokanserleri ve
Hodgkin hastalığı ile ilişkili olabilir. Bilinen bir otoantikor yoktur ve
paraneoplastik olmayan miyozit olgularında sıklıkla saptanan Jo-1 antikoru pek
çok paraneoplastik dermatomiyozit olgusunda saptanamamaktadır. İmmün-aracılı
bir vaskülopati olduğu düşünülen dermatomiyozitin tümör rezeksiyonu ve
immünsüpresif tedavilerden yarar görmesi mümkündür.
Diğer Sendromlar
Genellikle asetilkolin
reseptör antikorunun da pozitif saptandığı timoma olgularında gözlenen
myasthenia gravis, paraprotein ilişkili nöropatiler ve bir kanser eşliğinde
ortaya çıkan polimiyozit ve nekrotizan miyozit de PNH olarak kabul edilebilir.
Anlamı belirsiz monoklonal gamopati (MGUS) bazı kronik nöropatilerde
saptanabilir. Paraneoplastik nöropatiler multipl miyelom, plazmasitom, kronik
lenfositik lösemi ve lenfoma olgularında ortaya çıkabilir. Kronik inflamatuvar
demiyelinizan polinöropatiyi andıran demiyelinizan nöropati bulgularıyla
seyreden ve osteosklerotik miyelom ile beraber görülen polinöropati,
organomegali, endokrinopati, M proteini ve deri (“skin”) değişiklikleri
sendromu (POEMS) sıklıkla immünoterapiye dirençlidir. Diğer paraneoplastik
sendromlardan farklı olarak otoimmün mekanizmalardan çok vasküler endotelyal
büyüme faktörü (VEGF) gibi nörotoksik moleküllerin etkisiyle geliştiği
düşünülmektedir.
Merkezi Sinir Sistemini
Etkileyen PNH
Kanser olgularında, paraneoplastik
kökenli hastalık mekanizmaları limbik sistem, beyinsapı ve serebellum başta olmak
üzere çeşitli anatomik bölgeleri etkileyebilir.
Limbik Ensefalit
SSS’nin limbik
yapıları PNH olgularında en sık tutulan bölgelerdendir ve paraneoplastik
antikorların pek çoğu paraneoplastik limbik ensefalitte saptanabilir. Olgular
sıklıkla epileptik nöbet, amnezi ve davranış değişikliği bulguları ile
başvurur. MRG ve PET gibi görüntüleme yöntemleri ile mezial temporal lobda
sinyal değişiklikleri ve EEG’de temporal yavaş dalga veya epileptik deşarj sık
rastlanan bulgulardır. BOS incelemesi de genellikle inflamatuar bulgular verir.
Pek çok limbik ensefalit olgusunda bu tanı yöntemlerinin bir veya birkaçı
normal sonuçlanabilir veya limbik sistem dışı yapılar da tutulabilir.
Antikor testlerinin
sonuçlanması uzun sürdüğünden ampirik immünosüpresif tedaviye başlamak için
gerekli bulgular şunlardır: (i) Üç ayı geçmeyen bellek bozukluğu ve davranış
değişikliği hikayesi, (ii) Mezial temporal loblarda sinyal değişikliği, (iii)
Anormal EEG veya inflamatuar BOS bulgularından en az birinin saptanması, (iv)
Diğer sebeplerin dışlanması.
Ensefalomiyelit
SSS’nin farklı
bölgelerinin, arka kök, ganglion ve otonomik liflerin etkilenebildiği
inflamatuar bir hastalıktır. Ön planda hipokampus (limbik ensefalit), beyinsapı
(beyinsapı ensefaliti), diensefalon, serebellumun Purkinje hücreleri
(serebellar dejenerasyon), dorsal kök-ganglionu (duysal nöronopati), medulla
spinalis (miyelit) ve sempatik ve parasempatik sinir ve ganglionlar (otonomik
bulgular) tutulur. Anti-Hu ve KHAK en sık görülen antikor ve kanser tipidir.
Bunun dışında adrenal bez, prostat, nöroblastom ve KHAK dışı akciğer kanserleri
gibi diğer kanserlerle de görülebilir. Hu antikoru pozitif olgularda tabloya
saf duysal nöronopati de sıklıkla eşlik eder. Patolojik incelemede yaygın
lenfositik infiltrasyon ve CD8+ T hücre aracılı nöron kaybı saptanır. Anti-Hu
dışında anti-CV2 (KHAK, timoma), anti-Ma2 (testis kanseri), anti-amfifizin
(meme, KHAK), anti-LGI1 (KHAK, timoma) ve anti-NMDAR (over teratomu)
antikorları ile ilişkili ensefalomiyelit olguları da bildirilmiştir. EEG ve BOS
incelemeleri genelde patolojik sonuçlar verir. Genellikle progresyon hızlı ve
prognoz kötüdür.
Serebellar Dejenerasyon
Haftalar-aylar içinde
ilerleyen panserebellar sendrom, gövde ve taraf ataksisi, dizartri, horizontal
ve vertikal nistagmus ortaya çıkar ve kısa bir süre sonra tablo sabitleşir.
Ender olarak diplopi, işitme kaybı, yutma zorluğu, piramidal bulgular ve
polinöropati eşlik edebilir. Erken dönemde BT/MRG normaldir; geç dönemde
serebellar atrofi, bazen serebellum ak maddesinde hiperintensite görülebilir.
Histopatolojide yoğun ve selektif Purkinje hücre kaybı söz konusudur.
Serebellumda erken evrelerde yoğun inflamasyon bulguları görülürken, hastalığın
ileri evrelerinde inflamatuar hücreler kaybolur. Serebellar dejenerasyonda en
sık anti-Yo antikorları saptanır. Bu antikor hemen daima meme, over ve diğer
jinekolojik kanserlerle ilişkilidir. Nadiren lenfoma veya akciğer kanseri de
görülebilir. Daha seyrek olarak anti-Hu (KHAK), anti-Zic4 (KHAK), mGluR1 (lenfoma),
Homer-3 (lenfoma) ve anti-Tr (DNER) (lenfoma) ile ilişkili serebellar
dejenerasyon gelişebilir. DNER ilişkili serebellar dejenerasyon 20-40 yaş arası
erkeklerde daha sıktır ve genellikle nörolojik tutulum lenfoma tanısından sonra
ortaya çıkar. Diğer antikorların varlığında gelişen serebellar dejenerasyon
tedaviye dirençli olma eğiliminde olduğu halde DNER antikoru ilişkili bulgular
immünoterapi ile remisyona girebilir.
Stiff-Person Sendromu
ve Varyantları
Progresif rijidite,
refleks spazmlar ve lomber lordoz artışı ile seyreder. Bazen aksiyal kaslar
yerine ekstremitelerde baskın olabilir. Patolojik olarak “kronik spinal
internöronitis” söz konusudur. Sıklıkla (%80) non-paraneoplastiktir.
Paraneoplastik formu anti-amfifizin antikorları ile birlikte görülür ve
sıklıkla meme kanseri veya KHAK ile ilişkilidir. Paraneoplastik olmayan
olgularda glutamik asit dekarboksilaz (GAD) 65 ve glisin reseptör antikorları
saptanabilir. BOS genellikle inflamatuar özellikler gösterir. EMG’de istemli
kas kontraksiyonu yapılmazken bile sürekli motor ünite aktivitesi izlenir.
Semptomatik tedavi olarak benzodiazepinler, baklofen yararlı olabilir. Diğer
paraneoplastik sendromlardan farklı olarak tümör rezeksiyonu, kortikosteroid ve
IVIg ile tedavi genellikle mümkündür. Progresif ensefalomiyelit rijidite ve
miyoklonus (PERM) sendromu genellikle GAD65 antikoru ile görülür ve tümör
birlikteliği nadirdir. Bu sendromda irkilme (startle) refleksi ve beyinsapı
tutulumu ile uyumlu bulgular saptanabilir.
Opsoklonus Miyoklonus Sendromu
Opsoklonus sakkadik
stabilitenin bozulması sonucu ortaya çıkan istemsiz, aritmik, çok yönlü, yüksek
amplitüdlü konjuge sakkadik göz hareketleri olarak tanımlanabilir. Paraneoplastik opsoklonus-miyoklonus-ataksi (POMA) sendromunda opsoklonusa sıklıkla miyokloni, titubasyon
ve gövde ataksisinin baskın, taraf ataksisinin geri planda olduğu serebellar
sendrom eşlik eder. Çocuklarda olguların %50’sinde altta yatan nöroblastoma söz
konusudur ve diğer yandan nöroblastomalı çocukların %2’sinde opsoklonus miyoklonus
saptanır. Erişkinde ise %20-25 olguda paraneoplastik olarak ortaya çıkar. MRG
ve BOS incelemeleri genellikle normaldir. Son yıllarda yapılan çalışmalar
tutulan hedef sinir sistemi bölgesinin serebellumun fastigial çekirdeği
olduğunu göstermiştir. Bu gruptaki olguların önemli bir kısmında antikor
gösterilememektedir. Erişkinlerde olguların küçük bir kısmı anti-Ri antikorları
ile ilişkilidir. Altta yatan kanserler sıklıkla akciğer, meme ve diğer
jinekolojik tümörlerdir. Bu sendromla ilişkili antikorların günümüzdeki
yöntemlerle kolayca gösterilemeyen, muhtemelen hücre membranı ve aksonuna
bağlanan antikorlar olduğunu destekleyen çalışmalar vardır. Son yıllarda bazı
olgularda gama amino bütirik asit (GABA) reseptörü antikorları saptanması bu
görüşü desteklemektedir. Tümör rezeksiyonunun ardından yapılacak
kortikosteroid, IVIg, plazmaferez gibi tedavi yöntemleri ile remisyon
görülebilir.
Klasik PNH
Bu hastalık grubunun
karakteristik özellikleri, hücre içi antijenlere karşı gelişmiş ve klinik
bulguların gelişiminde rol oynamayan antikorlar, ağırlıklı olarak T hücre
aracılı mekanizmalarla gerçekleşen nöron kaybı, hastaların tamamına yakınında
kanser varlığı ve tedaviye direnç ve kötü prognozdur (Tablo 6).
Hu antikoru
sıklıkla KHAK (%90 kadarında) ve saf duysal nöronopati ile seyreder. Ayrıca saf
otonomik nöropati, limbik ensefalit, ensefalomiyelit ve serebellar dejenerasyon
da ortaya çıkabilir. Hu antikorunun nörolojik bulgusu olmayan KHAK olgularında
da %40 oranında pozitif çıkabildiği unutulmamalıdır.
Ma2 antikoru
pozitif olgularda limbik sistem, diensefalon, beyinsapı ve serebellar
bölgelerin tutulumu ile uyumlu bulgular görülür. Vertikal göz hareketi
bozuklukları, hipokinezi, rijidite, çene distonisi ile testisin germ hücreli
tümörleri özellikle anti-Ma2 ensefaliti ile ilişkilendirilmiştir. Ma2 antikoru
pozitif olgular immünoterapiye diğer onkonöral antikorlarla ilişkili
sendromlara kıyasla daha iyi yanıt verirler. Ma1 antikorları erkek ve kadın
olgularda çeşitli kanserlerin eşliğinde ortaya çıkar ve ataksi, oftalmopleji ve
farklı ensefalit bulguları ile seyreder. Ma2 antikoru pozitif olgulardan farklı
olarak Ma1 antikorlu hastalarda tedaviye yanıt kötüdür.
Tablo 6.
Hücre içi antijenlere karşı gelişmiş antikorlarla ilişkili klasik PNH
|
Antikor |
Cinsiyet
/ Yaş |
Sendrom |
Tümör |
|
Hu
(ANNA1); hedef nükleer HuD |
Erkek,
ortalama 60 yaş |
Duysal
nöronopati, ensefalomiyelit, serebellar dejenerasyon, otonomik nöronopati |
KHAK |
|
Ri
(ANNA2); hedef nükleer NOVA |
Kadın,
ortalama 60 yaş |
Serebellar
dejenerasyon, ensefalomiyelit, opsoklonus-miyoklonus |
Akciğer
veya meme |
|
ANNA3 |
Erkek
ve kadın, 8-80 yaş |
Nöropati,
miyelopati, beyin sapı limbik ensefalit |
Akciğer |
|
Ma2
(Ta); hedef nukleolar PNMA-2 |
Erkek,
ortalama 30 yaş |
Beyinsapı,
limbik ensefalit, serebellar dejenerasyon |
Testis |
|
Yo
(PCA-1); hedef sitoplazmik cdr2 |
Kadın,
tüm yaşlar |
Serebellar
dejenerasyon |
Meme,
over, diğer jinekolojik |
|
PCA-2 |
Az
sayıda hasta |
Serebellar
dejenerasyon |
Küçük
hücreli kanserler |
|
CV2
(CRMP-5) |
Erkek
ve kadın, ileri yaş |
Serebellar
dejenerasyon, miyelopati, saf duysal nöronopati, kore, retinopati |
Akciğer,
timus |
|
Amfifizin |
Erkek,
ortalama 60 yaş |
Stiff-person,
ensefalomiyelit |
Akciğer,
meme |
|
GAD65 |
Kadın,
30-80 yaş |
Limbik
ensefalit, serebellar dejenerasyon, stiff-person |
Çeşitli |
Yo antikoru
meme, over ve jinekolojik tümörlerle beraber ortaya çıkar ve kalıcı özürlülüğe
yol açan progresif serebellar sendrom ile ilişkilidir. Ri antikoru meme ve
akciğer kanserleri ile beraber saptanır ve beyinsapı başta olmak üzere çok
çeşitli beyin alanlarının tutulumu ile seyreden klinik bulgulara sebep olur.
CV2 antikoru akciğer ve timus tümörleri eşliğinde ortaya çıkan serebellum,
omurilik, bazal ganglionlar, periferik sinir ve retinanın etkilendiği
sendromlarla beraber görülür.
Sinaptik hücre içi
antijenler olan GAD65 ve amfifizine karşı antikorlarla ilişkili
sendromların tanınması da önemlidir. Tip 1 diabetes mellitus olgularında da
saptanabilen GAD65 antikoru ile ilişkili sendromlar stiff-person sendromu,
serebellar dejenerasyon ve limbik ensefalittir. GAD65 ve GABA-B reseptör
antikorlarının birlikteliği sık rastlanan bir durumdur. GAD65 antikorlarının
özellikle BOS örneklerinde ve yüksek düzeyde saptanmasının tanı için daha
anlamlı olduğu bildirilmiştir. GAD65 antikoru pozitif olguların sinir sistemi dokularında
T hücresinin baskın olduğu hücresel infiltrasyon saptanır. Olguların bir kısmı
immünoterapiye çok iyi yanıt verir. Son yıllarda ileri yaşlarda ortaya çıkan ve
sıklıkla altta yatan bir tümörün de bulunduğu bir GAD65 antikoru pozitif hasta
grubu tanımlanmıştır. Amfifizin antikorları meme ve akciğer adenokanserleri
olan paraneoplastik stiff-person ve ensefalomiyelit olgularında saptanır.
Nöronal Membran Antikorları
ile İlişkili PNH
Bu gruptaki PNH
olgularında altta yatan mekanizma hemen daima patojenik özelliğe sahip
antikorlardır (Tablo 7). Antikorların hücre yüzeyinde ifade edilen hedef
antijenlerine bağlanmak suretiyle klinik bulgulara sebep olduklarını düşündüren
klinik ve deneysel veriler bulunmaktadır. Hücre içi antikorları grubundan
farklı olarak tümör saptanma oranları farklılık gösterir ve tedaviye yanıt
oldukça iyidir. Remisyondan haftalar veya aylar sonra tekrarlayabilir. Sinir
sistemi örneklerinin patolojik incelemelerinde perivasküler plazma hücresinin
baskın olduğu seyrek hücre infiltrasyonu saptanır. Ayrıca dokuda IgG
birikimleri ve daha geri planda kompleman birikimi görülebilir. NMDAR antikoru
bu grubun en sık saptanan antikorudur. Ek olarak LGI1, CASPR2,
alfa-amino-3-hidroksi-5-metil izoksazol-4 propionik asit reseptörü (AMPAR), GABA-A
reseptörü, GABA-B reseptörü, glisin reseptörü, mGluR1, mGluR5 ve DNER
antikorları ile ilişkili sendromlar bu gruba dahildir.
NMDAR ensefaliti
NMDAR’nin NR1 alt
birimini hedef alan antikorlarla gelişir. Genellikle genç kadınlarda ve
çocuklarda ortaya çıkan NMDAR ensefaliti, olguların çoğunda ateş ve üst solunum
yolu infeksiyonu benzeri prodromal bulguların ardından gelişen psikotik
bulgularla başlar ve günler-haftalar içinde epileptik nöbet ve amnezi bulguları
eklenir. Bu bulguları sıklıkla kore, diskinezi ve distoni gibi hareket
bozuklukları, katatoni, mutizm, otonomik sistem bozuklukları ve hipoventilasyon
izler. MRG incelemesinde mezial temporal lob gibi limbik bölgelerin tutulma
oranı %30’un altındadır. BOS incelemesinde lenfositoz ve protein düzeyinde
artışın yanında olguların yaklaşık yarısında oligoklonal band saptanır.
EEG incelemesinde yaygın veya temporal yavaş dalga ve epileptik deşarjların
yanında olguların üçte birinde “extreme delta brush” bulgusu gözlenir. Over
teratomu, NMDAR ensefaliti olgularının %40-50 kadarında saptanır. Çocuklarda
tümör saptanma olasılığı daha düşüktür ve konuşma bozukluğu, distoni ve diğer
hareket bozuklukları ön planda görülür. Erken tanı ve tedaviye rağmen hastalık
%5-8 oranında mortalite ile sonlanır.
LGI1 ve CASPR2 ensefalitleri
Voltaj kapılı potasyum
kanalı kompleksinin alt birimleri olan LGI1 ve CASPR2 antijenlerine karşı
gelişen antikorlar ağırlıklı olarak periferik sinir hipereksitabilitesi ve
limbik ensefalopati bulguları ile seyreden klinik tablolara sebep olurlar. LGI1
ve CASPR2 ensefalitleri ileri yaşlarda ve erkeklerde biraz daha fazla ortaya
çıkar. LGI1 ensefaliti öncesinde, aynı taraf yüz, omuz ve üst ekstremite
kaslarının bazen sayıları yüzleri bulan ani kasılmaları ile karakterize
fasiobrakial distonik nöbetler gözlenir. Bunun dışında farkındalığın bozulduğu
veya korunduğu fokal ve jeneralize nöbetler de ortaya çıkabilir. Nöbetlere ek
olarak bellek bozukluğu, psikoz ve duygudurum değişiklikleri bildirilmiştir.
Tablo 7.
Nöronal membran antijenlerine karşı gelişmiş antikorlarla ilişkili
paraneoplastik hastalıklar
|
Antikor |
Cinsiyet
/ Yaş |
Sendrom |
Tümör |
|
NMDAR |
Kadın,
çocuk ve gençlerde |
NMDAR
ensefaliti |
Over
teratomu (%50) |
|
AMPAR |
Kadın,
ileri yaşlarda |
Limbik
ensefalit |
Akciğer,
meme, timus |
|
GABA-A
reseptörü |
Erkek,
ortalama yaş 20 |
Epilepsinin
ön planda olduğu ensefalit |
Hodgkin
lenfoma (nadir) |
|
GABA-B
reseptörü |
Erkek
ve kadın, ortalama 60 yaş |
Limbik
ensefalit, opsoklonus-miyoklonus |
KHAK
(%50) |
|
LGI1 |
Erkek
ve kadın, ortalama 60 yaş |
Limbik
ensefalit |
Timoma
(nadir) |
|
CASPR2 |
Erkek,
ortalama 60 yaş |
Nöromiyotoni,
Morvan sendromu, limbik ensefalit |
Timoma
(%40) |
|
Glisin
reseptörü |
Erkek
ve kadın, tüm yaşlar |
Stiff
person sendromu, PERM |
Hodgkin
lenfoma (nadir) |
|
mGluR1 |
Erkek
ve kadın, farklı yaşlarda 5 olgu |
Serebellar
dejenerasyon |
Hodgkin
lenfoma |
|
mGluR5 |
Erkek
ve kadın, farklı yaşlarda 2 olgu |
Limbik
ensefalit (Ophelia sendromu) |
Hodgkin
lenfoma |
|
DNER
(Tr) |
Erkek
ve kadın, tüm yaşlar |
Serebellar
dejenerasyon |
Hodgkin
lenfoma |
Diğer Ensefalitler
AMPAR ensefaliti,
AMPAR’nin GluR1 ve GluR2 alt birimlerini hedef alan antikorlarla gelişir.
Psikotik bulguların ilk ve baskın olduğu limbik ensefalit bulgularına sebep
olur. Kadınlarda biraz daha fazla ortaya çıkar ve çeşitli dokuların
adenokanserleri AMPAR ensefalitine eşlik edebilir.
GABA-B reseptör
ensefaliti nöbetlerin baskın bulgu olduğu limbik ensefalopati bulguları ile
seyreder. Olguların pek çoğunda KHAK görülür ve akciğer kanseri birlikteliği
ile ortaya çıkan paraneoplastik limbik ensefalit olgularında en sık saptanan
antikorlardandır. GABA-A reseptör ensefaliti status epileptikus, epilepsia
partialis continua ve diğer şiddetli epileptik nöbet tipleri ile ortaya çıkar.
T2 ağırlıklı MRG incelemesinde kortikal ve subkortikal bölgelerde yaygın
hiperintens lezyonlar saptanır.
mGluR5 antikoru hafif
konfüzyon ve oryantasyon bozukluğu, Hodgkin lenfoma ve immünoterapiye iyi yanıt
triadından oluşan Ophelia sendromu olgularında saptanır. Bu hastalarda ayrıca
bellek bozukluğu, psikoz ve duygudurum değişiklikleri olabilir. mGluR1 antikoru
Hodgkin lenfomanın eşlik ettiği serebellar dejenerasyon olgularında gözlenir.
Serebellumda mGluR1 ile aynı lokalizasyonda ifade edilen Homer-3 antikorları da
bazı serebellar dejenerasyon olgularında saptanmış ancak bu olgularda tümör
bulunamamıştır.
DNER (Tr) antikorları
Hodgkin lenfoması olan serebellar dejenerasyon olgularında en sık saptanan
antikordur. Pek çok PNH’dan farklı olarak altta yatan lenfomaya ait klinik
bulgular genellikle nörolojik bulgulardan önce ortaya çıkar. Lenfoma sıklığı
DNER antikor pozitif olgularda %90 civarındadır. Glisin reseptör antikorlarının
irkilme refleksinin ön planda olduğu PERM olgularında saptandığı
bildirilmiştir. Genellikle herhangi bir tümörü olmayan olgularda ortaya
çıkmakla beraber az sayıda Hodgkin lenfomalı ve adenokanserli olgu
bildirilmiştir.
Tedavi
PNH genellikle
hücresel (T hücre baskın) veya hümoral (antikor baskın) mekanizmalarla
gelişmektedir. Dolayısıyla bağışıklık sistemini baskılayıcı tedavi
yöntemlerinin önemi büyüktür. Hücre içi antijenlerine karşı gelişmiş
antikorlarla ilişkili sendromlarda tümör rezeksiyonunun yanında yüksek doz
kortikosteroid tedavisi uygulanır. Tedaviye dirençli olan ve kemoterapi
uygulanan olgularda siklofosfamid tedavisi yararlı olabilir. Bu grup PNH
antikor yanıtlarının geri planda olduğu ve T hücre aracılı yanıtların baskın
olduğu bir inflamasyonla seyrettiğinden IVIg ve plazmaferez gibi tedavi
yöntemleri yararlı olmayabilir. Aynı sebepten dolayı rituksimab tedavisinin de
rasyonel bir temeli yoktur ama B hücreleri antijen sunumundan da sorumlu
olduklarından dirençli olgularda denenmesi düşünülebilir. Steroid, IVIg ve
rituksimab gibi tedavi yöntemlerinin altta yatan kanseri kötüleştirici etkisi
olmadığı bildirilmiştir.
Nöronal membran
antikorları ile ilişkili PNH tedavisinde ilk basamakta yüksek doz
metilprednizolon ve IVIg (veya plazmaferez) uygulanması önerilmektedir. Bu
tedaviye yanıt vermeyen olgularda ikinci basamak tedavisi olan siklofosfamid ve
rituksimab tedavileri önerilmiştir. Bu gruptaki PNH olgularında saptanan
antikorların deneysel hayvan modeli (anti-NMDAR) ve/veya primer nöron hücresi
çalışmalarında (anti-NMDAR, anti-AMPAR, anti-LGI1, anti-glisin reseptörü)
patojenik etkileri gösterilmiştir. Ayrıca immünoterapi sonrasında serum ve
BOS’ta antikor düzeylerinin düşüşüne paralel olarak klinik bulguların da
düzelmeye başladığı bilinmektedir. Bu sebeple antikoru temizleyen IVIg ve
plazmaferez gibi yöntemlerin faydalı olması doğaldır. Aynı şekilde bu gruba ait
ensefalit olgularının patolojik incelemelerinde parenkimal, perivasküler ve
meningeal bölgelerde bol miktarda plazma hücresi biriktiği gösterilmiştir. B
hücrelerini hedef alan rituksimab tedavisinin bu sebeple tedaviye dirençli
olgularda yararlı olabileceği bilinmektedir. Ağır olgularda rituksimabın
birinci basamak tedavi olarak kullanılmasını önerenler de vardır. LGI1
ensefalitinde steroid tedavisinin diğer PNH sendromlarına göre daha etkili
olduğu bildirilmiştir.
NMDAR (%10-40), AMPAR
(>%50) ve LGI1 (%35) ensefalitlerinde remisyon sonrasında ortaya çıkan
nüksler tipiktir. Nüks riskinin yüksek olduğu PNH olgularında azatioprin gibi
bir ilaçla uzun süreli immünsüpresyon yapılması önerilmiştir ama bu konuda bir
görüş birliği bulunmamaktadır.
Bu bölümde yer alan tümöral
oluşumların birçoğuna ait makroskopik ve histopatolojik şekiller Sinir Sistemi
Semiyolojisi/Nörolojide Laboratuvar İncelemeleri/Nöropatoloji bölümünde bulunabilir.
Kaynaklar
1.
Abrey
L, Batchelor T, Fereri A, ve ark. Report of an international workshop to
standardize baseline evaluation and response criteria for primary CNS lymphoma.
J Clin Oncol 2005; 23: 5034-5043.
2.
Aminoff
MJ (editor). Neurology and general medicine (4. baskı). Churchill Livingstone Elsevier,
Philadelphia, 2008.
3.
Balm
M, Hammack J. Leptomeningeal carcinomatosis. Presenting features and prognostic
factors. Arch Neurol 1996; 53: 626.
4.
Banan
R; Hartmann C. The new WHO 2016 classification of brain
tumors—whatneurosurgeons need to know. Acta Neurochir 2017; 159:403–418
5.
Bataller
L, Kleopa KA, Wu GF, ve ark. Autoimmune limbic encephalitis in 39 patients:
immunophenotypes and outcomes. J Neurol Neurosurg Psychiatry. 2007;78:381.
6.
Berzero
G, Psimaras D. Neurological paraneoplastic syndromes: an update. Curr Opin
Oncol. 2018;30(6):359-367.
7.
Binks
SNM, Klein CJ, Waters P, Pittock SJ, Irani SR. LGI1, CASPR2 and related
antibodies: a molecular evolution of the phenotypes. J Neurol Neurosurg
Psychiatry. 2018;89(5):526-534.
8.
Bradley
WG, Daroff RB, Fenichel GM, Jankovic J (editor). Neurology in Clinical Practice
(5th Edition). Butterworth-Heinemann-Elsevier, Philadelphia 2008.
9.
Brandsma
D, van den Bent MJ. Molecular targeted therapies and chemotherapy in malignant
gliomas. Current Opinion in Oncology 2007; 19: 598-605.
10.
Dalmau
J, Tuzun E, Wu HY, ve ark.. Paraneoplastic anti-N-methyl-D-aspartate receptor
encephalitis associated with ovarian teratoma. Ann Neurol. 2007;61:25.
11.
DeVito
VT, Hellman S, Rosenberg SA. Cancer: Principles and Practice of Oncology 7th
Edition. Lippincot Williams and Willkins, Philadelphia 2005.
12. Epidemiology
of Brain Tumors Hiroko Ohgaki. Methods of Molecular Biology, Cancer
Epidemiology, vol. 472 © 2009 Humana Press, a part of Springer Science +
Business Media, Totowa, NJ Book.
13.
Grativvol
RS, Cavalcante WCP, Castro LHM, Nitrini R, Simabukuro MM. Updates in the
Diagnosis and Treatment of Paraneoplastic Neurologic Syndromes. Curr Oncol Rep.
2018;20(11):92.
14.
Graus
F, Delaterre JY, Antoine JC, Dalmau J, Giometto B, Grisold W, ve ark.
Recommended diagnostic criteria for paraneoplastic neurological syndromes. J
Neurol Neurosurg Psychiatry 2004; 75: 1135-1140.
15.
Gultekin
SH, Rosenfeld MR, Voltz R, ve ark. Paraneoplastic limbic encephalitis:
neurological symptoms, immunological findings and tumour association in 50
patients. Brain. 2000;123:1481.
16.
Hardell
L, Nasman A, Pahlson A, Hallquist A, Hansson Mild K. Use of cellular telephones
and the risk for brain tumours: A case-control study. Int J Oncol 1999; 15:
113.
17.
Honnorat
J, Joubert B. Movement disorders in autoimmune encephalitis and paraneoplastic
neurological syndromes. Rev Neurol (Paris). 2018;174(9):597-607.
18.
Kadan-Lottick
N, Sklusarek M, Gurney J. Decreasing incidence rates of primary central nervous
system lymphoma. Cancer 2002; 95: 193.
19.
Kan
P, Simonsen SE, Lyon JL, Kestle JR. Cellular phone use and brain tumor: a
meta-analysis. J Neurooncol 2008; 86: 71-78.
20.
Kun
LE, Gajjar A, Pollack IF. Pediatric brain tumors. İçinde: Perry MC (editör),
American Society of Clinical Oncology 1999 Educational Book. American Society
of Clinical Oncology, Alexandria, 1999.
21.
Lapointe
S, Perry A, Butowski NA. Primary brain tumours in adults. Lancet. 2018
4;392(10145):432-446.
22.
Lee
EQ. Nervous System Metastases From Systemic Cancer. Continuum (Minneap Minn)
2015;21(2):415–428
23.
Lynch
DR, Rattelle A, Dong YN, Roslin K, Gleichman AJ, Panzer JA. Anti-NMDA Receptor
Encephalitis: Clinical Features and Basic Mechanisms. Adv Pharmacol.
2018;82:235-260.
24.
Nayak
L; Pentsova E;Batchelor TT. Primary CNS Lymphoma and Neurologic Complications
of Hematologic Malignancies. Continuum (Minneap Minn) 2015;21(2):355–372
25.
Nolan
PC; DeAngelis LM, Neurologic Complications of Chemotherapy and Radiation
Therapy. Continuum (Minneap Minn) 2015;21(2):429–451
26. Ostrom
QT, Gittleman H, Truitt G, Boscia A, Kruchko C, Barnholtz-Sloan JS. CBTRUS
Statistical Report: Primary Brain and Other Central Nervous System Tumors
Diagnosed in the United States in 2011-2015. Neuro Oncol. 2018.
27.
Posner
J. Neurologic complications of cancer. FA Davis Company, Philadelphia, 1995.
28.
Ricken
G, Schwaiger C, De Simoni D, Pichler V, Lang J, Glatter S, Macher S, Rommer PS,
Scholze P, Kubista H, Koneczny I, Höftberger R. Detection Methods for
Autoantibodies in Suspected Autoimmune Encephalitis. Front Neurol. 2018;9:841.