EPİLEPSİ
Yazanlar:
Nermin Görkem ŞİRİN, Nerses BEBEK, Betül BAYKAN
Son güncelleştirme Tarihi: 25.03.2020
İçindekiler:
Epilepsi nöbeti ve epilepsi
hastalığının tanımlanması
Fokal başlangıçlı nöbetler
Fokal epilepsi nöbetlerinin
semiyolojik özellikleri: lateralizasyon ve lokalizasyon değerleri
Jeneralize başlangıçlı nöbetler
Duruma bağlı nöbetler
Epilepsi nöbetlerinde temel
mekanizmalar
Epileptogenez
Epilepsi
hastalığı
Epilepsi
hastalığı sınıflaması
Epilepsi sendromları
Epilepsili hastanın
değerlendirilmesi
Anamnez ve muayene
Laboratuvar incelemeleri
Elektroensefalografi
(EEG)
Nöropsikolojik
Değerlendirme
Bilgisayarlı
Tomografi (BT) ve Magnetik Rezonans Görüntüleme (MRG)
Fonksiyonel
Görüntüleme
Ayırıcı
tanı
Epilepsilerin genetiği
Epilepsi tedavisi
Dirençli epilepsiler ve
cerrahi yolla tedavi edilebilir epilepsiler
Status epileptikus ve
tedavisi
Epilepsinin biyolojik
yönleri ve sosyal etkileri
Epilepsili
hastalarda ani beklenmedik ölüm (SUDEP - Sudden unexpected death in epilepsy)
Epilepsi ve komorbidite
Giriş
Epilepsi
tüm dünyada ve ülkemizde sık görülen bir nörolojik hastalıktır.
Birbirinden farklı semptom ve bulgularla paroksizmal şekilde
seyreden, nöbetlerine farklı klinik bulguların eşlik ettiği
çok yönlü kompleks bir hastalık grubudur. Bu bölümde epilepsi nöbetleri ve
epilepsi hastalığının tanısı,
sınıflandırılması, etyolojisi ve tedavisinden
bahsedilecektir. Ayrıca epilepsi ile ilişkili durumların da
üzerinde durulacaktır.
EPİLEPSİ NÖBETİ VE EPİLEPSİ
HASTALIĞININ TANIMLANMASI
Uluslararası
Epilepsi ile Savaş Derneği (International League Against
Epilepsy-ILAE) epilepsi nöbeti ve epilepsi hastalığının
kavramsal olarak tanımlamasını ayrı ayrı
yapmıştır. Bu tanımlamada epilepsi nöbeti, beyin
içerisinde anormal, aşırı veya senkron nöronal aktiviteye
bağlı geçici semptom ve bulguların belirmesi olarak
belirtilmiştir. Epilepsi hastalığı ise süregelen
epilepsi nöbeti oluşturma yatkınlığıyla ve bu durumun
yarattığı nörobiyolojik, kognitif, psikososyal ve sosyal sonuçlar
ile karakterize bir beyin hastalığıdır. Epilepsi
hastalığından bahsetmek için en az bir epileptik nöbet
geçirmiş olmak şartı aranır. Son yıllar içinde
epilepsi hastalığını değerlendirirken yaşanan
sorunlar tanımlama içerisinde pratik klinik düzenlemeler
yapılmasını gerekli kılmıştır. Bu amaçla
2014 yılında ILAE bir rapor yayınlamıştır.
Epilepsi hastalığından, en az bir epilepsi nöbeti
varlığına ek olarak tekrarlayan nöbetleri oluşturmaya
yatkın bir durum eşliğinden bahsedilmektedir. Yeni klinik pratik
amaçlı tanımlamada aşağıdaki üç durumdan biri
varlığında epilepsi hastalığından bahsedilir:
1) Yirmi dört saat ara ile en az iki tetiklenmemiş
(spontan) veya refleks özellikli nöbet olması
2) En az bir tetiklenmemiş veya refleks özellikli
nöbet ve 10 yıl içerisindeki nöbet tekrarlama riskinin en az iki
tetiklenmemiş nöbet varlığındaki riske benzer olması
(en az %60)
3) Epilepsi sendromlarından birinin
varlığı (Bakınız: Epilepsi
Sendromları)
Tetiklenmiş
nöbetler, diğer adı ile akut
semptomatik nöbetler, metabolik veya toksik bir nedenle veya kafa
travması, serebrovasküler hastalık gibi nedenlerle öncesinde normal
olan beynin epilepsi nöbeti geçirme eşiğinin düşmesi sonucu akut
dönemde gelişir ve epilepsi hastalığı kapsamında
değerlendirilmezler. Ancak, epilepsi hastalığının
belirgin, süregiden etyolojik nedenleri bu kapsamla
karıştırılmamalıdır (örneğin, beyin tümörü).
Tahmin edileceği gibi bazı durumlarda tetikleyici
kavramının sınırları belirsiz olabilir. Bazı
epilepsi hastalarında tekrarlayan epilepsi nöbetlerinin
başlamasından önce bir tetikleyici olabilir (örn, kafa
travması). Tanımlamada bir diğer altı çizilen nokta refleks
nöbetlerdir. Aralıklı ışık uyaranı veya
sıcak su gibi uyaranlar ile tetiklenen nöbetler, öncesinde bir tetikleyici
olmasına rağmen bu grupta kabul edilmezler ve refleks özellikli
olarak nitelenirler.
Sadece
bir tetiklenmemiş nöbet varlığında epilepsi
hastalığından bahsedebilmek için 10 yıl içerisindeki nöbet
tekrarlama riskinin yüksek olarak belirlenmesi gerekmektedir. Bu durumlara son
birkaç ay içerisinde geçirilmiş serebrovasküler hastalık, epilepsi
nöbetine yol açma olasılığının yüksek olduğu
bilinen diğer yapısal lezyonlar ve EEG’de interiktal epileptiform
aktivitenin varlığı örnek olarak verilebilir (Tablo 1). İlk nöbet ile gelen
erişkin hastalarda yapılan çalışmalar nöbet tekrarlama
riskinin şu durumlarda artmış olduğunu bildirmektedir:
1) Akut bir beyin hasarı ile gerçekleşen
nöbetler (1-5 yıl içerisinde nöbet tekrarlama riski 2,55 kat
artmıştır)
2) EEG’de epileptiform anomali varlığı
(1-5 yıl içerisinde nöbet tekrarlama riski 2,16 kat
artmıştır)
3) Nörogörüntüleme ile epileptojenik bir lezyonun
gösterilmesi (1-4 yıl içerisinde nöbet tekrarlama riski 2,44 kat
artmıştır)
4) Noktürnal nöbet varlığı (1-4 yıl
içerisinde nöbet tekrarlama riski 2,1 kat artmıştır)
Nöbetler
zaman içinde her hasta için belli bir kalıpta, genellikle
kendiliğinden veya bazı tetikleyen faktörler zemininde tekrarlar.
Nöbetler arasında hasta genellikle normal yaşantısını
sürdürür. Nöbet aralıkları ve tipleri son derece değişken
olabilir. Ancak aynı hastada genellikle aynı veya belirli birkaç
nöbet tipi tekrarlama eğilimi gösterir. Epilepsi nöbet tipleri klinik
belirti ve bulgulara göre sınıflandırılırlar. Nöbet
sırasında izlenen klinik belirti ve bulgular epilepsi nöbetinin
semiyolojik özellikleri olarak adlandırılır.
Epilepsi
hastalığının varlığı ile antiepileptik
tedavi başlanılması kararı birbirinden ayrı
değerlendirilmesi gereken konulardır. Bazı epilepsi
hastalıkları iyi gidişli ve nadir nöbetlere neden
olduğundan tanı konulsa bile ilaç başlanılmayabilir.
Bazı akut semptomatik nöbetlerde epilepsi hastalığı
tanısı konulmasa da kısa süreli ilaç başlanabilir (örn,
nöbet ile prezante olan serebrovasküler hastalıklar).
ILAE
tarafından yayınlanan sınıflamada, epilepsi
hastalığının bitmesi iki koşulla
tanımlanmıştır. Bunlardan birincisi yaş ile
ilişkili epilepsi sendromlarında o sendrom için belirlenen yaş
sınırının geçilmesi ve ikinci koşulda hastanın en
az 10 yıldır nöbet geçirmediği ve 5 yıldır da antiepileptik
ilaç (AEİ) kullanmadığı durumlar olarak
belirtilmiştir.
Tablo 1. Epilepsi nöbetlerinin tekrarlama riskinin yüksek
olduğu bilinen bulgular (Kaynak 7 ve
15’ten esinlenerek).
|
Nörogörüntüleme bulguları
·
Hipokampal
skleroz
·
Kortikal
gelişim anomalileri
·
Fokal
kortikal displazi
·
Tuberoskleroz
·
Lizensefali
·
Heterotopi
·
Polimikrogiri
·
Hemimegalensefali
·
Şizensefali
·
Hipotalamik
hamartom
·
Tümörler
·
Düşük
“grade”li glial tümörler
·
Disembriyoblastik
nöroepitelyal tümörler (DNET)
·
Gangliogliomlar
·
Kortikal
yerleşimli serebrovasküler hastalıklar
·
Kavernom ve
arteriovenöz malformasyonlar*
·
Nörosistiserkozis
·
Porensefalik
kist
·
Travmatik
beyin hasarı
|
|
EEG bulguları
·
Epileptiform
deşarjlar
(jeneralize veya fokal diken ve keskin-yavaş dalga kompleksleri)
·
Temporal
aralıklı ritmik delta aktivitesi (TIRDA) (Temporal lob epilepsisi
varlığında)
|
*Venöz anomaliler, epileptojenik
lezyon olarak kabul edilmezler, epilepsi hastaları ile birliktelikleri
tesadüfi olarak kabul edilmektedir.
Epilepsi
Nöbetlerinin Sınıflandırılması
ILAE
tarafından 1981 yılında yapılan epilepsi nöbetlerinin
sınıflandırması son yıllara kadar
kullanılmaktaydı. Ancak bu sınıflamadaki güncel bazı
boşluklar ve aksaklıklar, epileptologları yeni bir nöbet
sınıflandırmasına yöneltti ve 2017 yılında yeni
bir epilepsi nöbetleri sınıflandırılması
yayınlandı. Bu sınıflamada, nöbetler öncelikle
başlangıçlarına göre gruplandırıldı: (Şekil
1)
1) Fokal başlangıçlı nöbetler
2) Jeneralize başlangıçlı nöbetler
3) Başlangıcı bilinmeyen nöbetler

Şekil 1. ILAE 2017 epilepsi nöbet sınıflaması. (Kaynak 8’den
Türkçeleştirilerek alınmıştır.)
Nöbetlerin
başlangıcına göre gruplanırken, fokal veya jeneralize
olduklarından en az %80 oranında emin olunması gerektiği
belirtilmektedir. Daha az oranda eminsek o zaman nöbetlerin,
başlangıcı belirlenemeyen olarak
sınıflandırılması önerilmektedir.
FOKAL BAŞLANGIÇLI NÖBETLER
Fokal başlangıçlı nöbetler, tek bir hemisferde, sınırlı veya daha
geniş bir bölgeden kaynaklanırlar. Fokal başlangıçlı
nöbetler, üç aşamada değerlendirilirler. İlk aşama
farkındalığın değerlendirilmesidir. Bu aşamada
nöbetler ikiye ayrılır: farkındalığın
korunduğu ve farkındalığın bozulduğu nöbetler.
Eğer nöbetin herhangi bir zamanında farkındalık bozulduysa
bu nöbet ‘fokal başlangıçlı farkındalığın
bozulduğu nöbet’ olarak sınıflandırılmalıdır.
Farkındalık durumu hasta ve hasta yakınlarından alınan
anamneze göre veya daha güvenilir şekilde nöbet sırasında
hastanın doktor veya sağlık personeli tarafından muayene
edilmesi ile belirlenir. Ancak bazı durumlarda hastanın
farkındalık durumu belirlenemeyebilir, o zaman nöbet ‘fokal ancak
farkındalık durumu belirlenemeyen’ olarak
sınıflandırılmalıdır. Ayrıca bir diğer
zorlayıcı durum, hastanın
farkındalığının bozulmasına rağmen nöbet
sonrasında bunu hatırlayamamasıdır. Böyle bir durumda hasta
yakınlarından bilgi alınmaya çalışılır ve
yine emin olunamazsa farkındalığı belirlenemeyen olarak
sınıflandırılmalıdır.
Fokal
başlangıçlı nöbetlerde ikinci aşama nöbet
sırasında izlenen ana klinik belirti ve bulguların incelenmesini
içerir ve iki ana grupta değerlendirilir. Birincisi motor semptomlar ve
ikincisi motor olmayan (nonmotor) semptomlardır.
Fokal
başlangıçlı nöbetler motor semptomlara göre
aşağıdaki gibi
sınıflandırılmıştır:
1) Otomatizmalar (amaçlı veya amaçsız
tekrarlayan hareketler)
2) Atonik (kas tonusunun kaybolduğu)
3) Tonik (devamlı kas kasılması,
katılığı)
4) Klonik (fokal ritmik sıçramalar)
5) Miyoklonik (düzensiz, ani fokal sıçramalar)
6) Epileptik spazm (gövdenin ekstansiyon ve kolların
fokal fleksiyon veya ekstansiyon postürü alması)
7) Hiperkinetik (pedal çevirme, ayağa kalkma,
koşma vb.)
Fokal
başlangıçlı nöbetler nonmotor semptomlara göre
aşağıdaki gibi
sınıflandırılmıştır. Bu semptomların
ayrıntılı alt grupları Tablo
2’de özetlenmiştir.
1) Otonom bulgular
2) Duraklama
3) Kognitif
4) Emosyonel
5) Duysal
Tablo 2. Nöbet sırasında ve sonrasında izlenen
davranışsal belirteçler (Kaynak 8’den
Türkçeleştirilerek alınmıştır)
|
Kognitif
·
Akalküli
·
Afazi
·
Dikkat
bozulması
·
Deja vu ve
jamais vu
·
Disosiasyon
·
Disfazi
·
Halüsinasyon
·
İlüzyon
·
Bellek
kaybı
·
İhmal
·
Zorlu
düşünce
·
Cevaplılık
halinin bozulması
|
|
Emosyonel
·
Ajitasyon
·
Korku
·
Öfke
·
Anksiyete
·
Ağlama
(dakristik)
·
Gülme
(jelastik)
·
Paranoya
·
Mutluluk
|
|
Otonom
·
Asistoli
·
Bradikardi
·
Ereksiyon
·
Yüzde
kızarma (flushing)
·
Gastrointestinal
·
Hiper/hipoventilasyon
·
Bulantı
veya kusma
·
Solukluk
·
Çarpıntı
·
Piloereksiyon
·
Solunum
değişiklikleri
·
Taşikardi
|
|
Otomatizmalar
·
Agresyon
·
Göz
kırpma
·
Baş
sallama
·
El
otomatizması (aranma, çekiştirme)
·
Oro-fasyal
·
Pedal
çevirme
·
Perseverasyon
·
Seksüel
·
Soyunma
·
Vokalizasyon/konuşma
·
Yürüme
|
|
Motor
·
Dizartri
·
Distoni
·
Eskrimci
pozisyonu
·
Figür 4
pozisyonu
·
Jacksonyen
yayılım
·
Paralizi
·
Versiyon
|
|
Duysal
·
İşitsel
·
Görsel
·
Vestibüler
·
Gustatuar
·
Sıcak-soğuk
hissi
·
Olfaktör
·
Somatosensoriyel
|
Epileptik
spazm ve atonik nöbetlerde farkındalık değerlendirilmesi
yapılmaz. Motor semptomlardan birkaç tanesi aynı nöbet içerisinde
bulunabilir, o zaman klinik tabloya en hakim olan bulgunun adı
kullanılarak sınıflandırma yapılmalıdır.
Eğer nöbet içerisinde motor ve nonmotor semptomlar sırası ile
izleniyorsa o zaman ilk izlenen hakim semptoma göre
sınıflandırılma yapılmalıdır. Örneğin
bir hastanın nöbeti bulantı ve yüzde kızarma ile
başlayıp ardından farkındalıkta etkilenme ile oral ve
ellerde otomatizma ile devam ettiyse bu nöbet ‘fokal
farkındalığın bozulduğu otonom nöbet’ olarak
sınıflandırılmalıdır. Fokal nöbetler,
sınıflandırılmanın birinci aşamasında
bulunan farkındalık durumu belirtilmeden motor veya nonmotor
semptomlu olarak da sınıflandırılabilirler. Motor ve
nonmotor semptoma göre sınıflandırdıktan sonra nöbetin geri
kalan kısmında izlenen bulgular serbest yazı şeklinde
eklenebilir. Örneğin yukarıda tarif edilen nöbet için, ‘fokal
farkındalığın bozulduğu otonom ve ardından oral
ve ellerde otomatizma ile giden nöbet’ şeklinde belirtilebilir.
Fokal
başlangıçlı nöbetlerin
sınıflandırılmasında üçüncü aşama nöbetin
yayılımı ile ilişkilidir ve tek bir özel durumu belirtmek
için sınıflamaya konulmuştur. Bu durum, fokal
başlangıçlı olup bilateral tonik-klonik nöbete dönüşen
nöbetler olarak sınıflandırılır. Bilateral kelimesinin,
her iki hemisfere yayılan nöbetlerde kullanılması
önerilmiştir, simetrik veya asimetrik olabilir.
FOKAL EPİLEPSİ
NÖBETLERİNİN SEMİYOLOJİK ÖZELLİKLERİ:
LATERALİZASYON VE LOKALİZASYON DEĞERLERİ
Beyin
fonksiyonlarının dinamik yapısı göz önüne
alındığında beynin anatomik olarak loblara bölünmesi
yetersiz kalmaktadır. Karmaşık entegrasyonları olan nöral
yollar yapay anatomik sınırlarla örtüşmediğinden epileptik
nöbetlerin klinik profillerinin belirlenmesinde anatomik bölümlemenin yeterli
bir ölçüt olamayacağı ve klinik belirtilerin çok değişken
olabilen yayılım şekilleri sonucunda ortaya
çıktığı unutulmamalıdır. Bu nedenlerle korteksin
çeşitli bölgelerinden benzer özellik gösteren nöbetler ortaya
çıkabilmekte ya da birbirinden farklı nöbetler aynı kortikal
odaktan kaynaklanabilmektedir. Bu karmaşık nöronal entegrasyon ve
yayılma paternleri konusunda bugünkü bilgilerimiz yetersiz kalmakta ve
derin elektrod çalışmaları ve ayrıntılı klinik
veriler ışık tutmaktadır. Aşağıda anatomik
lokalizasyonlara göre epileptik odakların lokalizasyonuna kısaca
değinilecektir.
Frontal Lob Kökenli Fokal Epilepsi Nöbetleri (Tablo 3)
Bugünkü
bilgilerimize göre beynin en gizemli ve büyük bölümü olan frontal loblardan
kaynaklanan bir nöbeti akla getiren başlıca semptomlar şunlardır:
1) Tonik ve postüral olabilen motor belirtiler
sıktır (%50-60). Özellikle farkındalık korunmuşken
tonik baş dönmesi frontal lob nöbeti için tipik kabul edilir. Bu
nöbetlerde zorlu baş dönmesi kontralateral frontal loba lateralizasyon
sağlar. Ancak başın dönmesinin lateralize edici değeri
diğer fokal motor tonik veya klonik nöbetlere göre belirgin şekilde
düşüktür.
2) Jaksonyen yayılım: Motor hareketlerin
komşu vücud kısımlarına ilerleyici şekilde
yayılması
3) Eskrimci postürü: Bir kol ekstansör postürde iken karşı
taraftaki kolda dirsek ve bilekten fleksör postürde tonik kasılma
(ekstansör kolun kontralateraline lateralizasyon sağlar)
4) Figür 4 pozisyonu: Bir kolda ekstansör tonik
kasılma ile karşı taraftaki kolda dirsekten fleksör tonik
kasılma ile “4” görünümü alma (ekstansör kolun kontralaterali epiletojenik
alan ile ilişkilendirilir)
5) Başlangıçta kompleks hareketler (örn, pedal
çevirme, koşma, tekme atma v.b.) şeklinde otomatizmalar sık
görülür.
6) Epileptik boşalım bilateral olduğunda
sıklıkla düşme eşlik eder.
7) Postiktal Todd paralizisi sık görülür.
8) Konuşmanın durması ve vokalizasyonlar
görülebilir.
9) Sıklıkla uyku sırasında ve bazen
küme halinde görülürler.
10)Kimi zaman nöbet tablosu histeri ile
karıştırılacak kadar atipik olabilmektedir.
Saçlı deriden kaydedilen yüzey EEG'sinin kimi
olgularda iktal dönemde bile negatif kalabildiği
unutulmamalıdır. Frontal
lobun "sessiz" bölgelerinden kaynaklanan nöbetler ancak komşu
yapılara veya diğer loblara yayılma gösterdikten sonra klinik
belirti vermektedirler.
Orbitofrontal
bölgeden kaynaklanan nöbetlerin unsinat fasikul veya girus singuli yoluyla
temporal loba yayılma göstermesi frontal nöbetlerin temporal nöbetlerle
karıştırılmasına yol açmaktadır. Yüzey EEG'si de
çoğu zaman bu ayırımı sağlamada yetersiz
kalmaktadır (Tablo 4).
Kallozal veya subkortikal yollarla karşı hemisfere hızlı
yayılım sonucu görülen sekonder bilateral senkroni nedeniyle bazen
idyopatik/genetik jeneralize epilepsilerle de
karıştırılabilmektedir.
İnsula, perioperküler bölgede derin
yerleşimli bir kortikal yapıdır. Frontal, temporal ve pariyetal
lob ile yoğun bağlantıları nedeniyle bu bölgelerden
kaynaklanan nöbetleri klinik olarak taklit edebilir. Bu bölgeden kaynaklanan
nöbetlerde semiyolojik olarak somatosensoryal aura, viserosensoryal veya
viserootonomik auralar, hipermotor nöbetler, işitsel veya görsel
halüsinasyonlar gibi geniş yelpazede klinik bulgu ve semptomlar
izlenebilir. İnsula bölgesi bu nedenle büyük taklitçi olarak kabul
edilmektedir.
Tablo 3. Frontal lobdan kaynaklanan nöbetlerin klinik özellikleri
|
Rolandik
bölge
(primer
motor alan)
|
Kontralateral fokal klonik
aktivite, somatotopik; Jacksonyen yayılım gösterebilir.
|
|
Dorsolateral
|
Zorlu düşünce, bilinçli
adversiyon, "psödoabsans" veya farkındalığın
kaybolduğu fokal nöbet, hızlı jeneralizasyon
|
|
SMA
(ek-süplemanter
motor alan)
|
Spesifik olmayan duysal aura,
bilinçli adversiyon ve tonik/distonik postür, eskrimci postürü, konuşma
durması, vokalizasyonlar
|
|
Frontopolar
|
Erken bilinç kaybı,
"psödoabsans", hızlı jeneralizasyon
|
|
Singulat
|
Korku, psödoabsans, erken el
hareketlerine ilişkin otomatizmalarla giden
farkındalığın kaybolduğu fokal nöbetler, jeneralize
tonik klonik nöbet
|
|
Orbitofrontal
|
Noktürnal kümeler halinde, ani
başlangıç, güçlü afekt (korku), tuhaf motor otomatizmalar (bimanuel,
bipedal), vokalizasyonlar (küfür, çığlık)
|
Temporal Lob Kökenli Fokal Epilepsi Nöbetleri
Fokal
nöbetlerin %50'den fazlasını oluşturan temporal lob nöbetleri
zengin ve ilginç semptomatolojisi nedeniyle üzerinde en çok araştırma
yapılan fokal epilepsi türünü oluşturur. Özgeçmişte febril nöbet
öyküsü ve pozitif aile öyküsüne sık rastlanır. Nöbetler genellikle
belli aralıklarla tekrarlamaktadır.
Tablo 4. Frontal ve temporal lob kökenli fokal nöbetlerin
ayrımı
|
|
Temporal
|
Frontal
|
|
Aura
|
sık, değişken, ama
tipiktir, başlangıç bölgesi hakkında ipucu verebilir
|
spesifik olmayan, müphem
sefalik duyumsamalar, zorlu düşünce
|
|
Süre
|
1-2 dakika
|
10-60 saniye
|
|
Sıklık
|
haftada/ayda bir çok kez
|
günde bir çok kez,
çoğunlukla kümeler halinde
|
|
Başlangıç
|
donarak durma, veya erken
oroalimentar otomatizmalar
|
vokalizasyon, korkulu yüz
görünümü
|
|
Otomatizma
|
basit, oroalimantar,
giysilerini çekiştirme şeklinde manuel
|
tuhaf, yarı-amaçlı,
kompleks, bimanuel, bipedal, seksüel
|
|
Vokalizasyon
|
basit, konuşma olabilir
|
tuhaf (çığlık, küfür)
|
|
Jeneralizasyon
|
nadir
|
Sık
|
|
Postiktal
|
konfüzyon, letarji, afazi, 30
dakika kadar sürebilir
|
minimal veya yok
|
Temporal
lob epilepsisini akla getiren başlıca nöbet semptomları
şunlardır: (Tablo 5)
1) Otonom ve/veya kognitif semptomlar ve bazı özel duysal
fenomenler, örneğin koku ve işitsel illüzyonlarla giden fokal
nonmotor nöbetler sık görülür.
2) Hemen her duysal modalitede basit veya kompleks
illüzyon ve halüsinasyon görülebilir. En sık olarak (%20) görsel illüzyon
ve halüsinasyonlara rastlanır. Oksipital bölgeden kaynaklananlarla
kıyaslanınca daha karmaşık yapıda oldukları
saptanmıştır.
3) Sıklıkla motor duraklama ile
başlayıp tipik oroalimanter otomatizmalarla devam eden ve
sıklıkla diğer otomatizmaların eklendiği fokal motor
otomatizmalı nöbet görülür.
4) Otonom bulgular viseral duyumsama şeklinde
görülebilir. En sık olan yükselen epigastrik duyumsamadır.
5) Kognitif bulgular ("déja vu", "jamais
vu" vb.) ve afektif semptomlar çok değişken formlarda
görülebilmekle birlikte her hasta için stabil olan bir nöbet paterni genellikle
vardır.
6) Konuşmanın durması, dizartri ve afazi
gibi konuşma bozuklukları görülebilir.
Temporal
lob epilepsisinin diğer bir önemli boyutu da bildirilmiş olan
interiktal kişilik değişiklikleridir.
Tablo 5. Lateral ve mezyal temporal nöbet başlangıç
ayrımı için kullanılan klinik özellikler
|
Mezyal
temporal
|
· Korku
· "Déja vu" ve "jamais vu"
· Duygulanımlar
· Olfaktor halüsinasyonlar
· Epigastrik duyumsama
· Otonom değişiklikler*
|
|
Lateral
temporal
|
· Basit duysal halüsinasyonlar (işitsel, vestibüler
veya gustatuvar)
· Reseptif afazi
· Fokal sensorimotor fenomenler
|
*Operküler/insüler başlangıçlı nöbetlerde
de görülebilir.
Pariyetal Lob Kökenli Fokal Epilepsi Nöbetleri
Pariyetal
lob kökenli nöbetler esas olarak kontralateral hemisferdeki odaktan kaynaklanan
somatosensoriyel nöbetlerden oluşur. Ancak nadir de olsa bilateral ve
ipsilateral nöbetler görülebilmektedir. Pariyetal lob kaynaklı fokal
nöbetleri düşündüren bulgular şunlardır:
1) Elementer paresteziler en sık görülen
somatosensoriyel nöbet semptomlarıdır. Pozitif fenomenler olarak
karıncalanma, elektriklenme, keçeleşme ve iğnelenme gibi
duyumsamalar sınırlı veya Jacksonyen tarzda olabilir. Negatif
fenomenler, örneğin hissizlik görülebilir.
2) Ağrı iktal bir semptom olarak görülebilir.
3) Nadiren termal algı değişikliği
şeklinde nöbetler olabilir.
4) Parasantral tutulumda bazı seksüel nöbetler
görülebilir. Bunlar temporal kökenli nöbetlerin tersine genellikle hoş
olmayan, korkutucu veya ağrılı şekildedir ve genellikle
kadınlarda görülürler.
5) Tat duyumsaması da temporal lob kökenli
nöbetlerin yanı sıra pariyetal operküler bölgeden kaynaklanabilir.
6) Vücudun belli bir bölümünü hareket ettirememe hissi
şeklindeki nöbetlerin suprasilviyen bölgedeki ikincil duysal alan kökenli
oldukları düşünülmektedir.
7) Beden imajı bozukluğu veya asomatognozi
şeklindeki fenomenlerin genelde nondominant hemisferden
kaynaklandığı düşünülmektedir.
8) Vertigo hissi de genelde inferior pariyetal lobdan
kaynaklanır.
Oksipital
Lob Kökenli Fokal Epilepsi Nöbetleri
Genellikle
görsel; negatif (iktal körlük, skotom, hemianopsi) veya pozitif
(ışıklar, renkler v.b.) elementer veya kompleks olabilen
belirtilerle giden nöbetlerdir. Bu fenomenler tek yanlı olduğunda
odağın genellikle kontralateralinde rastlanırlar:
1) Elementer görsel halüsinasyonlar ve kompleks görsel
halüsinasyonlar çok çeşitli formlarda nöbet semptomu olarak
karşımıza çıkabilirler. Epileptik orijinli
olduklarında genellikle kısa sürelidirler ve hasta bunların
gerçek olmadığının bilincindedir.
2) İktal körlük görülebilir ve migrenle
ilişkisi tartışılmaktadır.
3) Görsel illüzyonları da basit ve kompleks formlara
ayırmak mümkündür. Çok farklı şekillerde olabilirler.
4) Epileptik nistagmusun pariyeto-oksipital korteksten
kaynaklandığı gösterilmiştir. Gözlerin karşı
tarafa dönmesi, göz kırpma ve gözlerde hareket hissi oksipital lob nöbet
semptomları olarak görülebilmektedir.
5) Diğer loblara yayılım, özellikle
inferior longitudinal fasikül yoluyla temporal loba yayılım ve sekonder
jeneralizasyon sıktır.
Epilepsi
nöbetlerinin lokalizasyonu ve lateralizasyonu
tartışıldığında deşarjların hızla
yayılabildiği ve semptomların primer odağı değil,
yayılım bölgesini gösterebileceği göz önünde
tutulmalıdır.
FOKAL NÖBETLERDE
SEMİYOLOJİK BULGULARIN LATERALİZASYON DEĞERLERİ
Ekstremiteleri
belirli bir pozisyona getiren unilateral tonik aktivite, nöbeti kontralateral
hemisfere lateralize eder. Unilateral klonik aktivite de benzer şekilde
kontralateral hemisferi işaret eder. Yukarıda da bahsedildiği
gibi zorlu baş ve göz deviyasyonu kontralateral hemisfer ve özellikle
frontal göz alanı ile ilişkilidir. Unilateral distoni, nöbetin
kontralateral hemsifer ile ilişkili olduğunu belirtir. Temporal lob
epilepsilerinde unilateral distoniye karşı ekstremitede
otomatizmaların eşlik etmesi bu lateralizasyon bulgusunu güçlendirir.
Temporal lob epilepsilerinde distoni olmadan unilateral ekstremitede otomatizma
varlığı daha düşük duyarlılıkta olmakla birlikte
ipsilateral hemisferi işaret eder. İktal konuşma nondominant
hemisferi gösterir. Benzer şekilde iktal tükürme, kusma ve periiktal
dönemde idrar yapma ihtiyacı daha düşük lateralizasyon değerinde
de olsa nondominant hemisfer ile ilişkilidir. Tek taraflı göz
kırpıştırma fokal nöbetlerde ipsilateral hemisferi
gösterir. Bilateral tonik klonik nöbetlerde nöbet biterken, asimetrik tek
taraflı kloniler ile sonlanıyorsa kloniler ile ipsilateral hemisfere
işaret eder. İktal bulguların varlığının
yanında postiktal bulguların da lateralizasyon değeri
bulunmaktadır. Temporal lob nöbetlerinde postiktal tek elle burun silme
ipsilateral hemisferi gösterir. Postiktal afazi nondominant hemisfer ve Todd
paralizisi de kontralateral hemisferi işaret eder.
Nöbetlerde
izlenen semiyolojik bulguların lateralizasyon ve lokalizasyon
özelliklerini değerlendirirken nöbetin beynin bir bölgesinden
başlayıp diğer bölgelere yayıldığı
unutulmamalıdır. Nöbetin başlangıcında saptanan
semiyolojik bulguların, özellikle auraların, nöbetin beyin içerisinde
kaynaklandığı alanı (epileptojenik alan) belirlemeyi
sağlayacağı bilinmektedir. Nöbet
başlangıcının izlenemediği veya auranın
hatırlanamadığı durumlarda nöbetin geç dönem bulguları
nöbetin başladığı alandan çok yayıldığı
alan ile ilişkili olabilir. Bu durum hatalı lokalizasyon ve
lateralizasyona neden olabilir.
JENERALİZE BAŞLANGIÇLI NÖBETLER
Jeneralize başlangıçlı nöbetler, her iki hemisfer içerisindeki yaygın
ağlardan kaynaklanır. Burada farkındalık ile ilgili
değerlendirme sınıflama için kullanılmaz, bu gruptaki
nöbetlerin büyük çoğunluğunda farkındalık bozulur.
Jeneralize nöbetlerde, motor bulgular iki yanlı olarak başlar.
Jeneralize başlangıçlı nöbetler motor semptomların
varlığına göre motor ve nonmotor semptomlu (absans) olarak iki
grupta sınıflandırılmıştır. Bu
sınıflandırma Şekil
1’de görülmektedir.
Tonik-klonik
nöbetler devamlı kas kasılması ve bu kasılmayı takip
eden kloniler ile karakterize nöbetlerdir. Tonik nöbetlerde ise klinik olarak,
devamlı kas kasılması ve katılığı izlenir.
Klonik nöbetlerde ritmik sıçramalar tüm vücutta görülür. Miyoklonik
nöbetlerde, ritmik özellik göstermeyen ani sıçramalar izlenir.
Miyoklonik-tonik-klonik nöbetler, miyoklonik sıçramalar ile başlar ve
sonrasında tonik-klonik nöbete dönüşürler. Bu nöbetler, juvenil
miyoklonik epilepsili hastalarda izlenir.
Miyokolonik-atonik nöbetler, miyoklonik sıçramayı takiben kas
tonusunun azalması ile düşme atakları şeklinde görülürler.
Lennox-Gastaut sendromu gibi çocukluk çağında izlenen gelişimsel
ve epileptik ensefalopatilerde görülürler. Atonik nöbetler, kas tonusunun
kaybolması ile karakterizedir. Bu nöbetler birkaç saniye sürer ve
tonik-klonik nöbetlerden farkı bilincin kas tonusu kaybının
sonlanması ile saniyeler içerisinde yerine gelmesidir.
Tipik
absans nöbetlerinde bilinç kaybı ani başlar ve ani sonlanır. Hafif
motor otomatizmalar eşlik edebilir. Atipik absans nöbetlerinde ise, bilinç
kaybı tam olmayabilir; başlangıç ve bitişi genelde
sinsidir. Kas tonusu değişiklikleri daha sık görülür. Tipik ve
atipik absans ayırımı için EEG bulguları belirleyicidir (Tablo 6). Miyoklonik absans nöbetleri,
10-60 saniye süreli, miyoklonik sıçramaların eşlik ettiği
absans nöbetleridir. EEG’de 3 Hz jeneralize diken-dalga aktivitesi izlenir. Göz
kapağı miyoklonili absans nöbetlerinde, absans nöbetlerine göz
kapağında miyoklonik sıçramalar ve göz küresinin yukarı
deviyasyonu eşlik eder. Bu nöbetler sıklıkla göz kapama veya
ışık uyaranı ile tetiklenirler. Jeavons sendromu olarak da
bilinen göz kapağı miyoklonili absans epilepsi sendromunda görülürler
(Bakınız: Epilepsi sendromları).
Tablo 6. Tipik absans ve atipik absans nöbetlerin özellikleri
(Kaynak
17’den değiştirilerek hazırlanmıştır)
|
|
Tipik absans
|
Atipik absans
|
|
Başlangıç ve
bitiş
|
Ani başlayıp ani sonlanır
|
Başlangıç ve bitişi sinsidir
|
|
Kas tonusu
değişiklikleri
|
Nadir
|
Daha sık
|
|
Bilinç kaybı
|
Tam
|
Kısmi
|
|
Süre
|
Birkaç saniye-30 saniye
|
Saatler ve günler sürebilir
|
|
Mental gerilik
|
Beklenmez
|
Sık
|
|
Eşlik eden EEG bulgusu
|
3 Hz diken-dalga
|
<2,5 Hz diken dalga
|
|
EEG’de temel aktivite
|
Normal
|
Bozuk
|
|
Diğer nöbet tipleri ile
birliktelik
|
Nadir
|
Sık
|
Başlangıcı bilinmeyen nöbetler, nöbetin başlangıcına kimsenin
şahit olmadığı veya şahit olsa bile bilginin yetersiz
olduğu durumlarda kullanılır. Motor semptomlu, nonmotor
semptomlu (duraklama) veya sınıflandırılamayan olarak
gruplanırlar.
Başlangıcı
bilinmeyen nöbetler motor semptomlara göre aşağıdaki gibi
sınıflandırılmışlardır:
1) Tonik-klonik
2) Epileptik spazmlar
Eğer
epileptik bir nöbet olduğu net ancak nöbet bilgi yetersizliği
nedeniyle herhangi bir sınıflamaya konamıyorsa, o zaman
sınıflandırılamayan nöbet grubunda incelenmelidir. Ancak
geçirilen atağın epilepsi nöbeti olduğundan şüphe
ediliyorsa, sınıflandırılamayan nöbet grubuna
konulmamalıdır.
DURUMA BAĞLI NÖBETLER
Febril Nöbet
Febril
nöbet, çocukluk çağında en sık izlenen nöbettir. Toplum temelli
çalışmalar çocukların %2-5’ini etkilediğini bildirmektedir.
Febril nöbetler 6 ay ile 5 yaş arasında görülür. Ateş, nöbet
öncesinde bulunmayabilir ancak nöbet sonrası erken dönemde mutlaka
olmalıdır. Basit ve komplike olmak üzere ikiye ayrılır (Tablo
7). Basit febril nöbet
denebilmesi için nöbetin süresinin 15 dakikadan kısa olması,
bilateral tonik-klonik nöbet şeklinde başlaması, ilk 24 saat
içerisinde tekrarlamaması, nörolojik muayenenin normal olması ve
postiktal Todd parezisinin olmaması gereklidir. Komplike febril nöbetlerde
ise bu özelliklerin en az biri bulunur.
Tablo 7. Basit ve komplike febril nöbet özellikleri (Kaynak 4’ten
esinlenerek hazırlanmıştır)
|
|
Basit febril nöbet
|
Komplike febril nöbet
|
|
Süresi
|
15 dakikadan kısa
|
15 dakikadan uzun olabilir
|
|
Nöbet tipi
|
Jeneralize
|
Fokal veya jeneralize
|
|
24 saat içerisinde tekrarlama
|
Hayır
|
Olabilir
|
|
Fokal nörolojik defisit
|
Yok
|
Olabilir
|
|
Postiktal Todd parezisi
|
Yok
|
Olabilir
|
İlk
kez febril nöbet geçiren hastalar, 18 ay üzerinde ve basit febril nöbet ise ek
inceleme yapılmadan izlenebilir. Ancak 18 ayın altındaki
bebeklerde, ilk febril nöbette basit febril nöbet özellikleri de olsa lomber
ponksiyon incelemesi önerilmektedir. Ayrıca antibiyotik tedavisi
altında ilk kez febril nöbet geçiren bebeklere de nörolojik muayene
bulguları normal olsa da lomber ponksiyon önerilmektedir. Basit febril
nöbet tanısı konulan hastalara EEG ve nörogörüntüleme
yapılması gerekli değildir. İlk kez kompleks febril nöbet
geçiren hastalar etyolojinin araştırılması amacıyla
hastaneye yatırılmalı, laboratuvar incelemeleri, EEG,
nörogörüntüleme ve lomber ponksiyon ile incelenmelidir.
İlk
febril nöbet sonrası nöbetin tekrarlama riski %30-40 oranında
değişmektedir. On beş ay altında başlangıç,
ailede epilepsi veya febril nöbet öyküsü, sık ateşli hastalık
öyküsü ve febril nöbet başlangıcında düşük ateş
olması febril nöbetin tekrarlama riskini arttırmaktadır. Febril
nöbet geçiren çocuklarda epilepsi gelişme riski %1-1,5 olarak
saptanmıştır, bu oran normal popülasyonda yaklaşık
%0,5’tir. Febril nöbet sonrası epilepsi gelişme riskini arttıran
faktörler febril nöbet öncesi gelişim geriliği veya anormal nörolojik
durum, ailede ateşsiz nöbet varlığı ve komplike febril nöbet
varlığıdır. Komplike febril nöbet geçiren çocuklarda ise
ileri dönemde epilepsi gelişme riski %4-15 oranlarında
bildirilmektedir. Komplike febril nöbet üç komplike özellik taşıyorsa
(uzun, tekrarlayan ve fokal özellikli) o zaman bu riski %49’a çıkmaktadır.
Febril nöbetlerin antiepileptik ilaç (AEİ) ile tedavi edilmesinin epilepsi
gelişim riskini azalttığına yönelik bir kanıt
bulunmamaktadır. Basit febril nöbetlerin devamlı AEİ ile
tedavisi önerilmemektedir. Rektal veya oral diazepam ile profilaksi yapılabilir.
Kompleks febril nöbetlerde ise altta yatan akut bir nörolojik sendrom veya bir
epileptik ensefalopati (örn, Dravet sendromu) araştırılır.
Eğer sadece uzamış bir febril nöbet ise basit febril nöbet gibi
tedavi edilir.
Akut
Semptomatik Nöbet
Akut
semptomatik nöbetler, sistemik bir olayla (metabolik veya toksik) veya akut
beyin hastalığı ile yakın zamansal ilişki içerisinde
meydana gelmiş olan nöbetlerdir. Akut semptomatik nöbetlerin tekrarlama
olasılıkları epilepsi hastalığında olduğu
gibi artmamıştır. Ancak akut semptomatik nöbet sonrasında,
beyin ile ilişkili hastalık yatıştıktan sonra bu
hastalarda epilepsi hastalığı gelişme riski
artmıştır. Ayrıca, epilepsi hastalarında, akut
semptomatik nöbet de gelişebilir.
Akut
semptomatik nöbete neden olan akut beyin hastalığı toksik,
metabolik, enfeksiyöz, vasküler, inflamatuar veya yapısal bir etyolojiye
bağlı olarak oluşabilir. Akut beyin hastalığı ve
nöbet arası süre, hastalığa bağlı olarak
değişkenlik gösterebilir. Travmatik beyin hasarı, iskemik
serebrovasküler hastalık, hipoksik iskemik ensefalopati, intrakranial
cerrahi ve merkezi sinir sistemi (MSS) enfeksiyonları sonrasında ilk
7 gün içerisinde gelişen nöbetler akut semptomatik nöbet olarak
adlandırılır. Travma sonrası veya arteriovenöz
malformasyona sekonder parankim veya subdural hematomu olan hastalarda ve
bazı MSS enfeksiyonlarında bu süre uzayabilir.
Akut
semptomatik nöbete neden olan metabolik ve toksik nedenler arasında
hipo/hiperglisemi (özellikle ketoasidoz ile ilişkili), hipokalsemi,
hiponatremi, hipomagnezemi, kreatinin
yüksekliği, alkol çekilme sendromu, madde intoksikasyonu (kokain, MSS
stimülanları) sayılabilir.
EPİLEPSİ NÖBETLERİNDE TEMEL
MEKANİZMALAR
Hayvan
deneylerinde ve insanda yapılan çalışmalarda kortikal
nöronların membran potansiyellerinde ve ateşlenme şekillerinde
bazı karakteristik bozukluklar saptanmıştır. “Paroksizmal
depolarizasyon kayması (PDK)” olarak bilinen bu durumda membranı
depolarize eden postsinaptik potansiyelin anormal şekilde uzaması ve
büyümesi söz konusudur. Bunun sonucu olarak nöronlar gruplar halinde
ateşlenebilir ve etraflarındaki nöronları benzer şekilde
ateşleyebilecek bir kapasiteye ulaşırlar. PDK’nın eksitatör
nörotransmitterler olan glutamat ve aspartat ile inhibitör nörotransmitter gama
aminobitütik asit (GABA) sistemleri arasındaki
dengesizlikten kaynaklandığı ileri sürülmektedir. Bunun
dışında membranlardaki iyon kanallarındaki
bozuklukların da PDK’nın ortaya çıkmasında etkili
olduğu düşünülmektedir.
Epileptojenik
odak olarak adlandırılan bu bölgede "pacemaker" hücreler
yer almaktadır ve bu hücreler tam olarak bilinmeyen nedenlerle,
artmış uyarılma ve anormal ateşlenme özelliği
gösterirler, etraflarındaki hücreleri de bu ateşlenmeye ortak
edebilecek güçleri vardır. Sonradan katılan bu
nöronların miktarı; tablonun EEG'de bir interiktal (nöbet arası
dönem) dikenle sınırlı kalmasını ya da yeterli miktara
ulaşabildiğinde EEG'de ve klinikte nöbet aktivitesinin
oluşmasını belirler. Epileptik bir nöbet sırasında
beyindeki nöronların hipersenkron ve tekrarlayıcı aktivasyonu
söz konusudur. EEG'de görülen diken artmış eksitasyonu,
dikeni izleyen yavaş dalga ise inhibisyonu göstermektedir. Özetle, fokal
kortikal bir nöbet aktivitesinin oluşabilmesi için ilgili nöronlarda 2 temel
fizyopatolojik özellik (1- hipereksitabilite, 2- senkronizasyon) birlikte
bulunmalıdır. Nöbet aktivitesinin yayılması ise eksitasyon
alanını çevreleyen inhibitör nöronların inaktivasyonu (çevresel
inhibisyon alanının kaybı) ile gerçekleşmektedir.
Bugün
için eskiden kabul gören santrensefalik epilepsi, yani jeneralize ve santral
yerleşimli bir epilepsi jeneratörü olduğu kavramı
tartışmalıdır ve yerini korteksin ön planda rol
aldığı kortikoretiküler teoriye bırakmıştır.
Bazı nöbet tipleri için (örn, tipik absans nöbetleri) talamusta yer alan
T-tipi Ca kanallarının rolü kanıtlanmış olsa da, bütün
epilepsi nöbetlerinin kortikal mekanizmalarla tetiklendiği görüşü
giderek ağırlık kazanmaktadır.
Jeneralize epilepsilerde
beyinsapı retiküler formasyonundan, orta hat talamus nukleusları
üzerinden taşınan yaygın bir girdinin aşırı
uyarılmış durumdaki kortekse etkisi üzerinde durulmakta ve
bazı asendan biojenik aminlerin rolleri vurgulanmaktadır. Bazı
araştırmacılar ise tetikleyici bölgenin büyük
olasılıkla kortikal olduğunu ve anterograd veya retrograd yolla
senkron aktivitenin talamusa yayıldığını
savunmaktadırlar.
Nöbete
eşlik eden anormal deşarjların fizyolojisi konusunda bilgimiz
olmasına karşın epileptogenezden sorumlu hücresel mekanizmalar
halen bilinmemektedir. İstirahat membran potansiyelinin instabilitesine
neden olan primer bir nöronal membran defekti üzerinde durulmaktadır. Buna
neden olduğu düşünülen mekanizmalar; potasyum iletiminde bozukluk,
voltaja duyarlı kalsiyum kanallarında defekt veya ATPaza bağlı
iyon transportunda bozukluk olarak özetlenmektedir. GABAerjik inhibitör
sistemlerin primer defekti olasılığı veya eksitatör
nörotransmisyonda rol alan reseptörlerin duyarlılığı ve
düzenlenmesindeki olası defektler üzerinde de durulmaktadır.
Eksitatör nörotransmisyonla yakından ilişkili olan “mossy fiber”
sistemindeki morfolojik değişiklikler gösterilmiştir.
Epileptik
nöbetlerde paroksizmal deşarjlarla ilgili olarak bölgesel beyin kan
akımının arttığı uzun zamandan beri
bilinmektedir. Nöbet sırasında ATP azalırken AMP, ADP, laktik
asid gibi maddeler çoğalmaktadır. Yine hücre içi kalsiyumun
artmasıyla aktive olan fosfolipazlar serbest yağ asidlerinin
artmasına yol açmakta ve prostaglandinler de artış
göstermektedir. ADP ve prostaglandinlerin vazodilatasyondaki rolleri göz önüne
alınırsa, iktal dönemdeki bölgesel beyin kan akımının
artışını açıklamak kolaylaşmaktadır.
EPİLEPTOGENEZ
Epileptogenez,
tekrarlayıcı spontan nöbetlerin oluştuğu uzun süreli beyin
transformasyonudur. Normal bir beynin zaman içinde bir dizi
hücresel-moleküler, yapısal ve/veya fonksiyonel değişikliklere
maruz kalarak epileptik bir beyin haline dönüşmesi, kalıcı bir
şekilde ve spontan olarak nöbet oluşturabilme özelliği
kazanması sürecini ifade eder. Beynin fokal bir bölgesini (fokal epilepsi)
veya tüm beyni (jeneralize epilepsi) içerebilir. Epileptogenez mekanizması
ilerleyici bir süreçtir, başlangıç hasarını takiben sessiz
bir dönem oluşur. Takiben belli bir süre sonra spontan nöbetler ortaya
çıkar. Bu dönemlerde yaş, cins, genetik faktörlerin etkisiyle hücre
ölümü, aksonlarda filizlenme, sinaptik reorganizasyon, farklı tipteki
lokal reseptörlerin özelliklerinde değişiklikler meydana gelir. Tüm
bu süreç günler-aylar veya yıllar içinde gelişir (Şekil 2).
Geçmişte epileptogenezin kronik bir süreç olduğu
düşünülmekteydi. Son yıllarda yapılan çalışmalar akut
epileptogenezin de varlığını ortaya koymuştur. Akut
epileptogenez dakikalar veya saatler içinde gelişir ve geri dönüşlü olabilir.
Epileptogenez değişik mekanizmalarla oluşabilir. Genel olarak
bunlar genetik ve edinsel mekanizmalardır.
Genetik
mekanizmalar: Bugün için,
idyopatik/genetik epilepsi sendromlarıyla ilişkili olduğu
gösterilmiş çok sayıda iyon kanal alt ünitesi geni mutasyonu
vardır. Bu hastalıkların ortak özelliği voltaj veya ligand
kapılı kanal genlerindeki mutasyonlara bağlı
olmasıdır. Dolayısıyla epileptogenez sürecinin kanal
patolojilerine bağlı olduğu düşünülmektedir. Farklı
dokularda, kanal ekspresyonundan sorumlu spesifik genler vardır ve
inhibitör ve eksitatör ağlar kompleks bir iletişime yol açar. Aksonal
ileti voltaj kapılı kanallarla (aksiyon potansiyeli), sinyal iletimi
ise ligand kapılı kanallarla sağlanır (sinaptik
transmisyon). Ancak son yıllarda iyon kanallarından bağımsız
bazı genlerin de epilepsiye neden olabildiği, yani noniyonik
mekanizmaların varlığı genetik çalışmalarla kesin
olarak kanıtlanmıştır.
Edinsel
epileptogenez mekanizmaları: Semptomatik epilepsilerde de çevresel faktörlerin
yarattığı hücresel düzeydeki hasarın epilepsiye yol
açabilmesinin; kişinin genetik özelliklerine bağlı olduğu
düşünülmektedir. Fokal nöbetlerin çoğunluğu temporal lob
kaynaklıdır ve bu grupta en sık rastlanan nöropatolojik bulgu
hipokampal sklerozdur. Hipokampal skleroz süreci sinaptik ve aksonal
reorganizasyonda değişimler gösterir. Ancak gelişim
mekanizması tam olarak bilinmemektedir; çevresel ve genetik faktörlerin
etkisi olmakla birlikte net olarak
aydınlatılamamıştır.
Akut epileptogenez hayvan
deneyleriyle ortaya konmuştur. Bu amaçla uygulanan bazı stimuluslar,
kullanılan konvülzan ilaçlar ve iyon konsantrasyonları hipokampus,
amigdala ve neokortekste epileptiform boşalımlara yol açar.
Mekanizmanın N-metil D-aspartat (NMDA) reseptör aktivasyonu ve (α
Amino-3-hidroksi-5-metil-4-izoksazol-proprionik asit (AMPA)-NMDA sinaptik transmisyonundaki
artış sonucu kalsiyum akışı, GABA’erjik sinaptik
inhibitör transmisyonda azalma sonucunda eksitatör etkide artışla
ilişkili olduğu düşünülmektedir. Ayrıca nonsinaptik olarak
“gap-junction coupling”i, demir aracılı Ca osilasyonunda veya glutamat
salınımında değişimler, serbest oksijen radikallerinin
yayılımı diğer mekanizmalardır. Sonuçta, ilaca
yanıtlı epilepsi veya ilaca dirençli epilepsi gelişebilir ya da
epilepsi gelişmeyebilir.

Şekil 2: Epileptogenezin basitleştirilmiş
gelişim
şeması
EPİLEPSİ
HASTALIĞI
Epilepsi
sözcüğünün eski Yunan dilindeki "epilepsia"dan türediği ve
nöbet anlamına geldiği bilinmektedir. İnsanlık tarihi kadar
eski olan ve Hipokrat zamanından beri bilinen bu hastalığın
sınıflandırılması antik çağlardan beri
uğraşılan konulardan biridir. İ.Ö.175'de Galen, beyinden
kaynaklanan idyopatik nöbetlerden ve vücudun herhangi bir bölgesinden
kaynaklanan semptomatik nöbetlerden söz etmiştir. 19. yy sonlarında
sendrom yaklaşımı olguların yaş, cins, nöbet
özellikleri ile sınırlıyken, 20. yy ikinci yarısından
sonra EEG ve görüntüleme olanaklarının artması, 21. yy’da ise
insan genom çalışmaları ve teknolojik gelişmelerin
ışığında daha detaylı hale gelmiştir.
Epidemiyoloji
Epilepsi
hastalığının insidansı toplumdan topluma
değişmekle birlikte Avrupa ve Kuzey Amerika verilerinde yılda 20-50/100.000
olarak bildirilmektedir. Aktif epilepsi prevalansı ise 4-10/1000 olarak
verilmektedir. Yaşam boyu birikmiş insidens ise yaklaşık %3
olarak saptanır ki bu farklılık epilepsinin bazı hastalarda
geçici bir doğası olmasından kaynaklanmaktadır. Toplum
içerisindeki epilepsi hastalarının belirli bir andaki
sıklığının belirlendiği prevalans
çalışmalarında sıklık 2,2-44/1000 olarak
bildirilmiştir. Epilepsi insidensinin en yüksek olduğu iki dönem,
yaşamın ilk yılı ve 60 yaş sonrasıdır.
Epilepsi çocukluk ve ergenlik çağında en sık, erişkinlerde
ise beyin damar hastalıklarının ardından ikinci en sık
rastlanan nörolojik hastalık olarak belirmektedir. Erkeklerde ve
düşük sosyoekonomik sınıfta insidens ve prevalans daha
yüksektir.
EPİLEPSİ
HASTALIĞI SINIFLAMASI
Modern
sınıflama çalışmaları ortak bir terminoloji
oluşturarak iletişimi kolaylaştırma, eldeki tüm verileri
ortak havuzlarda toplayarak karşılaştırma ve tedavi
seçiminde bu verileri en doğru şekliyle kullanabilme isteğinden
doğmuş ve epilepsi ile ilgilenenlerin öncelikli sorunlarından
biri haline gelmiştir. Epilepsinin farklı ve benzer özellikleri olan
birçok hastalık grubunu içermesi nedeniyle sınıflama bize
sistemli bir yaklaşım sağlar.
Sonuç
olarak epilepsilerin sınıflanması:
• Kavrama, eğitim ve öğretim
• İletişim: Meslektaşlar arasında
aynı-ortak dili kullanmak
• Patofizyolojik açıdan ortak ve ayrı yönleri
saptamak
• Etyolojik yaklaşıma katkı
• Ortak bilimsel çalışmalar için gereklilik
• Prognoz ve tedavi hakkında yol gösterme
• Epilepsi nöbetlerinin sebebini anlamada ilerleme
sağlama açılarından önemlidir.
Nöbet
geçiren bir hasta klinisyenin karşına geldiğinde, ilk
yapılması gereken geçirilen paroksizmal olayın epilepsi nöbeti
olduğuna karar vermektir (Bakınız: Ayırıcı
tanı). Geçirilen atağın epilepsi nöbeti olduğundan emin
olduktan sonra epilepsi nöbeti
sınıflandırılmalıdır. Epilepsi nöbetinin
sınıflandırılmasından sonra epilepsi
hastalığı tanısı değerlendirilir. Epilepsi
hastalığı tanısı konulduktan sonraki aşama ise
epilepsi hastalığının
sınıflandırılmasıdır.
Uluslararası
epilepsi uzmanlarının 1960'lı yıllardan başlayarak bir
araya gelmeleriyle epileptik nöbetlerin ve epilepsilerin
sınıflandırılmasının ilk temelleri
atılmıştır. ILAE ilk olarak 1970'te epileptik nöbetler ve
epilepsi sınıflamalarını oluşturmuştur; uzun
yıllar süren çalışmaları sonucunda 1981, 2010 ve 2014
yıllarında epilepsi nöbet sınıflaması güncellenerek
2017’de son haline getirilmiştir (Şekil 1). Epilepsi hastalarında klinik seyir, prognoz, etyoloji ve
dolayısıyla tedavi yaklaşımı çok farklı
özellikler gösterebilir, bu nedenle yalnızca nöbetlerin
sınıflandırılması yetersiz kalmaktadır. Aynı
zamanda yeni belirlenen ve iyi tanımlanmış epilepsi
sendromlarının sayıları da giderek artmaktadır. Bu
nedenle yıllar içinde çabalar epilepsileri ve epileptik sendromları
sınıflama yönünde yoğunlaşmıştır. 1985'deki
ilk sınıflamayı 1989 yılında yapılan yeni
sınıflama izlemiş ve son halini 2017 yılında
almıştır (Şekil 3).
Şekil 3: ILAE 2017 epilepsi hastalığının
sınıflama şeması (Kaynak 17’den Türkçeleştirilerek
alınmıştır)
Yeni
epilepsi hastalığı sınıflaması 5 aşamadan
oluşur:
1) Epilepsi nöbeti tipi (Bakınız: Epilepsi
nöbetlerinin sınıflandırılması)
2) Epilepsi tipi
3) Epilepsi sendromu
4) Etyolojinin değerlendirilmesi
5) Komorbiditelerin değerlendirilmesi
Epilepsi tipi: İkinci aşama, epilepsi tipinin
değerlendirilmesidir ve 4 gruba ayrılır:
a) Fokal epilepsiler
b) Jeneralize epilepsiler
c) Kombine fokal ve jeneralize epilepsiler
d) Bilinmeyen
Fokal
epilepsili hastalarda, bir hemisferden kaynaklanan fokal nöbetler izlenir.
Fokal nöbetten bilateral tonik-klonik nöbete dönüş de görülebilir. EEG
bulgularında interiktal fokal epileptiform anomaliler veya fokal
yavaşlama görülür. Ancak EEG normal olabilir. Nörolojik görüntülemelerinde
fokal yapısal lezyonların görülmesi destekleyicidir.
Jeneralize
epilepsili hastalarda, jeneralize tipte nöbetler (absans, miyoklonik, atonik,
tonik, tonik-klonik nöbetler) izlenir. EEG’de tipik jeneralize epileptiform
deşarjlar görülür. Aile öyküsü destekleyici olabilir. Normal EEG’si olan
tonik-klonik nöbet geçirmiş bir hastada jeneralize epilepsi
varlığından bahsetmek için miyokloni nöbeti veya aile öyküsü
varlığı gerekmektedir veya tekrarlayan EEG incelemelerinde tipik
deşarjların gösterilmesi şartı aranır.
Kombine
fokal ve jeneralize epilepsili hastalarda, hem fokal hem jeneralize tipte
nöbetler bulunur. EEG bulguları da jeneralize ve/veya fokal özellikler
gösterebilir. Bu grubun örnekleri Lennox-Gastaut sendromu ve Dravet
sendromudur.
Epilepsi
tipi belirlenememişse, hasta bilinmeyen grubunda
sınıflandırılır.
Epilepsi
sendromu:
Sınıflamada üçüncü aşama epilepsi sendromunun belirlenmesidir.
Epilepsi sendromu belirlenirken klinik, elektrofizyolojik, laboratuvar ve
radyolojik bulgular bir arada değerlendirilir. Bunlar içerisinde nöbet
tipleri, başlangıç yaşı, nöbetlerin sirkadyen
dağılımı, nöbetleri tetikleyici faktörler, aile öyküsü, EEG
bulguları, eşlik eden sistemik nörolojik muayene bulguları,
nöbetlerin seyri ve prognoz, görüntüleme bulguları bulunur. Epilepsi
sendromu sınıflaması Tablo
8’te özetlenmiştir.
Tablo 8. Elektroklinik sendromlar ve diğer epilepsiler (ILAE 2010 ve
2017 epilepsi hastalığı sınıflaması
rehberlerinden alınmıştır)
|
Başlangıç
yaşına göre elektroklinik sendromlar
1) Yenidoğan dönemi
a) Kendini sınırlayan (benign)
yenidoğan nöbetleri
b) Kendini sınırlayan (benign) ailesel
yenidoğan epilepsisi
c)
Erken
miyoklonik ensefalopati
d) Ohtahara sendromu
2) İnfantil dönem
a) Gezici fokal nöbetler ile seyreden infantil
epilepsi
b) West sendromu
c)
İnfantil
miyoklonik epilepsi
d) Kendini sınırlayan (benign) infantil
epilepsi
e) Kendini sınırlayan (benign) ailesel
infantil epilepsi
f)
Dravet
sendromu
g) İlerleyici olmayan hastalıklar ile
ilişkili miyoklonik ensefalopati
3) Çocukluk dönemi
a) Febril nöbet artı (FN+) sendromları
(infantil dönemde başlayabilir)
b) Panayiotopoulos sendromu
c)
Miyoklonik-atonik
nöbetli epilepsi
d) Santrotemporal dikenli çocukluk çağı
epilepsisi
e) Otozomal dominant noktürnal frontal lob
epilepsisi
f)
Geç
başlangıçlı çocukluk çağı oksipital lob epilepsisi
(Gastaut tipi)
g) Miyoklonik absanslı epilepsi
h) Lennox-Gastaut sendromu
i)
Uykuda
devamlı diken ve dalga (CSWS) ile seyreden epileptik ensefalopati†
j)
Landau-Kleffner
sendromu (LKS)
k) Çocukluk çağı absans epilepsisi
l)
Göz
kapağı miyoklonili absans epilepsi
m) Fotosensitif oksipital lob epilepsisi
4) Ergen-erişkin dönem
a) Juvenil absans epilepsisi
b) Juvenil miyoklonik epilepsi
c)
Sadece
jeneralize tonik-klonik nöbetler ile giden epilepsi
d) Otozomal dominant işitsel özellikli epilepsi
e) Diğer ailesel temporal lob epilepsileri
5) Başlangıç yaşı
değişken sendromlar
a) Değişken odaklı ailesel fokal
epilepsi (çocukluk çağında erişkinliğe kadar)
f)
Refleks
epilepsi
g) Progresif miyoklonik epilepsi
|
|
Özel sendromlar
1) Hipokampal skleroz ile birlikte mezyal temporal
lob epilepsisi
2) Rasmussen sendromu
3) Hipotalamik hamartom ile ilişkili jelastik
nöbetler
4) Hemikonvülziyon-hemipleji sendromu
5) Yapısal ve metabolik bir durumun
varlığında veya yokluğunda herhangi bir kategoride
sınıflanmayan epilepsiler
|
|
Yapısal-metabolik nedenler
ile ilişkili epilepsiler
1) Kortikal gelişimsel malformasyonlar
2) Nörokütane sendromlar
3) Tümör
4) Enfeksiyon
5) Travma
6) Anjioma
7) Perinatal olaylar
8) İnme
|
|
Nedeni belirlenemeyen epilepsi
|
|
Epileptik nöbet ile seyreden
durumlar
1) Benign neonatal nöbetler
2) Febril nöbetler
|
†CSWS’ye uykuda elektrografik status epileptikus (ESES)
da denilmektedir.
Etyolojinin
değerlendirilmesi: Epilepsi
sendromu belirlendikten sonra etyolojik değerlendirmeye geçilir. Etyolojik
değerlendirme, epilepsi sınıflamasının her
aşamasında yapılmalıdır. Altı alt grupta
incelenir:
a) Yapısal
b) Genetik
c) Enfeksiyonlar
d) Metabolik
e) İmmünite
f) Bilinmeyen
Yapısal
etyolojiye bağlı epilepsilerde, beyinde epileptik nöbetlere neden
olduğu bilinen bir yapısal lezyon tespit edilir (Tablo 1). Bu yapısal lezyon edinsel veya genetik
kökenli olabilir. Edinsel yapısal lezyonlara iskemik inme veya serebral
apse örnek verilebilirken genetik olanlara ise, genetik defektlere
bağlı kortikal gelişim anomalileri (örn, tuberoskleroz)
verilebilir.
Genetik
etyolojili epilepsiler kromozom veya gen anomalilere bağlı
oluşabilir. Kromozom anomalileri, Tablo
9’te özetlenmiştir. Günümüze kadar, epilepsi
hastalığına neden olan birçok gen anomalisi tanımlanmıştır.
Bunlardan en önemlileri, Na, Ca, K kanalları, nikotinik, GABA ve glutamat
reseptörleri ile ilişkili genetik bozukluklardır. Genetik etyoloji
aile öyküsü ve genetik inceleme ile tespit edilir. Ayrıca juvenil absans
epilepsi veya juvenil miyoklonik epilepsi gibi iyi tanımlanmış
epilepsi sendromlarının da, genetik etyolojili olduğu kabul
edilir. Bu bilgilerimiz ikizler üzerinde yapılmış toplum
bazlı çalışmalara dayanmaktadır.
Tablo 9. Epilepsi hastalığı ile
birlikte görülen kromozom anomalileri (https://www.epilepsydiagnosis.org’dan
esinlenerek)
|
Kromozom anomalileri
·
15q13.3
mikrodelesyonu
·
18q-
sendromu
·
15.
kromozomun proksimal bölgesinin ters duplikasyonu
·
1p36
delesyonu
·
Angelman
sendromu
·
Down
sendromu (Trizomi 21)
·
Kleinfelter
sendromu (XXY)
·
Miller
Dieker sendromu (17p delesyonu)
·
Pallister
Killian sendromu (Tetrazomi 12p)
·
Ring 14
sendromu
·
Ring 20
sendromu
·
Trizomi 12p
·
Wolf-Hirschhorn
sendromu (4p delesyonu)
|
Çeşitli
enfeksiyonlar akut semptomatik nöbete neden olarak veya kronik dönemde epilepsi
hastalığının etyolojik nedenleri olarak
karşımıza çıkabilirler. Bakteriyel veya viral
menenjit/meningoensefalit, serebral toksoplazma, CMV enfeksiyonu, tüberküloz
menenjit/meningoensefalit, nörosistiserkoz, HIV, Zika virüs, subakut sklerozan
panensefalit bu grubun iyi bilinen örnekleridir. Enfeksiyonla ilişkili
durumlara yapısal lezyonlar da eşlik edebilir.
Metabolik
epilepsiler, bir metabolik bozukluğa bağlı gelişen
(sıklıkla genetik bir defekt sonucu) ve sendromun ana
parçasının epilepsi olduğu hastalık grubunu tanımlar.
Bu grup, bazı süreçlerin tedavi edilebilir özellikleri nedeniyle önem
kazanmaktadır (Tablo 10).
Tablo 10. Tedavi edilebilir metabolik epilepsiler (Kaynak 18’den değiştirilerek)
|
·
Biyotinidinaz
eksikliği
·
Serebral
folat eksikliği
·
Kreatin bozuklukları
·
Folik aside
yanıtlı nöbetler
·
Glikoz
taşıyıcı 1 (GLUT1) eksikliği
·
Mitokondriyal
bozukluklar
·
Peroksizomal
bozukluklar
·
Pridoksine
bağımlı epilepsi
|
İmmün
etyolojili epilepsiler arasında Rasmussen ensefaliti ve otoantikor
ilişkili sendromlar sayılabilir. Antikor ilişkili sendromlar,
son yıllarda tanımlanmış subakut başlangıçlı
limbik ensefalit ve nöbetler ile seyreden hastalıkları tanımlar.
Ayrıca bu antikorların kronik seyirli nedeni belirlenemeyen
epilepsilerde de tespit edilmesi etyoloji anlamında yeni bir grubun
doğmasına neden olmuştur. Bu grupta, anti-NMDA reseptör
ensefaliti, voltaj kapılı potasyum kanalı, anti-glutamik asit
dekarboksilaz (GAD), GABAB reseptör, AMPA reseptör antikoru
ilişkili ensefalitler, steroide cevaplı tiroid
hastalığı ile ilişkili ensefalopati (Hashimoto
ensefalopatisi) bulunur.
Komorbiditelerin
değerlendirilmesi: Epilepsi hastalık sınıflamasının son
aşaması komorbiditelerin değerlendirilmesidir. Epilepsi
hastalığı ile birlikte öğrenme güçlükleri, psikolojik ve
davranışsal bozukluklar, motor bulgular ve uyku bozuklukları
gibi çeşitli komorbiditelerin sıkça görüldüğü belirtilmektedir.
Yeni sınıflamada güncellenen terimler
Gelişimsel
ve epileptik ensefalopati (Tablo 11): 2010 yılında epileptik
ensefalopati olarak isimlendirilen sendromlar, 2017 yılındaki
sınıflamalarda gelişimsel ve epileptik ensefalopati olarak
tanımlanmıştır. Gelişimsel ve epileptik
ensefalopatiler, altta yatan patolojiye ek olarak epileptik aktivitenin
kendisinin kognitif ve davranışsal bozukluklara katkıda
bulunduğu durumlar için kullanılır. Epileptik ensefalopatiler,
her yaş grubunda izlenebilirler ancak infantil ve çocukluk
çağında çok daha sıktır. Sıklıkla genetik
etyolojili olmakla birlikte edinsel patolojiler (örn, hipoksik iskemik
ensefalopati veya inme) ile de ilişkili olabilirler. Epileptik
ensefalopatiler içerisinde Ohtahara sendromu, West sendromu, Dravet sendromu,
Lennox-Gastaut sendromu, CSWS ile seyreden epileptik ensefalopati,
Landau-Kleffner sendromu sayılabilir. Bu sendromlarda normal
gelişmiş iken ortaya çıkan gelişimsel gerileme mutlaka
izlenir. Epileptik aktivite ve/veya nöbetler gelişim geriliğinden
sonra, önce veya eş zamanlı başlayabilir. Bazı durumlarda,
epilepsi erken yaşlarda sonlanabilir, hafifleyebilir ancak gelişimsel
bozukluk devam edebilir. Bu nedenle son sınıflamada terminoloji,
epileptik ensefalopati yerine gelişimsel ve epileptik ensefalopati olarak
değiştirilmiştir.
2017 sınıflamasında benign yerine
kendini sınırlayan (self-limited) ve ilaca cevaplı
(pharmacoresponsive) terimlerinin kullanılması önerilmiştir.
Kendini sınırlayan sendromlar, kendiliğinden sonlanan epilepsi
sendromları için kullanılır. İlaca cevaplı olanlar
ise, ilaç ile nöbetleri kontrol altına alınan
sendromlardır.
İdyopatik (nedeni kendinden olan veya genetik olarak
belirlendiği varsayılan) terimi yeni sınıflamadan
çıkarılmıştır. Ancak, genetik jeneralize epilepsiler
içerisinde iyi tanımlanmış bir grup hastalık için
kullanılabileceği belirtilmektedir. Bu grupta, çocukluk
çağı absans epilepsisi, juvenil absans epilepsisi, juvenil miyoklonik
epilepsi ve sadece jeneralize tonik-klonik nöbetler ile giden genetik
jeneralize epilepsi bulunur. Bu gruba, eski terminolojide kullanılan
şekliyle idyopatik jeneralize epilepsiler de denilebileceği kabul
edilmiştir. Bu kitap bölümünde ‘idyopatik/genetik’ terimi
kullanılacaktır.
Semptomatik ve
kriptojenik terimleri
yeni sınıflamadan çıkartılmıştır. Semptomatik yerine, ‘yapısal epilepsi’ ve
kriptojenik yerine ‘nedeni belirlenemeyen’ denilmektedir.
Tablo 11. Gelişimsel
ve epileptik ensefalopatiler (Kaynak 17’den esinelenerek
hazırlanmıştır)
|
|
Başlangıç
yaşı
|
Ana nöbeti tipi
|
Kognitif sendrom
|
EEG
|
Etyoloji
|
|
Erken miyoklonik ensefalopati
|
0-2 ay
|
Miyokloni
|
Mental-motor retardasyon
|
Boşalım baskılanım
(Burst-supresyon)
|
Metabolik hastalıklar
|
|
Ohtahara sendromu
|
0-3 ay
|
Tonik spazm
|
Mental-motor retardasyon
|
Boşalım baskılanım
(Burst-supresyon)
|
Kortikal gelişimsel malformasyonlar
|
|
West sendromu
|
3-12 ay
|
İnfantil spazm
|
Mental-motor retardasyon
|
Hipsaritmi
|
Perinatal asfiksi
Kortikal gelişimsel malformasyonlar
|
|
Dravet sendromu
|
6 ay civarı
|
Hemiklonik nöbet
|
Mental-motor retardasyon
|
Temel aktivite yavaş,
jeneralize diken ve çoklu diken,
fokal diken,
fotosensitivite
|
SCN1A mutasyonu- %75
|
|
Miyoklonik-atonik nöbetli epilepsi
|
2-4 yaş
|
Miyoklonik, atonik
|
Mental-motor retardasyon
|
Temel aktivite yavaş,
jeneralize diken ve çoklu diken,
fotosensitivite
|
Nadir olgularda SCN1A ve SLC2A1 mutasyonları
|
|
Lennox-Gastaut sendromu
|
3-5 yaş
|
Tonik nöbetler,
atipik absans
|
Mental-motor retardasyon
|
Temel aktivite yavaş,
<2,5Hz jeneralize diken-dalga,
nonREM uykuda ritmik hızlı dikenler
|
Yapısal nedenler
|
|
Landau-Kleffner sendromu
|
3-10 yaş
|
Nöbet olmayabilir,
fokal nöbet,
atipik absans
|
Verbal işitsel agnozi,
dil problemleri,
davranışsal problemler
|
Temel aktivite normal,
uyku ile tetiklenen unilateral/bilateral posterior
temporal diken-dalga
|
Genetik-bilinmiyor
|
|
Uykuda devamlı diken dalga
(CSWS) ile seyreden epileptik ensefalopati
|
4-5 yaş
|
Nöbet olmayabilir,
hemiklonik nöbet,
status epileptikus
|
Dil problemleri,
davranışsal problemler
|
Temel aktivite uyanıklıkta normal,
uykuda CSWS
|
Genetik-bilinmiyor
|
|
Progresif miyoklonik epilepsi
|
Her yaş
|
Miyokloni
|
Mental retardasyon ve
serebellar bulgular
|
Temel aktivite yavaş,
jeneralize diken ve çoklu diken,
fotosensitivite
|
Unverricht-Lundborg hastalığı, Lafora
hastalığı,
Metabolik genetik diğer nedenler,
|
|
Rasmussen Sendromu
|
1-10 yaş
|
Epilepsia parsiyalis kontinua
|
Mental retardasyon ve hemiparezi,
|
Temel aktivite yavaş,
polimorfik delta aktivitesi,
multifokal diken-dalga
|
İmmünolojik
|
EPİLEPSİ
SENDROMLARI
Bu
bölümde seyir, prognoz, etyoloji, tedaviye yanıt bakımından çok
farklı davranabilen çeşitli epilepsi türleri arasında iyi
tanımlanmış, belirli epilepsi sendromlarından
bazıları hakkında kısaca bilgi verilecektir.
Sıralama Tablo 8’de yer alan sınıflama esas
alınarak yapılmıştır.
Başlangıç
yaşına göre elektroklinik epilepsi sendromları
Yenidoğan
Dönemi
Kendini
sınırlayan (benign) ailesel yenidoğan epilepsisi
Çok
nadir görülmekle birlikte tamamen selim olması ve genetik açıdan
mutasyonlarının belirlenmiş olması nedeniyle önem
taşımaktadır. Nöbetler, tamamen sağlıklı,
zamanında doğmuş bir yenidoğanda hayatın 4-7. gününde
başlar. Sıklıkla tek taraflı klonik nöbetler izlenir ve
farklı nöbetlerde taraf değiştirir. Nöbet sırasında
tonik kasılma, vokalizasyon, otonom bulgular veya otomatizmalar
izlenebilir, fokal başlangıçlı bilateral tonik-klonik nöbete
dönüş olabilir. Günde 20'ye dek varan sıklıkta nöbetler görülür
ve 4-6 ay içinde nöbetler sonlanır. Nörolojik açıdan normal
gelişen bu hastaların bir kısmında ileriki yaşlarda
febril nöbet ve sonradan epilepsi gelişebilir. Otozomal dominant (OD) ve
yüksek penetranslı olan bu hastalığın geninin 20.
kromozomda olduğu saptanmıştır. Ardından başka
bir grup tarafından 8. kromozomda da bir defekt gösterilmiştir.
Sorumlu genlerin her iki kromozomda yer alan farklı potasyum kanal genleri
(KCNQ2, KCNQ3) olduğu 1998'de
kanıtlanmıştır.
Kendini
sınırlayan (benign) yenidoğan konvülziyonları
Klinik
olarak kendini sınırlayan (benign) ailesel yenidoğan epilepsisi
ile benzerdir. Aile öyküsünün olmaması ile ayrılırlar.
Erken miyoklonik
ensefalopati
Gelişimsel
ve epileptik ensefalopati grubundadır. Bir öncül olay olmadan
yaşamın ilk 2 ayı içerisinde nöbetler başlar. Nöbetler
başlamadan hemen önce veya nöbetlerin başlaması ile birlikte
mental ve motor gelişim geriler, mikrosefali gelişir ve nörolojik
muayene anormal olur. Etyoloji olarak sıklıkla metabolik nedenlidir
(nonketotik hiperglisinemi, amino ve organik asidüriler, üre siklus defektleri,
mitokondriyal hastalıklar, Menkes hastalığı, Zellweger
sendromu, pridoksin ve pridoksal-5-fosfat ilişkili bozukluklar gibi), nadiren
yapısal ve genetik nedenler de tanımlanmıştır.
Sık, gezici (eratik) fragmente miyokloniler mutlaka olmalıdır.
Miyokloniler vücudun bir bölümünden diğerine doğru asenkron ve
rastgele bir şekilde yayılır. Diğer nöbet tipleri (fokal
nöbetler, tonik nöbetler, epileptik spazmlar) de izlenebilir. EEG’de burst
supresyon paterni görülür, ilerleyen aylarda multifokal dikenlerin
izlendiği hipsaritmi paternine dönüşür. Kötü prognozlu bir
sendromdur. Hastaların yaklaşık yarısı haftalar
içerisinde kaybedilir. Geri kalanında ise ağır mental ve motor
retardasyon gelişir. West sendromu ve Lennox-Gastaut sendromuna
dönüşebilir.
Ohtahara sendromu
Ohtahara
sendromu da gelişimsel ve epileptik ensefalopati grubundadır.
Nöbetler yaşamın ilk 10 günü-3 ayı arasında başlar.
Ana nöbet tipi tonik spazmlardır. Tonik spazmlar, jeneralize veya fokal,
1-10 saniye süreli, öne doğru fleksiyon postürü gelişen tonik
nöbetlerdir. Oldukça sık olarak izlenirler, tekli ve kümeler halinde
olabilirler. Ayrıca gezici fokal motor klonik nöbetler,
hemikonvülziyonlar, tonik-klonik nöbetler görülebilir. Miyokloniler nadirdir.
Etyoloji sıklıkla kortikal gelişimsel malformasyonlar ile
ilişkilidir (hemimegalensefali, Aicardi sendromu, porensefali, fokal
kortikal displazi). Metabolik hastalıklar ve genetik nedenler de
tanımlanmıştır. EEG’de burst supresyon paterni izlenir.
Burst fazında tonik nöbetler kaydedilir. Kötü prognozludur.
Hastaların yaklaşık yarısı kaybedilir. Kurtulanlar
ise, ilerleyen dönemde West sendromu ve Lennox-Gastaut sendromuna
dönüşürler.
İnfantil
Dönem
Gezici
fokal nöbetler ile seyreden infantil epilepsi
Bu
sendrom ortalama 3 aylıkken başlar. Nöbetler, fokal klonik
nöbetlerdir. Bu nöbetlere baş ve göz deviyasyonunun eşlik ettiği
duraklama nöbetleri eklenir. Status epileptikus (SE) sıktır. Fokal
nöbetler, belirli bir kortikal bölgeden başlar ve aynı nöbet
içerisinde başka bir kortikal bölgede izlenir, bu gezici özellik sendrom
tanısı için gereklidir. EEG’de temel aktivite bozuktur, interiktal
olarak multifokal diken aktivitesi görülür. De novo olarak Na ve K kanal
mutasyonları saptanmıştır. Prognoz kötüdür. Hastaların
bir kısmı kaybedilirken kalanlar ağır mental ve motor
retarde olarak yaşarlar.
West
sendromu
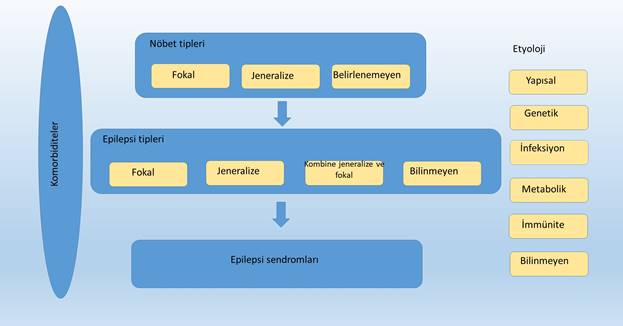
Bu tablonun tipik 3 ana belirtisi şunlardır:
1) İnfantil spazmlar (epileptik spazm)
2) Mental gerilik
3) EEG'de hipsaritmi denen yüksek amplitüdlü multifokal
odaklar, yaygın ve fokal yavaş aktivite ile giden kaotik tablo (Şekil
4)
Şekil 4. West sendromlu bir olguda hipsaritmi
örneği. EEG aktivitesinin yüksek amplitüdlü, multifokal ve kaotik
özellikte olduğu dikkat çekmektedir.
İnfantil spazmlar, gün içerisinde
1-30 kez kümeler halinde gelir ve her küme içerisinde 20-150 adet spazm
bulunabilir. Spazmlar kısa süreli, aksiyal ve ekstremite
kaslarını etkileyen bilateral tonik kasılmalardır.
Sıklıkla fleksör-ekstansör tiptedir, sadece fleksör veya nadiren sadece
ekstansör tipte olabilirler. Asimetrik özellikler izlenebilir. Spazmlar, uykuya
dalış ve uykudan uyanma ile artar. Bazıları taktil veya
sesli uyaran ile tetiklenebilir. Fokal nöbetler tabloya eklenebilir.
İnsidensi yaklaşık olarak 5000'de 1 olarak bildirilen bu
tablonun başlangıç yaşı 3-12 aydır; 3-7 aylar
arasında ve erkek çocuklarda nispeten sık rastlanır. West
sendromunda sık olarak saptanan etyolojik faktörler hipoksik-iskemik
ensefalopati (perinatal asfiksi), kortikal gelişimsel malformasyonlar (Aicardi
sendromu, tuberoskleroz, lizensefali gibi) ve MSS enfeksiyonlarıdır.
Moleküler genetik incelemelerin gelişimi ve yaygınlaşması
ile nedeni saptanamayan olguların önemli bir kısmında yeni
genetik mutasyonlar saptanmaktadır. Yapısal faktörleri, kromozom ve
gen anomalileri takip eder. West sendromu da gelişimsel ve epileptik
ensefalopati grubundadır. Mortalitesi %5’in altında olmasına
rağmen prognoz kötüdür. Nöbetlerin başlaması ile gelişim
geriliği tabloya eklenir. Hastaların yaklaşık 2/3’ünde
ilerleyen yaşlarda diğer nöbet tipleri tabloya eklenerek
Lennox-Gastaut sendromuna dönüşür. Normal mental ve motor gelişim
hastaların ancak %5-12’sinde izlenebilir. İnfantil spazmlar tedaviye
genellikle direnç gösterirler. Steroidler, tedavi mekanizması bilinmemekle
birlikte (ACTH veya prednizon) daha iyi sonuçlar verir. Özellikle tuberoskleroz
etyolojisi ile birliktelik olduğunda vigabatrin tedavisi ile olumlu
sonuçlar bildirilmektedir.
İnfantil
miyoklonik epilepsi
Nöbetler
6 ay-3 yıl arasında başlar. Erkekler daha sık etkilenirler.
Baş, göz küreleri, üst ekstremiteler ve diyaframı etkileyen
miyoklonik nöbetler ile karakterizedir. Miyokloniler, uykuya dalış ve
nonREM uykusunda belirginleşir. Aile öyküsü genellikle vardır ve nöbetler
kolayca kontrol altına alınır. Genellikle başka nöbet
tipleri eşlik etmez ama ergenlik döneminde bilateral tonik-klonik nöbet
görülebilir. Hastaların bir kısmında sık olmayan febril
nöbetler tabloya eklenebilir. Hastaların %20’sinde klinik ve elektrofizyolojik
ışık duyarlılığı izlenir. Ayrıca
hastaların bir kısmında nöbetler ses ve taktil uyaranla da
tetiklenebilir. Etyoloji genetik olarak kabul edilir. EEG’de temel aktivite
normaldir, interiktal EEG sıklıkla normal olarak izlenir. Nöbetler
sırasında jeneralize diken, çoklu diken-dalga aktivitesi görülür.
Kendini sınırlayan bir sendromdur. Nöbetler, başlangıçtan 6
ay-5yıl içerisinde sonlanır. Işık
duyarlılığı varlığında nöbetler devam
edebilir. Nörolojik muayene ve gelişim normaldir.
Kendini
sınırlayan (benign) ailesel ve ailesel olmayan infantil epilepsi
Doğum ve
yenidoğan dönemi sorunsuz olan hastada nöbetler 3-20 ay arasında
başlar ve 1 yaşında sonlanır. Duraklama, otomatizma,
baş ve göz versiyonu, kloniler ile karakterize fokal nöbetler izlenir.
Nöbetler hemikloni şeklinde olabilir, nöbetten nöbete taraf
değiştirebilirler ancak 3 dakikadan uzun sürmezler. Uzun süren
nöbetleri olan hastalarda Dravet sendromu ayırıcı tanıda
düşünülmelidir. Nörolojik muayene, mental ve motor gelişim normaldir.
EEG’de temel aktivite normaldir ve interiktal epileptiform anomali beklenmez.
Ailesel formu otozomal dominant geçişlidir. Hastaların
yaklaşık %90’nında 16. kromozomda bulunan prolinden zengin
transmembran protein 2 (PRRT2) geninde mutasyon izlenir.
Çarpıcı olarak bu genin mutasyonları aynı zamanda paroksizmal
kineziojenik diskinezi fenotipine de yol açmaktadır.
Dravet
sendromu
Genellikle
ilk yaş içerisinde sağlıklı bir bebekte nöbetler
başlar. Erkekler daha sık etkilenir. Erken infantil dönemde uzun
süreli febril nöbetler (çoğunlukla hemikonvülziyon şeklinde),
miyoklonik nöbetler, atipik absanslar ve fokal nöbetler görülür. Status
epileptikus sıktır. Febril SE olarak başlayabilir. Tonik
nöbetler ve epileptik spazmlar ise görülmez. Psikomotor gelişim ikinci
yıl içinde geriler ve oldukça belirgindir. Tablo ilaçlara dirençlidir.
Zaman içerisinde nöbetler sonlanır, ya da hafifler ve ağır
mental ve motor retardasyon geri dönüşümsüz olarak kalır, ataksi
tabloya eklenebilir. EEG’de temel aktivite bozuktur. İnteriktal olarak jeneralize
diken, çoklu diken-dalga aktivitesi ve fokal bulgular görülür. Klinik ve
elektrofizyolojik fotosensitivite izlenebilir. Göz kapama ile de deşajlar
tetiklenebilir. Hastaların yaklaşık %75’inde voltaj
kapılı Na kanalı tip 1 alfa alt ünitesinde (SCN1A)
mutasyonu saptanır ve çoğunlukla “de novo” mutasyondur, bilinen
başka farklı mutasyonlar da vardır. Na kanalı blokajı
yapan ilaçlar (karbamazepin ve fenitoin) nöbetleri kötüleştirebilir.
Lamotirijin de bazı olgularda tercih edilmez. Dirençli vakalarda
stripentol ve ketojenik diyet uygulanabilir.
İlerleyici
olmayan hastalıklar ile ilişkili miyoklonik ensefalopati
İlk
5 yaş içerisinde (sıklıkla 1. yaş) hastalık
başlar. Hastaların hepsinde hipotoni, ataksi ve kognitif gerilik ile seyreden
bir ensefalopati tablosu bulunur. Ana nöbet tipi miyoklonik SE
şeklindedir. Göz kapağı, yüz ve ekstremiteleri içeren gezici,
asenkron miyokloniler, absans nöbetleri sırasında düzenli ve senkron
hale gelir. Ayrıca fokal motor nöbetler, atonik nöbetler, jeneralize
tonik-klonik nöbetler izlenebilir. Bu sendromda tonik nöbetler görülmez.
Hastaların yarısında kromozom anomalileri (Angelman sendromu ve
Prader Willi sendromu) izlenir. Ayrıca Rett sendromu, yapısal beyin
anomalileri de saptanabilir. EEG’de temel aktivite anormaldir, interiktal
olarak multifokal diken-dalga aktivitesi görülür. Prognoz kötüdür.
Hastaların çoğunda nörokognitif gerilik kalıcı olur.
Çocukluk
Dönemi
Febril
nöbet artı (FN+) sendromları
İnfantil dönemde veya
çocukluk döneminde başlayan febril nöbetler ile karakterizedir.
Sıklıkla 6 ay-1 yaş arası başlar. Çok sayıda FN
geçirilir ve sıklıkla 6 yaşından uzun sürebilir.
Ateşsiz jeneralize nöbetler, absans, miyokloni, tonik, miyoklonik-atonik
nöbetler ve frontal veya temporal kökenli fokal nöbetler izlenebilir. Nörolojik
muayene ve gelişim normaldir. EEG’de temel aktivite normaldir.
İnteriktal olarak uyku ile belirginleşen jeneralize diken ve çoklu
diken-dalga aktivitesi izlenebilir. Otozomal dominant kalıtım gösterir
ve aynı aile içerisinde fenotip heterojen olabilir. Mutasyonlar 2 ve 19.
kromozomda SCN1A, SCN1B, SCN2A, GABRG2 ve PCDH19 genlerinde
tanımlanmıştır. Kendini sınırlayan bir
sendromdur. Nöbetler genelde ortalama 11 yaşlarında
sonlanır.
Panayiotopoulos
sendromu
Bu
tablo erken çocukluk çağında izlenen uzamış fokal otonom
nöbetler ile karakterize bir sendromdur. Sıklıkla 3-6 yaşlar
arasında başlar, büyüme gelişmesi normal olan çocuklarda
çoğunlukla gece uyku sırasında, başlıca bulantı
kusma yakınmalarını tonik göz deviyasyonu ile giden nöbetler
izler. Nöbet sırasında bilinç korunmuştur ve hasta
konuşabilir. Yüzde kızarıklık, solukluk, midriyazis,
miyozis, terleme, hipersalivasyon gibi otonom bulgular eşlik edebilir.
Nöbetler uzun süreli (%40 olguda en az 30 dakika) olabilmektedir. EEG’de
ışığa duyarlılık görülmez ancak fiksasyon-“off”
duyarlılığı belirlenir. EEG’de gözler kapatılınca
oksipital bölgede, tipik yüksek amplitüdlü sık sivri dalga dizileri
izlenir. İnteriktal olarak yer değiştirebilen multifokal
dikenler görülebilir, sıklıkla posterior bölgelerde
odaklaşırlar. Otonom SE gelişebilmekte ve bu olgularda
ayırıcı tanıda sorun yaşanabilmektedir. Kendini
sınırlayan bir epilepsi sendromudur ve otonom sistem epilepsisi
olarak ele alınmaktadır. Nöbetler başladıktan
yaklaşık 1-2 yıl sonra sonlanır. Hastaların
yarısı tüm hayatları boyunca en fazla 6 nöbet geçirirler.
Miyoklonik-atonik
nöbetli epilepsi (Doose sendromu)
Nadir
görülen bu sendromda normal bir çocukta aniden düşme nöbetleri
başlar. Başlangıç 2-4 yaşındadır.
Miyoklonik-atonik nöbetler mutlaka olmalıdır. Ayrıca miyoklonik,
atonik, absans, atipik absans ve tonik-klonik nöbetler olabilir. Absans
nöbetine kollarda ve yüzde miyokloni eşlik edebilir. SE sıktır.
Tonik nöbetler nadir görülür. Absans nöbetleri genellikle kısa sürelidir.
EEG'de temel aktivite bozuktur. İnteriktal olarak, jeneralize diken, çoklu
diken-dalga aktivitesi ve fotosensitivite görülebilir. Az sayıda hastada SCN1A and SLC2A1 genlerinde mutasyonlar saptanmıştır. Seyir ve
prognoz çok değişkendir. Bazı olgular remisyona girerken
bazıları Lennox-Gastaut sendromu ile kesişme gösterir.
Santrotemporal
dikenli çocukluk çağı epilepsisi (Rolandik epilepsi)
Çocukluk
çağının en sık görülen fokal epilepsisidir ve
çoğunlukla remisyonla sonlanır. Kendini sınırlayan
sendromlar içerisindedir. İdyopatik fokal epilepsilerin prototipi olan ve
“sensorimotor sistem” epilepsisi olarak kabul edilen bu tipik tablonun
tanınması çok önemlidir. Genellikle 4-10 yaşlarında nörolojik
açıdan normal bir çocukta başlar, erkeklerde biraz daha
sıktır. Tipik nöbet özellikleri yüzün bir yarısında
özellikle dil, boğaz ve dudaklarda uyuşma ve/veya taraflarda tek
yanlı motor bulgularla seyreder (frontopariyetal operküler nöbetler).
Farinks, larinks ve dil kaslarının tutulması nedeniyle
konuşma durur veya dizartrikleşir, tükürük artışı
eşlik edebilir. Bu nöbet sırasında hastanın bilinci
korunmuştur. Genellikle uyku sırasında gelen nöbetler bazen
jeneralize tonik klonik nöbete dönüşebilir. Tipik interiktal EEG bulgusu
tek yanlı, bazen iki yanlı ama birbirinden bağımsız ve
yer değiştirebilen yüksek amplitüdlü santrotemporal lokalizasyon
veren diken/diken-yavaş dalga aktivitesi şeklindedir ve bu aktivite
genellikle uykuda artma gösterir (Şekil 5). Nöbetleri genelde
seyrek olduğu için ve ergenlikte remisyonla sonlandığı için
bazı olgularda tedavi verilmeyebilir. Tedavi gereken durumlarda valproik
asidi yeğleyenler olduğu gibi karbamazepini üstün bulanlar da
vardır.
Bu
sendrom, atipik santrotemporal dikenli
çocukluk çağı epilepsisi formuna dönüşebilir. Bu atipik
formunda atipik absans ve negatif miyokloni ile giden fokal motor nöbetler
eklenebilir. Negatif miyokloni, bilinç kaybı olmadan kollarda ani kas
tonusunda azalma ile seyreden nöbetlerdir. Atipik form, gelişimsel ve
epileptik ensefalopati grubunda incelenir ve bu sendromda kognitif ve
davranışsal gerilik tespit edilir.
Şekil 5a. Rolandik epilepsi
tanısıyla izlenen bir olguda uyanıklık sırasında
sol santrotemporal bölgede faz dönmesi gösteren az sayıda keskin ve
keskin-yavaş dalgalar görülmektedir.

Şekil 5b. Aynı olguda uyku ile sol taraftaki
odağın sıklaşarak diziler oluşturduğu, amplitüdün
arttığı görülmektedir. Ayrıca sağ orta temporal
bölgede faz dönmesi gösteren bağımsız bir epileptojenik odak
daha tabloya eklenmiştir.
Otozomal
dominant noktürnal frontal lob epilepsisi
Noktürnal motor nöbetler ile karakterizedir. Nöbet
başlangıç yaşı değişkendir, hastaların büyük
kısmında nöbetler yirmili yaşlarda başlar. Nöbetler
kısa sürer (20-50 saniye) ve kümelenir. Nöbetler sırasında
tonik, distonik ve hiperkinetik motor bulgular izlenir. Hastaların bir
kısmında somotosensoriyel bir aura saptanır. Nöbetler bilateral
tonik-klonik nöbete dönüşebilir. EEG’de temel aktivite normaldir, uykuda
interiktal dönemde frontal ve frontotemporal diken aktivitesi görülebilir.
Prognoz değişkendir. Aynı aile içerisinde bile
değişebilir. Noktürnal parasomnilerden ve hareket
bozukluklarından ayırt edilmelidir. Otozomal dominant geçişli,
1. ve 20. kromozomlarda bulunan nöronal nikotinik asetilkolin reseptör alt
ünitelerini kodlayan genlerde (CHRNA4,
CHRNB2, CHRNA2) mutasyonlar saptanmıştır.
Geç
başlangıçlı çocukluk çağı oksipital lob epilepsisi
(Gastaut tipi)
Yaklaşık
8-10 yaşlarında başlar. Nöbetler sıklıkla gündüzleri görülür
ve başlıca görsel semptomlar bulunduğundan “görsel sistem
epilepsisi” olarak değerlendirilebilir. Nöbetler kısa sürelidir ve
bilinç kaybı olmaz. Nöbet sırasında elementer görsel
halüsinasyonlar veya körlük olabilir, baş ve göz deviyasyonu, göz
kırpıştırma eşlik edebilir. İktal ağrı
sıklıkla orbital bölgede izlenebilir. Gece izlenen nöbetler daha uzun
süreli olur ve bilateral tonik-klonik nöbete dönüşebilir. Otonom bulgular
burada sık değildir. EEG’de
temel aktivite normaldir, interiktal olarak göz kapama ile belirginleşen
bilateral oksipital dikenler izlenir (Şekil 6). Tedavi edilmezse
nöbetler görece sık olarak tekrarlar.

Şekil 6. Normal bir çocukta gelişen görsel belirtili
nöbetler nedeniyle yapılan bu EEG incelemesinde gözler açıkken normal
bir temel aktivite varken göz kapatmayı izleyerek iki yanlı oksipital
bölgelerde yüksek amplitüdlü sivri-yavaş dalga deşarjları
belirmektedir.
Miyoklonik
absanslı epilepsi
Nadir
rastlanan bu tabloda başlangıç genellikle 5-8 yaş
arasındadır. Tipik absanslara kol ve bacaklarda ritmik miyoklonik
atmalar eşlik eder. EEG özellikleri çocukluk çağı absans
epilepsisine benzerdir ancak zaman içerisinde temel aktivite bozulabilir.
Hastaların 2/3’ünde jeneralize tonik-klonik, atonik, absans ve atipik
absans nöbetleri eklenebilir. Tedaviye cevap çoğunlukla kötüdür ve
bazı olgularda diğer epilepsi tiplerine özellikle Lennox-Gastaut
sendromuna ilerleme görülebilir.
Lennox-Gastaut
sendromu
Bu
tabloyu, West sendromunun bir uzantısı gibi düşünmek mümkündür.
Başlangıç yaşı 1-7 yaştır (sıklıkla 3-5
yaş). Atipik absans, miyoklonik, tonik nöbetler, atonik ve tonik-klonik
nöbet tiplerinin birden fazlası bir arada görülür. Ana nöbet tipleri
atipik absans, tonik nöbetler ve atonik nöbetlerdir. Tonik nöbetler
sıklıkla nonREM uykusunda izlenir. Tonik ve atonik nöbetler sık
düşme ataklarına yol açar. Nöbetler dirençli seyreder. EEG’de
yavaş (<2,5Hz) diken-dalga görünümü tipiktir ve sıklıkla
atipik absans ile ilişkilidir (Şekil 7). EEG’de
başka bir tipik bulgu nonREM uykuda paroksizmal hızlı aktivite
veya ritmik hızlı dikenlerdir ve tonik nöbetlere eşlik
edebilirler. Olguların büyük kısmında belirgin motor-mental
gerilik söz konusudur. West sendromunun etyolojisinde söz edilen nedenlerin
çoğu, aynı zamanda Lennox-Gastaut sendromuna yol açabilir, neden
saptanamayan olgular da vardır. Lennox-Gastaut sendromunda etyolojik
nedenlerin %70’i yapısal kökenlidir. Bazı olgular West sendromundan
geçerek bu tabloya girerler. Nöbetler genellikle ilaç tedavisine dirençlidir.
Düşme nöbetleri özellikle kötü prognoza işaret eder, morbiditeye yol
açar ve AEİ’lere yanıt alınamayan bu tip bazı olgularda
düşmenin engellenmesi anlamında anterior kallozotomi girişimi
ile olumlu sonuçlar elde edilebilmektedir.

Şekil 7. Lennox-Gastaut sendromu için tipik olan
yavaş diken dalga aktivitesi ve yetersiz temel aktivite görülmektedir.
Landau-Kleffner
sendromu
Çok
nadir olan bu tablo konuşmayı normal olarak öğrenmiş olan
3-10 yaş arası bir çocukta konuşmanın kaybolması (edinsel
epileptik afazi) ve genellikle izleyen fokal motor ve jeneralize
tonik-klonik tipte epileptik nöbetlerle ortaya çıkar. Tamamen
sağlıklı çocukta ilk bulgu verbal işitsel agnozidir,
seslerin semantik anlamı kaybolur. Hasta kelime sağırlığı
içerisindedir. Takiben hasta konuşmasını da kaybeder. Nöbetler
hastaların 2/3’ünde izlenir, sıklıkla noktürnaldir. Fokal
nöbetlerin yanı sıra atipik absans ve atonik nöbet izlenebilir.
Jeneralize tonik nöbetler bu sendromda görülmez. Kognitif ve
davranışsal problemler de görülebilir. EEG bulguları tanı
koydurur (Şekil 8). EEG’de temel aktivite normaldir.
İnteriktal olarak uyku deprivasyonu veya uyku ile tetiklenen unilateral
veya bilateral temporal bölgelerde diken-dalga aktivitesi izlenir.
Patofizyolojisi aydınlatılamamış olan bu tabloda ilerleyen
yaşla birlikte genellikle nöbetlerde remisyon görülür. Ancak dil
problemleri hastaların çoğunda kalıcı olmaktadır.
Bazı hastalarda davranış problemleri ve iletişim
güçlüğü de izlenebilir. Gelişimsel ve epileptik ensefalopati grubunda
incelenmektedir. Tedavisinde klinik deneyimlerimize göre en etkili ilaç
steroidin yanı sıra klonazepamdır. Bazı olgularda uykuda
devamlı diken ve dalga aktivitesi (CSWS) görülebilir. CSWS tablosunun
kognitif, davranışsal ve dil fonksiyonlarını etkilediği
düşünülmektedir.

Şekil 8. Landau-Kleffner sendromu
tanısı ile izlenen 9 yaşındaki
erkek hasta; 7 yaşında başlayan jeneralize tonik
klonik nöbetlerine 7 ay sonra eklenen edinsel afazi nedeniyle başvurmuştur.
EEG incelemesinde uykuyla aktive olan, frontotemporal bölgelerde birbirinden
bağımsız diken-dalga deşarjları Şekil 8a. ile yavaş uyku sırasında
devamlı diken-dalga aktivitesi izlenmektedir Şekil 8b.
Uykuda devamlı diken ve
dalga (CSWS) ile seyreden epileptik ensefalopati
Bu sendrom
gelişimsel ve epileptik ensefalopatiler grubunda değerlendirilir.
Başlangıç yaşı 1-12 yaştır (sıklıkla
4-5 yaş). Hastaların yarısı ilk nöbete kadar normalken
diğer yarısında yenidoğan nöbetleri veya nörolojik
anomaliler bulunur. Nöbetler noktürnal karakterli ve hemiklonik SE
şeklindedir. Ayrıca fokal klonik nöbetler, miyokloni, absans ve
atipik absans veya jeneralize tonik-klonik nöbetler de izlenebilir. Tonik
nöbetler bu sendromda izlenmez. Nöbetlerden yaklaşık bir sene sonra
CSWS tespit edilir. CSWS izlenmesi ile nöbetler sıklaşır ve nöropsikolojik
kötüleşme başlar ve ilerler. Nöbetler her hastada görülmeyebilir.
Etyolojik olarak hastalarda yapısal lezyonlar, Rett sendromu veya
metabolik hastalıklar bulunabilir. EEG diagnostiktir.
Uyanıklıkta temel aktivite normalken uykuya dalma ile tüm uyku
süresinin yaklaşık %50-85’inde devamlı frontal bölgelerde
belirgin veya multifokal olarak izlenebilen diken-dalga aktivitesi
saptanır. İnteriktal EEG’de ise multifokal (frontotemporal,
santrotemporal bölgelerde) diken aktivitesi görülebilir. Tedavide CSWS saptandığında
ACTH başlanılması uzmanlarca önerilmektedir. Nöbet kontrolü için
valproat ve benzodiazepinler kullanılabilir.
Çocukluk
çağı absans epilepsisi (piknolepsi)
Tipik
absans nöbetleri ile giden bu tablo çocukluk çağı nöbetlerinin
yaklaşık %4'ünü oluşturur. Başlangıç yaşı
3-9 arasıdır, kızlarda biraz daha sıktır. Nöbetler her
çeşit mental aktivitenin aniden durması ve saniyeler sonra
kaldığı yerden devam etmesi şeklinde olur. Bu sırada
hastanın cevapsız ve hareketsiz olduğu, boş bir
şekilde baktığı gözlenir. Absans nöbetleri gün içerisinde
birçok kez tekrarlar (sayısı yüzleri bulabilir) ve 4-20 saniye
sürelidir. Sadece bilinç kaybı ile
seyreden nöbetlere basit absans; bilinç kaybı ile birlikte
hafif klonik, atonik, tonik ve otonom komponentlerin ve otomatizmaların
olduğu nöbetlere kompleks
absans nöbetleri denir. Çocukluk çağı absans
epilepsisinde diğer nöbet tipleri görülmez. Absans nöbeti
sırasında göz kapağı miyoklonisi beklenmez. Postiktal
konfüzyonun olduğu, 45 saniyeden uzun süren nöbetler için, fokal nöbet
açısından ayırıcı tanı
yapılmalıdır. Tipik absans nöbetlerinin EEG bulgusu bilateral,
genellikle düzenli ve simetrik 3 (2,5-4) Hz diken-dalga kompleksleri; nadiren
multipl diken-yavaş dalgalar şeklindedir; temel aktivite normaldir
(Bakınız: Sinir
Sistemi Semiyolojisi, EEG, Şekil 2). EEG’de
interiktal olarak oksipital intermitan ritmik delta aktivitesi (OIRDA)
izlenebilir. Absans nöbeti veya interiktal deşarjlar EEG’de hiperventilasyon
ile tetiklenir. Genellikle normal zeka düzeyinde olan bu hastalar sık
nöbetleri nedeniyle okul başarısında düşme gösterebilirler.
Tedaviye iyi cevap (%80 olguda tam kontrol) alınan bu tabloda uygun
ilaçlar sadece absans nöbeti olanlarda etosüksimid veya valproat, diğer
nöbet tipleri eşlik ettiğinde ise valproattır. Kendini
sınırlayan bir epilepsi sendromudur. Sıklıkla 12
yaşından önce nöbetler sonlanır. Ender görülen dirençli absans nöbetlerinde
levetirasetam, lamotirijin, etosüksimid veya diğer geniş spektrumlu
AEİ’ler birlikte kullanılabilir.
Göz
kapağı miyoklonili absans epilepsi (Jeavons sendromu)
Normal gelişimi olan bir çocukta 6-8
yaşlarında başlayan absans nöbetlerinin eşlik ettiği
ya da etmediği göz kapağı miyoklonileri ile karakterize bir
sendromdur. Nöbetler göz kapama ile tetiklenir. Göz kapağı
miyoklonileri ani, tekrarlayıcı, ritmik ve hızlı (4-6 Hz)
hareketlerdir. Nöbetler saniyeler sürer, sıklıkla 6 saniyenin
altındadır. Fotosensitivite mevcuttur. Febril nöbetler izlenebilir. İleri
yaşta genelde ışık uyarı ile tetiklenen jeneralize
tonik-klonik nöbetler görülebilir. EEG’de temel aktivite normaldir, 3-6 Hz
jeneralize diken, çoklu diken-dalga deşarjları izlenir.
Elektrofizyolojik olarak da göz kapama ve aralıklı
ışık uyaranı ile epileptiform deşarjların
tetiklendiği görülür (Şekil 9). Tedavi genelde ömür boyudur,
prognoz değişkenlikler gösterir. Bazı hastalar AEİ ile
kontrol altına alınabilirken bir grup hasta tedaviye dirençli
seyredebilir.

Şekil 9. Göz kapağı miyoklonili absans
epilepsili bir olguda göz kapamayı takiben ortaya çıkan 1,5-2 sn
süreli jeneralize diken-dalga deşarjları
Fotosensitif
oksipital lob epilepsi
Işık ile tetiklenen oksipital lob nöbetleri
ile karakterize 4-12 yaşlarında başlayan bir sendromdur. Mental
ve motor gelişi normal bir çocukta izlenir. Nöbetler, kısa süreli
(1-3 dakika) görsel semptomlar ile seyreder. Bunlar içerisinde renkli cisimler
veya körlük olabilir. Nöbetler uzun sürerse baş ve gözde deviyasyon,
bulantı-kusma ve otonom semptomlar nöbete eklenebilir. Nöbet bilateral
tonik-klonik nöbete dönüşebilir. Ayrıca miyoklonik ve absans nöbetleri
de nadiren eşlik edebilir. Tüm hastalar fotosensitiftir. EEG’de temel
aktivite normaldir, interiktal olarak oksipital dikenler ve posteriorda
belirgin jeneralize diken, çoklu diken-dalga aktivitesi izlenebilir. Prognozu
değişkendir ancak hastaların çoğunda hayat boyu tedavi
gerekir.
Ergen-Erişkin
Dönem
Juvenil
absans epilepsi
Çocukluk çağı absans
epilepsine oldukça benzerdir. Absans nöbetleri daha uzun süreli, gün içerisinde
sıklığı daha az ve görece daha hafiftir. 8-16 yaş
arası normal çocuk ve ergenlerde görülen bu tabloda nöbetler yıllar
içinde hafifler ama çocukluk çağı absans epilepsisi gibi remisyon
beklenmez. Hastaların neredeyse hepsinde jeneralize tonik-klonik nöbet
vardır ve 1/4’ünde ise miyokloni olabilir ama geri plandadır. EEG’de
temel aktivite normaldir, 3-4 Hz jeneralize diken, çoklu diken-dalga aktivitesi
izlenir (Şekil 10). Hiperventilasyon ile epileptik aktivite
tetiklenir. Nadiren fotosensitivite olabilir.

Şekil 10. 12 yaşındaki
olguda EEG’de izlenen, 7 saniye süreli, 4 Hz jeneralize, hemisfer ön
yarılarında belirgin multi-diken-dalga deşarjları
sırasında dalma ve gözlerinde aralanma ve
kırpıştırma görülmüştür.
Juvenil
miyoklonik epilepsi (Janz sendromu)
Genellikle
8-26 yaş arasında, en sık olarak 12-15 yaşlarında bir
hastada tipik olarak sabah uyandıktan sonra, şuuru yerindeyken,
yaygın, tekrarlayıcı, genellikle kollarda belirgin olan
miyoklonilerle başlar. Aniden uyandırılma ve uykusuzluk ile
tetiklenen bu nöbetler bazen hastanın elindekileri düşürmesine, nadiren
de düşmesine yol açar. Miyoklonik nöbetleri, sinirlilik, yorgunluk
yüzünden elde titreme, tik gibi açıklamalarla çoğu hasta önemsemez ve
bir doktora başvurmaz. Bu nedenle bu nöbetlerin anlaşılabilmesi
için ayrıntılı bir şekilde sorgulanması gerekmektedir.
Miyokloniler bazı hastalarda tek nöbet tipi olarak kalabildiği gibi
çoğu hastada bir yıl içinde yine benzer tetikleyici etkenlere
bağlı olarak ortaya çıkan jeneralize tonik-klonik nöbet tabloya
eklenir. Bu sendromda görülebilen üçüncü nöbet tipi olguların yaklaşık
1/3'ünde görülen ve genellikle daha erken yaşlarda ortaya çıkan
absans nöbetleridir. Bazı hastalarda nöbet sırasında bazı
fokal bulgular saptanabilir. Işık uyarana duyarlılık bu
sendromda özellikle kadın olgularda sık olarak görülmektedir.
Bu
idyopatik/genetik jeneralize epilepsi sendromu tüm epilepsilerin %4-10
kadarını oluşturmaktadır, buna karşın sık
olarak tanı konmasında gecikme ve güçlükler ortaya çıkar.
İlk tanı sorunu daha öncede belirtildiği gibi hastanın miyoklonileri
önemsemeyip genellikle ilk büyük nöbeti geçirince doktora başvurması
ve miyokloniler doktor tarafından sorulmazsa bahsetmemesi nedeniyledir. Bu
arada hasta çeşitli yanlış tanılar alabilir, örneğin
fokal bir epilepsi sanılabilir. Karbamazepin ve fenitoin tedavileri bu
sendromda olumsuz etki yaratarak absans ve miyoklonileri
arttırmaktadır. Doğru tanı konduktan sonra
yapılması gereken ilk önce hastanın nöbeti arttıran
faktörler açısından uyarılmasıdır. Bu faktörler
arasında uykusuzluk, ani uyanma, yorgunluk, alkol alımı, parlak
ışık uyarı, televizyon veya bilgisayar oyunları ve
nadir bazı olgularda bazı mental fonksiyonlar (praksi, okuma, oyun
oynama vb.) yer alır. Her hasta bu faktörlerin bir veya birkaçından
etkilenebilir. Kadın hastalar menstruasyon sırasında nöbet
artışından yakınabilirler.
EEG’de
tipik bulgu 3-6 Hz jeneralize çok diken-dalga deşarjları
görülmesidir, fotosensitivite de önemlidir (Bakınız: Sinir Sistemi Semiyolojisi, EEG, şekil
7, 8). Ancak 1/3 olguda ilk EEG normal bulunur. Kesin tanı için
EEG’nin tekrarlanması yararlıdır. Bazı olgularda EEG’de
asimetrik ve fokal anomalilere rastlanabilir ve deneyimsiz bir göz
tarafından yanlış sonuçlara varılabilir. Genetik olarak
ilişkili birçok mutasyon (EFHC1,
GABRA1, Ser1 gibi) tanımlanmakla birlikte, çoğu olguda köken
belirlenememektedir. Uzun takip çalışmaları 40
yaşından sonra miyoklonik nöbetlerin çoğu olguda ortadan
kalktığını veya hafiflediğini göstermiştir.
Psikiyatrik ek sorunları olan ve 3 nöbet tipi olan olgular %15
oranında görülebilen dirençli seyir açısından risk
taşırlar. Bu sendromda kullanılabilecek AEİ; valproat,
levetirasetam ve lamotirijindir. Ancak doğurkanlık çağında
bulunan kadın hastalarda teratojenite riski nedeniyle valproat
kullanılmamalıdır. Lamotirijin ise, bazı hastalarda
miyokloni nöbetlerini arttırabilir. Yaklaşık %80 olgu bu ilaçlar
ile tam olarak kontrol altına alınır. Ancak ömür boyu
sürdüğü bilinen bu hastalıkta ilaç kesme ve tetikleyici faktörlere
bağlı olarak nüksler görülmektedir. Son yıllarda sistem
epilepsilerinin prototipi olarak bildirilen bu tabloda fonksiyonel görüntüleme
yöntemleri ile talamo-kortikal hiperkonnektivite saptanmıştır.
Tedavide sorun yaşanan olgularda psikiyatrik sorunlara ve frontal tipte
kognitif bulgulara da dikkat çekilmiştir.
Sadece
jeneralize tonik-klonik nöbetler ile giden epilepsi
Genellikle
10-20 yaş arasında başlayan ve diğer idyopatik/genetik
jeneralize epilepsilerle ortak özellikler taşıyan bu sendromda
nöbetlerin %90'ı uyanma dönemleri ve uykusuz kalma ile ilişkilidir.
Akşamüstü dinlenme saatlerinde de nöbet görülebilir. Nöbetler alkol ile de
tetiklenebilir. Jeneralize tonik-klonik nöbet dışında nöbet tipi
gözükmez. EEG’de temel aktivite normaldir, interiktal dönemde jeneralize çoklu
diken-dalga aktivitesi izlenebilir. Fotosensitivite görülebilir. AEİ’lere
iyi yanıt verirler ancak ömür boyu tedavi edilmesi gerekir. AEİ
kesimi sonrası sık nüks görülür.
Otozomal
dominant işitsel özellikli epilepsi
Aile üyelerinde fokal işitsel nöbetler ile
seyreden otozomal dominant geçişli bir epilepsi sendromudur. Nöbetlerin
başlangıç yaşı oldukça değişkendir (4-40 yaş).
Mental ve motor gelişim normal olarak izlenir. Fokal duysal işitsel
semptomlarla giden veya afazi ile giden nöbetler izlenir. Nöbetler ses ile
tetiklenebilir. Nadir de olsa bilateral tonik-klonik nöbete dönüşebilir.
EEG sıklıkla normaldir, nadiren fokal anomaliler görülebilir.
İyi prognozlu bir sendromdur. Nöbetler ilaç ile kontrol altına
alınırlar ancak hastaların çoğu ilacı kesemez. Bu
tabloda son yıllarda LGI1 geni
başta olmak üzere farklı mutasyonlar
tanımlanmıştır.
Başlangıç Yaşı Değişken
Sendromlar
Değişken
odaklı ailesel fokal epilepsi
Yapısal
bir anomali olmadan aile bireylerinde farklı beyin bölgelerinden
kaynaklanan fokal nöbetler ile karakterize otozomal resesif bir sendromdur.
Nöbet başlangıç yaşı çocukluk çağından
erişkin döneme kadar uzayabilir. Fokal nöbetler izlenir. Frontal,
temporal, pariyetal veya oksipital lob kaynaklı olabilirler. Aile
bireylerinde nöbetin kaynaklandığı bölge, nöbet
sıklığı ve AEİ cevabı değişkenlik gösterir.
EEG’de temel aktivite normaldir, fokal epileptiform anomali izlenir. Bazı
olgularda kromozom 22’de bulunan DEPDC5
geninde mutasyon tespit edilmiştir. Beyin görüntülemelerinde özellik
saptanmamaktadır.
Refleks
Nöbetler
Refleks epilepsi tutarlı
şekilde duysal dış uyarana bağlı olarak gelişen
ve tekrarlayan epileptik nöbetler durumudur. Refleks nöbetler ise özgün bir uyarana karşı
daima veya hemen daima ortaya çıkan nöbetlerdir. Çeşitli refleks
epilepsi sendromları tanımlanmıştır, en sık
görüleni %75-80 oranıyla ışığa duyarlı
gruptur.
İlk
kez 1989 ILAE sendrom sınıflamasında, özel uyaranlarla ortaya
çıkan epilepsi tanımı konulmuştur. 2001 yılındaki
sınıflamada refleks nöbetleri ortaya çıkaran uyaranlar;
görsel uyaranlar (yanıp sönen ışıklar, paternler,
diğer görsel uyaranlar), düşünme, müzik, yemek yeme, praksi, somatik
duysal, propriyoseptif, okuma, sıcak su, “startle” olarak
belirtilmiştir. Refleks epilepsiler; idyopatik/genetik fotosensitif
oksipital lob epilepsisi, diğer görsel sensitif epilepsiler, primer
okuma epilepsisi ve “startle” epilepsisi olarak gruplandırılmıştır. Refleks
epilepsi sendromlarında görülen epileptik nöbetler fokal veya jeneralize,
erken veya geç latanslı, basit uyarana bağlı veya
karmaşık uyaranlara bağlı olabilir. Bu sendromlara spontan
nöbetler de eşlik edebilir. İzole refleks nöbet görüldüğünde bu
kişinin epilepsi tanısı almasını gerektirmez. Alkolün
bırakılması veya ateş yüksekliği gibi özel durumlarda
ortaya çıkmış olan nöbetler refleks nöbet değildir, duruma
bağlı nöbet olarak ele alınır.
Fotosensitivite: Klinik
olarak ışık uyaranlarla tetiklenen nöbetler refleks fotosensitif
nöbetlerdir. Epileptik hastaların yaklaşık %5’inde EEG’de
ışık duyarlılığı görülür Bu oran
idyopatik/genetik jeneralize epilepsilerde %21’e kadar yükselir.
İdyopatik/genetik jeneralize epilepsi sendromları içinde özellikle
juvenil miyoklonik epilepsi diğer sendromlara göre daha fazla
ışığa duyarlıdır. Fotosensitivitenin yaş ve
cinsiyete bağlı penetrans gösteren, otozomal dominant bir kalıtım
paterni olduğunu düşündüren bulgular vardır. Genetik mekanizması
çok sayıda araştırmaya rağmen ilişkili bazı
genler bulunmakla birlikte tam olarak açıklanmış değildir.
Aralıklı
ışık uyaranına karşı oluşan yanıta göre
ışığa duyarlılık 3 alt grupta incelenebilir:
- Sadece ışık
uyaranla nöbet geçirenler
- Işık uyaranla ya da
uyaran olmadan nöbet geçirenler
- EEG’de
ışığa duyarlılığı saptanan
asemptomatik kişiler
Işığa
duyarlılık belirgin olarak yaşa bağımlılık
gösterir ve en fazla görüldüğü yaşlar 10-15 arasındadır.
Kadınlarda 10-25 yaşlarında görsel uyaran ile ortaya çıkan
nöbet ve EEG’de fotoparoksizmal yanıt sık görülür. Kadın/Erkek
oranı 1,5-2 civarındadır. Fotosensitif epilepsinin tüm dünyada
tanınması Pokemon isimli çizgi film ile olmuştur. Bu yüksek
kontrastlı güçlü ışık uyaran içeren çizgi filmi 1997
yılında Japonya’da seyreden yaklaşık 685 çocuk nöbet
geçirme nedeniyle acillere başvurmuştur.
Işığa duyarlı refleks epilepsili
olgularda çeşitli ışık kaynakları rol oynayabilir. En
sık bildirilen ışık kaynağı TV’dir. TV ile
birlikte veya ayrı olarak bilgisayar ekranı, video oyunları da
uyaran olarak rol oynamaktadır. Güneş
ışığının direkt şekilde veya
ağaçlı bir yolda aralıklı yansıması, lamba
ışığı, eğlence yerlerinde yanıp sönen renkli
ve parlak ışıklar, paternler, fotosensitif olgularda diğer
uyaranlardır.
Patern duyarlılığı: Aralıklı
ışık uyaranının ardından en sık görülen
görsel tetikleyicidir. Patern duyarlılığının
fotosensitivite ile yakın ilişkisi bilinmekte ve patern-fotosensitif
birlikteliği %70’e varan oranda bildirilmektedir. Hastalar çizgili duvar
kağıtları, döşemeler, üniformalar, yürüyen merdiven,
ızgaralar, radyatörler, üst üste duran tabakları uyaran olarak
bildirmektedir. Paterne duyarlı hastalar ile ışığa
duyarlı hastalar arasında demografik ve klinik farklılık
bildirilmemiştir. Ailesel özellik sıktır.
“Startle”
epilepsi: Startle epilepsi ani, beklenmedik ses veya
somatosensoriyel uyarı ile tetiklenen nöbetlerle karakterizedir. Nöbetler
genellikle jeneralize toniktir, fakat fokal olabilir ve genellikle etyoloji
yapısal bir lezyon kaynaklıdır. Startle nöbetlerinin
epileptojenik bir fokusun proprioseptif uyaran ile aktivasyonuna
bağlı olduğu düşünülmektedir. Startle epilepsi, epileptik
kökenli olmayan startle (irkilme) reaksiyonlarından ayırt
edilmelidir.
Sıcak
su epilepsisi: Banyo sırasında sıcak suyun etkisiyle
oluşan epilepsi, sıcak su epilepsisi veya banyo epilepsisi olarak
adlandırılmaktadır. Avustralya, Japonya, İngiltere gibi
ülkelerden izole olgular bildirilmektedir. Buna karşılık Güney
Hindistan’dan geniş seriler bildirilmiştir. Bu bölgenin iklim
koşulları, banyo sırasında aşırı sıcak
su ile başın üstüne dökülerek yıkanılması ve genetik
özelliklerin etkisi vurgulanmaktadır. Bu nedenle toplumlar arası
kültürel farklılıklar, coğrafi özellikler, genetik faktörler ve
sosyal alışkanlıkların rolü tartışılmaktadır.
Sıcak su epilepsisi, özgün bir dış uyaran olan “sıcak su
ile yıkanma” sırasında ortaya çıkan fokal
farkındalığın bozulduğu nöbetlerle karakterize refleks
epilepsi türüdür. En sık erkek çocuklarda rastlanır, iyi seyirlidir,
aylar ya da yıllar içinde remisyon beklenir. Ancak 40 yaşından
sonra dahi ortaya çıkabilir. Diğer taraftan spontan nöbetler de
gelişebilir. Nöbetten yoğun haz duyma ve bilinç kaybı
oluşuncaya kadar kompulsif bir şekilde su dökünmeyi sürdürme yani
self-indüksiyon çoğu olgu için en dikkat çekici özelliklerinden biridir.
Nörolojik muayene normaldir, yapısal lezyon genellikle
bulunmamaktadır. Yurdumuzda Avrupa ülkelerine göre sık
rastlandığı görülen bu tabloda genelde temporal bölgede epilepsi
odağı dikkati çekmektedir ve genetik köken olduğunu
düşündüren ailesel olgular vardır. En sık banyo
sırasında sıcak suyun baştan aşağı
dökülmesiyle ortaya çıkar. Çok defa fokal belirtilere yol açan
nöbetlerdir. Uyaranın özellikleri kişiden kişiye
değişkenlik gösterebilir; soğuk su ile veya küvette yıkanma,
duş alma, hatta elini soğuk su dolu kovaya sokma, yağmur
damlasının vücuda değmesi vb. nöbet için uyarıcı
olabilir. Doktora az başvurulması ve banyodaki self-indüksiyonla
tetiklenen nöbetlerin gizlenmesi söz konusu olabilmektedir.
Okuma
epilepsisi: Okuma epilepsisi,
idyopatik/genetik ve daha az oranda semptomatik epilepsi olarak alt gruplara
ayrılır. Primer okuma epilepsisinde spontan nöbetler
olmaksızın sadece okuma ile ortaya çıkan çenede atma
kasılma şeklinde nöbetler görülür. Genellikle başlangıç
yaşı ergenlik dönemidir, %40-50’sinde ailesel özellik
görülmüştür ve juvenil miyoklonik epilepsili bazı olgularda
görülebildiğine dikkat çekilmiştir. Erkek/Kadın oranı
1,8’dir. Gelişim öyküsü, nörolojik muayene, interiktal EEG ve görüntüleme
tetkikleri normaldir. Nöbetlerin sessiz veya yüksek sesle okuma ile ortaya
çıkmasının dışında; 1/4’inde heyecanlı veya
tartışmalı bir konuşma sırasında da nöbet
görülebilir. Olguların bir kısmında yazı yazma
sırasında kullandığı kolda ya sıçrama ya da
kasılma olur (grafojenik epilepsi). Sekonder okuma epilepsisi olanlarda
ise spontan nöbetler ile birlikte okuma ve ışık uyaran ile
indüklenen nöbetler görülür. Oral miyoklonik atmalar yoktur ve interiktal EEG
anormaldir. Görüntülemelerde lezyon görülebilir.
Yemek
yeme epilepsisi: Çok nadir görülür. Epilepsisi olanlarda
prevalansı 1/1000-2000 olarak bulunmuştur, ancak Sri Lanka’dan hayli
yüksek değerler bildirilmiştir. Daha kısa latanslı yutma
ile tetiklenen ve uzun latanslı kompleks tetik gerektiren tipleri
bildirilmiştir. Genelde sol temporal bölgede aktif epileptiform anomali
saptanır ve özgeçmişte bir tetikleyici olay öyküsü vardır,
erkeklerde sıktır. Yemek yeme epilepsisi olan hastalarda spontan
nöbetler de görülebilir.
Müzikojenik
epilepsi: Müzik ile tetiklenen nöbetlerdir, çok nadirdir,
prevalansı 1/10.000.000 olarak tahmin edilmektedir ve tanıda güçlük
nedeniyle az tanı konduğu düşünülmektedir. Erkeklerde (%76) daha
sık görülmektedir. Başlangıç yaşı 11-39 arasında,
ortalama yaş 14’tür. Farklı müzik çeşitlerinde (caz, klasik
müzik, folk parçaları vb) kişiye göre değişen, sesin
yüksekliği, ortamdan etkilenme, afektif içeriği de olabilen
farklı uyaranlar bildirilmiştir, en sık sağ temporal
bölgeden kaynaklanmaktadır.
Düşünme
ve belli uzaysal görevlerle ortaya çıkan nöbetler: Uyaranlar;
matematik, yazı yazmak, resim yapmak, karar vermek, kağıt oyunu
oynamak, satranç vb oyunları oynamak ve düşünmektir. Çok ender
rastlanan tablolardır.
Progresif
Miyoklonik Epilepsiler (PME)
Başlangıç
yaşı erken çocukluktan erişkinliğe dek olabilen bu tablodan
altta yatan çeşitli, genetik geçişli nörodejeneratif süreçler
sorumludur (Tablo 12-14). Çok sayıda olan etyolojik nedenlerin tümü
seyrek görülür ve PME tüm epilepsilerin yaklaşık %1’ini
oluşturur. Hepsinin ortak klinik özellikleri genellikle postür, aksiyon
veya dış uyarılar (ışık, ses ve dokunma) ile
tetiklenen ağır miyoklonustur, buna jeneralize tonik-klonik ve
diğer nöbetler eklenir. Tedaviye dirençli, giderek şiddeti artan ve
genelde mental açıdan da yıkıma yol açan tablolardır.
Kognitif yıkım dışında başlıca serebellar
olmak üzere çeşitli nörolojik, oftalmolojik ve sistemik bulgular
görülebilir. EEG’de tipik olarak temel aktivitede yavaşlama ve jeneralize
epileptiform deşarjlar, ışık
duyarlılığı ve bazen fokal bulgular saptanır (Şekil
11). Klinik olarak başlangıçta juvenil miyoklonik epilepsi gibi
selim epilepsi sendromlarıyla karışabilir ama EEG’de temel
aktivite genelde bu tablolara göre belirgin yavaştır.
Miyoklonik
nöbetleri izlenen bir hastada aşağıdaki bulguların
varlığında PME düşünülmelidir:
1) Miyoklonik nöbetler zemininde motor etkilenme
2) Progresif kognitif yıkım
3) Serebellar bulgular
4) EEG’de temel aktivitede yavaşlama
5) Uygun AEİ tedavisine rağmen dirençli
miyoklonik nöbetler
Tablo 12. Progresif miyoklonik epilepsi nedenleri (https://www.epilepsydiagnosis.org’dan
esinlenerek)
|
Nörodejeneratif
hastalıklar
·
Unverricht-Lundborg
hastalığı
·
Lafora
hastalığı
·
Dentatorubropallidoluysian
atrofi
·
Aksiyon
miyoklonus-renal yetmezlik sendromu
·
Juvenil
nöroaksonal distrofi
·
Pantotenatkinaz
ilişkili nörodejenerasyon (PKAN)
·
Juvenil
Huntington hastalığı
|
|
Metabolik hastalıklar
·
Mitokondriyal
ensefalopati- çatlak kırmızı lifler "rugged red
fibers" (MERRF)
·
Sialidoz
(“cherry red spot”-miyoklonus sendromu)
·
Nöronal
seroid lipofuksinozislar
·
Gaucher tip
III-a
·
Niemann
Pick hastalığı
·
Tetrahidrobiyopiterin
eksikliği
|
|
İmmün etyolojiler
·
Çölyak
hastalığı
|

Şekil 11. Progresif miyoklonik epilepsi için tipik EEG
bulguları olan temel aktivitede yaygın yavaşlama,
ışığa duyarlılık ve jeneralize düzensiz çok
dikenli dalga deşarjları görülmektedir. Lafora olgusu olan bu hastada
ayrıca oksipital bölgede dikenler de dikkati çekmektedir.
Tablo 13. Miyoklonus, nöbetler, ataksi ve demansla giden klinik
sendromların ilişkisi ve kesişmesi
|
|
Nöbetlerle
giden progresif ensefalopatiler
|
Progresif
miyoklonik epilepsiler
|
Progresif
miyoklonik ataksiler
|
|
Erken
klinik bulgular
|
Kognitif etkilenme
|
Miyoklonik nöbetler
tonik-klonik nöbetler
|
Ataksi
miyoklonus
|
|
Eklenen
klinik bulgular
|
Nöbetler veya miyoklonus
(görece seyrek ve hafif)
|
Ataksi veya demans, progresyon
|
Demans, tonik-klonik nöbetler
hafif veya yok
|
|
Başlangıç
yaşı
|
Genelde erken çocukluk
|
Geç çocukluk veya
ergenlik dönemi
|
Tipik olarak erişkin
|
|
Ana
nedenler
|
Birçok kalıtımsal
metabolik hastalık
|
Unverricht-Lundborg
MERRF
Lafora Hastalığı
Nöronal Seroid Lipofuksinozlar
Sialidozlar
|
MERRF
Spinoserebellar dejenerasyon
Çölyak hastalığı
|
Tablo 14. Progresif miyoklonus epilepsilerinin bazı görece
sık nedenleri
|
Hastalık
|
Başlangıç
|
Klinik
özellikler
|
Tipik
laboratuvar özelliği
|
Genetik
tanı
|
Tedavi
|
|
Unverricht-
Lundborg
Hastalığı
|
6-18 yaş
|
Uyarana duyarlı
(ışık, ses, dokunma) kısıtlılık yaratan
ağır miyokloniler; ataksi hafif, JTK kontrolü görece kolay, kognitif
etkilenme hafif/ yok
|
Yok
Tüm PME lerde görülebilen EEG,
dev SEP ve diğer bulguları var
|
Otozomal resesif
21q22.3; EPM1
Sistatin B geninde mutasyon
(Dodekamer ekspansiyonu veya nokta mutasyonları)
|
Palyatif, semptomatik (valproik
asit, +klonazepam, yüksek doz pirasetam, levetirasetam, zonisamid,
topiramat).
Karbamazepin, fenitoin gibi
ilaçlardan kaçınılmalı!
|
|
Lafora
Hastalığı
|
10-19
|
Miyokloni, oksipital nöbetler,
atipik absanslar, atonik, fokal farkındalığın
bozulduğu nöbetler, ağır demans
|
Deri, karaciğer veya beyin
biyopsisinde ışık mikroskopisiyle Lafora cisimleri
|
Otozomal resesif
EPM2a ve b; Laforin ve malin genlerinde
-delesyonlar
-nokta mutasyonları
|
Palyatif, semptomatik (valproik
asit, +klonazepam, yüksek doz pirasetam, levetirasetam, zonisamid, topiramat)
Karbamazepin, fenitoin gibi
ilaçlardan kaçınılmalı!
|
|
MERRF
|
Her yaş
|
Serebellar ataksi, miyokloni,
miyopati, nöropati, işitme kaybı, demans geri planda, kısa
boy, optik atrofi, işitme kaybı
|
Kan veya BOS laktat á, anormal kas MR spektroskopisi
Kas biyopsisinde
kırmızı çatlak lifler (%90)
|
Maternal kalıtım,
mitokondrial DNA’da tRNA da nokta mutasyonları
|
Ampirik; antioksidan vitamin
kombinasyonları ve koenzimQ ve L-karnitin
Nöbet tedavisi için VPA’dan
kaçınılmalı
|
|
NSL
Geç infantil
|
2,5-4
|
Miyoklonik, tonik-klonik,
atonik, atipik absans nöbetleri
|
Deri, rektal/beyin biopsisinde
elektron mikroskopisiyle kurvilinear profiller
-Fibroblast/lökositlerde
azalmış/ saptanamayan TPP1 enzim akt.
|
11p15 Lizozomal proteaz nokta
mutasyonları, diğer
|
Semptomatik, palyatif
|
|
NSL
juvenil
|
4-10
|
Görme bozukluğu, demans,
dizartri, ataksi, ekstrapiramidal özellikler
|
Kanda vakuollü lenfositler,
deri biyopsisinde/lenfositlerde parmak izi profilleri
|
16p12 Membran protein 1 kb
delesyonu
-Nokta mutasyonları
|
Semptomatik, palyatif
|
|
NSL
Erişkin
(Kufs)
|
11-50
|
Demans, ataksi, ekstrapiramidal
bulgular, miyokloni
|
Garanüler osmiofilik
depositler/ Parmak izi profilleri
|
CLN6,
CTSF, DNAJC5, GRN gen mutasyonları
|
Semptomatik, palyatif
|
|
Sialidozlar
Tip I
Tip
II
|
8-20
10-30
|
Ağır miyokloni,
fundusta cherry-red spot, tonik-klonik nöbet, ataksi, dismorfizm, korneada
bulutlanma, hepatomegali, iskelet displazisi, öğrenme bozukluğu
|
İdrar kromatografide
artmış siyalil-oligosakkaritler,
-lökosit/ fibroblast
kültürlerinde lizozomal enzim defekti (ağır nöroaminidaz
eksikliği (tip I ve II) ve parsiyel b-galaktosidaz
eksikliği (tipII)
|
NEU1
|
Semptomatik, palyatif
|
|
DRPLA
|
Her yaş
|
Serebellar atrofi, ataksi, koreatetoz,
miyokloni, epilepsi, demans, psikiyatrik semptomlar
|
MRG bulguları destekleyici
|
12p13
-CAG ekspansiyon Anormal CAG
tekrarının gösterilmesi
|
Semptomatik
|
NSL: Nöronal seroid lipofuksinoz,
DRPLA: dentatorubropallidoluysian atrofi, PME: Progresif miyoklonik epilepsi,
BOS: Beyin omurilik sıvısı, MRG: Manyetik rezonans görüntüleme
Özel Sendromlar
Hipokampal skleroz ile birlikte mezyal temporal lob
epilepsisi (MTLE-HS)
Bu tipik tablonun ana özellikleri
şunlardır:
Öykü:
·
Hastaların çoğunun özgeçmişinde yukarda
ayrıntılı olarak anlatılmış olan komplike febril
nöbet geçirme öyküsü vardır. Bu durumun MTLE-HS tablosunun nedeni mi yoksa
aynı patolojik sürecin erken dönemdeki gösterisi mi olduğu halen
tartışmalıdır.
·
Ailede epilepsi öyküsü de yüksek oranda saptanmaktadır.
·
Başlangıç yaşı genellikle ilk onyılın son
yarısında olur.
·
Nöbetlerin ergenlik ve genç erişkinlik döneminde remisyona girmesinin
(lusid interval) ardından tekrar başlayıp ilaç tedavisine
dirençli duruma gelmesi tipiktir.
·
İnteriktal davranış değişiklikleri ve psikiyatrik
komorbid hastalıklar (sıklıkla depresyon) görülür.
Nöbet Özellikleri:
·
Fokal nöbet başlangıcında öncü belirti yani aura
genellikle vardır. Bu belirti MTLE-HS’de en sık olarak mideden
yükselen tuhaf bir his (rising epigastric sensation) şeklindedir. Bunun
dışında diğer otonom ve kognitif semptomlar (deja vu,
jamais vu vb.) emosyonlar (en sık korku), koku ve tat duyularına
ilişkin duyumsamalar da fokal nöbet olarak görülebilir. Fokal otonom,
kognitif veya duysal nöbetleri takiben farkındalığın
bozulduğu, aniden durma ve donmayı izleyen ağız
şapırdatma ve yutkunma, yalanma hareketleri gibi oroalimenter
otomatizmalar görülür. İktal deşarjın karşı
tarafındaki kolda distoni ve aynı taraftaki kolda çeşitli
otomatizmalar tipiktir. Süre genellikle 1-2 dakikadır.
·
Fokal başlayıp bilateral tonik-klonik nöbete dönüş görece
nadir görülür.
·
Postiktal dönemde genellikle oryantasyon bozukluğu, bellek
bozukluğu, odak dominant hemisferdeyse konuşma bozukluğu
görülür, postiktal dönem genellikle dakikalar sürer.
·
Diğer epileptik nöbet tipleri ve özel bir tetikleyici olmazsa genelde
SE tablosu görülmez.
·
Fokal motor veya spesifik duysal semptomlar ile (örn, görme kaybı veya
parestezi vb.) nöbetin başlaması MTLE-HS’de görülmez.
Nörolojik muayene:
·
Genellikle normaldir. Fokal nörolojik belirti tanıda kuşku
yaratmalıdır.
·
Bellek defektleri (genelde odak soldaysa sözel, sağdaysa görsel
modalitede) görülebilir, ancak belirgin ve yaygın ilerleyici kognitif
defektlerin varlığı görülmez.
·
Odağın karşı tarafındaki nazolabial telemin silik
olduğu bazı olgularda gözlenir (Emosyonel fasyal parezi).
EEG:
·
İnteriktal dönemde odak tarafında veya bazen iki yanlı
bağımsız ön temporal bölgede keskin dalga aktivitesi tipiktir.
Ayrıca aynı bölgede aralıklı veya devamlı ritmik ya da
düzensiz delta frekansında yavaş dalga aktivitesi görülebilir (Şekil
12a).
·
Yüzeyel EEG ile kaydedilen nöbet aktivitesi fokal (ön temporal bölgede) 5-7
Hz ritmik aktivite şeklinde başlar ve genellikle yayılım ve
frekans değişimleri gösterir (Şekil 12b).
·
EEG’de başka bölgelerde fokal diken varlığı ile
yaygın veya temporal dışı yavaş aktivite tanı ve
cerrahi tedavinin başarısı açısından kuşku
yaratmalıdır.
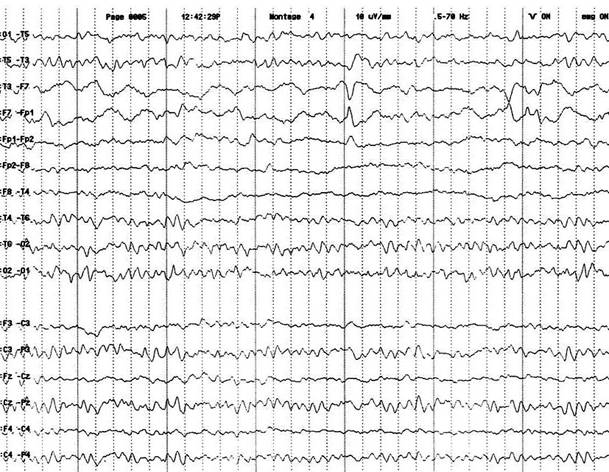
Şekil 12a. MTLE-HS sendromu tanısıyla izlenen bir
hastanın interiktal EEG’sinde sol ön temporal bölgede delta dalgaları
ve F7 elektrod pozisyonunda faz dönmesi gösteren keskin dalga aktivitesi
görülmektedir.
Şekil 12b. MTLE-HS
sendromlu başka bir hastanın preoperatif incelemesinde kaydedilen
sağ frontotemporal bölgedeki ritmik “build up” gösteren teta
dalgaları ve kas artefaktlarıyla şekillenen tipik iktal
aktivitesi izlenmektedir.
Görüntüleme:
·
Manyetik rezonans görüntüleme (MRG), epilepsi hastalarında
yapısal lezyonları görüntülemek amacıyla önerilen kurallara
uygun şekilde yapılmalıdır (Bakınız: Laboratuvar
incelemeleri-Bilgisayarlı Tomografi ve Manyetik Rezonans Görüntüleme).
·
MRG ile bir taraf hipokampusun daha küçük olduğu gözle ya da ölçülerek
(volümetrik analiz) saptanır (Şekil 13).
·
Ayrıca MRG ile ince kesitlerle incelendiğinde hipokampus
içyapısının bozulmuş olduğu ve T2 ve FLAIR
sekanslarında artmış sinyal özelliği görülür (Şekil
14).Bu durum bazen bilateral olabilir, normal insanlarla kıyaslama
gerekmektedir.
·
Bazen tüm temporal lobun daha küçük olduğu ve temporal boynuzun daha
geniş olduğu dikkati çeker.

Şekil 13. Sağ mezyal temporal yapılarda sola
kıyaslandığında belirgin atrofi dikkati çekmektedir.
Şekil 14. Sağ tarafta MRG’de T2 hiperintensitesi (ok) ve
solda izlenebilen normal hipokampal içyapının sağ tarafta
kaybı izlenmektedir.
Fonksiyonel görüntüleme:
·
PET tekniği daha üstün bir fonksiyonel inceleme yöntemi olmakla
birlikte pahalıdır. PET ile mezyal temporal bölgede, tüm temporal
lobu içine alabilen görece geniş ve bazen talamuslara uzanan
hipometabolizma görülmesi önemli bir destekleyici lateralizan veridir.
·
SPECT ise daha düşük görüntü kalitesine rağmen, görece ucuz
olduğu için ve özellikle iktal dönemde inceleme
yapılabildiğinden yardımcı bir yöntem olarak
kullanılmaktadır. SPECT ile interiktal dönemde odak bölgesinde
hipoperfüzyon ve iktal dönemde tipik hiperperfüzyon saptanır. Saptanan
hipoperfüzyon bölgesi genellikle tüm temporal lobu içine alacak şekilde
geniş bir alandadır.
Patofizyoloji:
·
Temporal lobun iç yan yapıları olan amigdala ve hipokampusda
sklerotik değişikliklerle karakterizedir. Tipik paternde hipokampusda
%30 oranından fazla hücre kaybı görülür.
·
Dual patoloji (eşlik eden patoloji) olarak heterotopi ve hamartomalar
eşlik edebilir. Mikrodisgenezis sıktır.
MTLE-HS tanısı klinik planda konduktan
sonra, iyi planlanmış bir MRG incelemesi ve video-EEG incelemeleriyle
nöbet kaydı yapılarak hastanın operasyona hazırlanması
söz konusudur. Hastaların nöropsikolojik testleri mutlaka detaylı
şekilde yapılmalıdır. Geçmişte WADA testi olarak
adlandırılan anjiografik olarak sodyumamital verilmesi ile bir
hemisferin geçici blokajı sonrasında çeşitli nöropsikolojik
testlerin uygulanarak hastanın postoperatif defektlerinin geçici olarak
belirlenmesi tekniği, günümüzde yerini nöropsikolojik değerlendirme
ve fonksiyonel MRI tekniklerine bırakmıştır. Tipik MTLE-HS
olgusunda invazif girişimlere genellikle gerek duyulmaz ama bazen
hipokampal sklerozun iki yanlı olması durumu güçleştirir ve
invazif tetkikler gerekebilir.
MTLE-HS tanısıyla incelemeleri sonucunda
operasyona verilen hastaların %80 kadarında anteromedial temporal lob
rezeksiyonu ya da selektif amigdalo-hipokampektomi girişimleriyle,
hastalar nöbetsiz kalmakta ya da çok belirgin oranda nöbetleri
azalmaktadır.
Rasmussen
sendromu
Rasmussen sendromu immün aracılı bir
epilepsi sendromudur. Nöbetler sıklıkla 5-6 yaşında
başlar. Başlangıç yaşı 1-10 arası
değişebilir. Nöbet başlangıcına kadar olan dönemde
mental ve motor gelişim normaldir. Fokal motor nöbetler, epilepsia
parsiyalis kontinua, fokal başlangıçlı bilateral tonik-klonik
nöbete dönüşen nöbetler ve fokal atonik nöbetler izlenebilir.
Epilepsia parsiyalis kontinua genellikle hayatın ilk on
yılında başlayan, ancak hemen her yaşta görülebilen bir
fokal epilepsi şeklidir. Hastada neredeyse devamlı bir şekilde
süren farkındalığın korunduğu fokal motor semptomlu
nöbetler (klonik) görülür. Genelde bu nöbetin başlaması Rasmussen
ensefaliti adını taşıyan, sadece bir hemisferi tutan kronik
bir ensefalitin bulgusu olarak görülür. Etyolojide bu kronik ensefalitten başka
en sık inme olmak üzere kortikal displazi, tümörler, vasküler lezyonlar
sorumlu olabilir.
Rasmussen ensefalitinde yavaş progresif bir
nörolojik kötüleşme izlenir. Hemiparezi, mental retardasyon ve dominant
hemisfer tutulmuşsa afazi olur. Hastaların %50’sinde ilk yıl
nörolojik kötüleşme ile birlikte nöbetler başlar. Radyolojik olarak
da yavaş progresif seyirli, genellikle tek yanlı serebral atrofi
tipiktir (Şekil 15). Patolojik olarak viral ensefalit
bulgularının olması tanıyı koydurur. Ancak Rasmussen
ensefaliti tanımlandıktan sonra birçok atipik olgu bildirimi
olmuştur. İlk nöbetler jeneralize tonik klonik nöbetler (%30),
jeneralize veya fokal motor SE (%20) şeklindedir. Tipik olgularda ilaçlara
dirençli tek yanlı fokal motor nöbetler ve/veya temporal lob nöbetleri ortaya
çıkar. En sık olarak yukarıda da belirtildiği gibi, fokal
motor SE olan “epilepsia parsiyalis kontinua” tablosu görülür. Atipik olarak
görece yavaş seyirli olan, erişkin yaşta başlayan veya
epilepsia parsiyalis kontinuanın miyoklonik formları ile ortaya
çıkan klinik bulgular olabilir. Kranial görüntülemede kontralateral atrofi
bazen beklenen kadar belirgin olmayabilir, bazen de bilateral tutulum
görülebilir. EEG’de etkilenen hemisferde en çok temporal ve santral olmak üzere
polimorfik delta frekansında yavaş dalgalar ve multifokal diken,
kesin-yavaş dalgalar görülür. Klinik nöbetin kontralateralinde
elektrografik nöbetler izlenebilir. Beyin omurilik
sıvısının (BOS) incelemesinde olguların
yaklaşık %50’sinde protein artışı ve lenfosit
ağırlıklı hücre artışı vardır. Oligoklonal
band hastaların %0-67’ sinde görülür. Glutamat reseptörü subunit 3’e
karşı oluşan antikor (anti-Glu3R) saptanabilmekle birlikte bu
hastalık için spesifik değildir ve tanı için artık
kullanılmamaktadır. Beyin biyopsisi sadece şüpheli olgularda
klinik ve radyolojik destek yok ise tanıyı doğrulamak için
yapılır. Nöropsikolojik testlerin tanı için değeri yoktur,
ancak takipte progresyonu göstermek için önemli yer tutar.
Ayırıcı tanıda unilateral epileptik nöbetlere yol açan
tablolardan kortikal malformasyonlar, Sturge-Weber sendromu, inme,
hemikonvülziyon-hemipleji-epilepsi sendromu, gliomatozis serebri gibi tümörler,
metabolik bozukluklar, MELAS (mitokondriyal
miyopati, ensefalopati, laktik asidoz, inme benzeri ataklar) veya inflamatuar durumlar
(vaskülit, HIV, meningoensefalit vb) düşünülmelidir. Tedavide AEİ
başlanır, ancak genelde dirençli nöbetlerdir. Beraberinde intravenöz
immünoglobulin, plazmaferez veya oral takrolimus verilir. Ayrıca
hemisferektomi/hemisferotomi bu olgularda önerilebilmektedir. Uzun dönem
immünoterapi şu an için gündemdedir.

Şekil 15a. Rasmussen ensefaliti olgusunda MR örneği:
15 yaşında erkek, 3 yıl önce başlayan jeneralize
konvülziyonlara, 2 yıl önce solda devamlı fokal motor nöbetler
eklenmiş. Sağ hemisferde frontal, temporal loblarda ve derin ak
maddede belirgin olmak üzere hacim kaybı dikkati çekmektedir.

Şekil 15b. Aynı hastanın EEG incelemesinde
sağ pariyetal bölgede sol hemisfere de yayılan nöbet aktivitesi
görülmektedir.
Hipotalamik hamartom ile
ilişkili jelastik nöbetler
Tuber
sinereum ve/veya mamiller cisimciklerde gelişen tümör benzeri
malformasyondur. Klinik olarak jelastik nöbetlerle karakterize epilepsi ve
erken puberte önemli özelliklerdir. Jelastik nöbetler yaşamın ilk
yıllarında hatta doğumla birlikte başlar. Mekanik ve
uygunsuz olarak atılan kahkahalar görülür. Genellikle kısa sürelidir
(10-30 saniye) ve nöbetlerin yarısında bilinç korunur. Pupil
dilatasyonu, yüzde kızarma gibi otonomik özellikler ve nefes tutma buna
eşlik eder. Bazen hipermotor, oroalimenter otomatizmalar, göz deviyasyonu,
tonik veya klonik fasyal kontraksiyonlar da jelastik nöbet sırasında
görülebilir. Hastaların yarısından fazlasında nöbetlerde
progresyon olur. Jeneralize tonik, atonik, tonik-klonik veya absans nöbetleri
eklenir, bazılarında Lennox-Gastaut sendromu gelişir.
Davranış ve kognitif problemleri sık görülür. Yüzeyel EEG’nin
iktal bulguları yanıltıcı olabilir. İnvazif video EEG
incelemesinde derinlik elektrodları iktal deşarjların
hamartomdan kaynaklandığını gösterir. Kranial MRG’de tipik
lokalizasyonda T2 ve FLAIR’de hiperintensite ve T1
ağırlıklı kesitlerde hipointensite olur. AEİ’ler
genellikle yetersiz kalır. Cerrahi olarak birçok farklı
yaklaşım vardır. Olgu sayısı az olduğundan karşılaştırma
çalışması bulunmamaktadır. ‘Gamma knife’ veya stereotaktik radyasyon
cerrahisi yaşı büyük hastalarda ve hamartomu küçük olanlarda tercih
edilmektedir.
EPİLEPSİLİ
HASTANIN DEĞERLENDİRİLMESİ
ANAMNEZ
VE MUAYENE
Epilepsi
nöbeti bir semptomdur. Altta yatan çok sayıda sebepten hangisinin sorumlu
olduğunu bulmak kimi zaman sadece iyi alınmış
ayrıntılı bir anamnezle (örn, tipik genetik geçişli
idyopatik/genetik jeneralize ve fokal epilepsi sendromlarında olduğu
gibi) mümkündür. Bazen MRG gibi en ayrıntılı yapısal
görüntüleme yöntemleri uygulanmış bir hastada kan kalsiyum düzeyi
bakılmadığı için gerçek etyoloji
anlaşılamayabilir. Epilepsi tanısı ve
değerlendirmesinde anamnezde hastanın perinatal öyküsü, gelişme
basamakları, kafa travması, MSS enfeksiyonu, ailede epilepsi ve
diğer sık görülen hastalıkların defalarca ve
ayrıntılı bir şekilde sorgulanması çok önem
taşımaktadır. Hastalığın başlangıç
yaşı da etyolojik açıdan önem taşır. Epilepsi nöbeti
beynin hemen her hastalığının sonucu olabileceği gibi
sistemik birçok hastalıkta ve iyatrojenik çeşitli nedenlerle de
epileptik nöbet oluşabileceği (intoksikasyonlar, postoperatif
metabolik anoksik nedenler, çeşitli ilaçların terapötik dozda bile
epilepsi eşiğini düşürmesine bağlı olarak vb.)
unutulmamalıdır. Bu nedenle çok ayrıntılı anamnez
alınmasını izleyerek detaylı bir nörolojik ve sistemik
muayene mutlaka yapılmalıdır.
LABORATUVAR
İNCELEMELERİ
Epilepsili
hastanın tanısında ve takibinde klinik değerlendirmenin
önceliği ve vazgeçilmez olduğu yadsınamaz bir gerçektir. Bununla
birlikte günümüzde çeşitli laboratuvar yöntemleri bu konuda hekime çok
yararlı bilgiler sunacak duruma gelmiştir.
Elektroensefalografi
(EEG)
İlk
olarak 1940'larda kullanılmaya başlayan bu yöntem bugün için de
epilepsi biliminin temel direğini oluşturmaktadır. EEG beyindeki
geniş bir nöron grubunun elektriksel aktivitesindeki dalgalanmanın
kayıtlanması ilkesine dayanmaktadır. Saçlı deriden
kayıtlanan potansiyellerin çoğu piramidal hücrelerdeki toplam
sinaptik potansiyellerin ekstrasellüler akımlarla ilişkisinin
sonucudur (Ayrıca bakınız: Sinir
Sistemi Semiyolojisi, Elektroensefalografi).
Klinik
pratikte EEG incelemelerinin çoğunluğu interiktal dönemde
kayıtlanmaktadır. EEG incelemesinde, zemin aktivitesindeki
değişiklikler (belirgin asimetri veya yavaşlama), yavaş
dalga aktivitesi, epileptiform deşarjlar (diken, keskin ve diken-dalga
deşarjları) kayıtlanır. Öte yandan, sulkusların
içerisinden, derin yapılardan, insular korteks gibi gömülmüş
bölgelerden yeterli şekilde aktivite kaydı yüzeyel EEG
elektrodlarına yansımaz. Bu nedenle normal bir EEG’nin epilepsiyi
dışlamayacağı unutulmamalıdır. İnteriktal
epileptiform deşarjlar (İED), epilepsinin göstergesidir. İEDler,
belirgin büyüklükteki fokal kortikal alanda meydana gelen membran
potansiyelindeki eksitatuar potansiyel değişimi sonucu oluşur ve
bu değişime, paroksizmal depolarizasyon kayması denilmektedir.
İED, yüzeyel EEG ile kayıtlanabilmesi için en az 6 cm2’lik
bir kortikal yüzeyde oluşması gerekmektedir.
Epilepsili
hastalarda tanıda, epileptik nöbet ve epilepsi sendromunun
sınıflamasında EEG incelemesinin rolü çok önemlidir. EEG'nin
epilepsili olgunun değerlendirilmesine başlıca
katkılarını 3 ana maddede özetlemek olasıdır:
- Klinik olarak konulmuş olan
tanının desteklenmesi ve doğru tanı konmasına
yardım
- Nöbet kaydı yapılabilirse veya
dolaylı bulgularla nöbet tipi ve buradan hareketle epilepsi
sendromunu belirlemesi
- Odağın lateralizasyon-lokalizasyonu
hakkında bilgi verebilmesi
Rutin EEG ilk nöbetle gelen
hastada en önemli testtir. Gözlemsel çalışmalar, ilk nöbet ile gelen
hastada, takip eden 2 yıl içerisinde nöbet tekrarlama oranını
%40-50 olarak bildirmektedir. EEG’de İED izlenen hastalarda, nöbet tekrar
etme riski normal popülasyona kıyasla iki katına
çıkmaktadır. İlk nöbet ile gelen hastalarda ilk EEG’de anomali
saptama oranı %32-59 arasında değişmektedir. Bu oran, EEG
incelemesi nöbetten sonraki ilk 24-48 saatte yapılırsa
artmaktadır. İlk nöbet ile gelen hastanın ilk EEG incelemesinde
zemin aktivite değişikliği veya fokal yavaş dalga
aktivitesi izlenirse nöbetin tekrar etme olasılığı
%40’ın üzerinde, İED izlenirse %60’ın üzerinde, EEG normal
tespit edilirse bu oran %30’un altında olmaktadır. 2017
yılında yayınlanan yeni epilepsi tanımına, %60
üzerinde tekrar etme riski bulunan ilk epileptik nöbet varlığı
da eklenmiştir. Epilepsili hastaların sadece %10’unda tekrar edilen
EEG incelemeleri normal olmaktadır. İdyopatik/genetik jeneralize
epilepsilerde bu oran düşükken, fokal nöbetli hastalarda daha
sıktır. Üç veya daha fazla kez tekrarlanan EEG incelemesinde,
İED saptama oranı yükselmekte ve %80-90’a ulaşmaktadır.
EEG’de patolojinin saptanmasında aktivasyon yöntemlerinin iyi
uygulanması esastır, gerekirse uyku kayıtları yapılmalıdır.
Bunun yanında, her EEG anomalisinin epilepsi ile eşdeğer
olmadığı unutulmamalıdır. Epileptik nöbet öyküsü
olmayan erişkinlerin %0,2-0,5’inde ve çocukların %3,5’inde EEG
incelemesinde İED görülebilir. Ayrıca beyin ile ilişkili hastalığı
olan (hipoksi, infarkt, tümör vb) ancak epileptik nöbeti olmayan
hastaların EEG incelemelerinde de anormalilik gözükebilir.
Anormal EEG bulguları, nöbet
tipi ve epilepsi sendromunun sınıflandırılmasında
yardımcı olur:
- Simetrik, senkron, hemisferlerin önlerinde
belirgin 3 Hz ve daha hızlı olan jeneralize diken ve
multidiken-yavaş dalga deşarjları, idyopatik/genetik
jeneralize epilepsi sendromları için oldukça spesifiktir. Bu
sendromlarda, EEG zemin aktivitesi postiktal dönem dışında
normaldir. Jeneralize diken ve multidiken-yavaş dalga deşarjları,
idyopatik/genetik jeneralize epilepsi sendromlarının tiplerine
özgü olmasalarda bazı sendromlar için ayırt edici özellikler
bulunur. Absans epilepsisinde ritmik, düzenli 3 Hz jeneralize
diken-yavaş dalga deşarjları görülürken, juvenil miyoklonik
epilepside 3Hz’den hızlı, fragmante multi-diken-yavaş dalga
deşarjları izlenir. Hiperventilasyon, çocukluk çağı
absans epilepsisinde deşarjları çoğunlukla tetikler.
Juvenil miyoklonik epilepside ise, uyku deprivasyonu ile deşarj
sıklığında artış ve aralıklı ışık
uyaranına karşı duyarlılık görülebilir.
- Temel aktivite yavaşlaması, 3Hz’den
daha yavaş jeneralize diken-yavaş dalga deşarjları,
jeneralize paroksizmal hızlı aktivite ve multifokal İED’ler
gelişimsel ve epileptik ensefalopatilerde görülen EEG bulgularıdır.
Jeneralize paroksizmal hızlı aktivite, sıklıkla
non-REM uykusunda görülür ve jeneralize tonik nöbet ile ilişkilidir.
- İdyopatik/genetik fokal epilepsilerden
Rolandik epilepside, yüksek amplitüdlü santrotemporal diken-yavaş
dalga deşarjları izlenir ve bu deşarjlar non-REM’de oldukça
sıklaşır. Diğer idyopatik/genetik fokal epilepsi
sendromlarından olan Panayiotopoulos sendromunda bilateral hemisfer
arka yarılarına lokalize olmuş yüksek amplitüdlü diken-yavaş
dalga deşarjları görülürken, Gastaut sendromunda uyku ile
tetiklenen oksipital diken-yavaş dalga deşarjları izlenir.
İdyopatik/genetik fokal epilepsi sendromlarında temel aktivite
normaldir ancak bu grupta dikkat edilmesi gereken nokta CSWS ile seyreden
ensefalopati tablosudur. Uykuda elektrofizyolojik SE olarak da
tanımlanan bu tablo için özellikle davranışsal ve kognitif
şikayetleri başlayan çocuklarda uyku EEG kayıtlaması
yapılmalıdır.
- Anterior temporal bölgede izlenen İED ve
temporal aralıklı ritmik delta aktivitesi olan TIRDA, MTLE’ye
işaret eder.
- Jeneralize yavaş dalga aktivitesi, diffüz
ak madde hasarı ile ilişkilendirilmiştir ve etyolojisi
nonspesifiktir. Toksik, metabolik, enfeksiyöz veya inflamatuar
ensefalopatilerde izlenebilir.
- Fokal yavaş dalga aktivitesi, subkortikal
ak maddeyi etkilemiş kortikal yapısal lezyon (infarkt, hematom,
tümör, ensefalit vb.) ile ilişkilidir.
EEG incelemesinde İED
varlığı ve sıklığı, klinik nöbet
sıklığı veya tedavi cevabı ile doğrudan
ilişkili değildir. Nöbetsiz olarak izlenen hastanın ilaç
kesiminde prognoz açısından bilgi verebilir. En az 2-4 yıl
nöbetsiz olan bir hastanın, EEG bulgularının kaybolmuş
olması ilaç kesimi sonrası nöbetsizlik olasılığı
arttırmaktadır. Epilepsi cerrahisi sonrası, İED
varlığı nöbet tekrarının habercisi olabilir.
Uzun süreli
video-EEG monitorizasyon,
epilepsi ayırıcı tanısı, epileptik nöbet ve sendrom
tipinin belirlenmesi veya doğrulanması, ayrıca epilepsi
cerrahisi öncesi elektroklinik nöbet kaydedilmesi amacıyla
yapılır. Epileptik olmayan psikojen nöbetler, hem epilepsi
ayırıcı tanısında hem de epilepsili hastalarda
komorbidite olarak klinik değerlendirmede zorluklara neden
olmaktadır. Video-EEG monitorizasyon, non-epileptik psikojen nöbet
dışında epilepsi ayırıcı tanısında
bulunan parasomni ve noktürnal hareket bozuklukları gibi
hastalıkların tanısında faydalıdır. Klinik ve
rutin elektroensefalografik değerlendirme epileptik nöbet ve sendrom
belirlenmesinde çoğunlukla yeterli olmaktadır ancak
sınıflandırılmanın yapılamadığı
durumlarda video-EEG monitorizasyon iktal ve interiktal değerlendirme
olanağı sağlar. Bunların yanında, epilepsi cerrahisi
öncesi epileptojenik odağın belirlenmesinde kullanılır. Bu
amaçla, interiktal EEG bulgularının yanında, video-EEG monitorizasyonla
iktal değerlendirmenin yapılması önemli bilgiler vermektedir.
Video-EEG monitorizasyon
incelemesinde nöbet kaydı yapılarak nöbet tiplerine göre tipik
elektrofizyolojik paternler saptanabilir (Tablo
15). Kural olarak, nöbet ile birlikte elektrofizyolojik temel aktivitede
bir değişiklik olmalı ve yeni bir ritmik aktivite ortaya
çıkmalı, bu ritmik aktivite hemisfer bölgelerine yayılmalı
ve değişiklik (evolusyon) göstermelidir. Nöbet ile birlikte görülen
tipik elektrofizyolojik paternler; amplitüd supresyonu (elektrodekremental patern),
amplitüdde ani yükselme (jeneralize diken-dalga), paroksizmal hızlı
aktiviteler, ritmik yavaş dalgalar ve ritmik diken aktivitesidir.
Ayrıca fokal nöbetlerde iktal deşarjların elektrofizyolojik
olarak lateralizasyonu ve lokalizasyonu yapılır. Bu şekilde
nöbetlerin elektrofizyolojik olarak kaynaklandığı beyin bölgesi
tespit edilebilir. Bu incelemeler aynı zamanda nöbet semiyolojisinin de
çok ayrıntılı analizine olanak sağlamaktadır (Şekil
16). Özellikle mezyal temporal lob, dorsolateral frontal lob nöbetlerinin
yüzeyel EEG ile tespiti, neokortikal temporal, mezyal frontal, oksipital lob
nöbetlerine kıyasla daha kolaydır. İkinci grup nöbetlerde intrakranial video-EEG monitorizasyonu
epileptik odak lokalizasyonunda faydalı bilgiler sağlar. Bunun
yanında dual patolojilerde ve ‘eloquent’ korteks komşuluğundaki
lezyonların cerrahi öncesi değerlendirilmesinde önemlidir.
İntrakranial video-EEG monitorizasyonunda subdural EEG elektrodları
ve/veya derinlik elektrodları cerrahi operasyon ile yerleştirilir. En
önemli avantajı, biyolojik sinyallerin kaydedilmesini engelleyen kemik
yapının ortadan kalkmasıdır. Bu sayede derin
yapılardan kaynaklanan deşarjların kayıtlanması mümkün
olabilmektedir. Elektrokortikografi, cerrahi sırasında korteks üzerine
yerleştirilen elektrodlar ile elektrofizyolojik kayıtlama yöntemidir.
Elektrokortikografi eşliğinde lezyon rezeksiyonu ile ilişkili
kapsamlı çalışmalara ihtiyaç vardır ancak ameliyathanede
‘eloquent’ korteksin fonksiyonel görüntülenmesinde kullanılmaktadır.
Son yıllarda epileptojenik odak tespitinde, görüntüleme destekli
bilgisayar arayüzü ve yüksek yoğunluklu EEG kullanılarak yapılan
elektriksel kaynak görüntüleme (electrical source imaging-ESI) epilepsi
cerrahisi ile uğraşan ileri merkezlerde başvurulan bir yöntem
olarak bildirilmektedir.
Tablo 15. Nöbet tiplerine göre elektrofizyolojik paternler
|
Nöbet tipi
|
Elektrofizyolojik
patern
|
|
Absans nöbeti
|
3 Hz jeneralize diken-dalga
|
|
Atipik absans
|
<2,5 Hz jeneralize
diken-dalga
|
|
Tonik nöbet
|
Elektrodekremental patern ve
onu takip eden jeneralize paroksizmal hızlı aktivite (Jeneralize
düşük voltajlı EEG, saniyeler sonra giderek amplitüdü artan 20-40
Hz hızlı ritmler, verteks ve ön bölgelerde daha yüksek amplitüdlü
olabilir)
|
|
Atonik nöbet
|
Jeneralize diken-dalga ve atoni
ile birlikte yüksek amplitüdlü yavaş dalgalar (Fotik stimülasyon ile
tetiklenirse miyoklonik-atonik nöbetler düşünülmeli, miyokloni varsa
multidiken de eşlik eder)
|
|
Miyoklonik nöbet
|
>3 Hz jeneralize diken-dalga
ve multidiken-dalga
|
|
Epileptik spazm
|
Yüksek voltajlı trifazik
keskin veya yavaş dalgayı takiben düşük amplitüdlü
hızlı ritimler veya jeneralize amplitüd supresyonu
|
|
Jeneralize tonik klonik nöbet
|
Jeneralize hızlı
ritmik dikenler (tonik fazda) ve küme (burst) halinde gelen diken-yavaş
dalga (klonik fazda)
|
|
Mezyal temporal lob
kaynaklı nöbet
|
Ritmik teta veya delta
aktivitesi (sıklıkla), evolusyon
|

Şekil 16. Çeşitli epilepsili olgularının,
tanısal veya preoperatif hazırlık amaçlı video-EEG
incelemeleri sırasında kaydedilen nöbet semiyolojisi
açısından tipik video kesitleri: Üst sırada en solda: Sağ
mezyal temporal lob epilepsisi (MTLE) olan hastada tipik başta sağa
doğru ipsiversiyon ve sağ elde ipsilateral otomatizmaya eşlik
eden kontralateral distoni; üst sıra ve ortada: Startle epilepsisi olan
bir hastada kaydedilen asimetrik tonik nöbet görünümü; üstte en sağda:
Frontal lob epilepsili bir hastada uyku sırasında ortaya çıkan
hipermotor tipte bacak otomatizmaları; Altta en sağda: Lennox-Gastaut
sendromlu bir olgunun uykuda ortaya çıkan tonik nöbeti; altta ortada:
MTLE’li bir olguda sekonder jeneralize tonik-klonik bir nöbetin ilk asimetrik
başlangıç anı; altta en solda nadir görülen bir absans nöbeti
tipi olan miyoklonik absans nöbeti geçiren bir olgunun omuzlarındaki
miyoklonik hareket.
Nöropsikolojik
Değerlendirme
Nöropsikolojik
değerlendirme, epilepsili hastalarda çeşitli amaçlarla
kullanılabilir. Bunlardan ilki hastanın mental durumunun belirlenmesidir.
Epilepsili hastaların normallere oranla kognitif bozukluklar
gösterebildikleri bilinmektedir. Bu defektler yapısal lezyonlarla bir
ölçüde açıklanmakla birlikte, gösterilebilen bir yapısal lezyonu
bulunmayan olgularda da kognitif defektlerin varlığına dikkat
çekilmiştir. İkinci olarak amaç AEİ tedavilerinin etkilerinin
gözlenmesidir. Eski AEİ’lerin, özellikle fenitoin ve barbitüratlar ve yeni
ilaçlardan özellikle topiramatın kognitif parametrelerde bozukluğa
yol açabildiği gösterilmiştir. Nöropsikolojik değerlendirmenin
bir diğer kullanım alanı ise epilepsi cerrahisi öncesi
değerlendirmedir. Epileptojenik alanın lateralizasyonu, cerrahi
sonrasında gelişebilecek kognitif bozuklukların öngörülmesi, dil
dominansının belirlenmesinde yol gösterici bilgiler verir.
Temporal
lob epilepsisinde, sol taraf bulguları sözel öğrenme/bellek ve
isimlendirmeyi içerirken, sağ tarafta görsel öğrenme/bellek
fonksiyonları bulunur. Frontal lob fonksiyonlarından dikkat, yürütücü
fonksiyonlar, çalışan bellek değerlendirilebilir ancak
lateralizasyon yapılması oldukça zordur. Pariyetal ve oksipital lob
için standart testler yoktur.
Cerrahi
sonrası gelişebilecek kognitif bozukluklar önemli bir sorundur ve
birçok belirleyicisi vardır. Fonksiyonel olarak sağlam ve çevresiyle
fonksiyonel bütünlüğü bozulmamış beyin dokusunun
çıkarılması daha kötü kognitif bozukluk ile sonuçlanır.
Bunun yanında, zaten kötü kognitif fonksiyon daha kötü
olamayacağı gibi cerrahi sonrası nöbet
sıklığının ve AEİ kullanımının
azalmasıyla kognitif fonksiyonlarda iyileşmeye neden olabilir. Bunun
yanında, çıkarılan lezyonun tarafı, büyüklüğü,
hastanın yaşı ve cinsiyeti, ameliyat sırasındaki
komplikasyonlar ve çevre dokularda meydana gelen hasar da cerrahi sonrası
kognitif bozukluğun belirleyicileridir.
Cerrahi
sonrasında istenmeyen kognitif bozukluk riskini attıran faktörler:
- Cerrahi öncesinde tamamen normal
nöropsikolojik test
- MRG lezyonunun olmaması
- Cerrahi öncesi çok düşük nöropsikolojik
performans (düşük rezervin göstergesi)
- Bilateral patoloji (örn, bilateral hipokampal
skleroz)
Epilepsili
hastaların ayrıntılı nöropsikolojik testlerle
değerlendirilmesi, takip ve tedaviyi yönlendirmek açılarından
çok yararlıdır. Ancak bu testleri değerlendirirken hastanın
kognitif fonksiyonlarını etkileyebilecek diğer durumların
(post-iktal dönem, kullanılan ilaçlar, eşlik eden psikiyatrik
hastalıklar) göz önünde bulundurulması gerekmektedir.
Epilepsili
olgularda kognitif fonksiyonu etkileyen faktörleri 7 noktada toplamak
mümkündür:
1) Etyolojiye ilişkin faktörler
2) Nöbete ilişkin faktörler
a.
nöbetlerin başlangıç
yaşı
b. nöbet sıklığı
c.
nöbet süresi
d. nöbet tipi
e.
EEG anomalisinin tipi
3) AEİ tedavisinin yan etkileri
4) Beyin fonksiyon bozukluğunun tarafı
5) Diğer yeti kayıpları ve nöropsikolojik
bozukluklar
6) Çevresel faktörler, aile yapısı
7) Emosyonel faktörler ve kişilik gelişimi
Bilgisayarlı
Tomografi (BT) ve Magnetik Rezonans Görüntüleme (MRG)
X
ışınlarını kullanan bilgisayarlı tomografi
yönteminin nöroloji pratiğinde yerini alması özellikle semptomatik
fokal epilepsiler açısından bir devrim niteliğinde
olmuştur. Günümüzde ise beyin anatomisini çok detaylı bir
şekilde gösteren MRG epilepsili hastalarda ilk tercih edilecek görüntüleme
yöntemi olarak BT'nin yerini almış durumdadır. Kranial BT
incelemesinde bulgu saptanmayan %20-30 hastada MRG pozitiftir (ayrıca
bakınız: Sinir Sistemi
Semiyolojisi, Nöroradyoloji).
İlk
epileptik nöbet ile hastaneye başvuran hastalarda, özellikle anormal
nörolojik muayene bulgusu, fokal başlangıçlı nöbet ve
predispozan faktör varlığında (kanama diyatezi, geçirilmiş
iskemik inme öyküsü, antikoagülan ilaç kullanımı, ateş ve
immunsupresyon varlığı vb.) ve nöbet tekrar ediyorsa daha
hızlı uygulanabilen beyin BT çekilmesi önerilmektedir. Beyin BT
intraserebral hematom, inme, tümör, abse, geniş yer kaplayıcı
lezyonların tespitinde faydalıdır. Beyin BT’de lezyon tespit
edilememiş ve nöbet tetikleyici faktörü olmayan hastalarda MRG
yapılması düşünülmelidir. Hiperakut iskemik inme, sinüs ven
trombozu, leptomeningial metastaz, düşük “grade”li tümörler, posterior
reversibl ensefalopati sendromu ve küçük metastatik lezyonların
gösterilmesinde MRG çok faydalı olabilir. İlk nöbetini geçiren
hastanın beyin görüntülemelerinde lezyon olması, nöbet tekrar etme
riskini yaklaşık 2 kat arttırmaktadır.
Epilepsi
tanısı konulmuş hastalarda, bazı özel sendromlarda
yapısal beyin görüntüleme yapılması gerekli değildir. Bu
özel durumlar, çocukluk çağının idyopatik/genetik jeneralize
epilepsi sendromları (örn, çocukluk çağı absans epilepsi,
juvenil miyoklonik epilepsi vb.) ve idyopatik/genetik fokal epilepsi
sendromlarıdır (örn, rolandik epilepsi, Panayiotopoulos sendromu
vb.). Kronik epilepsi hastalarında yapısal görüntüleme yöntemi olarak
MRG seçilmelidir. Glial tümörler, vasküler malformasyonlar, kavernoma,
hamartoma, lokalize atrofi ve nöronal migrasyon anomalileri gibi çeşitli
yapısal lezyonları göstermede MRG'nin BT'den üstün olduğu
saptanmıştır (Şekil 17 ve 18). Serebral
kalsifikasyonları gösterme açısından BT MRG'ye üstün konumdadır.
Fokal bilinci etkileyen nöbetlerin etyolojisinde %70-80 civarında görülen
hipokampal sklerozun MRG ile tespitinin mümkün olabildiği ortaya
konmuştur. MRG'nin radyasyon içermemesi, BT'ye oranla kemik
artefaktlarının olmaması nedeniyle mezyal temporal lobu ve arka
çukur yapılarını daha iyi görüntülemesi ve rekonstrüksiyonla her
planda görüntünün kolayca elde edilebilmesi diğer üstün
yanlarıdır.
MRG’nin
epilepsi protokolüne uygun yapılması sağlanmalıdır. Bu
protokol olabildiğince yüksek Tesla gücünde (en az 1,5 tercihen 3 Tesla)
bir cihazla, FLAIR, T1 ağırlıklı ve T2
ağırlıklı sekansları koronal, sagital ve aksiyal
planlarda görüntülemelidir. Ayrıca kortikal gelişim anomalileri için
üç boyutlu volümetrik görüntü elde edilebilecek şekilde ince kesitli
(1-1,5 mm), aralıksız T1A sekansları tüm planlarda içermelidir.
Vasküler malformasyonları görüntülemek için T2 Gradient Echo veya SWI
sekanslarının eklenmesi gereklidir. 2018 yılında ILAE
tarafından epilepsi hastalarına yönelik yapısal lezyonları
belirlemek için MRG protokolü uzlaşma raporu
yayınlanmıştır. Bu raporda, MRG incelemesinin yüksek
çözünürlüklü üç boyutlu (3D) 1 mm’lik kesitli aksiyel, koronal ve sagital T1 ve
FLAIR sekansları ve buna ek olarak iki boyutlu (2D) submilimetrik koronal
T2 sekanslarını içermesi önerilmektedir. Epilepsi hastalarında,
MRG iki amaçla çekilmektedir. İlki, yeni epilepsi tanısı
almış hastalarda yapısal lezyonların belirlenmesidir.
İkincisi ise, AEİ tedavisine dirençli hastalarda epilepsi cerrahisi
öncesi hazırlık amacı taşımaktadır.
Hipokampal skleroz, erişkin epilepsi hastalarında en sık cerrahi yapılan
lezyondur. MRG ile tanısı yüksek duyarlılık ve özgüllükle
konulabilmektedir. Bu hastalarda, inceleme protokolünde hipokampusun uzun
aksına paralel ince kesitli koronal planda görüntüler eklenmesi
önerilmektedir. MRG’de T1 ağırlıklı görüntülerde
hipokampusta atrofi ve T2/FLAIR ağırlıklı kesitlerde
hiperintensite görülmesi, hipokampal skleroz için tipiktir. Hipokampus atrofisi
volümetrik incelemeler ile ve sinyal değişikliği ise T2
relaksometri ile kantitatif olarak ölçülebilir. Hipokampüs
içyapısının bozulması görülebilir (Şekil 13 ve
Şekil 14). Lateral
ventikül temporal boynuzunda asimetri, anterior temporal lob, forniks veya
mamiller cisimciklerde atrofi daha az özgül bulgulardır. Hipokampal
skleroz ile ilgili dikkat edilmesi gereken durumlar, bilateral olabilmesi ve
dual patolojilerin eşlik edebilmesidir.
Fokal kortikal displazi, özellikle çocukluk çağı dirençli
semptomatik epilepsi sendromlarına yol açan, oldukça epileptojenik bir
kortikal gelişim anomalisidir (Şekil 17A). Kortikal
laminasyonun bozulması, displastik nöronlar ve balon hücreleri ile
karakterizedir. MRG’de tipik bulguları, korteks
kalınlığının artması, ak-gri madde
ayrımının bozulması, fokal atrofi, T2
ağırlıklı ve FLAIR sekanslarında sinyal
artışıdır. Fokal kortikal displaziler sulkusların
derinliklerinde yerleşmişlerse MRG’de izlenmeyebilir. Kortekste
kontrastı arttırmak amacıyla gri madde ve BOS sinyalini
baskılayan ‘double-inversion recovery’ sekansı eklenebilir.
Ayrıca ak-gri madde ayrımını belirginleştirmek
amacıyla ‘voxel based’ morfometri ve T2 relaksometri teknikleri
kullanılabilir. Patolojik olarak fokal kortikal displaziler 3 tipe ayrılır:
Tip 1’de displastik nöronlar veya balon hücreleri bulunmaz. Tip 2’de displastik
nörona balon hücreleri eşlik edebilir (Tip 2B-Taylor tipi displazi) veya
etmez (Tip2A). Tip 3’de başka lezyonun (hipokampal skleroz, infarkt,
tümör, vasküler malformasyon, travma, ensefalit vb.) eşlik ettiği fokal
kortikal displazi mevcuttur. Fokal kortikal displazi Tip 1’de fokal kortikal
atrofi, ak-gri madde ayrımının silikleşmesi ve anormal
yapıda sulkuslar gözükür. Fokal kortikal displazi Tip 2A’da belirgin
korteks kalınlığının arttığı küçük
alanlar, ak-gri madde ayrımının silikleşmesi ve subkortikal
bölgelerde silik T2 sinyal artışı izlenir. Fokal kortikal
displazi Tip 2B’de ise Tip 2A’ya ek olarak T2 ağırlıklı ve
FLAIR kesitlerinde subkortikal bölgelerde ventriküle kadar uzanan sinyal
artışı (‘transmantle’ belirtisi) görülür. Fokal kortikal
displazi Tip3’te MRG bulguları altta yatan lezyona göre
değişiklik gösterir. Epilepsi ile ilişkili diğer kortikal
gelişim anomalileri Tablo 16‘da
özetlenmiştir (Ayrıca bakınız: Sinir sisteminin
doğumsal hasarları ve gelişimsel hastalıkları).
Tablo 16. Kortikal gelişim anomalilerinde MRG
bulguları (Şekil 17 ve 18) (Kaynak 17’den
değiştirilerek)
|
Kortikal gelişimsel anomali
|
MRG bulgusu
|
|
Polimikrogri
|
Sığ sulkuslar ile
ayrılmış küçük giruslar (kortekste kalınlaşma gibi
izlenebilir)
Diğer malformasyonlar ile birlikte
görülebilir
Epilepsi ile ilişkili olmayabilir
|
|
Şizensefali
|
Kortikal yüzeyi ventrikül ile birleştiren
yarık
Açık veya kapalı olabilir
Epilepsi ile ilişkili olmayabilir
|
|
Lizensefali-agiri-pakigiri
|
Girus ve sulkus sayısında azalma,
sığ sulkuslar, büyük giruslar ve kalınlaşmış
korteks
Agiri: hiç girus olmaması, pakigiri: az
sayıda girus olması
|
|
Hemimegalensefali
|
Serebral hemisferin tümü veya bir
kısmının hamartomatöz büyümesi
Hemisferik genişleme, ipsilateral ventrikül dilatasyonu,
ak maddede sinyal değişiklikleri
|
|
Heterotopi
|
Ektopik nöron kümeleri
Subepandimal, fokal subkortikal
veya bant heterotopi şeklinde
|
|
Tuberoskleroz
|
Subependimal kalsifiye nodüller
Kortikal tuberler-hamartomlar (infantil dönemde
T1 ağırlıklı kesitlerde hiperintens, T2
ağırlıklı kesitlerde sınırları silik
hipointens lezyonlar, çocukluk döneminde T2 ağırlıklı
kesitlerde hiperintens olur)
|

Şekil 17. Epilepsiye
sık neden olan kortikal gelişimsel bozukluk örnekleri: A:
Fokal kortikal displazi, B: Nodüler heterotopi, C: Bilateral
(bir yanda açık dudaklı diğer yanda kapalı dudaklı
tipte) şizensefali, D: Bilateral perisilviyen
displazi E: Hemimegalensefali, F: Çift
(double) korteks sendromu.

Şekil 18. Nörogörüntüleme
yöntemleri ile çeşitli epilepsi olgularında gösterilmiş olan ve
epilepsiye yol açtığı düşünülen lezyonlar A: Mezyal
temporal lob epilepsisi tanısıyla izlenen bir olguda ok ile
işaretli olan mezyal temporal sklerozu düşündüren hiperintens görünüm
B: Semptomatik fokal epilepsili
olguda kavernom ile uyumlu heterojen lezyon, C: Tuberoz
sklerozlu bir olguda izlenen multifokal kortikal tuberler, D: Lipoid
proteinoz tanılı hastada BT’de diagnostik bilateral mezyal temporal
kalsifikasyonlar, E: Sturge-Weber sendromu tanılı olguda
BTde tipik oksipital lob yerleşimli kalsifikasyon, F: Herpes
simpleks ensefaliti olgusunda tipik mezyal temporal yerleşimli lezyon
Sturge-Weber Sendromu, nadir görülen kapiller-venöz malformasyonlarla
karakterize bir nörokütane hastalıktır. Posterior pariyetal ve
oksipital bölgelerde leptomeningeal veya dural anjiomatozis izlenir. Beyin BT
incelemesinde, parietooksipital bölgelerde kalsifikasyon, atrofi ve üzerindeki
kalvaryumda kalınlaşma vardır. MRG’de ise, posterior bölgelerde
atrofi, leptomeningeal kontrast tutulumu, internal serebral venlerde
genişleme ve koroid pleksusta asimetrik büyüme görülür (Şekil
18D).
Düşük dereceli beyin tümörleri içersinde fokal epilepsi ile birlikte en sık
görülenler gangliogliom, oligodendrogliom ve disembriyoplastik nöroepitelyal
tümörler (DNET)’dir. Sıklıkla temporal lobda bulunurlar. Düşük
dereceli beyin tümörleri, kitle etkisi yaratmayan, çevresinde ödem olmayan, T1
ağırlıklı sekanslarda hipointens T2
ağırlıklı sekanslarda ise hiperintens lezyonlar olarak
görünürler, kontrast tutulumu değişkendir ve genellikle izlenmez.
Oligodendrogliom ve DNET, beyin BT’de kalsifikasyon içerebilir (Şekil
19).

Şekil 19. Dirençli
epilepsi nedeniyle izlenen bir olguda sağ temporal bölgede
disembriyoblastik nöroepitalyal tümör (DNET) düşünülen MRG lezyonu
Arteriovenöz malformasyonlar (AVM), kapiller yatağı oluşturmadan arter ve
venin anormal bağlantısıdır. Sklerotik beyin dokusu ile
çevrilidirler. AVM görüntülemesinde altın standard konvansiyonel
anjiografidir. MRG’de AVM etrafındaki gliozis T2
ağırlıklı ve FLAIR kesitlerinde hiperintens gözükür.
Kavernomlar, immatür, multilobüle, çevresi endotel ile kaplı dilate
damarlardan oluşan yapılardır. Tipik görüntüsü, mısır
patlağı şeklindedir. Multikistik, farklı yaşlarda kan
elemanları içermesi nedeniyle değişik sinyal intensiteleri
barındırır. MRG’de Gradient Echo sekansında hipointens
hemosiderin halkası izlenir.
Ensefalomalazi, lökomalazi veya porensefali terimleri hayatın erken döneminde beyinde
meydana gelen destrüktif lezyonlar için kullanılırlar. MRG’de gliozis
dokusu T2 ağırlıklı ve FLAIR sekanslarında hiperintens
ve T1 ağırlıklı sekanslarda hipointens olarak izlenir.
MRG’de
görülen lezyonun EEG ve klinikle korelasyonu yapılmalıdır. Her
MRG lezyonu epileptik nöbete yol açmaz. Bulguların kolay gözden
kaçabileceği ve tekrar tekrar incelenmesi gerekliliği
unutulmamalıdır. Ayrıca epileptik nöbetlerin bir kısmı
da yapısal lezyonlardan kaynaklanmaz. Diğer önemli bir nokta ise
kimi zaman geçici BT veya MRG anomalilerine rastlanabilmesidir. Bu görüntülerin
fokal nöbet veya SE aktivitesi sırasındaki ödeme, perfüzyona veya
bölgesel kan beyin bariyeri yıkımına bağlı
olabileceği düşünülmektedir.
Yeni
gelişen ilaç seçeneklerine rağmen epilepsi hastalarının
üçte biri medikal tedaviye dirençlidir. Bu hastalarda epilepsi cerrahisi tedavi
seçeneği olarak gündeme gelmektedir. Epilepsi cerrahisi
hazırlığında ilk basamak yapısal lezyonları
görüntülemek amacıyla epilepsi protokolüne uygun MRG görüntülemesinin
yapılmasıdır. İster temporal ister ekstratemporal lob epilepsisi
olsun MRG lezyonu olan hastaların, MRG negatif olanlara kıyasla
cerrahi sonrası nöbetsizlik olasılıkları iki kat daha
yüksek olmaktadır. Bu oranlar yapısal görüntülemenin önemini ortaya
koymaktadır. Gelişmiş ülkelerde epilepsi cerrahisi merkezlerinde,
yapısal lezyonları saptamak amacıyla 7 Tesla MRG
kullanımı onaylanmıştır. Seçilmiş vakalarda da
fonksiyonel görüntüleme yapılır. Fonksiyonel görüntüleme
yöntemlerinden aşağıda ayrıntılı bahsedilecektir.
Fonksiyonel
Görüntüleme
SPECT (Single photon emission computed tomography): SPECT
incelemesi, nöbetin hemen öncesinde, nöbet sırasında veya hemen
sonrasında beyin perfüzyonu ile ilişkili bilgiler verir. Geniş
klinik kullanım olanaklarıyla epilepsili hastaların
incelenmesinde BT, MRG, EEG ile birlikte klinik bilgilere katkısı
olabileceği düşünülmektedir. Epileptojenik odağın
lokalizasyonu açısından kullanılan hiçbir yöntem tek
başına tam güvenilir değildir ve bu çeşitli yöntemlerin
kombinasyonunu gerektirmektedir. SPECT bu multidisipliner yelpaze içinde
epileptik odak belirlenmesinde, invazif tanı yöntemlerine geçmeden önce
katkı sağlayabilecek tamamlayıcı bir yöntemdir. Özellikle
MRG negatif olan hastalarda, iktal SPECT ile saptanan hiperperfüzyon ile odak
lokalizasyonu açısından çok yararlı bilgiler elde edilir.
İktal
SPECT verisini işlemek amacıyla çeşitli programlar
kullanılmaktadır. MRG destekli SISCOM (substraction ictal SPECT
co-registered to MRI) incelemesinde interiktal ve iktal SPECT datası
birbirinden çıkartılarak nöbet sırasında aktif olan epileptojenik
zon tespit edilebilmektedir. STATISCOM (statistical ictal SPECT co-registered
to MRI) ise, aynen SISCOM’a benzer ancak analize ek olarak verilerin
normalizasyonu ve kontrol grubu ile kıyaslanma basamaklarını da
içerir.
Temporal
lob epilepsili MRG negatif vakaların %40-70’i ve ekstratemporal lob
epilepsili MRG negatif hastaların %35-55’inde bu yöntemlerle lokalizasyon
yapılabilmektedir. İktal SPECT, %65-90 oranında EEG
odağıyla uyumlu hiperperfüzyon gösterir, interiktal dönemde ise
aynı bölgede hipoperfüzyon gözlenir. İnteriktal SPECT interiktal
epileptiform aktivitenin oluşturduğu iritatif alanın
belirlenmesinde de kullanılabilir.
SPECT,
beyin kan akımının technetium-99m-hexamethylpropylenamine oxime
(99mTc-HMPAO) veya başka işaretleyiciler ile görüntülenmesidir.
99mTc-HMPAO beyin içerisine alımı 30 saniye ile 1 dakika
içerisindedir, 6 saat boyunca stabil kalır. İntraselüler glutatyon
ile etkileşir, hidrofilik hale geçer ve kan-beyin bariyerini geçemez. Bu
nedenle iktal kayıt yapmak daha kolaydır, bu noktada PET'e üstündür.
İktal SPECT’de önemli olan enjeksiyonun yapılma zamanı ve
nöbetin süresidir. Enjeksiyon, nöbet başlangıcına en yakın
zamanda yapılmalıdır. Tam bir iktal değerlendirmede
epileptojenik odakta hiperperfüzyon, çevresinde hipoperfüzyon görülür. Otuz
saniyeden kısa süren nöbetlerde maddenin kan-beyin bariyerinden geçmesi
için yeterli zaman olmaz, uzun süren nöbetlerde ise epileptojenik
odağın yanında yayılım alanlarında da perfüzyon
görülür.
PET (Positron
emission tomography): PET incelemesinde, beyin
metabolizmasının bir göstergesi olarak glikoz
kullanımının ölçülmesi amacıyla 18 florodeoksiglikoz (FDG)
kullanılır. FDG-PET incelemesinde interiktal olarak epileptojenik
odağın işareti, azalmış glikoz
metabolizmasıdır (Şekil 20). Genellikle epileptojenik
odaktan daha geniş bölgede hipometabolizma görülür. Bunun nedeni
epileptojenik odak çevresindeki nöronların inhibisyonudur. Cerrahi tedavi
düşünülen vakalarda, özellikle MRG negatif veya MRG ve EEG bulgularının
uyumsuz olduğu vakalarda kullanımı önemlidir. Temporal lob
epilepsi hastalarında FDG-PET’de hipometabolizma, %86-90 oranında iyi
prognoz ile ilişkili bulunmuştur. Ekstratemporal lob epilepsilerinde
(%52-67), temporal lob epilepsisine kıyasla daha az duyarlıdır.
MR negatif olgular (%36-79), pozitiflere (%73-90) göre cerrahi sonrası
daha kötü sonlanım ile ilişkili bulunmuştur. FDG’nin beyinde
tutulum süresinin kısa olması sebebiyle (30-40 dakika), iktal FDG-PET
incelemesi oldukça güçdür. Yapılabilirse fokal nöbet sırasında
epileptojenik odakta bölgesel glikoz metabolizması artış ile
karakterizedir. İnteriktal PET sırasında farkedilmeyen nöbetler,
incelemenin yanlış yorumlanmasına neden olur. Talamus ve
ekstratempolar alanlara yayılan hipometabolizma kötü prognoz belirtisi
olarak kabul edilmektedir.
Jeneralize
epilepsilerde PET kullanımı rutinde yapılmaz. Çalışma
verileri, sık absansların varlığında serebral glikoz
metabolizmasında yaygın artış görüldüğünü
göstermektedir. Absans statusta ise beyin glikoz metabolizmasında azalma
olur. Absans sırasında O-15-işaretli su kullanılan PET
incelemelerinde, beyin kan akımında global olarak özellikle talamusta
artış vardır. İnfantil spazmlı MRG negatif olan
çocukların FDG-PET incelemeleri altta yatan fokal kortikal displazi ve
diğer kortikal malformasyonlar açısından yol gösterici
olabilmektedir.
FDG
ve O-15 işaretli su molekülleri haricinde reseptör PET incelemeleri de son
yıllarda kullanılmaya başlanılmıştır. 11C
ve 18F-flumazenil (FMZ), santral GABAA reseptörüne
bağlanan bir ligandtır. FMZ-PET incelemelerinde, GABAA
reseptörlerine düşük bağlanma tespit edlilebilir. Epileptojenik
odağın belirlenmesinde kullanımı henüz çelişkilidir.
Neokortikal ekstratemporal lob epilepsilerinde faydalı olabileceği
bildirilmiştir. FMZ-PET, epilepsi patofizyolojisi konusunda ve medikal
tedaviye dirençli vakaların belirlenmesinde faydalı bilgiler
sağlayabilir. α-11C-methyl-L-tryptophan (AMT)-PET, tuberosklerozlu
çocuklarda epileptojenik kortikal tuberlerin belirlenmesinde kullanılmış
ve umut vaadeden sonuçlar elde edilmiştir.
Şekil 20. Epilepsili bir hastanın PET inceleme
örneği: 15 yaşındaki kadın hasta 3 yaşından beri
tedaviye dirençli fokal bilinç kaybının eşlik ettiği
nöbetler geçirmektedir. MRG‘si tamamen normal olan hastanın sol
temporofrontal bölgede hipometabolizma gösteren PET bulguları (oklar), EEG
bulgularına destek oluşturmuş ve invazif incelemenin planlanmasını
sağlamak açısından çok değerli olmuştur.
Magnetoensefalografi (MEG): Kortikal piramidal hücrelerin dendritlerinde oluşan
postsinaptik senkronize akımların manyetik alanını ölçmek
amacıyla kullanılır. Son yıllarda manyetik alan ve MRG verisi
kullanılarak manyetik kaynak görüntülemenin (magnetic source imaging-MSI)
epilepsi cerrahisi öncesi epileptojenik odak lokalizasyonunda
kullanılabileceği bildirilmektedir. İntrakranial elektrod
pozisyonlarının belirlenmesinde yardımcı olarak
kullanılır.
Fonksiyonel MRG (fMRG):
fMRG tekniği beyin içerisindeki fokal kan oksijen seviyelerinin zaman
ekseni içerisinde görüntülenmesine dayanır. Kan oksijen seviyesi
bağımlı (BOLD-blood oxygen level dependent) sinyalleri kaydeder.
BOLD sinyalleri, nöronların sinaptik girdilerini yansıtır.
İki şekilde analiz edilir. İlkinde belirli bir görev
sırasında BOLD sinyalleri kaydedilerek, dinlenim durumu BOLD sinyali
ile kıyaslanarak, belirli bir görevle ilişkili (task-based)
lokalizasyon yapılır. İkincisinde ise bağlantısallık
yani konnektivite değerlendirilir. Beyin haritaları kullanılarak
ilgili bölgeyi (region of interest-ROI) oluşturan voksellerin ortalama
BOLD sinyali beynin geri kalanı ile ya da yine belirli bir bölge ile karşılaştırılabilir.
Bu şekilde, ilgili beyin bölgesinin beynin geri kalanı ile
bağlantısallığı tespit edilmiş olur ve buna tohum
tabanlı analiz (seed-based) denilmektedir. Bir diğer analiz yöntemi
ise dinlenim durumunda beynin BOLD sinyalini kaydederek (resting state fMRI)
birbiri ile ilişkili ağların incelenmesidir. fMRG
bağlantısallık analizleri, epilepsinin bir odak
hastalığından çok, bir ağ (network)
hastalığı olduğunu desteklemektedir. Halen deneysel olan bu
çalışmaların artması, epilepsinin patofizyolojisinde önemli
bilgiler edinmemizi sağlayacaktır.
Epilepside fMRG kullanımı şöyle
özetlenebilir:
·
Kritik (eloquent) korteksin belirlenmesi
·
Odak lokalizasyonu ve EEG-fMRI
·
Nöral ağların belirlenmesi
Kritik korteksin belirlenmesi: Kritik korteksin belirlenmesi, özellikle önemli
fonksiyonları yerine getiren beyin dokusu yakınındaki bir
lezyonun rezeksiyonu planlanıyorsa, önemlidir. Son 10 yıl içerisinde
fonksiyonel görüntülemedeki gelişmeler eşliğinde ‘eloquent’
korteksin belirlenmesi birçok gelişmiş epilepsi cerrahi merkezinde
fMRG ile yapılmaktadır (task-based fMRI).
Konuşma
merkezinin lateralizasyonu, fMRG görüntüleri alınırken hastaya sözel
akıcılık, kelime türetme ve semantik karar verme ödevleri
verilerek gerçekleştirilir. Aktivasyon izlenerek lateralizasyon indeksi
belirlenir ve konuşma merkezi sağ, sol veya bilateral olarak
belirlenir. Çalışmalarda, fMRG ile konuşma merkezi
lateralizasyonu WADA testi ile %80-90 oranında benzerlik göstermektedir ve
WADA testi yerine kullanılabileceği önerilmektedir.
Epizodik
bellek testleri de fMRG ile yapılabilir. Sözel ve görsel bellek
kayıpları ve cerrahi sonrası bellek bozukluklarının
öngörülmesinde çalışmalar olumlu sonuçlar vermiştir ancak
standart testler ve daha geniş çalışmalar ile kullanımının
yaygınlaşması gereklidir.
Motor
korteksin fMRG ile belirlenmesi, parmak veya topuk vurma testleri ile
gerçekleştirilir ve kortikal stimülasyon verileri ile uyumludur.
Odak lokalizasyonu ve EEG-fMRG: Tohuma bağlı analiz (seed-based) ile
epileptojenik alanın ipsilateral ve kontralateral beyin bölgeleri ile
bağlantısallık analizini yapmak mümkündür. Çeşitli
çalışmalarda epileptojenik alan ile ipsilateral ve kontralateral
beyin bölgeleri arasında bağlantısallığın
sağlıklı kontrollere kıyasla değişiklik
gösterdiği bildirilmiştir. Bu değişiklik sağ ve sol
hemisfer lezyonlarında da farklılık göstermektedir.
Eş
zamanlı EEG ve fMRG verinin kaydedilmesi mümkündür. Bu şekilde
interiktal ve iktal aktivite tetikli BOLD sinyalleri analiz edilebilir ve odak
lokalizasyonu yapılabilir. Sınırlı sayıda
çalışma, EEG tetikli fMRG incelemesinin, fokal epilepsili hastalarda
epileptik deşarjların lokalizasyonunu bulmakta
kullanılabileceğini bildirmiştir.
Nöral ağ yaklaşımı: Epilepsi hastalarında yapılan
çalışmalar, dinlenim durumu ağlarında değişiklik
olduğunu ortaya koymaktadır. Epilepsi hastaları ile
ilişkili nöropsikiyatrik etkilenme ile ilişkili olabileceği
söylenmektedir.
Difüzyon ağırlıklı görüntüleme (DTI) ile ak madde yollarının yapısal
görüntülenmesi yapılmaktadır. Bu incelemede, su molekülünün içinde
bulunduğu ortam içerisindeki difüzyon özellikleri
değerlendirilmektedir. Su her yöne eşit miktarda difüze
olduğunda buna izotropik difüzyon denilmektedir ve gri madde içerisindeki
difüzyonu bu şekildedir. Oysa su, ak maddede aksonların içerisinde
tek bir yönde diffüze olur, bu koşulda anziotropik difüzyondan bahsedilir.
Fraksiyonel anizotropi, DTI
kesitlerinde ak madde içerisinde suyun difüzyonunu ölçen bir parametredir ve ak
madde içerisindeki aksonal kaybın ve aksonal yapının
bütünlüğü ile ilgili bilgi verir. DTI kesitlerini değerlendirmek için
kullanılan bir diğer yöntem ak madde haritalarıdır ve traktografi olarak
adlandırılır. Bu yöntemle ak madde yollarının anatomik
lokalizasyonu belirlenmektedir. Temporal lob cerrahisi öncesinde optik
radyasyonun ve frontal lob cerrahisi öncesinde kortikospinal traktusun
belirlenmesi amacıyla traktografi kullanılmaktadır ve bu sayede
postoperatif görsel veya motor defisitlerin oluşma riski
azaltılabilir.
AYIRICI TANI
Epilepside tanı
öncelikle klinik olarak konur ve dolayısıyla geçirilen atakların
çok ayrıntılı olarak hasta ve görenler tarafından tarif
edilmesine dayanır. Sık olarak karşımıza
çıkacağı şekilde eğer anlatılanlar şüpheli
kalırsa ve laboratuvar bulguları gereken desteği
sağlayamazsa yanlış bir etiket yapıştırmaktansa
sadece gerekli tüm araştırmaları yapıp izlemek daha
doğru bir yaklaşımdır. Hatalı konmuş “epilepsi”
tanısı hastayı, ciddi biçimde çok boyutlu (ömür boyu epilepsi
hastası olarak damgalanmak, doğru tedavinin gecikmesi, uzun süre
AEİ’lerin maddi yükü ve yan etki olasılıkları, ehliyet
alamamak gibi çeşitli sosyal boyutlar vb.) sıkıntılarla
karşı karşıya bırakmak demektir. Bu nedenlerle
epilepsi tanısı koyarken ayırıcı tanıya çok önem
vermek gerekmektedir (Tablo 17). Tablo
17’de sunulanlar sadece bir özet niteliğinde olup tüm
olasılıkları içermekten uzaktır. Epilepside nöbet
tiplerinin ne kadar çok çeşitli ve değişken bulguları
olduğu düşünüldüğünde ayırıcı tanı
yelpazesinde yer alan hastalıkların da ne kadar çok ve çeşitli
olacağı açıktır. Nerdeyse tüm nörolojik, psikiyatrik ve
sistemik hastalıklar belli aşamalarda ayırıcı
tanıya girmektedir (Ayrıca bakınız: Sinir
Sistemi Semiyolojisi, Epizodik Bilinç
Bozuklukları).
En sık
karşılaşılan ve sorun yaratan diğer
olasılıklar senkop ve nonepileptik psikojen ataklar veya sık kullanılan ama
tartışmalı boyutları olan eski adıyla yalancı
(psödo-) nöbetlerdir (Tablo 18). EEG tanıda yardımcı
olabilir ama yeterli bilgi sahibi olmayanlar tarafından
yorumlandığında ciddi yanılgılara da yol açar.
Örneğin yavaş dalgalar gibi spesifik olmayan bozuklukların
epilepsi lehine yorumlanması sık rastlanan bir
yanılgıdır. Bunun dışında epilepsisi olmayan
normal bireylerde %5'e varan oranda epileptiform bulguların
görülebildiği ve tersine epilepsi olgularında ilk EEG'nin yarıya
yakın oranda normal bulunabildiği unutulmamalıdır.
(Ayrıca bakınız: Sinir Sistemi Semiyolojisi, EEG).
Psikojen tabloların ayrımı deneyimli uzmanlar için bile çok zor
olabilmekte ve dahası iki tablo sıklıkla bir arada
olabilmektedir. Hiçbir bulgunun tek başına kesin tanı koydurucu
olmadığı unutulmamalıdır. Auralı migrenin
ayırıcı tanıda olması dışında çok nadir
olarak migren aurası sırasında nöbetlerin tetiklenebileceği
de bilinmelidir.
Tablo 17. Epilepside
ayırıcı tanı
|
A) Çocukluk Çağı
- Senkoplar
- Siyanotik nefes tutma atakları
(şiddetli ağlamaya bağlı veya istemli nefes tutma)
- Kalıtsal ve edinsel metabolik nedenlere
bağlı bilinç kaybı ve/veya kasılma, titreme
- Migren (konfüzyonel durum, baziler migren,
auralı migren)
- Kardiyak ritim bozuklukları (özellikle
supraventriküler taşikardi)
- Tikler
- Titreme atakları (yenidoğan
döneminde)
- Çocukluğun benign miyoklonisi
- Nonepileptik psikojen ataklar
- Psikiyatrik kökenli diğer durumlar ve
Münchausen sendromu (ebeveynlerin katkısı)
- Benign paroksizmal koreatetoz
- Diğer paroksizmal hareket
bozuklukları
- Gece korkuları (pavor nocturnus)
- Hipnagojik miyokloniler
- Uykuda çeşitli hareket bozuklukları,
konfüzyonel uyanma, parasomniler
- Enürezis
- Mastürbasyon
- Gastroözofageal reflü
- Hiperekspleksia, artmış irkilme
reaksiyonu
- Stereotipiler
|
|
B) Erişkin dönem
-refleks:
postüral, valsalvaya bağlı, miksiyona bağlı vb
-kardiyak:
disritmi (kalp bloğu, taşikardi vb); valvüler (en sık aort
stenozu); kardiyomiyopati; şantlı hastalıklar vb
-perfüzyon
yetmezliği: hipovolemi, otonom yetmezlik
-psikojenik
(yalancı) nöbet
-panik
atak
-hiperventilasyona
bağlı
- Geçici iskemik atak (düşme atakları,
“limb-shaking” ataklar başta olmak üzere)
- Migren (baziler ve diğer auralı
tipler)
- Meniére hastalığı
- Narkolepsi/katapleksi
- REM uyku davranış bozukluğu
- Huzursuz bacak sendromu
- Geçici global amnezi
- Akut konfüzyonel durumlar
- Hipoglisemi başta olmak üzere metabolik
nedenler
- Fizyolojik miyoklonus
- Paroksizmal hareket bozuklukları (egzersizle
tetiklenen ve diğer)
|
Tablo 18. Epilepsi, senkop ve
nonepileptik psikojen atak ayrımında ipuçları
|
|
Senkop
|
Epilepsi
|
Nonepileptik
Psikojen Atak
|
|
Postür
|
Ayakta (hemen daima)
|
Her postür
|
Her postür
|
|
Terleme
|
Sık
|
Seyrek
|
Değişken
|
|
Renk
|
Beyaz
|
Morarma
|
Değişken
|
|
Başlangıç
|
Yavaş (presenkop)
|
Ani/aura
|
Değişken
|
|
Uykuda olma
|
Çok seyrek
|
Sık
|
Yok (uyanınca)
|
|
Yaralanma
|
Sık
|
Sıkça (yanma)
|
Çok seyrek
|
|
Yalnızken olma
|
Olası
|
Olası
|
Çok nadir
|
|
Kasılma
|
Seyrek
|
Tipik
|
Atipik değişken
|
|
İdrar inkontinansı
|
Seyrek
|
Sık
|
Çok seyrek
|
|
Bilinçsiz süre
|
Saniyeler
|
Dakikalar
|
Genelde yok veya tersine çok uzun
|
|
Düzelme
|
Hızlı
|
Yavaşça
|
Değişken
|
|
Postiktal konfüzyon
|
Çok seyrek
|
Sık
|
Yok/seyrek
|
|
Ağlama
|
Yok
|
Yok
|
Sık
|
|
Sıklık
|
Seyrek
|
Değişken
|
Genelde sık
|
|
Arttıran faktörler
|
Açlık, heyecan, sıkıntı, sıcak, ayakta uzun
kalma
|
Uykusuzluk, stres, tipik refleks uyaranlar
|
Stresli olay, kalabalık, kötüye kullanılma
|
|
Pelvik hareket
|
Yok
|
Seyrek
|
Sık
|
|
Asenkron hareket
|
Yok
|
Seyrek
|
Sık
|
|
Yuvarlanma
|
Yok
|
Seyrek
|
Sık
|
|
Stereotipik atak
|
Seyrek
|
Sık ve tipik
|
Seyrek
|
|
Göz açmaya direnç
|
Yok
|
Seyrek
|
Sık
|
|
İndüklenebilme
|
Yok
|
Yok
|
Sık
|
|
Dil ısırma
|
Yok
|
Sık (yan taraf)
|
Seyrek (ucu)
|
|
Eşlik eden öykü, durum
|
Kalp hastalığı, hızlı boy uzaması,
presenkoplar
|
Psikiyatrik tablolar, migren
|
Fibromiyalji ve somatoform tablolar
|
|
EEG iktal bulgu
|
Yok
|
Çoğunlukla
|
Yok
|
|
İnteriktal bulgu
|
Yok veya yavaş aktivite
|
Sık
|
Yok/seyrek
|
EPİLEPSİLERİN
GENETİĞİ
Epilepside genetik eğilimin önem
taşıdığı Hipokrat döneminden beri bilinmektedir. Tüm
epilepsilerin %40-60'ının etyolojisinde genetik faktörler rol
oynamaktadır. Özellikle idyopatik/genetik epilepsi sendromlarında
aile öyküsünün pozitifliği semptomatik epilepsilere oranla daha
belirgindir. Genel olarak toplumdaki epilepsi prevalansı %0,5 iken örnek
olarak idyopatik/genetik absans nöbetleri olan anne veya babanın
çocuğunda epilepsi olasılığı %9 oranında
bildirilmiştir. Epileptik olguların akrabalarında epilepsi
olasılığının artmasının yanısıra,
%50'ye varan oranlarda EEG anomalisi varlığı da
bildirilmektedir.
Özellikle son yıllarda genetik biliminde
kaydedilen teknolojik ve biyoenformatik ilerlemeler sonucunda epilepsi
genetiği ile ilgili bilgiler giderek artmaktadır. 1980’lerin
ortalarından bu yana İnsan Genom Projesi ile insan
DNA’sının yapısı, düzeni ve işlevleri konusunda birçok
bilgi hazır hale gelmiştir. Toplumdaki hastalıkların
çoğu bir veya daha fazla gen ile çevresel faktörlerin etkileşimini
içeren kompleks etyolojiye bağlı olarak gelişir. Bazen tek bir
gendeki mutasyon hastalığı oluşturmaya yeterli olurken,
bazen farklı birkaç gendeki değişimler veya çevresel faktörler
genetik yatkınlığa eklendiği zaman hastalık
gelişir. Toplumda sık görülen diğer hastalıklar gibi
epilepsi sendromlarının bazıları Mendel tipi kalıtım
özelliği göstermekle birlikte çoğunlukla kompleks kalıtım
özelliği görülür.
Epilepsiye genetiğin katkısı klinik
olarak uygun tasarlanmış aile çalışmaları ve moleküler
genetik çalışmalardan elde edilmiş, tanı testlerinin (örn, SCN1A ve Dravet sendromu) temelini
oluşturmuştur. Genetik geçişli semptomatik epilepsiye yol açan
mutasyon ve kromozomal defektler daha erken tanımlanmış, nadir
idyopatik/genetik epilepsilerle ilişkilendirilen mendelyen geçişli
hastalıklarda birden çok gen tespit edilmiştir.
Çoğunlukla basit kalıtım
özelliği gösteren, epilepsinin fenotipin sadece bir kısmını
oluşturduğu ve farklı nörolojik bulguların birarada
olduğu 200’den fazla tek gen hastalığı bilinmektedir. Bu
hastalıklar çoğunlukla semptomatik epilepsilerdir ve tüm epileptik
sendromların yaklaşık yüzde birinden sorumludur (tuberoskleroz,
subkortikal band heterotopisi, PME vb.). Epilepsili olguların
yaklaşık yarısını idyopatik/genetik jeneralize ve
fokal epilepsiler oluşturur. 2010 yılına kadar ‘idyopatik
jeneralize epilepsi’ olarak bilinen hastalık grubu son gelişmelerin
ışığında ILAE tarafından yayımlanan son
sınıflandırmayla birlikte genetik jeneralize epilepsi olarak
isimlendirilmeye başlanmıştır. Bu isimlendirme
değişikliğine asıl dayanak, özellikle nedeni
anlaşılamayan belirli yaş gruplarına özgü epilepsi
sendromlarının temelinde başlıca genetik nedenlerin
yattığının düşünülmesidir. Yıllar içerisinde bu
hastalıklarda özellikle ailesel olgularda başlıca voltaj
kapılı gen mutasyonları ağırlıklı olarak
saptanmıştır. Ancak bu tanım juvenil miyoklonik epilepsi ve
PME hastalıkları arasında fark gözetmemekte, bu da akıl
karışıklıklarına yol açabilmektedir. Bu nedenle
sıklıkla idyopatik/genetik ifadeleri birarada
kullanılmaktadır.
İdyopatik/genetik epilepsi
sendromlarının çoğunluğu poligenik “kompleks”
kalıtım paterni’’ gösterir. Aynı ailede fenotipik varyasyonlar
görülebilir: Bir olgu çocukluk çağı absans epilepsili, diğeri
juvenil miyoklonik epilepsili olabilir. Aynı aile ağacında
farklı idyopatik/genetik jeneralize epilepsi formlarının bir
arada bulunması, ortak bir genetik temel olduğunu
düşündürmektedir. Bazı genlerin nöbet tipine, bazı genlerin
diğer fenotip özelliklerine yol açtığı düşünülebilir.
İyon kanalları nöronal eksitabilitede majör rol oynamakta ve genetik
epileptojenik değişikliklerin ana nedeni olarak görünmektedir.
Ayrıca idyopatik/genetik fokal epilepsiler, kortikal malformasyonlar, PME
gibi hastalıklarda kanal dışındaki genlerde mutasyonlar yer
almaktadır. Birçoğunun tam olarak etkisi bilinmemekle birlikte
nöronal uyarılabilirlik artışına yol açan veya inhibisyonu
azaltan mutasyonlar oldukları düşünülmektedir.
Epilepside genetik araştırmalar son
yıllarda bilim adamlarının gözde uğraşı
olmuştur. Fare deneyleri ve insanda yapılan çalışmalar ile
epilepsilerin moleküler genetiği konusunda ilerlemeler kaydedilmektedir.
Avrupa Birliği 6. çerçeve projesi kapsamında ülkemizin de içinde yer
aldığı EPICURE (Fonksiyonel Genomik ve Epilepsi Nörobiyolojisi),
takiben tüm dünyada daha geniş katılımlı Epi25 projeleri
buna önemli bir örnek oluşturmaktadır. Epilepside hastalığa
neden olan moleküler mekanizmaların ve genetik faktörlerin belirlenmesi,
tanımlanan veriler ışığında yeni tedavi
yaklaşımlarının geliştirilmesi özellikle ilaca
dirençli epilepsilerde, ilaca direnç mekanizmalarının açığa
kavuşturulması açısından önemlidir.
Genetik çalışmalar; hastalıkların
kesin tanısı (bazen klinik ve laboratuvar bulguları yetersiz
kalabilir), prenatal tanı ve taşıyıcıların
tanınması, sağlık ve hastalıkta genetik faktörlerin
rolü, diğer faktörler ile etkileşimi, kalıtsal hastalıklara
yatkınlıkların ortaya konulması, ilaçlara direnç ve cevap,
epilepsilerin sınıflandırılması, fizyopatolojinin
anlaşılması, farklı tanı yöntemleri ve yeni tedavi
önerilerine ışık tutmaktadır. Gendeki mutasyona bağlı patolojinin
fonksiyonel incelemesi, hedefe yönelik tedaviyi oluşturmak için zorunludur.
İlaç metabolizmasından sorumlu genler toplumlararası, hatta
bireysel farklılıklar gösterebilmekte, bazı olgularda ilaca
dirence yol açabilmekte ve hastanın tedavi seçeneklerini
değiştirebilmektedir. Bu alanda yapılan araştırmalar
farmakogenetik çalışmaları oluşturmaktadır.
İnsan epilepsilerinin genetik temellerinin
araştırılmasında şu basamaklar söz konusudur:
- İlk önce
araştırılan klinik sendromun net olarak belirlenmesi ve
uygun ailelerden DNA örneklerinin toplanması gerekmektedir.
Başarılı sonuç elde edebilmek için çok sayıda
etkilenmiş bireyi içeren büyük birkaç aile ya da küçük ama homojen,
çok sayıda ailelerin aile ağaçları (pedigri)
belirlenmelidir. Bu klinik çalışmalarla kalıtım tipi
belirlenmeye çalışılır. Genetik incelemelerin
yapılacağı materyal olarak genellikle kandaki lökositlerden
elde edilen DNA kullanılmaktadır. Bu amaçla her bireyden 10 ml
EDTA’lı tüpe (genellikle mor kapaklı) alınan kan oda
ısısı veya +4 C’de en kısa sürede DNA izolasyonunun
yapılacağı laboratuvara ulaştırılır.
- Bu materyal üzerinde
uygun moleküler genetik incelemenin seçilmesi (bağlantı analizi,
tüm genom asosiyasyon çalışmaları (GWAS), tüm ekzom
sekanslama (WES), tüm genom sekanslama (WGS) gibi yöntemlerle ilgili bölge
içindeki hatalı gen saptanır, mutasyonlar belirlenir)
- Gen
haritalaması ve mutasyonların belirlenmesi
- Gen ürünlerinin ve
hücresel görevlerinin tanımlanması
- Hedefe yönelik
tedavilerin belirlenmesi
Epileptik nöbetlerin fenotipin yalnızca bir
parçasını oluşturduğu, ek olarak birçok nörolojik bulgu saptanan
ve seyrek görülen çok sayıda klinik tablo içinden
bazılarının basit (mendelyen) geçiş gösterdiği ve
genleri saptanabilmiştir. Bunların başlıca örnekleri PME
ana nedenleri arasında yer alan Unverricht-Lundborg
hastalığı (sistatin B geninde mutasyonlar) ve Lafora cismi
hastalığı olarak verilebilir. Bir diğer ilginç grup da
ailesel subkortikal band heterotopisi (doublecortin mutasyonları) ve
ailesel periventriküler heterotopi (filamin 1 mutasyonları) gibi
çeşitli gelişimsel kortikal malformasyonun yer aldığı
gruptur.
Ailesel özellik gösteren çoğu epilepsi türünde
kalıtım şeklini tanımlamak kolay olmamaktadır. Bu
olgularda birden fazla gen aynı epilepsi sendromuna yol açabilmekte
(poligenik), edinsel ve çevresel faktörler birlikte rol oynayabilmektedir (multifaktöryel).
Ayrıca aynı epileptik sendroma birden fazla genin neden olması
(genetik heterojenite) ve bir genin birbirinden farklı epilepsi
fenotiplerine yol açması (fenotipik heterojenite) da genetik temelin
aydınlatılmasını güçleştiren özelliklerdir. İdyopatik/genetik
epilepsiler, özellikle idyopatik/genetik jeneralize epilepsiler genellikle bu
grupta yer almaktadır.
Kromozomal anomali ve nonkonvülzif status
epileptikus (NKSE) ile seyreden ilginç bir tablo halka kromozom 20’dir. Ana klinik özelliği epilepsi olan nadir
bir kromozomal anomalidir. Kromozomun iki kolunun füzyonu söz konusudur.
Translokasyon veya distal parçaların delesyonu olabilir. Kısa kolun
delesyonu epilepsiye yol açmazken, uzun kolun terminal delesyonu epilepsiye yol
açar. Uzamış bir konfüzyonel durumdan oluşan sık epileptik
nöbetler bir saati nadiren aşar, her gün ya da en azından her hafta
ataklar tekrarlar. Ek motor nöbetler olabilir. İktal EEG‘de uzun süreli,
bilateral paroksizmal yüksek amplitüdlü yavaş dalgalar ve seyrek dikenler görülür.
Nörolojik muayene tümüyle normaldir. Nöro-görüntülemede normal sonuçların
yanı sıra bazı olgularda ilişkisinden emin olunamayan MRG
bulguları görülmüştür. Tedaviye dirençli seyir sıktır.
Mozaisizme dikkat çekilmiştir (lenfositlerdeki halka 20 oranı 10-100%
arasındadır).
Basit kalıtım paterninin rol
oynadığı idyopatik/genetik epilepsilerden olan otozomal dominant
noktürnal frontal lob epilepsisi (ilk bulunan gen, 1995), benign ailesel
yenidoğan konvülziyonları (ilk başarılı
"linkage", 1989) ve FN plus sendromlarının gen
mutasyonları saptanmıştır. Otozomal dominant noktürnal
frontal lob epilepsisi yeni genetik araştırmalarla ortaya konan
kalıtsal fokal epilepsi sendromları için prototipik bir örnektir.
Erken çocukluktan erişkinliğe dek hemen her dönemde görülebilir, en
sık 10 yaş civarında başlar. Nöbetler hemen tümüyle uykuda
görülür. Sorumlu genlerin nöronal asetilkolin reseptörünün subünitelerini
kodladığı anlaşılmıştır.
Bu gelişmelere karşın kompleks
kalıtım paterninin rol oynadığı düşünülen ve geniş
bir grubu oluşturan idyopatik/genetik jeneralize epilepsilerin ancak
bazılarında gen lokalizasyonları saptanabilmiştir.
İdyopatik/genetik jeneralize epilepsili hastalarda
hastalığın başlangıcı, seyri, tedaviye
yanıtı, süresi gibi özellikler sendromun tek bir alt tipinde dahi
önemli farklılıklar gösterebilmektedir. Örneğin juvenil
miyoklonik epilepside hastaların belli bir kısmının (1/3)
ışığa duyarlı olduğu bilinmektedir.
Fotosensitiviteden bir endofenotip olarak yararlanmaya yönelik
çalışmalar da yapılmaktadır. Bugün için saptanmış
bulunan epilepsi genlerinin bazıları Tablo 19’da
görülmektedir. Genetik faktörler, farklı mekanizmalar ile nöronal
hiperksitabilite ve anormal senkronizasyona neden olabilmektedir.
Kalıtım monojenik olduğunda saptanan mutasyon
hastalığın gelişimi için yeterlidir. Monojenik olan
epilepsi sendromları %1-2’yi oluşturmakta, çoğunlukla çocukluk
çağı absans epilepsisi, juvenil miyoklonik epilepsideki gibi kompleks
kalıtım özelliği görülmektedir. Giderek artan sayıda
idyopatik/genetik epilepsilere neden olan, genellikle voltaj veya ligand
kapılı iyon kanallarıyla ilişkili mutasyonlar
bulunmuştur (Tablo 20).
Selim ailesel yenidoğan konvülziyonlarında KCNQ2, KCNQ3, KCNQ5 ve FN plus
sendromunda SCN1A, GABRG2 mutasyonları
bazı örnekleri oluşturmaktadır. Epilepsilerle ilişkili
kanal patolojileri dışında da genler
tanımlanmıştır. Henüz geni bilinmeyen klinik tablolarda
bağlantı analizi (linkage analysis) araştırma amaçlı
olarak kullanılabilir.
Son yıllarda çeşitli lokalizasyonlarda
mikrodelesyonlar ve kopya sayısı değişimleri
saptanmış ve epilepsi ile ilişkisinin üstünde
durulmaktadır. AKT, ARX, CDKL5, DCX ve Frajil X sendromu ile ilişkili
olan FMR1 geni gibi birçok gende
kanalopatiler dışında kalan gen anomalilerine örnek olarak
gösterilebilir. Refleks epilepsiler ise mekanizmaları tam olarak
aydınlatılamamış bir grup olmakla birlikte
idyopatik/genetik fotosensitif epilepsinin NEDD4-2
geniyle ilişkili olduğu düşünülmektedir ve son yıllarda
genetik araştırmaların yoğunlaştığı
noktalardan biri konumuna gelmiştir.
Sorumlu geni saptanmış olan genetik
epilepsi sendromlarının tanısı moleküler genetik testler
kullanılarak doğrulanabilmektedir. Bunlara örnek olarak PME (EPM1,
EPM2, MERRF), selim ailesel yenidoğan konvulziyonları, otozomal
dominant noktürnal frontal lob epilepsisi ve FN plus sendromları örnek
olarak verilebilir. Henüz geni bilinmeyen klinik tablolarda artık
günümüzde GWAS, yeni jenerasyon sekanslama (NGS-yeni nesil dizileme)
araştırma amaçlı olarak kullanılabilir.
Tablo 19. Semptomatik epilepsiye yol açtığı
bilinen bazı tek gen bozuklukları
|
Otozomal dominant
kalıtım özelliği gösterenler:
·
Tuberoskleroz: 9q34 (TSZ1) hamartin ve 16p13.3 (TSZ2) tuberin genleri
·
Nörofibromatozis: 17q11.2 (NF1) nörofibromin ve NF2 22q12.2
·
Ailesel kavernöz anjiom: CCM1 7q11.2-q21, CCM2 7p15-p13, CCM3
3q25.2-q27
|
|
Otozomal resesif
kalıtım özelliği gösterenler: (Çoğunluğu
kalıtsal metabolik hastalıklar)
·
Unverricht-Lundborg hastalığı: EPM1 21q22.3 kromozomda
sistatin B geni
·
Lafora hastalığı: EPM2A 6q24 tirozin fosfataz ve EPM2B
6p22.3 NHRLC1 gen mutasyonları, diğer eklenen yeni mutasyonlar
·
Juvenil nöronal seroid lipofusinoz 16p12 yerleşimli CLN3 geninde
mutasyon
·
Gaucher hastalığı tip III-1q21- glukoserebrosidaz
enzimini kodlayan gende mutasyon
|
|
X’e bağlı
kalıtım gösterenler:
·
Ailesel subkortikal band heterotopisi: Xq22.3-q23-Doublecortin
mutasyonları
·
Ailesel periventriküler heterotopi: Xq28-filamin 1 mutasyonları
·
Frajil X sendromu: Xq27.3
|
|
Mitokondriyopatiler:
·
MERRF: 8344 nükleotid çiftinde A-G geçiş mutasyonu
·
MELAS: 3243 nükleotid çiftinde A-G geçiş mutasyonu
|
MERRF; Mitokondriyal ensefalopati- çatlak
kırmızı lifler "rugged red fibers, MELAS; mitokondriyal
miyopati, ensefalopati, laktik asidoz, inme benzeri ataklar.
Tablo
20. İdyopatik/genetik
epilepsilerde iyon kanallarıyla ilişkili genler
|
Sendrom
|
Gen
|
Gen Fonksiyonu
|
|
Otozomal
dominant noktürnal frontal lob epilepsisi
|
CHRNA4 CHRNB2 CHRNA2
|
Asetil kolin
reseptörü alt ünitesi
|
|
Jeneralize
epilepsi febril nöbet artı
|
SCN1B SCN1A
|
Sodyum
kanalı alt ünitesi
|
|
Selim
neonatal/infantil ailesel konvülziyonlar
|
SCN2A
|
Sodyum
kanalı subünitesi
|
|
Selim ailesel
neonatal nöbetler
|
SCN1A
|
Sodyum
kanalı alt ünitesi
|
|
Dravet
Sendromu
|
SCN1A
|
Sodyum
kanalı alt ünitesi
|
|
Çocukluk
çağı absans epilepsisi ve febril nöbet
|
GABRG2
|
GABAA
reseptörü alt ünitesi
|
|
Çocukluk
çağı absans epilepsisi ve febril nöbet
|
CLCN2
|
Klor
kanalı alt ünitesi
|
|
Febril nöbet
ve temporal lob epilepsisi
|
GABRA1
|
GABAA
reseptörü ünitesi
|
|
Lateral
temporal lob epilepsisi
|
LGI1
|
Fonksiyonu
bilinmiyor
|
|
Otozomal dominant
juvenil miyoklonik epilepsi
|
GABRA1
|
GABA reseptör
alt ünitesi
|
|
Juvenil
miyoklonik epilepsi
|
EFHC1
|
R tip
kalsiyum akımı
|
|
Juvenil
miyoklonik epilepsi
|
Ser 1A reseptörü
|
|
|
Erken
başlangıçlı absans epilepsisi
|
GLUT1
|
Glikoz
transport geni
|
MOLEKÜLER
GENETİK İNCELEMELER
Aşağıda son yıllarda moleküler
genetik alanındaki gelişmeler ışığında
kullanıma girmiş yeni yöntemlerden söz edilecektir.
Mikroarray, gen ifade profillerinin
araştırılması, mutasyon taraması kromozomların
haritalanması, polimorfizim, mikrodelesyon ve kromozomal
aberasyonların tespiti için kullanılan bir genom araştırma
yöntemidir. Mikroarray tekniğinin temelinde birbirinin eşi olan DNA
moleküllerinin melezleşmesi işlemi yatmaktadır. Bu yöntemde
katı bir yüzeye sıralı bir şekilde oluşturulmuş
DNA kümeleri kullanılır. Bir mikroarray çipinde bu kümelerden
onbinlerce bulunabilir ve herbiri, prob denilen ve genellikle 20-100 nükleotid
uzunluğunda olan DNA molekülleri içermektedir. Problar bilinen bir gen ya
da kromozom dizisine eş olarak üretilmiştir. Mikroarray’lerin en
önemli avantajları, aynı anda birçok gen ile ilgili bilgi
alınması, hızlı sonuç elde edilmesi, az sayıda deney
yapılması ve otomasyona dayalı bir sistem olduğu için insan
kaynaklı hataların ortaya çıkma ihtimalinin düşük
olmasıdır. Gen ifade profili için öncelikle çalışılan
konuya göre farklı dokular, koşullar ya da zamanlar baz alınarak
hastalıklı dokudan ve kontrol dokusundan örnek alınır. Gen
ifade analizi karşılaştırmalı bir testtir, bu nedenle
kontrol dokusunun seçimi veri analizi açısından çok önemlidir.
Ayrıca, yüksek çözünürlükleri sayesinde mikroarrayler sadece
hastalığa neden olan değişiklikleri tespit etmekle kalmaz
aynı zamanda toplumda bulunan genetik farklılıkları da
ortaya çıkarır.
GWAS; ’’sık görülen hastalıklar, sık
bulunan varyantlar’’ hipotezine dayanmaktadır. Genel olarak toplumda
%30-40 sıklığında görülen sık varyantların
birliktelikleri hastalıklara yatkınlığa, risk
artışına yol açmaktadır. GWAS otomatize tek nükleotid
polimorfizmi (single nucleotide polymorphism-SNP) genotiplemesi yapan SNP array
cihazları ile yapılmaktadır. SNP alleleri floresan hibridizasyon
yöntemiyle belirlenmekte, böylece kromozomal delesyon, dublikasyon ve
örneğin kopya sayısı farklılıklarını (copy
number variation-CNV) saptayabilmektedir. Bu yöntemle idyopatik/genetik
jeneralize epilepsili olgularda 15q13.3 mikrodelesyonu bildirilmiştir.
İdyopatik/genetik jeneralize epilepsi ile ilk geniş GWAS
çalışması 1527 hasta ve 2461 kontrol olgusunda yapılmış
ve iki lokusta 2p16.1 and 17q21.32 anlamlı ilişki
saptanmıştır. Subgrup analizi absans epilepside 2q22.3ve juvenil miyoklonik epilepside 1q43 lokusunda anlamlı bağlantı göstermiştir. Bunu
izleyerek giderek artan sayıda çalışma yapılmış
ve kesişen-farklılaşan veriler elde edilmiştir.
Genetik alanında günümüzün gözde ve
çarpıcı yaklaşımı olan NGS analizi (yeni nesil
dizileme), daha önce kullanılan Sanger dizi analizi yöntemine oranla çok
daha hızlı, düşük maliyetli DNA ve RNA dizi belirlemesi
sağlayarak genetik ve moleküler biyoloji alanında yakın zamanda
bir devrim yaratmıştır. NGS sayesinde genetik alanında
önemli atılım sağlanmış, özellikle de genetik
taramaların daha geniş çaplı ve yaygın olarak gerçekleştirilebilmesinin
önü açılmıştır. Bu sayede genomik bilgi
ışığında kişisel tıp çağının
eşiğine gelinmiştir. NGS yöntemi sonucunda yüksek miktarda genom
verisi elde edilmektedir. Artan genetik verilerin kıymetlendirilmesini
sağlayacak bilişim ve biyo-informatik yaklaşımlar günümüzün
en gözde ve önem arz eden başlıkları arasına girmiştir. Kullanıma girmesinden itibaren yüzlerce
yeni gen saptanmasına yol açmıştır. Epilepsi için en önemli
etkisi ağır sporadik epileptik ensefalopati alanında
olmuştur. WES ve WGS hastalık mutasyonlarının
saptanmasında en etkili yöntemlerdir.
Epilepsiler özelinde
bakıldığında tanı ve araştırma için
üretilmiş ticari kitlerin ve gen panellerinin sayıları her geçen
gün fazlalaşmaktadır. Mevcut durumda 100-400 gen içeren NGS panelleri
klinisyenler ve genetik uzmanları tarafından kullanılmakta,
hastalığa gen düzeyinde tümleşik bir yaklaşım mümkün
olabilmektedir. NGS ile birden çok gende
aynı anda hasta ya da hasta popülasyonu düzeyinde, mevcut olabilecek gen
varyasyonlarının analizi mümkün olabilmektedir. Uzun yıllar
alacak araştırma süreçleri NGS yöntemi ile günler ve haftalar
içerisinde tamamlanabilmektedir Tanımlanan gen varyasyonlarının
hem hasta fenotipi hem de diğer varyasyonlarla ilişkisinin
belirlenmesi, hastalığın gen ölçeğinde incelenmesinde çok
önemli kapıları aralamış, epilepsilerde önem arz edecek yeni
aday gen ve gen ağlarının belirlenmesini mümkün hale
getirmiştir.
Epileptogenezin nörokimyası ile ilişkili
altta yatan mekanizmalara yönelik spesifik tedavilerin geliştirilebilmesi
gelecekteki hedefi ve umudu oluşturmaktadır. Genetik defektlerin
bulunması ve mutant iyon kanallarının elektrofizyolojik
özelliklerinin belirlenmesi yeni tedavi stratejilerini gündeme getirmektedir.
Özellikle defektif protein farmakolojik hedef haline gelebilir. Mevcut
AEİ’lerin öngörülen etki mekanizmaları arasında iyi bilinen
örnekleri voltaj kapılı Na kanallarını bloke eden fenitoin,
karbamazepin veya lamotirijin veya GABA’erjik sinaptik inhibisyonu
arttıran benzodiazepin, barbitüratlar, tiagabin veya vigabatrindir. Hayvan
modellerinden yola çıkılarak gen tedavisinin insanda
uygulanabileceği düşünülmektedir. Nöropeptid-Y ve Galamin ile ilgili
önemli ipuçları vardır ve bunlar nöbet baskılanması
sağlamaktadırlar. Ancak daha birçok teknik aşamanın
sağlanması gerekmektedir. Başarılı gen tedavisi için
hedef nörona transgenin taşınabilmesi gerekir. Yeni geliştirilen
vektörler nörona çekim, toksisitenin olmaması, nöronda persiste etmesi
sonucunda transgenin uzun süreli dayanıklılığını
sağlayarak birçok basamağın başarıya
ulaşmasını sağlamıştır. Ancak bu alanda faz
1 çalışmaların başlamasına epilepside henüz çok zaman
vardır.
Kuşkusuz sorumlu gen saptandıktan sonra bu
genin fonksiyonunu anlamak, dolayısıyla genetik
danışmanlık, gen terapisi yaklaşımlarını
uygulamak, amaçlanan hedeflerdir.
EPİLEPSİ
TEDAVİSİ
Epilepsi tedavisinde ilk basamak,
tanının doğru konması ve doğru tedavinin
belirlenmesidir. Daha önce belirtildiği gibi ilaçla tedaviye gerek olup
olmadığının belirlenmesi önem taşır. Tedaviye
gerek görüldüğünde nöbet şekli, sendrom özellikleri ve kısmen de
etyolojiye göre hangi ilacın verilmesi gerektiği belirlenir.
Tanı doğru olsa da verilen ilacın uygun olmaması durumunda
istenen başarılı yanıtın alınamaması
nedeniyle takiplerde yanlış olarak olgunun dirençli epilepsisi
olduğu düşünülebilir. Epilepside genel olarak verilen tek
başına veya kombinasyon halinde uygun iki adet AEİ ile nöbet
kontrolü %65-75 civarındadır. Tedavi alanlarda 2 yıl nöbetsiz
kalma oranı %74,7 olarak bildirilmiştir. İlaç
tedavisine cevapsız olduğu düşünülerek epilepsi cerrahisi
merkezlerine gönderilen olguların video-EEG monitorizasyon incelemeleri
sonunda %15 kadarının “yalancı nöbet” tanısı
aldığını anımsamak ayırıcı tanı ve
doğru tanının önemini vurgulamak açısından
anlamlıdır.
AEİ tedavisi en az 4-5
yıl süren, erken veya geç dönemde çıkabilecek, olası hafif veya
ağır yan etkilerin görülebileceği bir tedavidir. Hastanın
bireysel özellikleri iyi belirlenmelidir. Hastanın tedaviye
göstereceği bireysel farklılıklar, tedaviye uyumu, uzun süreli
takip gerekliliği konunun önemini göstermektedir. Ayrıca komorbid hastalıkları
ve bu hastalıkları için kullanması gereken ilaçların da
rolü unutulmamalıdır.
İyi bir AEİ’den
beklenen özellikler yan etkisinin olmaması, iyi bir
biyoyararlanımının olması, basit lineer kinetiğe sahip
olması, ilaç etkileşimlerinin olmaması, proteinlere az veya hiç
bağlanmaması, ilacı metabolize eden sistemleri etkilememesi,
günde 1 veya en fazla 2 kerede kullanılabilmesi ve maliyetinin düşük
olmasıdır. Teratojen olmaması ve farklı formüllerde alınabilmesi
de önemlidir (şurup, kapsül, tablet vb). Karaciğerde
metabolize olan ve enzim indükleyicisi olan AEİ’ler, birbirleriyle ve
diğer ilaçlarla etkileşmektedir. Bu özellikler eski kuşak
ilaçlarda daha belirgindir. Genel olarak yeni kuşak ilaçlar bu amaçla geliştirilmiştir
ve daha az etkileşim gösterirler. Gabapentin, pregabalin, levetirasetam ve
lakozamid hemen hemen hiç etkileşime girmeyen ilaçlar olarak öne
çıkmaktadır. Geçtiğimiz 20 yılda lamotirijin,
okskarbazepin, levetirasetam, vigabatrin, klobazam, topiramat, felbamat,
gabapentin, tiagabin, zonisamid, pregabalin, lakozamid gibi pek çok ilaç
dünyada ve ülkemizde kullanıma sunulmuştur. Yeni kuşaktan
başlıca beklenti yüksek etkinlik, yani daha az yan etki ve daha çok
etkili olmaları iken, yeni AEİ’lerle de birçok sorunun var
olduğu ve devam ettiği görülmektedir. Yine de yan etkilerin
varlığında yerine konabilecek diğer tedavi
olasılıklarının olması hekimi pratikte
rahatlatıcı bir unsurdur.
Bu gelişmelere rağmen
yeni AEİ’ler epilepsinin zorlu tedavisinde, arzu edildiği kadar etkin
olmaktan henüz uzaktır. Yeni kuşak AEİ’lerin eskiler kadar iyi
tanınmamaları ve beklenmedik yan etkilere rastlanma
olasılığı akılda tutulmalıdır. Ayrıca
ülkemiz koşullarında etkinliği bilinen ve daha ucuz olan
tedavilerin yerini almadan önce, titizlikle değerlendirilmeleri
gerekmektedir.
Antiepileptojenik
bir ilaç idealde epilepsili hastalarda kalıcı remisyon
sağlamalı, epilepsinin progresyonunu ve ilaca direnç
gelişimini durdurmalı, risk grubundaki hastalarda
epilepsinin başlamasını önlemelidir. Ancak AEİ’lerin,
epileptogenezi yani epilepsiyi oluşturan temel mekanizmaları
engelleyemediğini, sadece kullanıldıkları süre için
nöbetleri azaltabildiğini veya ortadan kaldırabildiğini bilmek
tedaviyi belirlemek açısından önemlidir. İlaca direnç
mekanizmasının, nöbetlerin nörobiyolojik sonuçlarının ne
olduğu halen belirsizliğini korumaktadır. Ancak nöbetlerin
önlenmesiyle nöronal ölümün önlenme sürecine katkı da potansiyel
antiepileptojenik etki olarak kabul edilebilir.
AEİ
tedavisinin başlanıp-başlanmamasının tartışılabileceği
klinik durumlar şöyle sıralanabilir:
· İlk nöbet
· Çok seyrek nöbetler (5-10 yılda bir nöbet gibi)
· Çok hafif nöbet (sadece deja-vu vb.)
· Profilaksi amaçlı kullanım (posttravmatik,
postoperatif dönemlerde)
· Özel durumlara bağlı nöbetler (uykusuzluk,
alkol alımı, aşırı yorgunluk, ateş ile tetiklenen
nöbetler)
· Akut semptomatik nöbetler
· Yalnızca uykuda olan hafif semptomlu nöbetler
· Dış uyaranlara bağlı refleks
nöbetler
İlk
epileptik nöbet ile başvuran bir hastada tedaviye başlama konusu
bugün için halen tartışmalı bir konudur. İlk nöbetin,
kronik bir epilepsi hastalığına bağlı ortaya
çıkan ilk epileptik nöbet mi yoksa altta yatan metabolik veya nörolojik
bir olay ile ilişkili ‘akut semptomatik bir nöbet’ mi olduğu
belirlenmelidir.
Akut
semptomatik nöbet tanısı konulmuşsa antiepileptik tedavi
başlamadan altta yatan nedene yönelik tedavi verilmelidir. Metabolik veya
toksik neden düzeltilmesine rağmen nöbet geçirmeye devam eden veya altta
yatan neden kontrol altına alınamayan hastalarda nöbetlerin devam
etmesinin yaratacağı risk göz önüne alınarak AEİ tedavi
başlanabilir. Bu tedaviye altta yatan neden ortadan kalktıktan sonra
3-6 ay devam edilir. Ancak nörolojik bir olayla ilişkili bir nöbet söz
konusu ise (örneğin herpes ensefalitine bağlı nöbet, nöbet ile
başvuran serebrovasküler hastalıklar gibi), nöbet tekrar riski yüksek
olması sebebiyle AEİ tedavinin hemen başlanması önerilir.
Bir
tetikleyici olmayan geçirilen ilk nöbette ilaç başlama kararını
etkileyen en önemli belirteç nöbetin tekrarlama riskidir. İlk nöbet ile
başvuran hastada nöbetin tekrarlama riski özellikle ilk yılda olmak
üzere ilk 2 yılda en yüksektir (%21-45). Hemen tedavi başlanan hasta
grubunda, tedavi başlanmayan gruba göre yeni nöbet riski ilk 1-2 yıl
için daha az olmasına rağmen, uzun süreli remisyon
açısından 3-5 yıl sonra belirgin bir fark görülmemektedir. Nöbet
tekrarı açısından belirlenmiş olan en önemli risk faktörleri
olarak, akut bir beyin olayı sonrasında gerçekleşen nöbetler,
EEG’de epileptiform anomali, nörogörüntüleme ile epileptojenik lezyonun
gösterilmesi sayılmaktadır. Hastanın başvuru
sırasındaki nöbet sayısının birden fazla olması,
SE şeklinde başvuru, ailede epilepsi öyküsü, epilepsi nöbetinin tipi,
hastanın yaşı ve cinsiyetinin ilk nöbet ile gelen hastada nöbet
tekrar etme riskini arttırdığı ile ilişkili güvenilir
veri yoktur. İlk nöbet ile gelen hastada başlanan AEİ
tedavisi %7-31 oranında yan etkiye neden olabilir ve bu yan etkiler hafif
ve geçici yan etkilerdir.
Nöbet
tekrarının getireceği fiziksel (yaralanma, kaza-sakatlık
vb.) ve psikososyal (iş kaybı, mahcubiyet duygusu,
dışlanma vb.) zararlar ile tedavinin olumsuz etkileri (sistemik
ve/veya psikiyatrik yan etkiler, ilaç etkileşimleri, doz ayarlama, uyum
güçlükleri) iyi değerlendirilmeli ve denge
oluşturulmalıdır. Burada hastanın meslek ve yaşam
koşulları da belirleyici rol oynamaktadır. Örneğin
yalnızca gece uykusunda nöbet geçiren bir ev hanımı ile
ışıklı ortamlarda nöbet geçiren bir televizyon spikerinin
epilepsi tedavisi aynı bağlamda ele alınamaz.
Bazı
olgular ömürleri boyunca tek bir nöbet veya uzun yıllar arayla seyrek
olarak nöbet geçirmektedirler. Bir epileptik nöbetin tekrarlama riski ilk 3 ay
içinde en yüksektir. İlk yıl içinde tekrarlamadığında
bir nöbetin tekrarlama riski düşmektedir. Bu nedenle bazı durumlarda
uzun sürecek, yan etkileri olabilecek ve belki de gereksiz olan bir tedaviye
başlamadan önce epilepsinin doğal seyrini izlemek akılcı
olabilir. Ancak bu durumda da hasta ayrıntılı olarak etyolojik
açıdan araştırılmalı ve mutlaka izlemeye
alınmalıdır. Önemli bir diğer nokta da hastaya durumun
anlatılması ve verilecek karara katılımının
sağlanmasıdır. Bazı çok iyi seyirli, hafif ve
yaşamı etkilemeyen ve nöbetleri sadece uyku sırasında olan
sendromlarda (örn, çocukluk çağının selim santrotemporal dikenli
epilepsisi) hastanın ve ailesinin görüşü de alınarak tedavi
yapılmayabilir. Burada önemli noktalardan biri de düzenli, yıllar
süren bir tedaviye uyum sağlamak açısından hastanın iş
birliğinin mutlaka sağlanmasıdır. Özel durumlara
bağlı nöbetler eğer tekse tedavi verilmeyebilir.
Bugün
için posttravmatik ya da postoperatif dönem gibi riskli zamanlarda AEİ
verilmesini destekleyen veri yoktur, ancak özellikle beyin cerrahları
tarafından erken dönem risklerini önlemek için verilmektedir.
Başlanmış profilaktik amaçlı tedavinin de mutlaka dikkatle
izlenmesi ve rasyonel bir şeması olması gerektiğini
unutmamak gerekir. Benzer şekilde saptanmış toksik/metabolik
nedene bağlı nöbetler tedavi gerektirmez, eğer
tekrarlamış ve AEİ başlanmışsa da genellikle 3-6
ay tedavi yeterli sayılabilir. İzole refleks nöbetlerde uyarıdan
uzak durarak kontrol yoluna gidilebilir. Spontan nöbetler eklenirse ya da uyarıyı
kontrol etmek yeterli değilse nöbet tipine göre tedavi başlanır.
Ancak
hastalar yanlızca uykuda nöbet geçirseler dahi ani beklenmedik ölüm
(sudden unexpected death of epilepsy patients-SUDEP) açısından risk
taşıdıkları için dikkatlice değerlendirilmelidir.
Özellikle bilateral tonik-klonik nöbet geçiren ve ilaca dirençli hastalara
mutlaka AEİ tedavi başlanılmalıdır.
Epilepside
ilaç tedavisinin temel ilkeleri şu şekilde özetlenebilir:
- Hasta ile tedavinin
gerekliliği ve riskleri konusunda konuşularak uzun süreli bir
izleme planı yapılmalı
- Altta yatan bir neden varsa
(örn, intrakranial tümör/arteryovenöz malformasyon) veya komorbid
başka bir hastalığı varsa epilepsi ile birlikte tedavi
edilmeli
- Sendroma ve nöbet tipine
uygun seçilen tek ilaç ile tedavi uygulanmalı (monoterapi ilkesi)
- En düşük etkili dozla başlanmalı
ve ilacın yan etkisi en az olmalı veya hiç olmamalı
- Doz tam nöbet kontrolü
sağlanana ya da yan etkiler görülene dek arttırılmalı
- İlaç kan düzeyi, toksik
etki veya tedaviye uyumsuzluk şüphesi olursa kontrol edilmeli ve
bunun dışında nöbet kontrolü sağlanmış
hastada sadece kan düzeyine bakılarak doz değişikliği
yapılmamalı
- Kullanılacak diğer
ilaçlarla (örn, antibiyotikler, oral kontraseptifler vb.) ilacın
etkisinin kaybolabileceği ya da toksik düzeye ulaşabileceği
hastaya mutlaka belirtilmeli
- İlacın aniden
kesilmesi, unutulmasının riskleri ve ilaç alımındaki
uyumsuzluk nedeniyle doğabilecek riskler hakkında
bilgilendirilmeli
- İlk ilaca yanıt
alınmazsa ikinci uygun seçim ile monoterapi; buna da yanıt
alınmazsa uygun ilaç kombinasyonuna gidilmesi veya diğer tedavi
seçenekleri (epilepsi cerrahisi, vagal sinir stimülasyonu vb.)
düşünüleceği konusunda hasta bilgilendirilmeli
- Sadece ilaç
alımının yetmeyebileceği, bu nedenle nöbeti
tetikleyecek durumlardan uzak durulması konusunda bilgilendirilmeli
(uykusuzluk, alkol alımı vb.)
- Epilepsi nöbetlerinin
tedavisi ile özellikle psikiyatrik hastalıkların (depresyon,
anksiyete vb.) gerileyip kaybolabileceği ve sosyal entegrasyonun
tekrar sağlanacağı anlatılmalı
- Tedavi
şartlarının hastanın yaşam kalitesini yükseltmeye
yönelik olduğu unutulmamalı (örn, nöbet kontrolü
sağlanmasına rağmen yan etkilerin hastayı kötü durumda
bırakmasının engellenmesi gibi)
İLAÇ SEÇİMİ
AEİ
seçimi için ilk önce hastanın nöbetlerinin ve epilepsi tipinin
sınıflanması gerekir. İlaç seçiminde rol oynayan en önemli
faktörler etkinlik, toksik etkilerin olmaması veya daha gerçekçi
yaklaşımla en az yan etki ve ucuzluk olarak özetlenebilir.
Bunların yanı sıra kullanım kolaylığı
(örneğin günde tek doz kullanma) önemli bir faktördür. Komorbiditesi olan
hastalarda kullanılan diğer ilaçlar ile etkileşim AEİ
seçiminde bir diğer önemli faktördür. İleri yaş grubunda
özellikle çoklu ilaç kullanımı nedeniyle ilaç etkileşimleri esas
sorunu yaratırken doğurganlık çağındaki
kadınlarda teratojenite ön planda değerlendirilmektedir.
Hastanın karaciğer veya böbrek hastalığının
olması ve hatta diyalize giriyor olması ilaç seçiminde
değişikliklere yol açar ve bazen de doz ayarlamasının
yapılması gerekir. Hastanın sigorta sorunları ilaç teminini
zorlaştırabilir, böyle durumlarda en ucuz ilaç tercih
edilmelidir.
Yeni
başlangıçlı epilepsili hastalarda ilaç başlanılmaya
karar verildiyse ilk olarak monoterapiler denenmelidir. Eğer monoterapiye
de yanıt alınmazsa bu ilaçların kombinasyonları hastaya
verilebilir. Ancak dirençli epilepsilerde en çok yapılan hata monoterapi
denenmeden gereksiz polifarmasi veya yüksek doz ilaç
kullanımıdır. İlaç çok çabuk titre edilerek
yükseltilmemelidir. Ayrıca daha önce yüksek doz denenmiş ilaçlar veya
aynı kombinasyondaki ilaçlarda ısrarcı olmamak, farklı
kombinasyonda düşük dozda ilaç vermek yan etkilerin ortaya
çıkmasını önler. Yüksek dozda ortaya çıkabilecek nöbetlerin
de önüne geçilmiş olur.
Antiepileptik
İlaçlar
Bu
grupla ilgili ana özellikler Tablo 21 ve 22’de kısaca
özetlenmiştir. Miyokloni ve absans nöbetleriyle seyreden sendromlarda
geniş spektrumlu ilaçların örneğin valproat veya lamotirijinin
ilk seçenek olduğu ve yanlış tedaviyle örneğin karbamazepin
veya fenitoin kullanıldığında bu nöbetlerin
artabileceği bilinmektedir. Bu durum tedavi seçiminde doğru
sınıflamanın önemini açıkça ortaya koymaktadır.
Günümüzde
kullanılmakta olan yerleşmiş AEİ’lerin tüm olgularda tam
nöbet kontrolü sağlamaması ve yan etkilerin olması yeni
AEİ’lerin geliştirilmesine neden olmaktadır. Yeni AEİ’lerın
etki mekanizmaları (iyon kanallarının blokajı, GABAerjik
transmisyonu artırmaları, eksitatör transmisyonun antagonizması)
temelde, daha önceki bazı ilaçların mekanizmalarıyla büyük
ölçüde çakışmaktadır (Tablo 23). Bununla birlikte,
moleküler düzeyde meydana getirdikleri etkiler kesin olarak bilinmemektedir. Bu
ilaçların bazıları, metabolik transformasyon ve düşük
etkileşim potansiyeli (levetirasetem, vigabatrin ve gabapentin) gibi
özelliklerle eskilerden ayrılmaktadır; bazıları ise
yoğun bir şekilde metabolize olmakta ve ilaç etkileşimlerinde
yaygın bir rol oynamaktadır (felbamat, lamotirijin). Bazı yeni
antiepileptikler aktivite spektrumu açısından, sadece fokal
nöbetlerin tedavisinde kullanılırken (okskarbazepin, vigabatrin,
pregabalin, gabapentin gibi); bazıları ise hem fokal hem de
jeneralize nöbet tipleri üzerinde etkinlik göstermektedir (levetirasetam,
lamotirijin, topiramat, zonisamid ve felbamat). Ayrıca, bu ilaçların
infantil spazmlar (özellikle vigabatrin) ve ilaca dirençli epilepsilerin tedavisinde
ve nöbet sıklığının düşürülmesinde de etkili
oldukları görülmektedir.
Yeni
AEİ’lerin yerleşmiş diğer antiepileptiklere oranla en büyük
şanssızlıkları yoğun araştırmalar sonucu
geliştirilmelerine bağlı olan çok yüksek maliyetleridir. Bunun
dışında eskiden beri kullanılan ilaçların olası
tehlikeli yan etkilerinin zaten biliniyor olması oysa yeni
antiepileptiklerle bilinmeyen tehlikeli yan etkilere rastlama
olasılığı da yerleşmiş antiepileptiklere tercih
edilmelerini engellemektedir. Yerleşmiş antiepileptiklere dirençli
veya yan etki görülen hastalarda yeni antiepileptikler kullanılabilir.
Türkiye’de yeni antiepileptiklerden lamotirijin, vigabatrin, okskarbazepin,
topiramat, gabapentin, pregabalin, levetirasetam, zonisamid, lakozamid piyasada
bulunmaktadır. Klobazam, Türk Eczacılar Birliği
Yurtdışı İlaç grubunda bulunmaktadır ve özel bir rapor
ile Türk Eczacılar Birliği tarafından
yurtdışından getirilebilmektedir. Örneğin stripentol,
Dravet sendromunda valproat ve klobazama ek olarak kullanılmak üzere onay
almış yeni bir AEİ’dir ve bu sendroma özel olarak
getirilebilmektedir. Bazı yeni ilaçların da önümüzdeki yıllarda
pazarda yerini alması beklenmektedir.
Yeni
tanı almış epilepsili hastaların ilk monoterapisinin
belirlenmesi amacıyla, randomize kontrollü çalışmaların bir
derlemesi yapılarak 2013’te bir ILAE rehberi
yayınlanmıştır. Erişkin
başlangıçlı fokal epilepsili hastalar üzerinde yapılan
randomize kontrollü çalışmalar ve metaanalizler sonucunda
karbamazepin, fenitoin, levetirasetam ve zonisamid ilaçlarının
etkinliği açısından sınıf A düzeyinde (kesin)
kanıt bulunmaktadır. Valproat için sınıf B düzeyinde
(muhtemel) kanıt olmakla birlikte karbamazepin ile
karşılaştırmalı çalışmalarda jeneralize ve
fokal başlayıp bilateral tonik-klonik nöbete dönüşen nöbetlerde
etkinliği benzer ancak fokal nöbetlerde karbamazepine kıyasla
düşük etkinlikte olduğu bildirilmiştir. Ayrıca valproat ile
son yıllarda bildirilen yüksek teratojenite riski nedeniyle,
doğurganlık çağındaki kadın epilepsili hastalarda
kullanımından kaçınılması gerekmektedir (Ayrıca
bakınız: Epilepsi ve Gebelik).
Gabapentin, lamotirijin, okskarbazepin, topiramat, vigabatrin ve fenobarbital
ile sınıf C düzeyi (olası etkinlik) kanıt
bulunmaktadır. Klonazepam ve pirimidon ile nöbet kontrolü
açısından etkin olma potansiyelinden bahsedilebilir (sınıf
D düzeyi kanıt). İleri yaş hastalar ve çocuk yaş grubunda
yeni başlangıçlı fokal epilepsi hastalarının
monoterapi etkinliğini değerlendiren çalışma
sayısı azdır. Elde bulunan çalışmalarda edinilen
veriler ileri yaş grubunda fokal epilepsili hastalarda lamotirijin ve
okskarbazepinin etkinliğini göstermektedir, karbamazepin ile olası
etkinlik bildirilmiştir. Topiramat ve valproat ise bu grupta etkin olma
potansiyeli bulunan ilaçlardır. Çocukluk yaş grubunda ise etkinliği
çalışmalar ile kesin olarak kanıtlanmış tek ilaç
okskarbazepindir. Bu yaş grubunda olası etkinlikten bahsedilen
ilaçlar ise karbamazepin, fenitoin, fenobarbital, topiramat, valproat ve
vigabatrindir. Bu gibi rehberler ve metaanalizler değerli olmakla birlikte
hasta özelinde uzman deneyimlerine dayanan farklı seçimler de
yapılabilmektedir.
Absans
nöbetleri olan çocuk yaş grubunda valproat ve etosüksimid kesin etkilidir
(kanıt düzeyi sınıf A). Lamotirijin ise olası etkili olarak
belirtilmiştir. Absans epilepsisinde levetirasetam, zonisamid ve topiramat
ikinci seçenek olarak kabul edilir. Olgu serilerinden edinilen veriler
gabapentin, karbamazepin, fenitoin, fenobarbital, okskarbazepin ve tiagabinin
absans nöbetlerini arttırabileceğini göstermektedir. Buna ek olarak
juvenil miyoklonik epilepsili hastaların da gabapentin, karbamazepin,
fenitoin, okskarbazepin, tiagabin ve vigabatrin ile absans, miyokloni ve hatta
jeneralize tonik-klonik nöbetlerinin artabileceği bildirilmiştir.
Juvenil miyoklonik epilepside lamotirijinin miyoklonileri
arttırabileceği de düşük kanıt düzeyinde olmakla birlikte
belirtilmektedir.
AEİ’lerin
vücuttan atılım yerleri karaciğer ve/veya böbrek olduğundan
karaciğer veya böbrek yetmezliği olan hastalarda antiepileptik tedavi
seçimi değişmektedir. Karaciğer hastalığı olan ve
karaciğer testlerinde yükseklik saptanan hastalarda hepatotoksisite yan
etkisi olan ilaçların yüksek dozlarından kaçınılması
gerekir. Bu ilaçlar arasında karbamazepin, fenitoin, fenobarbital ve
lamotirijin sayılabilir. Ayrıca tamamen karaciğerden metabolize
olan ilaçlar karaciğer yetmezliğinde vücutta birikerek toksisiteye
neden olur. Bu grupta fenitoin, karbamazepin, valproat, rufinamid ve tiagabin
bulunur.
Tamamen
ya da büyük çoğunlukla değişmeden böbrek üzerinden atılan
ilaçların böbrek yetmezliğinde kullanılmasından
kaçınılmalı ya da mutlaka kullanılması gerekiyorsa doz
ayarlaması yapılmalıdır. Bu grup içerisinde gabapentin,
pregabalin, vigabatrin, topiramat ve levetirasetam bulunur. Tamamen veya
ağırlıklı olarak karaciğerden metabolize olan
ilaçların böbrek yetmezliğinde kullanımlarında doz
ayarlamasına gerek yoktur.
Bir
kısmı karaciğerde metabolize olan ve bir kısmı
böbrekten değişmeden atılan ilaçların hem böbrek hem de
karaciğer yetmezliğinde kullanımlarına dikkat edilmelidir.
Bu grup içerisinde okskarbazepin, eslikarbazepin ve lakozamidin ağır
böbrek yetmezliğinde doz ayarlaması gerekmektedir. Böbrek
yetmezliğinde lamotirijin, zonisamid, fenobarbital ile ilgili yeterli veri
olmadığından dikkatli kullanılmalıdırlar.
Lamotirijin, okskarbazepin
karaciğer dışı dokularda da metabolize olabildikleri
için karaciğer yetmezliğinde serum düzeyleri değişmez.
Diyalize
giren hastalarda proteine yüksek oranda bağlanan ilaçlar tercih
edilmelidir. Bu durumda birinci seçenek fenitoin olmakta ancak yan etki profili
nedeniyle kullanılamadığı durumlarda valproat veya
karbamazepin tercih edilmelidir. Bu ilaçlar ile diyaliz sonrası ek doz
verilmesi gerekmez. Diyaliz sonrası eliminasyon riski en yüksek olan
ilaçlar levetirasetam, etosüksimid, gabapentin, pregabalin, lakozamid ve
topiramattır. Diyaliz hastasında bu ilaçların
kullanımında diyaliz öncesi veya sonrası 4 saat içerisinde ek
doz verilmesi önerilir. Ek doz miktarı hastaya göre değişir,
diyaliz öncesi ve sonrası ilaç düzeyi bakılarak karar verilebilir.
Topiramat, etosüksimid, levetirasetam ve lakozamid için bu ek doz kabaca günlük
toplam dozun %50’si, gabapentin ve pregabalin için %100-200’ü
kadardır.
Başka
hastalıklar nedeniyle çoklu ilaç kullanan özellikle ileri yaş grubundaki
hastalarda ilaç etkileşimi az olan levetirasetam, pregabalin, gabapentin,
topiramat, lakozamid tercih edilebilir.
Psikiyatrik
ve nörolojik komorbiditesi olan hastalarda AEİ tercihi bu durumlar göz
önüne alınarak yapılmalıdır. Örneğin duygudurum bozukluğu
olan hastalarda valproat, lamotirijin ve karbamazepin tercih edilebilir. Dürtü
kontrol bozukluğunda benzer şekilde karbamazepin daha iyi bir seçenek
olabilir. Migren ile epilepsi birlikteliğinde topiramat, valproat veya
lamotirijin her iki hastalığa birden etki gösterdiği için ön
sıralarda tercih edilebilir.
AEİ
seçiminde uzman görüşleri ve kanıta dayalı veriler bir arada
değerlendirildiğinde şu sonuçlara varılabilir:
1) İdyopatik/genetik jeneralize epilepsi
hastalarında birinci basamak monoterapi valproat, levetirasetam,
lamotirijindir. Ayrıca bu gruba ek olarak yeni AEİ’lerden zonisamid
ve topiramat da eklenmiştir. Absans epilepside etosüksimid birinci basamak
kabul edilir.
2) Miyoklonik epilepsilerde birinci seçenek
levetirasetam, valproat, lamotirijindir. Zonisamid ve topiramat da miyoklonik
epilepsilerde kullanılabilir.
3) İdyopatik/genetik jeneralize epilepsili
doğurganlık çağındaki kadınlarda birinci basamak
lamotirijin veya levetirasetamdır. İkinci seçenek zonisamid olabilir.
4) Fokal epilepsili hastalarda birinci basamak
monoterapide karbamazepin, okskarbazepin, levetirasetam, zonisamid tercih
edilir. İkinci basamak ilaçlar arasında lamotirijin, topiramat,
valproat, vigabatrin, gabapentin ve fenobarbital bulunur. Fenitoin kanıt
düzeyi yüksek olmasına rağmen bu grupta ilk basamak olarak
kullanımını yan etkiler nedeniyle pratikte kaybetmiştir.
Benzer şekilde yan etkileri nedeniyle fenobarbital ve vigabatrin de tercih
edilmez.
5) Yaşlı hastalarda levetirasetam, pregabalin,
gabapentin, lamotirijin ve lakozamid tercih edilir.
6) Beyin tümörü olan hastalarda, sağkalımı
uzattığı yönünde veriler olması sebebiyle levetirasetam ilk
seçenektir. Bunun yanında diğer onkolojik hastalarda kemoterapi ile
etkileşimi engellemek amacıyla levetirasetam, lamotirijin veya
lakozamid tercih edilebilir.
7) Böbrek yetmezliği olan ancak diyalize girmeyen
fokal epilepsili hastalarda karbamazepin ve jeneralize epilepsili hastalarda
valproat ilk seçenektir. Ağır böbrek yetmezliği yoksa fokal
epilepsili hastalarda okskarbazepin veya lakozamid kullanılabilirken,
jeneralize epilepsili hastalarda lamotirijin kullanılabilir.
8) Böbrek yetmezliği olan ve diyalize giren fokal
epilepsili hastalarda karbamazepin ve jeneralize epilepsili hastalarda valproat
kullanılabilir. Bir diğer seçenek her iki grup için de lamotirijin
veya fenitoin olabilir.
9) Karaciğer yetmezliği olan jeneralize ve
fokal epilepsili hastalarda ilk seçenek levetirasetamdır. Ayrıca
fokal epilepsili hastalarda lakozamid, gabapentin veya pregabalin
kullanılabilir.
Antiepileptik İlaçların Etki Mekanizmaları
(Tablo 21)
Fenobarbital, postsinaptik membranda bulunan GABAA reseptörünün
açık kalma süresini uzatır ve bu sayede hücre membranında
hiperpolarizasyona neden olur. Fenitoin, karbamazepin ve okskarbazepin, voltaja
bağımlı Na kanal blokeridir. Etosüksimid, talamusta bulunan T
(transient) tipi kalsiyum kanal inhibisyonu yapar. Valproatın, birden
fazla mekanizması vardır, beynin GABAerjik inhibitör aktivitesini
arttırır, Na kanal blokajı yapar ve beyin içerisinde birçok
reseptörü etkileyerek eksitabiliteyi azaltır. Topiramat, AMPA/kainat
reseptör antagonizması, GABA aktivitesinin artırılması,
voltaj kapılı Na kanal blokajı yapar. Zonisamid, T-tipi kalsiyum
kanal blokajı, voltaj kapılı Na kanal blokajı yaparken,
topiramat ve zonisamidin ayrıca karbonik anhidraz inhibitörü özellikleri
bulunur. Lamotirijin, yüksek voltaj aktivasyonlu kalsiyum kanal inhibisyonu ve
voltaj kapılı Na kanal blokajı yapar. Levetirasetam, sinaptik
vezikül protein 2A’ya bağlanır, nöronal hiperaktiviteye neden
olabilecek nörotransmitter düzeylerini azalttığı
düşünülmektedir. Felbamat, yüksek voltaj aktivasyonlu kalsiyum kanal
inhibisyonu, voltaj kapılı Na kanal blokajı, NMDA
antagonizması, GABA’nın arttırılmasında görev
alır. Benzodiazepinler, GABA ile ilişkili inhibisyonu fasilite eder.
Gabapentin ve pregabalin, voltaj aktivasyonlu kalsiyum kanalının
alfa-2-delta subunitini inhibe ederek, hücre içine kalsiyum girişini
azaltır ve hipereksitabilite durumlarında ilişkili
nörotransmitter salınımını azaltır. Vigabatrin, gama aminobitürik
asit transaminazın geri dönüşümsüz inhibitörüdür. Bu enzim,
GABA’nın yıkımından sorumludur. Diğer Na kanal
blokerlerinin hızlı inaktivasyonundan farklı olarak lakozamid,
voltaj bağımlı Na kanalının yavaş inaktivasyon
fazını arttırır. Eslikarbazepin, voltaj kapılı Na
kanalının kompetetif blokeridir, ayrıca lakozamid gibi voltaj
kapılı Na kanalının yavaş aktivasyonu üzerinden de
etkilidir. Ezogabin (retigabin), KCNQ potasyum kanalının pozitif
allosterik modulatörüdür ve K kanalının açık kalmasını
sağlar, ancak olumsuz etkileri nedeniyle kullanılmamaktadır.
Tiagabin, GABA geri alım inhibisyonu yapar. Rufinamidin Na
bağımlı aksiyon potansiyellerinin ateşlenmesini
azalttığı düşünülmektedir. Voltaj kapılı Na
kanalı tip 1 alfa subunitinin (SCN1A) aktivasyonunu inhibe eder.
Ayrıca deneysel modeller, voltaj kapılı Na kanalının
inaktivasyon fazını uzattığını göstermiştir.
In vitro çalışmalarda, yüksek doz ile insan rekombinan metabotropik
glutamat reseptör subtip 5 (mGluR5) üzerinde inhibitor etkiye neden olur.
Perampanel ise AMPA reseptörünün non-kompetetif
antagonistidir ve AMPA reseptörüyle ilişkili sinaptik geçişin
azalmasına neden olur.
Tablo 21. Antiepileptik ilaçların farmakokinetikleri (Kaynak 1’den
değiştirilerek)
|
İlaç
|
Emilim*
(Biyoyararlanım)
|
Proteine
bağlanma
|
Yarılanma
süresi (saat)
|
Atılma
yolu
|
Özellikler
|
|
Karbamazepin
|
6-12 (%75-85)
|
%70-80
|
·
24-45 (tek
doz)
·
8-24
(kronik)
|
· Hepatik metabolizma (%95)
· aktif metabolit (karbamazepin-10,11-epoksid)
|
· Enzim indüksiyonu
· Otoindüksiyon
|
|
Na-Valproat
|
3-5 (%100)
|
%88-92
|
·
7-17
(chrono formu dışında)
|
· Hepatik metabolizma (%95)
· aktif metabolit
|
· Enzim inhibisyonu
· Otoindüksiyon
· Konsantrasyona bağlı protein
bağlanması
|
|
Fenitoin
|
4-8 (%90)
|
%90-93
|
·
9-40
|
· Doyurulabilen hepatik metabolizma
|
· Enzim indüksiyonu
· Konsantrasyona bağlı
yarılanma
|
|
Fenobarbital
|
6-12 (iyi)
|
%48-54
|
·
72-144
|
· Hepatik metabolizma (%75)
·
%25’ideğişmeden
idrarla atılır
|
· Enzim indüksiyonu
|
|
Klonazepam
|
4 (%80-90)
|
%80-90
|
·
30-40
|
· Hepatik metabolizma
|
-
|
|
Okskarbazepin
|
1-3 (iyi)
|
%40
|
·
1-3,7
·
8-10
(monohidroksi derivesi)
|
·
Hepatik
metabolizma (p450 aktivitesini kullanmaz)
|
· Zayıf enzim indüksiyonu
· Majör metaboliti 10-hidroksi karbazepin
· Daha az ilaç etkileşimi
|
|
Eslikarbazepin†‡
|
2-3 (iyi)
|
<%40
|
·
13-20
|
·
Hepatik
metabolizma (%50) ile aktif metabolitine (S-licarbazepine) dönüşür
·
%50’si
değişmeden idrarla atılır
|
· Zayıf enzim indüksiyonu
|
|
Lamotirijin
|
1-3 (iyi)
|
%50-55
|
·
10-15
|
·
Hepatik
metabolizma (%90)
·
Gliküronidasyon
ile temizlenir ve idrar ile atılır
|
· VPA ile yarılanma ömrü artar
· Enzim indükleyiciler ile yarılanma ömrü
azalır
|
|
Levetirasetam
|
1-1,5 (>%95)
|
<%10
|
·
7-8
|
·
%66 idrar
ile değşmeden atılır
·
%34 inaktif
bileşenlerine hidroliz olur
|
· İlaç etkileşimi yok
|
|
Topiramat
|
1-4 (iyi)
|
%9-17
|
·
20-24
|
·
%70 idrar
ile değişmeden atılır
·
%30 hepatik
metabolizma
|
· Enzim indükleyiciler ile yarılanma ömrü
artar
|
|
Pregabalin
|
1 (iyi)
|
0
|
·
6
|
·
Değişmeden
idrarla atılır
|
· İlaç etkileşimi yok
|
|
Gabapentin
|
2-3 (düşük)
|
0
|
·
5-7
|
·
Değişmeden
idrarla atılır
|
· İlaç etkileşimi yok
· Doza bağımlı emilim
|
|
Tiagabin‡
|
0,5 (iyi)
|
%96
|
·
5-9
|
·
Hepatik metabolizma
(%90)
|
· Enzim indükleyiciler ile yarılanma ömrü
azalır
|
|
Felbamat‡
|
1,5-3 (iyi)
|
%25
|
·
20-23
|
·
Hepatik
metabolizma (%50)
·
%50
değişmeden idrarla atılır
|
· Enzim inhibisyonu
|
|
Zonisamid
|
5-6 (iyi)
|
%40-60
|
·
50-68
|
·
Hepatik
metabolizma (%70)
|
· Enzim inhibisyonu ve indüksiyonu yapmaz
· İlaç etkileşimi yok
· Zayıf karbonik anhidraz inhibitörü
|
|
Lakozamid
|
0,5-4 (iyi)
|
<%15
|
·
14-16
|
·
%40’ı
idrar ile değişmeden atılır
·
Karaciğerde
inaktif metabolitlerine dönüşür
|
· İlaç etkileşimi az
|
|
Vigabatrin
|
1-2 (iyi)
|
0
|
·
10.5 (genç
erişkin)
·
5-6
(infant)
|
·
İdrar
ile değişmeden atılır
|
· Zayıf enzim indüksiyonu
|
|
Rufinamid T‡
|
4-6 (iyi) Yemek ile birlikte
alındığında emilim iyi.
|
%26-35
|
·
6-10
|
· Hepatik metabolizma ile inaktif metabolitine
hidroliz olur ve idrar ile atılır
|
· Valproat ile atılımı azalır
ve plazma seviyesi %70 oranında artar
|
|
Ezogabin
(Retigabin) ‡
|
1,5-1,8 (%60)
|
%80
|
·
7-11
|
·
Hepatik
metabolizma (%64)
·
%36’sı
idrar ile değişmeden atılır
|
· Aktif N-asetil metabolitine dönüşür ve
sonrasında glukronide olur.
|
|
Perampanel‡
|
0,15-2 (iyi)
|
%95
|
·
48-105
|
·
Hepatik
metabolizma
|
· Karbamazepin ve fenitoin ile perampanelin
klirensi 2-3 katına çıkar.
|
|
Etosüksimid
|
1-3 (%90-95)
|
0
|
·
20-60
|
·
Hepatik
metabolizma (%80)
·
%20
değişmeden idrarla atılır
|
· Çocuklarda daha hızlı klirens
|
*Tmax
değerleri, saat (tek doz sonrası maksimum plazma konsatrasyonuna
ulaşma süresi)
†Okskarbazepinin monohidroksi
derivesi. ‡Türkiye’de
bulunmayan ilaçlar
Tablo 22. Antiepileptik
İlaçların Endikasyonları ve Dozları (Kaynak 1’den değiştirilerek)
|
İlaç
|
Endikasyonları
|
Başlama
dozu
|
Ortalama
doz (gün)
|
Kullanıldığı
devam dozları
|
Doz
aralığı
|
Preparat
tipi
|
Hedef
düzey
|
|
Karbamazepin
|
·
Fokal nöbet
·
JTKN
|
200mg
|
600-800mg
|
400-1600mg
|
Günde 2-3*
|
·
Oral
|
4-12mg/L
|
|
Na-Valproat
|
·
Fokal nöbet
·
JTKN
·
Absans
·
Miyokloni
·
LGS/infaltil
spazm
·
Status
epileptikus
|
500mg
|
1000mg
|
500-3000mg
|
günde 1-2*
|
·
Oral
·
İv
|
50-100mg/L
|
|
Fenitoin
|
·
Fokal nöbet
·
JTKN
·
Status
epileptikus
|
200mg
|
300mg
|
100-400mg
|
Günde 2-3
|
·
Oral
·
İv
|
10-20mg/L
|
|
Fenobarbital
|
·
Fokal nöbet
·
JTKN
·
Status
epileptikus
|
60mg
|
120mg
|
60-240mg
|
Günde 1-2
|
·
Oral
·
İv
|
10-40mg/L
|
|
Klonazepam
|
·
Fokal nöbet
·
JTKN
·
Absans
·
Miyokloni
·
LGS/infaltil
spazm
·
Status
epileptikus
|
1mg
|
4mg
|
2-8mg
|
Günde 1-2
|
·
Oral
·
İv
(ülkemizde yok)
|
yok
|
|
Okskarbazepin
|
·
Fokal nöbet
|
300mg
|
600-1200mg
|
300-2400mg
|
Günde 2-3*
|
·
Oral
|
15-35 mg/L
|
|
Eslikarbazepin
|
·
Fokal nöbet
|
400mg
|
800mg
|
400-1600mg
|
Günde 1-2
|
·
Oral
|
-
|
|
Lamotirijin
|
·
Fokal nöbet
·
JTKN
·
Absans
·
Miyokloni
(değişken)
·
LGS/infantil
spazm
·
Status
epileptikus
|
50mg
|
200-300mg
|
200-600mg
|
Günde 2-3
|
·
Oral
|
2-20 mg/L
|
|
Levetirasetam
|
·
Fokal nöbet
·
JTKN
·
Absans
·
Miyokloni
·
Status epileptikus
|
500mg
|
1500mg
|
1000-4000mg
|
Günde 2-3
|
·
Oral
·
İv
|
-
|
|
Topiramat
|
·
Fokal nöbet
·
JTKN
·
LGS/infaltil
spazm
|
25mg
|
300mg
|
100-400mg
|
Günde 2-3
|
·
Oral
|
-
|
|
Pregabalin
|
·
Fokal nöbet
|
75mg
|
300mg
|
150-600mg
|
Günde 2-3
|
·
Oral
|
-
|
|
Gabapentin
|
·
Fokal nöbet
|
300mg
|
1200mg
|
900-4800mg
|
Günde 2-3
|
·
Oral
|
-
|
|
Tiagabin
|
·
Fokal nöbet
|
4mg
|
24mg
|
36-48mg
|
Günde 2-3
|
·
Oral
|
-
|
|
Felbamat
|
·
Fokal nöbet
·
JTKN
·
LGS/infantil
spazm
|
1200 mg
|
1200-2400mg
|
1200-2400mg
|
Günde 2-3
|
·
Oral
|
-
|
|
Zonisamid
|
·
Fokal nöbet
·
JTKN
·
Absans
·
Miyokloni
|
100mg
|
300mg
|
200-600mg
|
Günde 2
|
·
Oral
|
10-40 mg/L
|
|
Lakozamid
|
·
Fokal nöbet
|
100mg
|
200-400mg
|
200-600mg
|
Günde 2
|
·
Oral
·
İv
|
-
|
|
Vigabatrin
|
·
Fokal nöbet
·
LGS/infaltil
spazm
|
1000mg
|
3000mg
|
1000-6000mg
|
Günde 2
|
·
Oral
|
-
|
|
Rufinamid
|
·
Fokal nöbet
·
JTKN
·
LGS/infaltil
spazm
|
200mg
|
1000mg
|
400-3200mg
|
Günde 2
|
·
Oral
|
-
|
|
Ezogabin (retigabin)
|
·
Fokal nöbet
|
200mg
|
600mg
|
600-1200mg
|
Günde 3
|
·
Oral
|
-
|
|
Perampanel
|
·
Fokal nöbet
·
JTKN
|
|
|
|
|
·
Oral
|
-
|
|
Etosüksimid
|
·
Absans
|
500mg
|
750mg
|
500-1000mg
|
günde 1-3
|
·
Oral
|
40-100mg/L
|
*od-bd sadece kontrollü
salınım formları ile
Tablo 23. Antiepileptik İlaçların Etki
Mekanizmaları
|
İlaç
|
Na kanalı
|
Ca kanalı
|
GABA sistemi
|
Glutamat reseptörü
|
K kanalı
|
SV2A
|
|
Fenitoin
|
+
|
|
|
|
|
|
|
Karbamazepin
|
+
|
|
|
|
|
|
|
Lamotirijin
|
+
|
HVA
|
|
|
|
|
|
Zonisamid
|
+
|
T tipi
|
|
|
|
|
|
Fenobarbital
|
|
HVA
|
GABAA reseptörü
|
AMPA
|
|
|
|
Benzodiazepin
|
|
|
GABAA reseptörü
|
|
|
|
|
Vigabatrin
|
|
|
GABAT inhibisyonu
|
|
|
|
|
Tiagabin *
|
|
|
GABA geri alım inhibisyonu
|
|
|
|
|
Etosüksimid
|
|
T tipi
|
|
|
|
|
|
Valproik asit
|
+
|
T tipi
|
GABA artımı
|
+
|
|
|
|
Topiramat
|
+
|
|
GABAA
|
KA / AMPA
|
|
|
|
Gabapentin
|
|
VAα2δ
|
GABA artımı
|
+?
|
|
|
|
Pregabalin
|
|
VAα2δ
|
GABA artımı
|
|
|
|
|
Levetirasetam
|
|
|
|
|
|
+
|
|
Felbamat*
|
+
|
HVA
|
GABAAres
|
NMDA
|
|
|
|
Okskarbazepin
|
+
|
|
|
|
|
|
|
Eslikarbazepin*
|
+
|
|
|
|
|
|
|
Lakozamid
|
+
|
|
|
|
|
|
|
Rufinamid*
|
+
|
|
|
|
|
|
|
Ezogabin (retigabin)*
|
|
|
|
|
+
|
|
|
Perampanel*
|
|
|
|
AMPA
|
|
|
HVA: (high voltage activated)
yüksek voltaj aktivasyonlu, GABAT: GABA transaminaz, NMDA: N-metil D-aspartat,
KA: kainik asit, AMPA: αamino-3-hidroksi-5-metil-4-izoksazol-proprionik
asit, VA: (voltage activated) voltaj aktivasyonlu, SV2A: sinaptik vezikül protein
2A
*Ülkemizde bulunmamaktadır.
Son
yıllarda özellikle tedaviye dirençli gelişimsel ve epileptik
ensefalopatili pediyatrik yaş grubunda kanabinoid tedavisi ile nöbet
sıklığında belirgin azalmanın izlendiği
gösterilmiştir. Bu durum kanabinoidlerin antiepileptik özelliklerinin,
güvenilirlik ve etkinliklerinin araştırılmasına yol
açmıştır. Endokanabinoidler, nöronal eksitabilitede azalmaya
neden olurlar ve K kanallarını açıp Ca kanallarını
kapatarak nörotransmitter salınımını azaltırlar. Ancak
dışarıdan verilen kanabinoidler bu etkileri kullanmadan nöbet
azaltıcı etki gösterirler. Tam etki mekanizması bilinmemekle
birlikte Na ve Ca iyon kanalları üzerinden etki ettiği
düşünülmektedir. Dravet sendromu ve Lennox Gastaut sendromlu çocuklarla
yapılan randomize kontrollü çalışmalarda antiepileptik
etkinliği gösterilmiştir. Ancak karmaşık farmokokinetik
özellikleri, yetersiz etkinlik ve güvenilirlik çalışmaları ve
yasal konular sebebiyle günümüzde kullanımı
sınırlıdır. Kanabinoidler ile her geçen yıl artan
çalışmalar ile toplanan veriler bu sorulara cevap verecek ve
antiepileptik tedavi listesinde yerini belirleyecektir.
Antiepileptik İlaçların Yan Etkileri (Tablo
24-26)
AEİ
yan etkileri beş alt başlık altında gözden geçirilebilir:
1)
İlk grup
reaksiyonlar, akut reaksiyonlar, ilaç etki mekanizması ve doz ile
ilgilidir. Burada en çok MSS üzerine etkiler görülür. Bu yan etkiler içinde
kognitif etkilenme kişinin sosyal ve iş yaşamını en
çok etkileyen önemli bir problemdir. Bütün AEİ’ler kognitif etkilenme
yapabilir. Polifarmasi, yüksek dozda kullanım veya AEİ serum
düzeyinin yüksek olması riski arttırır. Yerleşmiş
AEİ arasında pirimidon ve fenobarbital ve yeni AEİ’lerden
topiramat kognitif etkilenmeyi en fazla yapan ilaçlardır. Akut doza veya
konsantrasyona bağlı toksisite genellikle spesifik
olmayan bir ensefalopati şeklinde seyreder, nistagmus, ataksi, dizartri,
konfüzyon, sersemlik dikkati çeker. Bu sırada genellikle ilaç kan düzeyi
normal sınırların üstünde bulunur. Bu tabloya hemen tüm
yerleşmiş antiepileptikler yol açabilir, en az valproat ile
bildirilmiştir. Fenitoin istemsiz hareketlere yol açabilir. Çocuklarda
fenobarbital ve benzodiazepinlere bağlı olarak davranış
bozuklukları oluşabilmektedir. Bu tip yan etkilerin özelliği doz
azaltılması ile iz bırakmadan kaybolmasıdır.
2)
İdyosenkratik etki
mekanizmalarıyla ilişkilidir (immünolojik, genetik veya diğer
mekanizmalar). Akut idyosenkratik toksisite, dozdan
bağımsız gelişir. Fenitoin ve karbamazepin başta olmak
üzere çoğu ilaç ile görülebilen hafif makülopapüler deri döküntüleri bunun
en sık görülen örneğidir. Çok ağır seyreden eksfolyatif
erüpsiyonlar veya Stevens-Johnson sendromu nadir ancak çok tehlikeli bir
dermatolojik yan etkidir, fenitoin, fenobarbital karbamazepin ve lamotirijinle
görülebilmektedir. Bu tabloya ateş, hepatotoksisite, lenfadenopati
eşlik edebilir. Karbamazepine bağlı bir diğer ciddi ancak
çok seyrek idyosenkratik yan etki aplastik anemidir. Yeni AEİ’lerden felbamatın
1/2000 oranında aplastik anemiye yol açtığının
sonradan anlaşılması üzerine kullanımı
sınırlanmıştır. Özellikle küçük çocuklarda valproata
bağlı akut fatal karaciğer yetmezliği bildirilmiştir. Tablo
24’de bazı idyosenkratik etkiler belirtilmiştir. Tedaviye
başlandıktan sonraki ilk haftalar veya aylarda görülür.
3)
Kronik veya
gecikmiş yan etkiler ise aylar ve yıllar içinde görülmektedir. Altta
yatan mekanizmalar anlaşılamamıştır hemen her sistemi
ilgilendirebilir. Yüksek doz kullanımı ve ilaç kombinasyonları
genellikle kümülatif olan bu tip yan etkiyi arttırıcı rol oynar
örneğin fenitoine bağlı dişeti hiperplazisi, barbitürata
bağlı omuz-el sendromu. Bilinen biyokimyasal veya metabolik
mekanizmalar sonucu vitamin D katabolizmasını hızlandıran
enzim indükleyen AEİ’ler osteomalazi yapmaktadırlar. Endokrin ve
üreme organlarında örneğin overlerde polikistik
değişiklikler, yüksek testosteron düzeyi ve menstrüel düzensizlikler
görülebilir. Önemli diğer sorun ise AEİ’lerin kilo üzerine etkisidir.
Topiramat, felbamat ve zonisamid kilo kaybına neden olurken, valproik
asit, pregabalin, gabapentin, vigabatrin ve daha az oranda karbamazepin kilo
alımı ile ilişkilidir. Kozmetik istenmeyen yan etkiler bir
yandan da hastaların kompliyansının azalmasına veya ilacı
bu nedenle tamamen bırakmasına neden olabilir. Kilo alımı
özellikle diabetus mellitusu ve kalp hastalığı olanlarda ciddi
riskler yaratabilir.
4)
Bu grup yan etki olarak ikinci
jenerasyona etki (ör: teratogenez) ve karsinojenik etki ele alınır.
AEİ ile karsinojenik etki bilinmemektedir. Ancak teratogenez konusu son
yıllarda geniş çalışmalarda ele
alınmıştır. Gebelik ve epilepsi bölümünde ele alınacak
olan bu konuda bilinen, tüm AEİ’lerin olası teratojenik etkileri
olmasına karşın bunun özellikle düşük doz
kullanıldığında ve monoterapi ile düşük olduğudur.
5)
Son olarak da ilaç etkileşimleri
incelenir ve Tablo 24’de özetlenmiştir.
Tablo 24. Antiepileptik ilaçların bazı yan etkileri
|
Akut (doza bağımlı)
|
İdyosenkratik
|
Kronik
|
|
Ataksi
|
Aplastik anemi
|
Bağ dokusu
hastalıkları: diş eti hiperplazisi, yüz hatlarında
kabalaşma, Dupuytren kontraktürü
|
|
Görme bulanıklığı, çift görme
|
Glokom
|
Endokrin hastalıklar:
Tiroksin düzeyinde azalma, kortizol ve seks hormon metabolizmasında
artma
|
|
Kognitif etkilenme
|
Karaciğer toksisitesi
|
Ig A eksikliği, ilaca bağlı sistemik lupus eritematozus
|
|
Duygudurum/davranış bozukluğu
|
Cilt döküntüsü
|
Saç dökülmesi
|
|
Baş dönmesi
|
Diğer
immün-aracılı reaksiyonlar
|
Hirsutizm, akne
|
|
Yorgunluk
|
|
Nefrolitiyazis
|
|
Gastrointestinal semptomlar
|
|
Görme alanı defekti
|
|
Başağrısı
|
|
Vitamin D/folat eksikliği
|
|
Hiponatremi
|
|
Kilo alımı, kilo kaybı
|
|
İnsomni
|
|
Osteomalazi
|
|
Uykuya eğilim
|
|
Bellek başta olmak üzere kognitif bozukluklar
|
|
Tremor
|
|
Davranış problemleri
|
Tablo 25. Yerleşmiş ve Yeni Antiepileptik İlaçların
Bazı Ciddi ve Diğer Yan Etkileri
|
AEİ
|
Ciddi yan etki
|
Diğer yan etki
|
|
Karbamazepin
|
·
Aplastik
anemi, hepatotoksisite, Steven-Johnson sendromu, lupus benzeri tablo, ilaç
hipersensitivite sendromu
|
· Sersemlik, ataksi, diplopi, bulantı, yorgunluk
hissi, lökopeni, döküntü, trombositopeni, hiponatremi, davranış
bozukluğu, tikler, kognitif bozukluk
· Uzun dönem kullanımda kilo alımı
ve osteoporoz
|
|
Etosüksimid
|
·
Kemik
iliği depresyonu, hepatotoksisite, lupus, psikoz, Steven-Johnson
sendromu, otoimmun tiroidit
|
· Bulantı, gastrik iritasyon,
başağrısı, döküntü, insomni, yorgunluk, ataksi,
sersemlik, davranış değişiklikleri
|
|
Felbamat
|
·
Aplastik
anemi (ölümcül), hepatik yetmezlik, Steven-Johnson sendromu
|
· Anoreksi, kusma, insomni,
başağrısı, uykuya eğilim, kilo kaybı
|
|
Klonazepam
|
-
|
· Sedasyon, dizartri, sersemlik, ataksi, nistagmus,
tolerans, “rebound” etki
|
|
Gabapentin
|
-
|
· Uykuya eğilim, sersemlik, ataksi, yorgunluk
hissi, nistagmus, kilo artışı, miyoklonus, çocuklarda
davranış bozukluğu
· Ödem (yaşlılarda)
|
|
Lamotirijin
|
·
Steven-Johnson
sendromu ya da toksik epidermal nekroz, hipersensitivite sendromu, akut
hepatik yetmezlik /multiorgan yetmezliği
|
· Sersemlik, ataksi, insomni,
başağrısı, diplopi, görme
bulanıklığı, bulantı, kusma, döküntü, ajitasyon,
tremor
|
|
Levetirasetam
|
-
|
· Bulantı, başağrısı,
sersemlik, yorgunluk, uykuya eğilim, davranış
değişikliği, iritabilite, depresyon
|
|
Okskarbazepin
|
·
Döküntü
|
· Bulantı, kusma, ataksi,
başağrısı, sersemlik, yorgunluk, hiponatremi*
|
|
Eslikarbazepin
|
·
Döküntü
|
· Bulantı, kusma, ataksi, başağrısı,
sersemlik, yorgunluk, hiponatremi†
|
|
Fenobarbital
|
·
Hepatotoksisite, bağ
dokusu ve kemik iliği bozukluğu, Steven-Johnson sendromu, ilaç
hipersensitivite sendromu
|
· Sedasyon, “rebound”, tolerans, ataksi, nistagmus,
döküntü, depresyon, çocuklarda hiperaktivite, öğrenme güçlüğü
· Uzun süreli kullanımda osteoporoz, Dupuytren
kontraktürü, plantar fibromatozis, donuk omuz
|
|
Fenitoin
|
·
Aplastik
anemi, hepatik yetersizlik, Steven-Johnson sendromu, lupus benzeri tablo,
psödolenfoma, retroperitoneal fibroz, serebellar atrofi, ilaç
hipersensitivite sendromu
|
· Sedasyon, ataksi, nistagmus, diplopi, döküntü,
anoreksi, bulantı, makrositoz
· Uzun süre kullanımda osteoporoz, akne,
dişeti hipertrofisi, hirsutizm, anemi, periferik nöropati
|
|
Tiagabin
|
·
Stupor,
absans epilepside nonkonvülzif status ve ensefalopati
|
· Bulantı, başağrısı,
sersemlik, yorgunluk, güçsüzlük, emosyonel labilite, depresyon
|
|
Topiramat
|
·
Böbrek
taşı, dar açılı glokom
|
· Bulantı, başağrısı,
sersemlik, yorgunluk, kognitif yavaşlama, kelime bulma güçlüğü,
parestezi, kilo kaybı
· Hiperamonyemi (valproat kullanımı ile
birlikte)
· Metabolik asidoz, oligohidroz ve hipotermi
(çocuklarda)
|
|
Valproat
|
·
Hepatotoksisite
(<2 yaş çocukta ve polifarmaside belirgin), hiperamonyemi, lökopeni,
trombositopeni, pankreatit
|
· Bulantı, kusma, saç dökülmesi, tremor,
amenore, kilo artışı, konstipasyon, polikistik over sendromu,
· Yaşlılarda uzun süreli kullanımda
geri dönüşümlü parkinsonizm, yürüme bozukluğu, demans, beyin
atrofisi
|
|
Zonisamid
|
·
Böbrek
taşı, aplastik anemi, Steven-Johnson sendromu, toksik epidermal
nekroz
|
· Bulantı, başağrısı,
sersemlik, ataksi, yorgunluk, parestezi, döküntü, konsantrasyon
azalması, kognitif yavaşlama, kilo kaybı, depresyon, psikoz
· Metabolik asidoz, oligohidroz ve hipotermi (çocuklarda)
|
|
Vigabatrin
|
·
Kalıcı
görme alanı defekti
|
· Sersemlik, yorgunluk, ataksi, kilo
artışı, ödem, halsizlik, psikoz, iritabilite,
davranış değişiklikleri
|
|
Pregabalin
|
-
|
· Karaciğer enzimlerinde geçici hafif
yükselme, uyuklama, yorgunluk hali, baş dönmesi ve kilo
artışı, miyoklonus
· Ödem (ileri yaşta)
|
|
Lakozamid‡
|
-
|
· Sersemlik, bulantı, ataksi, diplopi,
uyuklama, hareketlerde yavaşlama
|
|
Rufinamid
|
·
QT
aralığında kısalma
|
· Sersemlik, yorgunluk, uykuya eğilim,
başağrısı, kusma
|
|
Ezogabin
(retigabin)
|
·
QT aralığında
uzamaya neden olan ilaçlarla birlikte alındığında QT
mesafesi takip edilmeli
|
· Sersemlik, yorgunluk, uykuya eğilim,
konfüzyon, görme bulanıklığı, tremor, kilo
alımı, üriner retansiyon, bulantı, uzun süreli kullanımda
deri, retina ve tırnaklarda mavimsi pigmentasyon
|
|
Perampanel
|
-
|
·
Sersemlik,
yorgunluk, uykuya eğilim, başağrısı, görme
bulanıklığı, ataksi
· Agresyon (12 mg üzerindeki dozlarda)
|
AEİ: antiepileptik ilaç
*Karbamazepine
kıyasla daha yüksek hiponatremi riski bulunur.
†Okskarbazepine
kıyasla hiponatremi riski daha düşüktür.
‡Yan etkiler diğer Na kanal blokorleri ile
birlikte alınırsa belirgindir.
İlaç etkileşimleri açısından
bakıldığında genelde yeni AEİ’lerle önemli bir sorun
olmaz iken yerleşmiş AEİ’lerin birçok ilaç ile etkileşime
girdiği bilinmektedir. Tablo 24’te
bilinen bazı ilaç etkileşimleri gösterilmiştir. Önemli ve
sık etkileşimlerden biri oral kontraseptiflerledir (OK).
Karbamazepin, fenitoin, fenobarbital, okskarbazepin, felbamat ve >200mg/g
dozda topiramat bu ilaçların metabolik temizlenmesini arttırmakta
yani doğum kontrolünü bozmaktadır. Lamotirijinin ikili etkisi
vardır. OK’lerin klirensini arttırırken diğer yandan
OK’lerde lamotirijinin klirensini arttırır. Yeni AEİ’lerden
levetirasetam, gabapentin, tiagabin, pregabalin ve zonisamidin OK’lerle
etkileşimleri yoktur. Hastaya anti-gribal ilaçlar örneğinde
olduğu gibi gereksiz şekilde ve doktoruna danışmadan ilaç
kullanmaması öğütlenmelidir.
Karbamazepinden, okskarbazepine geçiş gerekir ise 2:3 oranındadır.
İlaç
etkileşimleri birkaç mekanizma ile gerçekleşir. Bunlar farmakokinetik
etkileşimler; enzim indüksiyonu veya inhibisyonu, emilim, proteine
bağlanma ve taşıyıcı proteinlerin rolüdür. Ayrıca
ilaçlar etki bölgelerinde de farmakodinamik mekanizmalar ile
etkileşebilirler. Bu etki sinerjistik ve antagonistik olabilir. Enzim
indükleyici olan AEİ (fenitoin, karbamazepin ve fenobarbital), diğer
AEİ’lerin kan düzeylerini azaltırlar ve bu durum ilacın etkinliğinin
azalması ile sonuçlanır. Bu ilaçlarla beraber
kullanıldıklarında doz arttırılması ve
yarılanma ömrü kısa olan ilaçların gün içerisinde daha sık
aralıklarla alınması gerekebilir. Ayrıca enzim indükleyici
ilacın kesilmesi ile diğer ilaçların serum konsantrasyonu
artabilir ve toksik etkilere neden olabilirler. Enzim inhibisyonu yapan
valproat, fenobarbital ve lamotirijinin metabolizmasını azaltarak
yarılanma ömürlerini uzatır. Toksik yan etkiler nedeniyle bu ilaçlar
beraber kullanıldıklarında doz azaltımı
yapılması gerekir. Valproat ile lamotirijin arasında sinerjistik
etki de bulunur. Valproat, karbamazepin ile birlikte
kullanıldığında, aktif metabolitinin
metabolizmasını azaltarak toksik etkiye neden olabilir. Valproat ile
etosüksimid absans epilepsilerinde ve valproat ile karbamazepin fokal
epilepsilerde sinerjistik etki gösterir. Na blokajı yapan ilaçlar beraber
kullanıldığında, etkinliğin artmasından çok yan
etkilerin artmasına neden olurlar. AEİ ile diğer ilaçların
etkileşimi iki yönlü olarak değerlendirilebilir. Bazı ilaçlar
AEİ düzeylerini değiştirebileceği gibi Tablo 26’da özetlendiği gibi AEİ de bazı
ilaçların etkinliğini arttırabilir ya da azaltabilir. Oral
kontraseptifler, lamotirijin, okskarbazepin ve valproatın serum
konsantrasyonlarını azaltırlar. Antiviral ve antiretroviral
ajanlar lamotirijin düzeylerini azaltabilirler. AEİ, antiviral,
antikoagulan, oral kontraseptif ve antineoplastik ajanların
etkinliğini azaltabilirler, bu nedenle bu ilaçları kullanan hastalarda
ilaç etkileşimi en az olan AEİ’ler tercih edilmelidir. Enzim
indükleyici ilaçlar, oral kontraseptif ilaçların etkinliğini
azaltır.
Tablo 26. Antiepileptik
ilaçların diğer ilaçlar üzerine etkisi
|
AEİ
|
Etkisini azaltır
|
Etkisini arttırır
|
Öneri
|
|
Benzodiazepin
|
-
|
-
|
· Sedatif ilaçlarla sedasyon etkisi artar
|
|
Karbamazepin
|
Antikoagulan, kinidin, OK,
kortikosteroid, siklosporin A, folat, dijital glikozidler, donezepil,
doksisiklin, felodipin, mebendazol, metadon, kas gevşeticiler, nifedipin,
nimodipin, prasikuantel, teofilin, verapamil
|
Diltizem
|
· AEİ ve MAO inhibitörleri ile lityum
nörotoksisitesi artabilir
|
|
Felbamat
|
OK
|
Varfarin
|
·
Spesifik
inhibitör veya indükleyici etkileşim olabilir.
· CBZ metabolitini, valproat, fenobarbital,
fenitoin arttırabilir.
|
|
Gabapentin
|
-
|
-
|
· Diğer ilaçlarla etkileşimi yoktur, oral
kontraseptifleri etkilemez.
· Antasitlerle birlikte
alındığında biyoyararlanımı %20 oranında
düşer.
|
|
Lamotirijin
|
OK
|
OK kesilmesi
|
·
OK
kullanımına dikkat edilmeli
·
VPA ile
yarılanma ömrü artar. Enzim indükleyiciler ile yarılanma ömrü
azalır
|
|
Levetirasetam
|
-
|
-
|
· Hepatik olmayan hidroliz ile kısmen
metabolize olur
|
|
Okskarbazepin
|
Felodipin, OK, nifedipin,
nimodipin
|
+
|
·
AEİ ve
MAO inhibitörleri ile lityum nörotoksisitesi artabilir
· Diğer Na kanal blokeri AEİ ile yan
etkileri (sersemlik, ataksi, diplopi) arttırabilir.
|
|
Eslikarbazepin
|
Varfarin, OK
|
|
·
CBZ ile
verildiğinde, plazma konsantrasyonunu azaltabilir.
·
Diğer
Na kanal blokeri AEİ ile yan etkileri (sersemlik, ataksi, diplopi)
arttırabilir.
|
|
Fenobarbital Pirimidon
|
Antikoagulan, kinidin, OK
kortikosteroid, siklosporin A, diltizem, dijital glikozidleri, donezepil,
doksisiklin, felodipin, folat, griseofulvin, haloperidol, mebendazol,
metadon, metoprolol, nifedipin, nimodipin, metronidazol, kas
gevşeticiler, prasikuantel, propanolol, takrolimus, tamoksifen,
teofilin, verapamil
|
-
|
· AEİ ile metotreksat toksisitesi artabilir
|
|
Fenitoin
|
Antikoagulan, kinidin, OK
kortikosteroid, siklosporin A, dijital glikozidleri, donezepil,
doksisiklin, folat, mebendazol, metadon, kas
gevşeticiler, nifedipin, nimodipin,
metronidazol, praksikuantel, teofilin, verapamil
|
Diltizem, disulfiram
|
· AEİ ve MAO inhibitörleri ile lityum
nörotoksisitesi artabilir
|
|
Pregabalin
|
-
|
-
|
· Antidiyabetik tiyazolidindiyon birlikte
kullanıldığında egzersizlere dikkat edilmeli
|
|
Tiagabin
|
-
|
-
|
· Diğer AEİ’larla ve simetidin, digoksin,
teofilin, varfarin veya OK ile etkileşmez.
· PB, KBZ veya FT, TGB gibi sitokrom-P450
izoenzimlerini indükleyen ilaçlarla alındığında daha
hızlı metabolize olur.
|
|
Topiramat
|
OK (TPM>200mg/g)
|
-
|
-
|
|
Vigabatrin
|
-
|
-
|
· İlaç metabolize edici sistemleri
etkilememekle birlikte AEİ’lerle dinamik etkileşimi vardır.
|
|
Valproik asit
|
İtrokonazol
|
Antikoagulan, lopinavir,
lorazepam, nimodipin, paroksetin, amitriptilin, nortriptilin
|
·
Valproik
asit antikoagulasyonu arttırabilir
· CBZ metabolitini, lamotirijin, fenobarbital ve
rufinamidi arttırabilir.
|
|
Zonisamid
|
KBZ, FB ve
FT yarılanma ömrünü
azaltır.
|
-
|
· Diğer ilaçlarla indüklenebilir veya inhibe
olabilir, ancak Zonisamid’in diğer AEİ’ler üzerine etkisi yoktur.
|
|
Perampanel
|
OK (Perampanel ≥12 mg)
|
-
|
-
|
|
Rufinamid
|
OK
|
-
|
-
|
OK: oral kontraseptifler,
AEİ: antiepileptik ilaç
AEİ Kan Düzeylerinin İzlenmesi
AEİ kan düzeylerinin izlenmesi erişkin hastalarda
rutin olarak önerilmemektedir. Her hastanın terapötik ve toksik
aralığı farklıdır, bu nedenle ilaç dozuna
yalnızca kan düzeyine göre karar verilmemelidir. Ancak bazı
durumlarda ilaç kan düzeylerinin takibi fayda sağlar. İntoksikasyon
bulguların varlığı, gebelik ve diyaliz öncesi ve
sonrası gibi terapötik doz aralığının
değişebileceği bu durumlarda ilaç kan düzeylerinin takibi
yararlıdır.
Tedavinin Sonlandırılması
AEİ’lerin
kronik kullanımında yan etkileri olabileceğinden ve bazı olgularda
remisyonların görülmesi mümkün olduğundan çocukluk çağında
son nöbetten 2 yıl, erişkinlikte ise 4-5 yıl sonra ilaç dozu
yavaşça azaltılarak kesilebilir. Ancak burada da tedaviye
başlama konusunda olduğu gibi, öncelikle riskleri anlatarak
hastanın görüşünü almak rol oynar. Juvenil miyoklonik epilepsi gibi
bazı sendromlarda nüksün çok yüksek oranda (yaklaşık %80)
olduğu unutulmamalıdır. Yapılan çalışmalar, ilaç
kesiminin hastalarda nüksü 2 kat oranında
arttırdığını göstermiştir. Bunun yanı
sıra nükse etkili olan diğer faktörler arasında epilepsinin
süresi, erken yaşlardan beri nöbetlerin olması, remisyon süresi,
remisyona kadar geçen nöbetli süre, fokal başlangıçlı nöbetler,
farklı nöbet tiplerinin birlikte bulunması, EEG’de fokal anomalinin
bulunması, AEİ kesimi sırasında veya kesilirken EEG
anomalilerinin artması, bilateral tonik-klonik nöbet
varlığı, miyokloni olması, nörolojik muayenenin anormal
olması, IQ<70 olması, kranial görüntülemede anormal bulgunun
olması sayılabilir. İlaç kesimi sırasında nöbet
tekrarlama riski az olan grup ise idyopatik/genetik epilepsisi olanlar,
jeneralize nöbet geçirenler, aile öyküsü olmayanlar, normal nörolojik muayene
ve normal EEG’si olanlardır. En doğru yaklaşım genel ilaç
kesim kriterleri yerine her hasta için özgün koşulları
değerlendirerek hareket etmektir. İlaç kesimi sonrası ilk bir
yıl içinde, özellikle ilk 3 ayda nöbet olma riskinin yüksek olduğunu
bilinmeli ve hiçbir zaman nöbet olma riskinin sıfır olmadığı
hastaya anlatılmalıdır.
DİRENÇLİ
EPİLEPSİLER VE CERRAHİ YOLLA TEDAVİ
EDİLEBİLİR EPİLEPSİLER
Dirençli
Epilepsinin Tanımı
Epilepsili
hastaların yaklaşık üçte birinde uygun AEİ
kullanımına karşın nöbetler kontrol altına
alınamamaktadır ve bu gruba “ilaç tedavisine dirençli epilepsi”
denir. ILAE tarafından 2010 yılında ilaç tedavisine dirençli
epilepsi tanımı güncellenmiştir. Bu tanımda, hasta iki
aşamada değerlendirilir: 1) mevcut kullanılan AEİ’nin
başarısız olma durumu 2) ilaç tedavisine dirençli epilepsi
hastalığı saptanması. Bir AEİ tedavisinin
başarısız olma durumu, farkındalığın
korunduğu nöbetler dahil olmak üzere nöbetlerin devam etmesidir. İlaç
tedavisine cevaplı epilepsilerden, nöbetsizlik süresi en az 12 ay veya
AEİ eklenmesinden önceki en uzun nöbetsizlik süresinin en az 3 katı
süre boyunca nöbet olmadığı durumlarda bahsedilir ve bu iki
süreden hangisi daha uzunsa o hasta için o süre geçerli kabul edilir. Dirençli
epilepsi hastalığından bahsedebilmek için en az iki farklı
uygun AEİ’nin monoterapi veya kombinasyon halinde maksimum tolere
edilebilir dozda kullanılmasına rağmen tedavinin
başarısız olması gerekmektedir. Bazı durumlarda hasta
ilaç tedavisine cevaplılık durumu belirlenemeyen olarak
sınıflandırılır, örneğin nöbetsizlik süresi 12
aydan kısa ise ya da en az iki AEİ denemesi yapılmadıysa bu
tanım geçerlidir. Eğer nöbetsizlik sağlandıktan sonra hasta
yeniden nöbet geçirirse süre sıfırlanır ve hastanın tek
ilaç kullandığı durumda ilaç tedavisine cevapsızlık
durumu belirlenemeyen olarak sınıflanır. Bu hastada bir ikinci
ilaç ile de nöbetsizlik sağlanamazsa o zaman dirençli epilepsiden
bahsedilir.
Dirençli
epilepsi tanısı konulduktan sonra hasta cerrahi tedavi,
nörostimülasyon tedavisi ve diyet tedavisi açısından
değerlendirilmelidir. Bu arada bir yandan da medikal tedavi düzenlemesi de
yapılır.
Epilepsi Cerrahisi
Dirençli
epilepsi hastaları sayıları giderek artan epilepsi
merkezlerinde, epilepsi cerrahisi adayı olarak değerlendirilmelidir.
Ülkemizde de epilepsi cerrahisi konusunda çalışmalar epilepsi
merkezlerinde sürdürülmektedir. Ayrıca nöbetlere bağlı
yaşam kalitesinin bozulması da epilepsi cerrahisi adayına karar
verirken önemli bir kriterdir.
Epilepsinin
yarattığı yeti kaybı kişinin geçirdiği nöbetlerin
sayısı ve şiddetiyle ölçülür. Bunun dışında altta
yatan patoloji, eşlik eden nörolojik ve nöropsikolojik sorunlar, AEİ
toksisitesi, sosyal ve aile ilişkileri de kişinin cerrahi adayı
olmasında önemli etkenlerdir. Kişi ve ailesiyle
ayrıntılı şekilde konuşarak cerrahi tedavi konusunda
bilgilendirme yapılır. Epilepsi cerrahisinde deneyimli bir beyin cerrahının
elinde morbidite ve mortalite oranı düşüktür. Yarar ve risk
oranına bakarken cerrahi riske karşılık olarak kontrol
altına alınamayan nöbetlere bağlı yaralanma, boğulma,
intihar girişimi, SUDEP gibi risklerin de olduğu bilinmelidir. Yarar
ve risk oranının her olgunun özel bulgularına göre
değerlendirilmesi en uygunudur.
Epilepsi
cerrahisi uygulamaları iki gruba ayrılabilir. İlki rezektif
cerrahi olarak da adlandırılan epileptojenik beyin dokusunun
belirlenmesi ve cerrahi olarak çıkarılmasıdır. Diğer
cerrahi tedavi seçenekleri fonksiyonel hemisferotomi ve korpus kallozotomidir.
Hipokampal
sklerozla ilişkili mezyal temporal lob epilepsisinde cerrahi tedavinin
medikal tedaviye üstünlüğünün gösterilmesi ile bu hastaların erken
dönemde cerrahiye yönlendirilmesi önerilmektedir. Bu gruba iyi
tanımlanmış çıkarılabilir epileptojenik lezyonu tespit
edilen hastalar da eklenir. Bu bahsedilenlerin dışında ise
yapılmış birçok randomize olmayan çalışmada epilepsi
cerrahisinden sonra nöbetsiz kalma oranının arttığı veya
yaşam kalitesinin yükseldiği bilinmekte ve tavsiye edilmektedir.
Epilepsi cerrahisi için uygun adayın belirlenmesinde cerrahi öncesi
değerlendirme önem taşır. Cerrahi öncesi değerlendirilmede
esas amaç epileptik nöbetlerin kaynaklandığı beyin bölgesinin
(epileptojenik alan) tanımlanmasıdır. Bu amaçla çeşitli
değerlendirmeler yapılır:
1) Nöbet semiyolojisi: Nöbet sırasında izlenen
klinik bulgular epileptojenik alanın lateralizasyonu ve lokalizasyonu ile
ilgili faydalı bilgiler verir (Ayrıca Bakınız: Fokal epilepsi
nöbetlerin semiyolojik özellikleri). Ancak bazı nöbetler
başlangıçlarında klinik bulgu vermez ya da müphem bulgulara
neden olup nöbet yayıldıktan sonra klinik bulgu verir. Nöbetin
semiyolojik özellikleri hasta öyküsünden alınabileceği gibi video-EEG
monitorizasyon incelemesinde nöbet kaydı yapılarak daha güvenilir
olarak da elde edilir.
2) İnteriktal ve iktal EEG bulguları: Rutin EEG
ve video EEG monitorizasyon incelemesinde İED’lerin kaydı
yapılır. Ayrıca video EEG monitorizasyon incelemesinde kaydedilebilen
nöbetler sırasında yüzeyel EEG kaydı ile iktal aktivitenin
başlangıcı belirlenir. Ancak bazı nöbetlerde nöbet
başlangıcından ancak uzun bir süre sonra iktal aktivite yüzeyel
EEG’de kaydedilebilmektedir. Bu gibi durumlarla iktal EEG
yanıltıcı sonuçlar verebilir. Ayrıca ekstratemporal lob
nöbetlerinde iktal EEG daha az bilgi vericidir. Bazı çalışmalar
interiktal EEG bulgularının iktal bulgulara kıyasla cerrahi
sonrası nöbetsizlik durumu daha iyi yansıttığını
bildirseler de halen iktal EEG bulguları önemini yitirmemiştir.
3) Nörogörüntüleme bulguları: Epilepsi protokolüne
uygun çekilen beyin MRG’de epilepsi nöbetlerine neden olan epileptojenik lezyon
tespit edilmeye çalışılır. Diğer sık
kullanılan nörogörüntüleme yöntemi kranial PET’dir. İnteriktal PET’de
epileptojenik alanda hipometabolizma izlenir. Bir diğer nadir
kullanılan yöntem ise iktal SPECT’tir. Nöbet sonrası kısa
uygulama penceresi ve işaretleyici maddenin temin zorluğu nedeniyle
sık olarak kullanılamamaktadır. Epileptojenik alanda iktal
hiperperfüzyon izlenir. İnteriktal dönemde hipoperfüzyon eşlik eder.
4) Nöropsikolojik değerlendirme: Özellikle temporal
lob epilepsili hastalarda bellek fonksiyon bozukluğunu göstermek
amacıyla kullanılır. Bazı olgularda lateralizasyon
değeri olsa da esas kullanım amacı cerrahi sonrası
gelişebilecek bellek sorunlarının belirlenmesidir.
Bu
değerlendirmelerin sonunda semiyolojik, elektrofizyolojik, nöropsikolojik
bulgular birbiri ile uyumluysa (aynı bölgeyi gösteriyorsa) ve MRG’de
aynı bölgede epileptojenik özellikli lezyon tespit edildiyse hasta
epilepsi cerrahisi için iyi bir adaydır; cerrahi sonrası nöbetsizlik
oranı yüksektir. Genel olarak temporal lob epilepsili hastaların
cerrahi başarısı ekstratemporal lob epilepsisinden ve MRG’de
lezyon izlenen hastalarınki nonlezyonel epilepsi hastalarından daha
iyidir. Bu bulgular eşliğinde hasta epilepsi cerrahi merkezinde
geniş bir ekip tarafından değerlendirilmelidir. Bu ekip
içerisinde epilepsi uzmanı, epilepsi cerrahisi konusunda uzman beyin
cerrahı, nöroradyolog, nükleer tıp uzmanı, deneyimli
psikiyatrist ve nöropsikoloji alanında uzman psikolog
bulunmalıdır. Eğer yukarıda belirtilen maddeler
arasında epileptojenik alanın belirlenmesi konusunda uyumsuzluk
varsa, o zaman hasta intrakranial video EEG monitorizasyon ile
değerlendirilmelidir. İntrakranial video EEG monitorizasyon
incelemesinde subdural ve derinlik elektrodları bir hipotez
doğrultusunda (epileptojenik alan hipotezi) hastaya göre düzenlenerek
kullanılır.
Kombine
fokal ve jeneralize epilepsi sendromlarında, epileptojenik alanın
önemli kortikal fonksiyon alanlarının üzerinde olduğu durumlarda
ve birden fazla epileptojenik odağın varlığında
rezektif epilepsi cerrahisi uygun değildir. Bu durumda korpus kallozotomi
veya hemisferektomi gibi diğer cerrahi girişimlerde bulunulabilir
veya nörostimülasyon uygulanabilir (Şekil 21).
Korpus kallozotomi, sıklıkla korpus kallozumun ön 2/3’ünü içine alınacak
şekilde yapılır. Faydalı olduğu nöbet tipi ani
düşme ataklarıdır. Nöbetlerin hızlıca bilateral
yayılmasını engellemek amaçlanmaktadır. Bilateral
tonik-klonik nöbetlerde de etkinliği bildirilmiştir.
Sıklıkla gelişimsel ve epileptik ensefalopatili hastalarda
tercih edilir.
Hemisferektominin
temel amacı, bir hemisferin neredeyse tümüne yayılmış olan
epileptojenik alanın beynin diğer bölgeleri ile
bağlantısını kesmektir. Bu teknik yüksek komplikasyon riski
olması sebebiyle günümüzde kullanılmamakta ve yerine fonksiyonel hemisferotomi
yapılmaktadır. Burada amaç beyin dokusunu canlı bir şekilde
bırakmak ancak beynin geri kalan bölgeleri ile ak madde
bağlantılarını yok etmektir. İnfantil hemiplejik
epilepsi, bir hemisfer içerisinde yaygın kortikal displazi,
hemimegalensefali, hemikonvülziyon-hemipleji sendromu, Rasmussen ensefaliti,
Sturge-Weber sendromu, konjenital porensefali veya perinatal serebrovasküler
olaylara bağlı dirençli epilepsili hastalar fonksiyonel hemisferotomi
için adaydır. Yapılan çalışmalarda fonksiyonel
hemisferotomi, hastaların %60-90’ında nöbetsizlik
sağlayabilmektedir. Hastaların büyük çoğunluğunda cerrahi
sonrası sağlam hemisfer fonksiyonlarında kognitif açıdan
iyileşme izlenir.
Epilepside Nörostimülasyon
Epilepsi
hastalarında tedavi amaçlı kullanılan nörostimülasyon teknikleri
vagal sinir stimülasyonu (VNS), derin beyin stimülasyonu (DBS), kapalı
devre veya cevaplı nörostimülasyon (closed loop veya responsive
stimulation), ardışık transkranial sinir stimülasyonu ve
transkranial direk akım stimülasyonu gibi farklı yöntemlerdir.
Epilepsi hastalarında nörostimülasyon uygulamaları nöbetsizlik
sağlamaktan çok, nöbet sıklığını ve bilateral
tonik klonik nöbetleri azaltmayı hedeflemektedir. VNS ve DBS için
açık devre nörostimülasyon (open loop neurostimulation) da denilmektedir.
Açık devre nörostimülasyonda cihazın içerisinde önceden
programlı şekilde hedef dokuya stimülasyon verilir. Kapalı devre
sistemlerde ise, cihaz epileptik aktiviteyi tespit eder ve buna cevap olarak
stimülasyon verir.
Vagal
Sinir Stimülasyonu (VNS): İlaca
dirençli epilepsisi olan ancak epilepsi cerrahisi açısından uygun
bulunmayan kişilerde uygulanır. Tüm yaş gruplarında, tüm
epilepsi nöbet tiplerinde ve sendromlarında kullanımı
onaylanmıştır. VNS’nin tam etki mekanizması
bilinmemektedir. Hayvan deneylerinde antikonvülzan etkisinin kortikal
ritimlerin desenkronizasyonu ile oluştuğu belirtilmektedir, insan
EEG’si üzerinde görülebilir değişiklikler meydana getirmemektedir.
Cihaz
sol N. vagusa genel anestezi veya servikal lokal anestezi altında
yerleştirilir. Sağ taraftaki vagusda daha fazla miktarda kardiyak eferent
dallar olduğu ve bradikardi yapabileceği için VNS uygulanması
tercih edilmez. İki spiral elektroddan biri vagus sinirine diğeri ise
infraklaviküler bölgeye konacak olan pile takılır. Deneyimli ellerde
iki saatten kısa süren basit bir cerrahi işlemdir. Operasyon
komplikasyonları olarak vagus siniri veya karotis zedelenmesi ya da
enfeksiyon olabilir. Stimulatör dışarıdan programlanabilir ve
istenirse pil kapatılabilir veya supraklaviküler bölgeye magnetin
tutulması ile etkisi arttırılabilir. Özellikle aurası olan
hasta nöbetini durdurmak için magneti kullanabilir. VNS iyi tolere edilebilen
ve çok nadir yan etki veya komplikasyon olabilen güvenilir bir alternatif
tedavidir. VNS uyarı ayarı, yüksek veya düşük stimülasyon olarak
yapılabilir. Uyarı verildiği ‘on’ ve verilmediği ‘off’
dönemlerinden oluşur. ‘On’ dönemi genellikle 30 saniye, ‘off’ dönemi ise
yüksek stimülasyonda 5 dakika ve düşük stimülasyonda 3 saate
ayarlanır. Uyarı şiddeti (0-3,5 mA), süresi (100-500 mikrosaniye)
ve frekansı (1-30 Hz) da istenilen protokole göre belirlenir. VNS
takıldıktan sonra istenilen uyarı ayarlarına
çıkılması haftalar sürer ve etkinlik bundan sonra
başlamaktadır. VNS uyarısı hasta tarafından
hissedilir. Bu nedenle gerçek anlamda bir plasebo grubu (sham uyarı)
yapılamaz. Yan etkiler açısından bakılınca en sık
ses kısıklığı, öksürük, parestezi, dispne, vokal kord
paralizisi ve lokal enfeksiyon görülür. VNS uyku apne sendromunu
kötüleştirebilir. Kardiyak aritmi ve astım bronşiyalesi
olanlarda dikkatli kullanılmalıdır.
Derin
Beyin Stimülasyonu: Anterior talamusun derin beyin stimülasyonu dirençli
epilepsi tedavisinde son yıllarda bazı ülkelerde
kullanılmaktadır. Mekanizması bilinmemekle birlikte klinik ve
deneysel çalışmalarda anterior talamusun epileptik nöbetlerin
yayılımında rolü olduğu ve anterior talamusun elektriksel
uyarımı veya anterior talamusta lezyon
oluşturulmasının nöbet sonlanması
sağladığı bildirilmiştir. En sık yan etki
anterior talamusun Papez devresinin ana çekirdeği olması sebebiyle
unutkanlık ve parestezilerdir. Nadiren asemptomatik intrakranial kanama
vakaları bildirilmiştir. Rezektif cerrahi sonrası nöbetleri
devam eden veya VNS tedavisinden fayda görmeyen hastalarda da
başarılı sonuçlar belirtilmiştir. Ayrıca santromedial
talamus ve hipokampus için DBS de az sayıda hastada denenmiş ve nöbet
sıklığını azaltabileceği söylenmiştir.
Kapalı
devre nörostimülasyon (cevaplı-‘responsive’ nörostimülasyon): 2013 yılında ruhsatlanan kapalı devre
nörostimülasyon cihazının (Neuro Pace®) temel prensibi
beyin içerisinde epileptik aktiviteyi tespit etmek ve buna cevap olarak
aktiviteyi baskılamak amacıyla elektriksel stimülasyon vermekten
oluşmaktadır. İntrakranial olarak yerleştirilen ve
kafatasına sabitlenen bu cihaza iki adet intrakranial strip veya derinlik
elektrodu bağlıdır. Bu elektrodlar hastanın epileptojenik
alanının üzerine yerleştirilir. Aynen intrakranial video EEG
monitorizasyonda olduğu gibi EEG kaydı yapar, hastanın iktal
elektrografik paterni cihaza tanıtılır ve cihaz bu paterni
farkettiği anda aynı elektrodlar aracılığıyla
önceden programlandığı protokol ile elektriksel
uyarıyı verir. Uzun süreli çalışmalarında 1
yıllık nöbet azaltma oranı %44 ve 2 yıllık %53 olarak
bildirilmiştir. En sık yan etki işlem sırasında
intrakranial kanama olup yaklaşık %5 oranındadır. Hafif yan
etkiler başağrısı, parestezi, implant bölgesinde
ağrı olarak tanımlanmıştır. Diğer
nörostimülasyon tekniklerinden farkı, hastanın öncesinde intrakranial
EEG monitorizasyonu ile epileptojenik alanının belirlenmiş
olması gerekmektedir. Kapalı devre nörostimülasyonun uygun
olabileceği adaylar birden fazla epileptojenik odağı olan veya
tek epileptojenik odağı önemli kortikal fonksiyon
alanlarının üzerinde olması veya derin yerleşimli
olması sebebiyle rezektif cerrahiye uygun olmayan epilepsili hastalardır.
Ayrıca başarısız rezektif cerrahi geçiren hastalar da bu
tedavi için uygun olabilirler. Henüz bu yöntem ülkemizde kullanımda
değildir. Pahalı ve deneyimli personel gerektirmesi ve hasta için
zahmetli bir süreç olması olumsuz yönleridir.
Diğer nörostimülasyon
yöntemleri: Repetetif transkranial magnetik stimülasyon (rTMS), epilepsi hastalarında terapötik amaçlı
olarak yaklaşık 20 yıldır
çalışılmaktadır. Az sayıda hasta ile yapılan
randomize kontrollü çalışmalar, İED’lerin rTMS ile belirgin
olarak azaldığını göstermektedir. Ancak, nöbet
sıklığını azaltıcı etkisi ile ilgili
sonuçlar çelişkilidir. rTMS ile fokal epilepsisi olan, epileptojenik
odağın korteks yüzeyinde olduğu (örneğin neokortikal
epilepsi ve fokal kortikal displazi) ve uyarının odak üzerine
verildiği olguların daha belirgin fayda gördüğü iddia
edilmektedir. Epilepsi nöbetini tetikleyebileceği için yüksek
frekanslı uyarılar önerilmez. En sık yan etkisi
başağrısı ve geçici halsizliktir. Transkranial direk akım stimülasyonu (tDCS), kortikal aktivite
üzerine etkisi olan non-invazif bir yöntemdir. Az sayıda randomize
kontrollü çalışma ve vaka serilerinde dirençli fokal ve jeneralize
epilepsisi olan hem erişkin hem de çocuk yaş grubunda faydalı
etkilerinden bahsedilmektedir. En sık yan etkileri lokal
rahatsızlık hissi ve başağrısıdır. Son
yıllarda uygulanan bir diğer nörostimülasyon tekniği de
trigeminal sinir stimülasyonudur. Fokal dirençli epilepsi hastalarında
bilateral trigeminal sinirin oftalmik dalı uyarılarak
yapılmaktadır ve olumlu sonuçlar olduğu bildirilmektedir. Bu son
yöntemler henüz deneme aşamasında olup rutin kullanıma
girmemiştir.
EPİLEPSİDE DİYET TEDAVİSİ
Ketojenik
Diyet: Alternatif tedavi olarak özellikle çocuk yaşta ilaca
dirençli epilepsilerde ketojenik diyet tedavisi önerilmektedir. Dört çeşit
ketojenik diyet bulunur: klasik ketojenik diyet, modifiye Atkins diyeti, orta
zincirli trigliserit diyeti ve düşük glisemik indeks diyeti. Temelinde bu
diyetler, yüksek yağ, düşük karbonhidrat ve protein içeriklidir.
Açlıkla ortaya çıkan ketozisin nöbetleri
baskıladığı görüşüyle ortaya konmuş bir tedavi
yöntemidir. Mekanizması tam olarak bilinmemektedir. Yurdumuzda yeterli deneyime
sahip diyetisyenlerin olmaması nedeniyle çok sınırlı bir
kullanımı vardır.
Klasik
ketojenik diyette, diyetteki yağın, karbonhidrat ve proteine
oranı 4’e bir (4:1) olmalıdır ve kalorinin %90’ı
yağdan alınmalıdır. Orta zincirli trigliserit diyetinde
günlük kalorinin %40-60’ı (hastanın tolere edebileceği
sınıra kadar verilir) orta zincirli trigliseritlerden oluşur.
Orta zincirli trigliseritlerin ketozis oluşturma oranı uzun
zincirlilere kıyasla daha fazla olduğu için diyetteki protein ve
karbonhidrat oranı arttırılabilir. Modifiye Atkins diyeti,
yüksek yağ ve düşük karbonhidrattan oluşur ve protein ve kalori
kısıtlaması yapılmaz. Düşük glisemik indeks diyetinde
ise karbonhidrat kısıtlaması yapılmadan düşük glisemik
indeksli karbonhidratlar tercih edilir. Ketojenik diyet tipi hasta ve
bakım sağlayanlara göre özel olarak belirlenmelidir ve
kontrendikasyon olan durumlar dikkatle uzmanlarca elenmelidir.
İki
yaşından küçük çocuklarda gelişimi olumsuz etkileyebileceği
için önerilmez. Erişkin ve ergenlerde kullanımı ile
ilişkili bilgiler sınırlıdır. Nörostimülasyonda
olduğu gibi epilepsi cerrahisi için uygun aday olamayan ve en az iki
AEİ tedavisine rağmen nöbetleri devam eden hastalarda (dirençli
epilepsi) kullanılır. Ağır gelişimsel ve epileptik
ensefalopatilerde daha erken başlanabilir. 2018 yılında
yayınlanan ILAE ketojenik diyet konsensusunda 7 sendrom için ketojenik
diyetin erken dönemde başlanılması önerilmiştir. Bunlar
glikoz taşıma eksikliği (GLUT-1), pirüvat dehidrogenaz
eksikliği, miyoklonik-atonik nöbetli epilepsi, infantil spazm,
tuberoskleroz, Dravet sendromu ve gastrostomi tüpü olan epilepsili
hastalardır. Bu grup içerisinde GLUT-1 eksikliği ve pirüvat
dehidrogenaz eksikliğinde ketojenik tedavi birinci basamak olarak kabul
edilmektedir. Bu gruba ek olarak ketojenik diyetin genel etkisinde daha fazla etkinlik
görülmesi beklenen hastalar Angelman sendromu, kompleks-II mitokondriyal
bozukluklar, ateşli hastalıkla ilişkili epilepsi sendromu
(febrile infection related epilepsy syndromu-FIRES), Ohtahara sendromu ve süper
dirençli SE’dir. Ketojenik diyet kullanan hastalarda kullanımından
kaçınılması gereken bir AEİ yoktur. Ketojenik diyetlerin
uygulanması için çeşitli rehberler bulunmakla birlikte, uygulama
güçlüğü, taşıdıkları riskler ve kontrendikasyonlar
açısından deneyimli bir diyetisyen ve ekip çalışması
ile yönetilmelidir.

Şekil 21. Epilepsi hastalığında tedavi
algoritması. *Ketojenik tedavi glikoz taşıma eksikliği
(GLUT-1) ve pirüvat dehidrogenaz eksikliği (PDH) ile giden epilepsilerde
birinci seçenek tedavidir.
STATUS
EPİLEPTİKUS VE TEDAVİSİ
Bir epileptik
nöbetin alışılmıştan uzun sürmesi veya hasta düzelip
eski nörolojik durumuna dönmeden çok sayıda nöbetin arka arkaya
tekrarlamasına SE adı verilir. SE için iki farklı zaman
noktası belirlenmiştir; 1. zaman noktası yani (t1), tedaviye
kadar geçen süreye, 2. zaman noktası (t2) ise uzak dönem sonuçların
belirdiği süreye işaret etmektedir. Buna göre SE, anormal
şekilde uzamış ve acil tedavi gerektiren nöbetler ile
karakterize, nöbetleri sonlandıran mekanizmaların yetersizliği
ve/veya nöbetleri başlatan mekanizmaların neden olduğu klinik
durumdur. Uzak dönem etkisi olarak, SE süresi ve tipine bağlı şekilde
nöronal ölüm ve nöronal ağlarda değişiklikler meydana
gelebilmektedir. Jeneralize konvülzif SE (JKSE) için t1 süresi 5 dakika, t2
süresi 30 dakika olarak tanımlanırken yeterli kanıta dayalı
bilgi olmamakla birlikte NKSE’nin fokal formu için t1 süresi 10 dakika, t2
süresi >60 dakika, jeneralize formu (absans status epileptikus-ASE) için t1
süresi 10-15 dakika olarak bildirilmiş, ancak burada uzak dönem
komplikasyona yol açan t2 süresinin bilinmediği rapor edilmiştir.
SE insidensinin
yüzbinde 20-50 civarında olduğu tahmin edilmektedir; hemen her yaş
grubunda görülebilmekle birlikte, infantil dönem ve geç erişkinlik dönemi
olmak üzere iki farklı yaş döneminde sıklaşma gösterir. SE
tablosunun yaklaşık yarısı daha önce epilepsili olmayan
olgularda görülürken, bir epilepsi sendromu ile birliktelik gösteren SE,
hastaların %65’inde ve klinik tablonun başlangıcında
görülür. Tüm epilepsili olguların %1-16 kadarı en az bir kez SE
tanımına uyan atak geçirirken, SE’nin yaklaşık %13
oranında tekrarladığı bilinmektedir. SE
tablolarının %31-43’ü dirençli SE haline dönüşmektedir ve
mortalite ortalama %20’dir; ne yazık ki bu oran artan teknik olanaklara
rağmen yıllar içinde azalmamıştır. SE’nin süresi ve
altta yatan neden, prognoz ve mortaliteyi belirleyen en önemli faktörlerdir.
Eşlik eden komorbid durumlar ve sistemik komplikasyonlar da prognoz
üzerinde olumsuz etkiye sahiptir.
Tüm epileptik
nöbet tipleri SE’ye dönüşebilir; en sık rastlanan şekilleri
JKSE; fokal bilinç kaybı olmayan nöbetler ya da eski adı ile basit
parsiyel SE; miyoklonik SE (MSE) ve NKSE’dir. JKSE mortalite ve morbiditesi en
yüksek tablodur; bu nedenle
JKSE’nin hızlı tanı ve tedavisi çok önemli bir dahili ve
nörolojik acil durumdur. Etkin tedavinin zaman kaybetmeden başlanması
önemlidir. Çalışmalar hastane öncesi tedavinin önemini ortaya
koymuştur.
SE’nin etyopatogenetik mekanizmaları hala tam
olarak anlaşılabilmiş değildir. Çok sayıda moleküler
ve hücresel süreçler rol oynasa da SE’nin oluşum mekanizmasının
temelinde azalmış inhibisyon ve artmış eksitasyonla
şekillenen nöbeti sonlandıran mekanizmaların yetersizliği
söz konusudur. En önemli inhibitör transmitter olan GABA reseptörleri aynı zamanda SE
tedavisinde en etkili AEİ’lerin de (benzodiazepinler gibi) etki
yerleridir. SE’nin devam etmesinde rol oynadığı düşünülen
eksitatör nörotransmitterler ise glutamat, aspartat ve asetilkolindir. NMDA
reseptörleri ile ilişkili kalsiyum (Ca+2) kanalları SE’nin patogenezinde önemlidir. SE süresi uzadıkça
GABA reseptörlerinin sayısı ve duyarlılığı
azalırken, eksitatör glutamat reseptörlerinin (NMDA ve AMPA tipi)
sayısı hücre yüzeyinde hızla artar. Dolayısıyla SE’nin
ilerleyen evrelerinde tedavinin hedefi değişmekte ve GABA
agonistlerinin etkisi azalırken, NMDA reseptör antagonistleri tedavide
daha etkili hale gelmektedir. İlerleyen süreçte ortaya çıkan eksitatör
ve inhibitör nöropeptid değişiklikleri aşırı
uyarılmış ortamın devamlılığına neden
olmaktadır. Daha sonra da epileptogenez ve SE’nin yol
açtığı nöronal hasarda önemli rol oynayan hipokampal hücre
DNA’sında metilasyon, mikro-RNA değişiklikleri gibi epigenetik
değişiklikler meydana gelmektedir. Eksitotoksisite, nekroz, apoptoz
ve mitokondriyal fonksiyon bozukluğu nöronal
hücre hasarı ve hücre ölümüne yol açmaktadır. Çeşitli
deneysel çalışmalarda uzayan nöbetlerin hipertermi, hipotansiyon ve
hipoksiye neden olarak talamus, hipokampus ve neokortekste hasara yol
açtığı gösterilmiştir. Bu bölgeler aynı zamanda SE sonrası kalıcı
hasarların eksenini oluşturmaktadır.
Kan beyin bariyeri
(KBB) bozukluğu ile ilişkili olarak ortaya çıkan inflamasyon ve
serebral damar yapısı bozuklukları da epileptogeneze katkı
sağlamaktadır. KBB bozukluğu sonucunda biriken albumin
astrositlerde “transforming growth factor” (TGFß) yolunu aktive ederek
nöronal uyarılabilirliği arttırmakta, bunun sonucunda
inflamasyon, sinaptogenez ve epilepsi nöbetleri meydana gelmektedir. SE’nin
oluşturduğu sistemik metabolik bozuklukların da nöron
hasarı oluşumuna önemli katkısı bulunmaktadır. Hem
deneysel çalışmalar hem de nöronal hasarın gösterisi olan serum
nöron-spesifik enolazın (NSE) yüksekliğini gösteren klinik
çalışmalar NKSE tablolarının da nöronal hasar ve hücre
ölümüne neden olduğu görüşünü desteklemektedir. Yeni deneysel ve
klinik çalışmalar ışığında SE sürecinde
etken olan tüm etyopatogenez basamaklarının daha iyi
anlaşılması, şu anda klinik öncesi
çalışmaları süren allopregnanolon, kanabinoidler ve bilinen
AEİ yeni deriveleri gibi olası farklı mekanizma hedefli yeni
tedavi basamaklarının da gelişmesini sağlayacaktır.
Etyoloji
SE için etyolojik
faktörler akut ve kronik olarak iki ana gruba ayrılır.
Çalışmalarda farklı oranlar bildirilmekle birlikte akut
semptomatik nedenler daha sık görülmektedir ve kronik nedenlere göre daha
fazla mortalite ve morbiditeye sahiptir. SE, öncesinde nöbet öyküsü olmayan
hastalarda beynin çeşitli akut metabolik veya yapısal
hastalıkları sonucu tetiklenebilir. Akut elektrolit
bozuklukları, böbrek yetmezliği, sepsis, sinir sistemi
enfeksiyonları, inme, ilaç zehirlenmeleri, hipoksi ve kafa travması
da SE’nin diğer sık nedenleridir. Epilepsi öyküsü olan hastalarda AEİ
uyumsuzluğu, ani AEİ kesme en sık görülen ve mortalitesi görece
en az olan durumdur. Sendroma uygun olmayan AEİ kullanımı, araya
giren bir enfeksiyon ve uygun olmayan tedaviler başta olmak üzere
diğer ek tetikleyici faktörlerin varlığı ve kötü seyirli bazı
epilepsi sendromlarının doğal seyri sırasında
tekrarlayan SE görülmesi, kronik epilepsili hastalarda görülen SE
tablolarının diğer sık karşılaşılan
nedenlerini oluşturmaktadır. Günlük pratikte sefalosporin, kinolonlar
ve bazı diğer antibiyotiklerle ortaya çıkan SE tabloları
ile de sık karşılaşılmaktadır. Bazı
immunoterapi ve kemoterapi ilaçları geri dönüşümlü ensefalopati
tablolarına neden olur ve karşımıza özellikle NKSE olarak
çıkarlar. Psikotrop ilaçlar da bazen yüksek dozlarda SE’ye neden
olabilirler. İkiz çalışmalarında, SE konkordans
oranının monozigotlarda dizigotlara göre anlamlı şekilde
yüksek olduğu bildirilmiştir. Epilepsi ile ilişkili çok
sayıda gen tanımlanmış olmasına rağmen sadece SE
ile birliktelik gösteren ‘pür status epileptikus geni’ tanımlanmamıştır.
Nonkonvülzif Status Epileptikus (NKSE)
NKSE
davranışlar ve mental durumda açıklanamayan bir
değişiklik, konfüzyon, hatta komaya kadar varabilen ciddi uyku
eğilimi gibi klinik bulgulara EEG’de devamlı nöbet aktivitesinin
eşlik ettiği tablo olarak tanımlanır. Fokal ve jeneralize
formları bilinmektedir ve giderek artan bilgi birikimi ile teknolojik
gelişmeler sonucunda NKSE tanımı birbirinden oldukça farklı
birçok tabloyu kapsayacak şekilde genişlemiştir. Klinik
bulguları çok değişken olabildiğinden ve tanısal net
bir ipucu bulunmadığından, tanısı için akla gelmesi ve
yüksek oranda kuşku duyularak EEG incelemesi yapılması
gereklidir. NKSE’nin gerçek insidensinin belirlenebilmesi, NKSE’yi tanımlama
farklılıkları, EEG’de tanı güçlükleri ve farklı
çalışmalarda incelenen grupların yaş gibi farklı
özelliklerine bağlı olarak güçtür ve yaklaşık olarak
yılda 1,5-18,3/100000 olarak tahmin edilmektedir. Yoğun bakım
ünitesinde (YBÜ) koma tablosunda yatan hastalarda ise NKSE prevalansı
olarak %8-27 gibi oranlar dikkati çekmektedir. NKSE hastanın ilk
başvuru nedeni olabilir ama çoğu olgu önceden epileptiktir.
Jeneralize konvülziyonları izleyerek NKSE gelişebilir, nedeni
bulunamayan olgular olabileceği gibi etyolojide psikotrop ilaç
kullanımı, klorokin, ifosfamid, propofol, baklofen, seftazidim gibi
ilaçlar, hipokalsemi, hiponatremi gibi metabolik problemler, kronik alkolizm,
metrizamid, amitriptilin intoksikasyonu, hipertiroidizm, benzodiazepinlerin ani
bırakılması, paraneoplastik sendrom, jeneralize epilepsilerde
karbamazepin gibi hatalı AEİ kullanımı, intravenöz kontrast
madde kullanımı, trombotik trombositopenik purpura gibi çok
sayıda neden olabilmektedir. Bir ucunda sadece dikkati
dağınık polikliniğe yürüyerek gelen, psikiyatrik semptomlar
sergileyen veya epilepsi tanılı olguların diğer ucunda ise
komada anoksi nedeniyle yüksek mortalitesi olan hastaların aynı
çatı altında toplanmasının getirdiği güçlükler
açıktır.
NKSE
tek bir hastalık değildir ve günümüze dek yaygın kabul
görmüş bir tanımlaması ve sınıflaması da yoktur.
Son zamanlarda bu alanda en
kayda değer gelişmelerden biri ILAE komisyonunca yapılan yeni SE
sınıflamasıdır. Yeni SE sınıflamasının
diğer bir yeni özelliği de 4 aksdan oluşan bir klinik tanı
araştırma ve tedavi yaklaşımı çerçevesi önermesidir.
Bu akslar (1) semiyoloji (2) etyoloji (3) EEG
karşılıkları (4) yaş şeklindedir.
Yeni
sınıflamada bazı tabloların şu an için NKSE yönünden
belirsiz olduğu ifade edilerek “sınır sendromlar” olarak ele
alınmıştır. Bu tablolar arasında epileptik ensefalopatiler,
evolusyon göstermeyen (monoton görünümlü periyodik lateralize epileptiform
deşarjlar gibi) epileptiform paternlerle giden koma, epileptik olgularda
davranışsal bozukluklar (örn, psikoz) ve akut konfüzyonel durumlarla
birlikte giden epileptiform EEG bulguları sayılmıştır.
Landau-Kleffner sendromunda afazik status, juvenil
absans epilepsisinde ASE, Creutzfeldt-Jakob hastalığında NKSE ve
geç yaşamın de novo (ve bazen tekrarlayan) ASE tablosu da diğer
başlıklardır.
Halka 20 ve
diğer karyotip anormallikleri, Angelman sendromu, Dravet sendromu, bazı
metabolizma hastalıkları, mitokondriyal hastalıklar veya kortikal gelişimsel bozukluklar gibi çeşitli
genetik etyolojilerle de NKSE karşımıza çıkabilmektedir.
ASE ile fokal NKSE
ayrımı genelde güçtür. Fokal form daha siklik bir patern gösterirken
ASE daha devamlıdır, ASE'de tam cevapsızlık görülmez,
fokalde ise bir dönemde cevapsızlık genellikle vardır. Fokal
NKSE’de otomatizmalar daha belirgin, zengin ve uzundur. Anksiyete, agresyon,
korku vb. fokal NKSE’de daha sıktır. Tedaviye cevap her iki tablo
için etyoloji ve tipe bağlıdır. Fokal NKSE genelde temporal lob
kökenli olarak düşünülür ama ekstratemporal, özellikle de frontal lob
kökenli olduğu kanıtlanmış olgular vardır.
ASE iyi prognozlu olarak
bilinmektedir ve tedaviye yanıtı hızlıdır.
İdyopatik/genetik jeneralize epilepsi seyrinde, belirleyici
özelliğini ve ana nöbet tipini tekrarlayan absans status
ataklarının oluşturduğu ve mevcut sınıflamada net
bir yeri olmayan atakları geçiren olgular tarafımızdan ve
başka gruplarca tanımlanmıştır. Atipik ve tipik absans
status tablolarını da ayırmak kimi zaman çok güçtür. Ancak bu
iki tablonun etyoloji ve prognozları çok farklı olduğu için
ayırma gayreti anlamlı görünmektedir. Atipik absans statusu sekonder
jeneralize epilepsilerde veya Lennox-Gastaut sendromunda görülen
uzamış atipik absans nöbetidir. Tipik absans statusundan
farkları mental retardasyon, gelişme geriliğinin eşlik
etmesi, anormal interiktal EEG saptanması, değişik nöbet
tipleriyle birliktelik, 3-6 Hz’den yavaş (ya da daha hızlı)
diken-dalga paroksizmleri, tablonun başlaması ve bitişinin sinsi
olmasıdır.
NKSE tablosunun
genişleyen ve giderek bulanıklaşan klinik spektrumu yanı
sıra EEG açısından bazı sorunlar da tanısında
güçlüklere yol açmakta ve EEG bulgularının yorumlanması
özellikle kritik hastalığı olan olgularda ve epileptik
ensefalopatiler gibi zaten çok aktif epileptifom EEG bulgusu olan özel
durumlarda uzmanlar için bile soru işaretleri yaratmaktadır. Bu
nedenle düzenlenen uluslararası toplantılar sonrasında epileptik
ensefalopatisi olan ve olmayan olgularda farklı şekilde “Salzburg
Konsensus Ölçütleri” geliştirilmiştir. Epileptik
deşarjların frekansı (saniyede 2,5 Hz’ den büyük veya küçük), ritmik
delta/teta aktivitesi ile intravenöz anti-epileptik ilaç (AEİ) ile klinik
ve EEG’de düzelme ve tipik spasyotemporal evolusyon paternleri de dikkate
alınmaktadır. Bu yeni net kriterlerin doğru tanı
koyulmasında yardımı olması umulmaktadır. YBÜ’de devamlı EEG monitorizasyonunun her olguda
gerekip gerekmediği, NKSE saptandığı takdirde ne ölçüde
agresif tedavi gerektiği tartışılan konulardır. NKSE
tedavisi için doğru bir bakış açısı, aynen tanı
için olduğu gibi, NKSE tablosunun tipine ve etyolojisine göre
yaklaşımdır.
Nonkonvülsif Status Epileptikusun
Ayırıcı Tanısı
· Diğer nöbetle ilişkili
değişmiş mental durum nedenleri
o Uzamış postiktal konfüzyon
o Periyodik lateralize epileptiform deşarjlar
(PLED) ile ilişkili değişmiş mental durum
·
Ensefalopatiler
o Toksik-metabolik
o Hipoglisemi
o Mitokondriyal ensefalopatiler
o Hiperamonyemi
o İlaç intoksikasyonu
·
Post-travmatik
·
Vasküler
·
İskemik
·
İnflamatuvar
·
Psikiyatrik sendromlar
o Disosiyatif reaksiyonlar
o Akut psikoz
o Histerik konversiyon reaksiyonu
o Katatoni
Sonuçta NKSE birçok nörolojik
ve psikiyatrik tablonun ayırıcı tanısında her zaman
akla gelmesi ve geniş klinik spektrumu ile iyi tanınması gereken
bir tablodur. NKSE olguları klinik durumlarına göre
değerlendirilmeli, EEG bulguları ve tanı kriterleri uzmanlarca
yorumlanmalı, etyoloji ve birlikte bulunabileceği durumlar dikkatle
araştırılmalıdır. Mekanizmaları,
tanımı, tanı kriterleri, tedavi yaklaşımları ve
prognoz konusunda araştırmalara ihtiyaç duyulan bu tabloda komada ve
ağır hastalığı olan grupla gerçekten epileptik mekanizmaların
rol oynadığı grup arasında ayırım
yapılmalıdır. Bu aşamada bizlere düşen hastaları
en iyi şekilde izleyip, ayrıntılı olarak incelemek ve bir
yandan da tedavilerini rasyonel olarak ilaç yan etkilerini de göz önüne alarak
planlamaktır.
Tedavi
Yaklaşımı
SE tedavisi nöbet aktivitesinin damar içi yolla verilen
AEİ ile sonlandırılması, destekleyici bakım, nöbet
nüksünün önlenmesi, tabloyu tetikleyen nedenlerin düzeltilmesi,
komplikasyonların önlenmesi ve gelişen komplikasyonların
tedavisi gibi farklı basamakları içerir. Destekleyici bakım ve nedene yönelik incelemeler
erken ve hızlı nöbet tedavisini asla geciktirmemelidir. Tedavideki
gecikme direnç gelişiminin en önemli nedenlerinden biridir.
Hastanın
kendisinden veya yakınından bilinen epilepsi hastası olup
olmadığı, varsa kullandığı AEİ’ler ve
dozları, tedavisinde aksama ve/veya araya giren başka bir tedavi olup
olmadığı öğrenilmelidir. Ardından da metabolik
bozukluk, intoksikasyon, enfeksiyon veya yeni bir yapısal lezyon
varlığı gibi SE’ye yol açabilecek nedenler hızla ve
planlı şekilde araştırılmalıdır.
Hipoglisemi, elektrolit bozukluğu gibi nedenler hızlıca
saptanmalı ve düzeltilmelidir. AEİ kullananlarda bakılabiliyorsa
ilaç serum seviyesine bakılmalıdır. Nöbetler hızla durup
hasta açılsa bile gereğinde diğer tedavi basamaklarına
geçilip, etyolojik araştırmalara ve hastayı izlemeye devam
edilmelidir. SE tedavi edildiği takdirde en az 24 saat daha nüks
olabileceği düşünülerek hasta gözlem altında tutulmalıdır.
SE tedavisi tüm diğer acil durumlar gibi hava
yolu, solunum ve dolaşım gibi yaşamsal fonksiyonların
sağlanması ile başlar. Solunum yetmezliğini engellemenin en
iyi yolu nöbetlerin durdurulmasıdır. Havayolunun açık tutulması
hipoksi nedeniyle SE’nin şiddetlenmesini engellemek açısından da
çok önemlidir. Çoğu durumda noninvazif pozitif solunum desteği
yeterli olmasına rağmen, genellikle tam güvenli değildir çünkü
hem uzayan nöbetlerin etkisi hem de ilaç tedavileri nörolojik
fonksiyonları baskılar. Solunum yetmezliği olan hastalarda ise
entübasyon sonrası mekanik ventilasyona başlamak gereklidir. Bazen
solunum yetmezliği gelişmeden önce solunum baskılanması
yapma eğilimi olan ajanlarla tedaviye karar verildiğinde entübasyon
gerekmektedir. SE tablosundaki hastalar entübasyonu
kolaylaştırıcı nöromüsküler blok yapan ajanlara
sıklıkla gereksinim gösterirler, oysa bunların
kullanımları istenen bir durum değildir. Kısa etki süresi
olan nöromüsküler blok yapan ajanları (vekuronyum veya rokuronyum bromid
gibi) kullanmak, kas aktivitesinin ilaçla durdurulması nöbetleri
tanımayı güçleştirdiğinden tercih edilmelidir. Neostigmin
(50-70 mikrog/kg) azalmaya başlayan nöromüsküler bloğu eski haline
çevirir. Süksinilkolin kullanımından ise nörolojik hastalıklarda
bu ilaca bağlı ciddi hiperpotasemi gelişme riski nedeniyle
kaçınılmalıdır.
Hava yolu takılmasının ardından
uygun damar yolu açılmalı ve hemen gerekli kan testleri için örnekler
alınmalıdır. İyi bir IV yol kan örneklerinin alınması,
sıvı ve elektrolit dengesinin sağlanması ve ilaçların verilmesi
için vazgeçilmezdir. Kardiyolojik açıdan izlem bu hastalarda
gelişebilen yaşamı tehdit eden aritmilerin tanınması
ve tedavisine olanak sağlar. Eğer hastada hipotansiyon varsa, volüm
replasmanı başlanmalı ve vazoaktif ajanların
kullanımı düşünülmelidir. Buna karşın eğer
hastada hipertansiyon varsa, kan basıncı agresif şekilde tedavi
edilmemeli ve beklenmelidir. Çünkü SE’yi durdurmak için uygulanan ilaçlar
genelde hipotansiyon yapma eğilimindedir.
Eğer kan
glikoz konsantrasyonu düşükse, hastanın diyabetik olduğu
biliniyorsa ya da SE nedeni henüz bilinmiyorsa 50mL %50’lik dekstroz (veya
1mL/kg) 100 mg tiaminle birlikte damar içi yolla verilmelidir, çünkü
hipoglisemi genellikle çok daha ağır sekel bırakabilir ve
dirençli nöbetlerle seyredebilir. Hipogliseminin fokal motor, afazik ve görsel
belirtili olabilen fokal nöbetlerle kendini gösterebildiği
unutulmamalıdır. Öte yandan hipoglisemi uzamış SE’nin bir
komplikasyonu olarak da gelişebilir.
SE’li hastalarda
fenitoin kullanılması düşünüldüğünde infüzyon
sıvısı olarak serum fizyolojik solüsyonu gerekli olduğundan
bir damar yolu daha açılmalıdır ve fenitoinin glikoz içeren
solüsyonlarda çökerek etkisini kaybettiği dikkate
alınmalıdır. Kan pH’sı ölçülmeli ama yaşamı
tehdit eden boyutlara ulaşmadıkça tedavi edilmeye
çalışılmamalıdır, çünkü SE’ye bağlı asidozun
kalıcı hasar oluşturduğuna dair bilgi yoktur ve fazla
bikarbonat verilmesi metabolik alkaloza neden olabilir.
SE’de
sistemik komplikasyonlar:
·
Hipoksi
·
Laktik asidoz
·
CO2 narkozu
·
Hiperkalemi
·
Hipoglisemi
·
Hipertansiyon izleyerek ileri
dönemde hipotansiyon ve şok
·
Kardiyak aritmi
·
Pulmoner ödem
·
Akut tubuler nekroz
·
Aspirasyon pnömonisi
·
Lökositoz
·
Otonomik disfonksiyon
Yapılması Gereken Tetkikler
·
Damar yolu açıldıktan sonra kan gazı,
glikoz, elektrolitler, karaciğer ve böbrek fonksiyon testleri, kalsiyum,
magnezyum, tam kan sayımı ve AEİ düzeyleri için kan örneği
alınmalıdır.
·
Bu tetkiklerle nedenin
aydınlatılamadığı durumda örneğin toksikolojik
tetkikleri yapabilmek için 5 ml serum ve 50 ml idrar örneği
saklanmalıdır.
·
Miyokard infarktüsü, ileti blokları, aritmi
tanısı için EKG çekilmeli, troponin değerlerine
bakılmalıdır.
·
Olası aspirasyon tanısı için akciğer
grafisi çekilmelidir.
·
Etkin tedavinin başlamasını geciktirmesine
izin vermeden, klinik tabloya göre gerekirse beyin görüntülemeleri ve lomber
ponksiyon planlanmalıdır.
Konvülziyonlar kontrol altına alındıktan
sonra hastanın öyküsü ve ilk veriler SE’nin nedenini
açıklamıyorsa hızla ve kolay uygulanabilmesi nedeni ile
öncelikli olarak kranial BT tercih edilebilir ancak SE’nin etyolojisinin
aydınlatılmasında kranial MRG, BT’ye göre daha üstündür. Erken
dönemde kranial MRG’de en fazla hipokampus, neokorteks, korpus kallozum ve
posterior talamusta görülen, inflamasyon veya iskemiyi taklit eden, nöbetlere
bağlı bazı sinyal değişiklikleri geri dönüşümsüz
beyin hasarı olarak değerlendirilmemelidir. SE’nin etyolojisinde
inflamatuar veya enfeksiyöz MSS hastalığından
şüpheleniliyorsa lomber ponksiyon yapılmalıdır. Nöronal
otoantikorların araştırılması da bu grup hastalarda
son yıllarda giderek artan öneme sahiptir.
SE takibinde düzenli nörolojik muayene kontrollerinin
yanısıra nabız dakika sayısı, kan basıncı ve
vücut ısısı monitorizasyonu yapılmalıdır. EKG,
biyokimya analizleri, kan gazı, pıhtılaşma faktörleri, kan
sayımı ve AEİ düzeyleri takip edilmelidir. İlk basamak
AEİ’lere cevap vermeyen dirençli SE hastalarının takip ve
tedavisi YBÜ’de yürütülmelidir.
JKSE tanısı için EEG monitorizasyonuna primer
olarak ihtiyaç yoktur. Hızlı ve etkin tedavi vakit kaybetmeden
başlanmalıdır. Ancak JKSE sonrası bilinci açılmayan
veya primer nörolojik hasar olsun olmasın herhangi bir nedenle
açıklanamayan, devam eden veya dalgalanma gösteren ensefalopati ve koma
tablolarında NKSE veya “subtle” SE akla gelmeli, en azından
yatakbaşı EEG incelemesi yapılmalı, hatta
yapılabiliyorsa hastalar devamlı EEG monitorizasyonu ile
izlenmelidir.
SE’nin modern tedavi
protokolleri evre temelli bir yaklaşımı esas alır, SE’ye
yaklaşım algoritması Şekil 22’de yer
almaktadır.
1. Erken SE: Nöbetlerin başlayarak
sıklığının ve şiddetinin arttığı
dönemdir. Beş dakikadan uzun süren JTKN erken SE olarak
tanımlanır.
2. Yerleşmiş SE: Hastanın
aralarda açılmadan, nöbetlerin arka arkaya veya devamlı hale
geldiği, 10 dakikadan uzun, 30 dakikadan kısa süren SE tablolarıdır.
3. Dirençli SE: “Sinsi SE” veya “stupor
SE” olarak da isimlendirilen bu evrede SE’nin süresi uzadıkça konvülzif
fenomen azalmakta, diğer deyişle elektromekanik ayrışma
meydana gelmektedir. Hastanın komada olduğu bu evre “benzodiazepin ve
diğer bir AEİ tedavisine dirençli SE” olarak da tanımlanır.
4. Süper dirençli SE: YBÜ’de izlenen,
maksimum dozda IV anestezik tedaviye rağmen 24 saatten uzun süren SE ile
karakterizedir. Malign SE olarak da tanımlanır.
Benzodiazepinlerin yan
etkileri benzerdir, sedasyon, baş dönmesi, güçsüzlük, solunum depresyonu
ve hipotansiyon görülür. Erken tedavi edilen SE’lerin %80’i sonlanırken, 2
saat ve daha çok gecikildiğinde %40’dan azında
başarılı olunabilmiştir

Şekil 22. Status epileptikus tedavi algoritması
Dirençli ve süper
dirençli SE tedavisinde kullanılan ajanlar yüksek doz barbitüratlar
(pentobarbital, tiyopental ve fenobarbital), yüksek doz benzodiazepinler
(midazolam), propofol, ketamin ve diğer anesteziklerdir. Bu ilaçları
karşılaştıran çok az çalışma vardır, bu
konudaki literatür daha çok retrospektif olgu serileri ve kontrol grubu olmayan
çalışmalardan oluşur. Kardiyovasküler ve metabolik
komplikasyonlar en az midazolam, en fazla barbitüratlar ile görülür.
Tedavi
başarılı olduğu takdirde, nöbetler baskılandıktan
uygun bir süre sonra (12-24 saat), ilacın dozu dikkatle azaltılarak
nüks olup olmadığı izlenmelidir.
Yukarda adı
geçen sık kullanılan anestezikler dışında izofluran
(inhalasyon), etodimat (IV) ve ketamin (IV) de kullanılabilir.
İyonotropik glutamaterjik AMPA reseptörleri üzerinden etkili
topiramatın IV formu bulunmamasına rağmen oral yüksek doz
kullanımının (çocuklarda 2-25 mg/kg/gün, erişkinde 900-1600
mg/gün) çeşitli olgu serilerinde dirençli SE tedavisinde etkin
olabileceği bildirilmiştir. Süper dirençli SE’de denenen diğer
ilaçlar ise lakozamid (IV), paraldehid (rektal, IM), lidokain IV, klormetiazol
(IV), ve CBZ (rektal)’dir. Kortikosteroidler, IV immunoglobülinler, plazma
değişimi ve diğer seçeneklerden oluşan immun tedaviler
dirençli ve süper dirençli SE tablolarında voltaj kapılı
potasyum kanalı ve NMDA reseptörlerine karşı olanlar başta
olmak üzere diğer nöronal oto-antikorların saptanması ve
epileptogenezde inflamasyonun rolünün net olarak gösterilmesi nedeni ile
herhangi bir kesin tanımlanmış immunolojik hastalık olmasa
bile giderek daha fazla kullanılmaktadır. Hangi tedavinin ne kadar
süre ile uygulanması gerektiğine dair bir uzlaşma olmamakla
birlikte 5 gün, 1 gr/gün IV prednizolon sonrası 1 hafta, 1 mg/kg/gün
sonrası 2 gün içinde değişiklik yoksa 0,4 g/kg/5 gün IV
Immunoglobulin (IVIG) ya da plazma değişimi tedavisi önerilmektedir.
Olgu bildirileri
şeklinde medikal tedavi uygulamalarına yanıtsız dirençli SE
tedavisinde iyi seçilmiş olgularda bazı epilepsi cerrahisi
girişimleri (fokal rezeksiyon veya multipl subpiyal transseksiyon gibi),
nörostimülasyon tedavileri ve ketojenik diyet önerilmektedir.
Yukarda sözü
edilen tedavi tartışması JKSE için geçerlidir. Buna karşın diğer sık
karşılaşılabilen SE formlarında
aşağıdaki noktalar göz önüne alınmalıdır:
a) Bilinç
kaybı olmayan fokal SE de hızla tedavi edilmesi zorunlu bir tablo olmakla
birlikte bu durumda daha az yoğun tedaviye gerek duyulur. Çünkü bu
tablonun önemli nöron hasarı oluşturduğunu düşündüren ve
kanıtlayan kesin veri bulunmamaktadır. Bir diğer neden de bu tip
nöbetlerin AEİ’ler ile tedaviye çok yüksek oranda dirençli olması ve
nöbetleri tamamen yok etmeye yönelik çabaların ilaçlara bağlı
yan etkileri ortaya çıkarmasıdır. Hastanın korunmuş
olan bilincinin AEİ yan etkileri nedeniyle bozulması ve genel durumu
kötü değilken YBÜ gerektiren ve daha fazla risk içeren bir tabloya girmesi
de olumsuz bir faktördür. Tedavi basamakları genel SE tedavisi ile
aynıdır ama direnç görüldüğünde ileri basamaklara geçme konusu
hastanın genel durumu göz önüne alınarak belirlenir.
b) MSE eğer
kronik epilepsi sürecinde gelişmişse miyokloni kontrolünde
başarı sağlayan ilaçlarla (damar içi klonazepam, fenobarbital,
valproat, levetirasetam gibi) kontrol altına alınabilir. Buna
karşın, anoksiye bağlı ensefalopati tablosunun
komplikasyonu olarak beliren MSE ise standart AEİ tedavilerine oldukça
dirençlidir. Bu durumda klonazepam ve valproat gibi ilaçlar
kullanılmalıdır, fenitoinin miyokloniler üzerinde olumsuz etkisi
olabileceği unutulmamalıdır. Sık olarak da yetersiz gözlem
sonucu MSE, JKSE ile karışabilmektedir ve bu nedenle yanlış
tedavi uygulanması söz konusu olabilir. SE tedavisinde EEG
bulgularının yönlendiriciliği konusundaki görüşler halen
tartışmalı olarak kalmıştır. Önerimiz EEG’nin
mümkün olduğu ölçüde izlenmesinin uygun olacağı ve temel
aktivitenin yavaş, izoelektrik hat üzerinde veya baskılanım-boşalım
şeklinde olmasına bakılmaksızın nöbetler önlenene dek
kullanılan ilacın titre edilmesi yönündedir.
c) NKSE için standart SE tedavisi uygulanır. Nüks görülen olgularda NKSE
tipine uygun AEİ (valproat veya karbamazepin gibi) önerilir. Tipik ASE’de
tedaviyle veya tedavisiz prognoz iyidir. Nüks durumlarında da mortalite ve
morbidite bildirilmemiştir. Atipik ASE ise tedaviye daha zor cevap verir.
Mortaliteye etkili faktörler arasında yaş,
etyolojik neden gibi faktörler değiştirilemez. İlk seçenek olarak
seçilen AEİ de mortalite açısından çok büyük fark
yaratmamaktadır. Oysa SE süresi etkin ve hızlı tedavi ile
kısaltılabilir. Bu nedenle erken ve etkin tedavi mortalitenin
azaltılması açısından en önemli faktördür.
Sonuç olarak SE en ciddi nörolojik acil durumlardan
biridir. Tanı yöntemleri, tedavisinin sadece IV uygun dozlarda AEİ
uygulaması ile yapılabileceği kuralı ve takip
yaklaşımı çok iyi bilinmelidir. Çalışmaların
SE’li hastalarda klinik nöbetler sonlandıktan sonra bile %20 oranında
elektrografik olarak nöbetlerin sürebileceğini gösterdiği
unutulmamalıdır. Prognoz ve mortaliteyi belirlemek için mevcut
skorlamaların daha geniş hasta serilerinde deneneceği prospektif
çalışmalara ihtiyaç vardır.
EPİLEPSİNİN
BİYOLOJİK YÖNLERİ VE SOSYAL ETKİLERİ
Epilepsili hastanın normal
bir hayat sürdürmesi bazı basit noktalara dikkat edilerek çoğu zaman
mümkündür. Ancak toplumdaki genel eğitimsizlik bazen bu hastaların
hayatını gereksiz şekilde zorlaştırmakta ve verimsizleştirmektedir.
Tüm adımlar, hastanın mümkün olduğunca normal bir hayat sürmesi,
yani eğitim, evlenme, meslek sahibi olma ve boş zamanlarını
değerlendirme gibi çeşitli sosyal olayları normal bir
şekilde sürdürmesi amacıyla atılmalıdır.
Epilepsi ve Gebelik
Epilepsi ve gebelik birçok yönden
etkileşim gösterir. Gebelik ve çocuk doğurma epileptik
kadınların büyük çoğunluğunda ciddi bir soruna yol açmaz.
Yapılan çalışmalarda epilepsili kadınların sezeryan ve
doğum ile ilişkili komplikasyonlarda artmış riski
olmadığı bildirilmekteyken sigara kullanan epilepsili
kadınlarda erken doğum riski artabilir. Ayrıca epilepsili
kadınlarda düşük doğum ağırlıklı bebek
oranı bazı çalışmalarda yaklaşık 2 kat
artmaktadır.
Çocuk sahibi olmak isteyen
epilepsili bir kadında ilk önce tanının doğru olup
olmadığı ve AEİ kullanımının gerekli olup
olmadığının belirlenmesi gereklidir. Hastaya ve eşine
olası konjenital malformasyonlar hakkında sağlıklı
bilgi verilmelidir. AEİ kullanımı normal popülasyonda %2-5
civarında olan konjenital malformasyon riskini yaklaşık 2-3 kat
arttırmaktadırlar (%7-11). Risk politerapiyle ve artan dozlara
paralel olarak yükselmektedir. Gebelik daha gerçekleşmeden, nöbetlerin iyi
bir şekilde kontrol edilebildiği minimal AEİ dozunun
ayarlanması ve gereksiz AEİ kombinasyonlarından kaçınarak,
mümkün olduğunca monoterapiye geçilmesi uygun adımlardır.
Özellikle nöral tüp defekti başta olmak üzere majör malformasyonlardan
korunmak için tüm hastalara gebelik öncesinde 5 mg günlük folik asid
replasmanı önerilmektedir. Folik asit replasmanı, gebelik öncesinde başlanılmalı
ve 1. trimesterin sonuna kadar devam edilmelidir. Majör konjenital
malformasyonlar kozmetik, medikal ve cerrahi öneme sahip
malformasyonlardır. Valproatın monoterapi ve özellikle politerapide
majör konjenital malformasyon riskini diğer ilaçlara kıyasla bir
miktar daha fazla arttırdığı bilinmektedir. Valproat, daha
çok nöral tüp defekti, yarık damak ve dudak, fasyal dismorfizm ve
hipospadiasa neden olur. Ayrıca kardiyak, renal, ürogenital ve ekstremite
defektleri de izlenebilmektedir. Fenitoin ve karbamazepin yarık dudak ile,
fenobarbital kardiyak malformasyonlar ile daha sık
ilişkilendirilmektedir. Gebelikte AEİ kullanmayan epilepsili
kadınların çocuklarında kognitif bozukluk açısından
fark izlenmemektedir. Ancak gebeliğinde valproat, fenitoin ve fenobarbital
kullanan epilepsili kadınların çocukları ile yapılan
çalışmalar bu çocuklarda kognitif bozukluk olabileceğini
göstermektedir. Bu etki valproat kullanan gebelerde daha fazla gibi
gözükmektedir. Gebeliğinde AEİ kullanan annelerin çocuklarında otizm
riski valproat başta olmak üzere lamotirijin, okskarbazepin kullananlarda
artış göstermektedir. Son yıllarda yapılar
çalışmaların metaanalizinde valproatın olumsuz etkileri
üzerinde belirgin şekilde durulmaktadır. Gebeliğinde valproat
kullanan annelerin çocuklarında 3 kat fazla otizm spektrum
hastalıkları, 5 kat fazla otizm geliştirme riski olduğu
belirtilmiştir. Ayrıca dikkat eksikliği ve hiperaktivite
bozukluğu görülme oranı bu çocuklarda artmaktadır. Anne
karnında valproat maruziyeti olan çocukların okul öncesi dönemde
%30-40’a varan oranlarda yürüme ve konuşma geriliği, hafıza
problemleri, dil ve konuşma zorlukları gibi gelişimsel
bozukların izlendiği çalışmalarda ortaya konulmuştur.
Diğer AEİ’lere kıyasla bu çocukların zeka seviyeleri daha
düşük izlenmektedir. Bunun yanında %11 oranında majör konjenital
malformasyona (nöral tüp defekti ve yarık damak) yol açmaktadır, bu
oran normal popülasyonda %2-3 oranında izlenmektedir. Valproat dozu
arttıkça majör ve minör malformasyon riskleri artmaktadır.
Avrupa Tıp Kurumu (European
Medical Agency-EMA) 2015 yılında valproat kullanımı ile
ilişkili bir rapor yayınlamış ve bu raporda gebe veya gebe
kalma potansiyeli olan kadın ve kız çocuklarında diğer
medikal tedavilerin yetersiz kaldığı veya yan etki nedeniyle
kullanılamadığı durumlar haricinde valproat
kullanımının kısıtlanmasını önermiştir.
Tedavi açısından valproatın tek seçenek olduğu hastalarda
ise etkili doğum kontrol yöntemlerinin kullanılması gereği
vurgulanmıştır. Valproat başlanılması planlanan
hastalara ise yukarıda bahsedilen risklerin ayrıntılı bir
şekilde anlatılması ve aydınlatılmış onam
alınması zorunlu hale gelmiştir. Valproat kullanırken gebe
kalan hastalar için ise alternatif tedavilerin gözden geçirilmesi
önerilmiştir. EMA tarafından yayınlanan bu öneriler
sonrasında ülkemizde de Sağlık Bakanlığı
tarafından hastanelere ve ilgili uzman hekimlerine doğurganlık
çağındaki kadınlar ve kız çocuklarında valproat
kullanımı ile ilgili uyarıları içeren bilgilendirme
yazısı gönderilmiştir.
Monoterapide yüksek doz AEİ
kullanımı majör malformasyon riskini büyük oranda
arttırırken, politerapide doza bağımlılık
belirgin değildir. Politerapi kullanan gebelerde yapılan
çalışmalarda özellikle topiramat ve valproat ile majör malformasyon
riski büyük oranda artmaktadır. Topiramatın monoterapide teratojenite
riski politerapiye kıyasla düşük bulunmuş ancak bu durumun
nedeni tam olarak anlaşılamamıştır. Monoterapide,
politerapiye oranla topiramatın daha düşük dozlarda
kullanılması suçlanmıştır. Bazı çalışmalarda
topiramatın ilk trimesterde maruziyetinin yarık damak malformasyon
riskini arttırdığına dair bilgiler bulunmaktadır.
Politerapide lamotirijin ve levetirasetam kullanımı majör
malformasyon açısından diğer kombinasyonlara kıyasla en
güvenlisi gibi gözükmektedir.
Hastanın gebelik süresince
nöbet sıklığı açısından dikkatle ve sık
kontrollerle izlenmesi gerekir, çünkü gebelik sırasında AEİ kan
düzeylerinde dalgalanmalar olmaktadır. Hastanın jinekologuyla
nörologunun ilaç tedavisi, gebeliğin gidişi ve doğum
sırasındaki tedavi düzenlemeleri açısından sıkı
bir işbirliği içinde olmaları önerilmelidir. Fetal riskler
nedeniyle anne adayının aniden AEİ kesmesi %50 oranında
nöbet nüksü ve daha da tehlikelisi SE riskini beraberinde getirir. Bu nedenle
anne adayının bilinçlendirilmesi ve böyle bir durumun bebeğe
AEİ’den daha fazla zarar verebileceği, hatta annenin de SE
sırasında yaşamını yitirebileceğinin
anlatılması gerekmektedir.
Epilepsili gebelerde nöbetlerin
veya SE’nin sıklığında artış ile ilgili
yapılan çalışmalar yetersizdir. Kontrol grubunun
olmadığı bir çalışmada, sadece 1/3 olguda nöbet
sıklığında artış olduğu bildirilmiştir.
Gebelik sırasında ilaç
kan düzeylerinin değişebildiği bilinmektedir. Özellikle
lamotirijin kan düzeyi 3. trimesterde belirgin derecede düşer ve bu nöbet
sıklığında artışa neden olur. Lamotirijinin kan
düzeyinin gebelik boyunca izlenmesi önerilir. AEİ kan düzeyi azalan veya
nöbet sıklığında artış olan gebelerde ilaç
dozları arttırılır. Gebelik sonrası, ilaç düzenlemesi
ile ilişkili yeterli veri yoktur. Bu hastalarda hasta özelinde ve
kontrollü bir şekilde eğer arttırılmış ise ilaç
azaltımı yapılabilir. Lamotirijin, dışında
okskarbazepin ve levetirasetamın da ilaç düzeylerinin gebelik ile
değişebildiği ve nöbet sıklığında
artışa neden olabildiği bildirilmiştir.
Epilepsili kadınların
yenidoğan bebeklerinde hemorajik komplikasyon riskinin (yaşamın
ilk 24 saatinde herhangi bir yerden kanama riski) artması ile
ilişkili yeterli veri bulunmamaktadır. Ülkemizde zaten yenidoğan
bebeklerin hepsine doğumda K vitamini verilmektedir.
Fenobarbital, pirimidon,
fenitoin, karbamazepin, levetirasetam, valproat muhtemelen plasentadan klinik
olarak önemli miktarlarda geçer. Gabapentin, lamotirijin, okskarbazepin ve
topiramat ise olasılıkla klinik anlamda önemli miktarda plasentadan
geçer.
Levetirasetam ve pirimidon, anne
sütüne muhtemelen önemli miktarda geçer. Gabapentin, topiramat, lamotirijin ise
olasılıkla anne sütüne önemli miktarlarda geçer. Valproat,
fenobarbital, fenitoin ve karbamazepin anne sütüne önemli miktarlarda geçmez.
Ancak annenin aldığı AEİ’lerin bebeğe olumsuz etkileri
bilinmemektedir. Bu nedenle bebeği izleyerek emzirmeye izin verilmelidir.
Epilepsi ve
Yaşlılık
Yaşlı hastalarda
başlıca serebrovasküler hastalık, demans ve neoplastik
hastalıklar nedeniyle epileptik nöbetler görülebilir. Yaşlı
hastaları değerlendirirken öncelikle ayırıcı tanı
dikkatle yapılmalıdır. Kardiyak aritmi, ortostatik senkop gibi
kardiyak etyolojiler ayırt edilmelidir. Yaşlı hastalarda
AEİ seçiminde de bu yaş grubunda çoklu ilaç kullanımı
dikkate alınarak ilaç etkileşimi az olan ilaçlar tercih edilmelidir.
Ayrıca AEİlerin bazı yan etkileri yaşlılarda daha
sık görülür. Bunlar içerisinde; valproat yaşlılarda
Parkinsonizme neden olmaktadır ve okskarbazepin ile özellikle diğer
diüretik ilaçlarla beraber kullanıldığında hiponatremi
sık görülmektedir. Pregabalin ve gabapentin ile de ödem görülebilmektedir.
Yaşlı hastalarda AEİ kullanımı ile düşmelerin sıklaşabileceği
bildirilmiştir. Bu nedenle hastanın sosyal çevresi düzenlenmelidir.
Epilepsi ve Meslek Seçimi
Epilepsi ani bilinç kaybı ve
hareketlerde kontrol kaybı nedeniyle bazı meslekler
açısından risk oluşturur. Örneğin yüksekte
çalışma, tehlikeli olabilecek iş makinelerinin
kullanımı, araba kullanma gerektiren meslekler epileptik
hastaların tam nöbet kontrolü sağlanmadan seçmemeleri gereken
mesleklerdir. Bazı sendromlarda (örneğin juvenil miyoklonik epilepsi)
uykusuzluk nöbet riskini arttırdığı için uyku düzenini bozan
meslekler ve vardiyalı gece çalışması sakınca
doğurabilir. Fotosensitivite saptanan olgular parlak ışık,
TV, bilgisayar ekranının nöbet riski yaratma
olasılığı nedeniyle meslek seçiminde
uyarılmalıdırlar, ancak bu durumun epileptiklerin sadece ufak
bir bölümü için geçerli olduğu da göz ardı edilmemeli ve her hastaya
gereksiz sosyal hayatını kısıtlayıcı öneriler
getirilmemelidir. Epilepsili bir çocuğun ciddi bir mental geriliği
olmadığında normal okullara devam etmesi ve başarısı
ölçüsünde eğitimini sürdürmesi ve yüksek öğrenim gerektiren birçok
mesleği bile sorunsuz bir şekilde yürütmesinin mümkün olduğu
unutulmamalıdır. Bu açıdan gereksiz sosyal çekinceler veya
korkular nedeniyle ailelerin epilepsili çocukların öğrenime devam
etme isteklerini engellememeleri öğütlenmelidir. Aşırı
koruyucu tavır ilerde hastanın sosyal açıdan izole
kalmasına, sağlıklı sosyal ilişkiler
oluşturamamasına ve eğitim eksikliği nedeniyle iş
bulması açısından da sorunlar yaşamasına neden
olabilmektedir. Ancak epilepsili hastanın iş bulma
açısından yaşadığı en büyük sorun toplumun bu
hastalığa karşı bilinçsiz ve bilgisiz
yaklaşımının sürmesi nedeniyle olmaktadır. Bu nedenle
ülkemizde birçok epilepsili hasta niteliklerine uygun iş bulamamakta,
hastalıklarını saklamak zorunda kalmakta ve iş yerinde
nöbet geçirince işten atılma stresiyle yaşamaktadırlar. Bu
açıdan halkın ve işverenlerin hastalığın gerçek
doğası hakkında bilgilendirilmeleri ancak çok geniş
çaplı eğitim adımlarının atılması ile mümkün
olabilecektir.
Epilepsi ve Askerlik
12/10/2015 tarihli Bakanlar
Kurulu kararı ile belirlenen Türk Silahlı Kuvvetler Sağlık
yeteneği yönetmeliğine göre A grubu askerliğe elverişli, B
ve D grubu askerliğe elverişli olmayan ve C grubu hastalık
nedeniyle 1 yıl erteleme olarak değerlendirilir. Buna göre epilepsi
hastalarının değerlendirilmeleri aşağıdaki
maddelere göre yapılır:
A) 2. Klinik ve laboratuvar bulguları normal olup
EEG’de belirgin bulgular (fokal veya jeneralize diken veya keskin dalga
kompleksleri, fokal veya jeneralize yavaş aktivite) gösterenler
(Bayılma öyküsü olmayan ancak nonspesifik EEG anormalliği bulunanlar
sağlam kabul edilir)
3. Anamnez ve klinik bulguları ile kesin epilepsi
tanısı konulamayan, spesifik EEG bulgusu olmayan paroksizmal
bayılmaları olan hastalar
4. Spesifik EEG veya görüntüleme bulgusu olmayan ancak
anamnezinden veya tıbbi belgelerinden nöbetlerinin seyrek olduğu
anlaşılan epilepsili hastalar.
B) 1. Nöroloji uzmanı tarafından nöbeti
gözlenen veya klinikte yattığı dönemde yapılan video
kayıtlarına göre nöroloji uzmanınca epilepsili olduğu
anlaşılan tüm hastalar
2. Anamnezi epilepsi ile uyumlu olan ve bu
tanıyla takip ve tedavi edildiğini belirten, nöroloji uzmanına
kesin fikir veren tıbbi belgeleri olan hastalar
3. Anamnezi epilepsi ile uyumlu olup EEG’sinde
spesifik bulgu (multipl diken dalga, sık ortaya çıkan lateralize veya
jeneralize diken, keskin-yavaş dalga kompleksi) olan hastalar
C) A, B ve D dilimlerinde yer alan hallerin tedavi ve
nekahat halleri
D) 1. İlaca dirençli epilepsi nöbetleri olan
hastalar
Epilepsi ve Araç Kullanımı
Epilepsi nedeniyle sosyal
açıdan hastanın yaşadığı en önemli
sıkıntılardan biri de, ne zaman geleceği belli olmayan
nöbetler nedeniyle hastanın nöbetsiz döneminde rahatça yürütebileceği
bazı aktivitelerin nöbet anında yaşam tehlikesi
yaratmasıdır. Araba kullanmak bunlar arasında gündelik
yaşam ve iş bulma açısından belki de en önemlisidir.
Ülkemizde epilepsi hastalarına bazı koşullar
sağlandığı zaman sadece birinci grup sürücü belgesi
sınıfları verilebilir. Bu koşullar aşağıda
sıralanmıştır:
1) Bilinç kaybı ile giden nöbet geçiren epilepsili
hastaların sürücü belgesi alabilmesi için 6 aylık aralarla
kontrollerinin yapıldığı, 5 sene boyunca nöbet
geçirmediklerinin ve AEİ kullanmadıklarının belgelenmesi
gerekmektedir. Bu koşulları sağlayan epilepsili hastalar
sağlık kurulunda değerlendirilirler ve sürücü belgesi
almaları uygun görüldüğü takdirde kontrol süresinin raporda
belirlenmesi gereklidir.
2) Direksiyon başında tekrarlama
olasılığı olmayan, fark edilebilir bir tetikleyici ile
refleks nöbet geçiren epilepsili hastalar nöroloji uzmanının
kanaatine göre sürücü belgesi alabilirler.
3) İlk ve tek semptomatik olmayan epilepsi nöbeti
geçiren hastaların ehliyet alabilmesi için 6 aylık aralarla
kontrollerinin yapıldığı, 3 sene boyunca nöbet
geçirmediklerinin ve AEİ kullanmadıklarının belgelenmesi
gerekmektedir.
4) Sadece uykuda nöbet geçiren hastaların sürücü
belgesi alabilmesi için 6 aylık aralarla kontrollerinin
yapıldığı, 5 sene boyunca nöbet geçirmediklerinin ve
AEİ kullanmadıklarının belgelenmesi gerekmektedir. Bu
koşullar sağlandığında hasta sağlık
kurulunda değerlendirilebilir.
5) Bilinci veya hareket etme yetisini etkilemeyen nöbet
geçiren hastaların sürücü belgesi alabilmesi için 6 aylık aralarla
kontrollerinin yapıldığı, 5 sene boyunca nöbet
geçirmediklerinin ve AEİ kullanmadıklarının belgelenmesi
gerekmektedir. Bu koşullar sağlandığında hasta
sağlık kurulunda değerlendirilebilir.
6) Epilepsi cerrahisi geçirmiş hastalar 1. maddede
yazıldığı gibi değerlendirilir.
7) Epilepsi hastaları ambulans, resmi veya ticari
araç kullanamazlar.
Epilepsi hastalarında
sağlık kurullarında ayrıntılı nörolojik inceleme,
EEG ve nörogörüntüleme yapılır. Epilepsi hastalığı
nedeniyle sürücü belgesi alamayan veya sürücü belgesi alması belirli
koşullara bağlanan hastalar, Sağlık
Bakanlığınca elektronik sistem üzerinden Emniyet Genel
Müdürlüğüne bildirilir. Elektronik altyapısının
olmadığı durumlarda yazılı olarak bildirim
yapılır. Hastada bilinç kaybının
yaşandığı ancak epilepsi tanısının
konulmadığı durumlarda nöroloji uzmanınca bilinç
kaybının sürüş esnasında tekrarlama riskine göre
değerlendirilir. Bu kurallar 29 Aralık 2015 tarihli resmi gazetede
İçişleri Bakanlığı tarafından yayınlanan
yönetmelik ile belirlenmiştir.
Epilepsi ve
Mortalite
Epilepsili hastalarda mortalite
hızı normal popilasyona göre 2 kat artmıştır. Mental
retardasyon ve epilepsi varlığında mortalite en yüksektir. Nöbet
sıklığı mortalite hızını etkilememektedir.
Beyin tümörü, neoplastik hastalık ve serebrovasküler hastalık
varlığında mortalite artmaktadır.
Epilepsili
Hastalarda Ani Beklenmedik Ölüm (SUDEP - Sudden unexpected death in epilepsy)
Epilepsiye
bağlı ölümlerin içinde en önemlisini oluşturmaktadır,
nedeni bilinmemektedir. Diğer ölüm nedenleri nöbet
sırasındaki yaralanma, boğulma ve SE’ye bağlı
ölümlerdir. Çok merkezli çalışmalar yapılarak SUDEP
etyopatogenezi belirlenmeye çalışılmaktadır. Çoğu
olguda tanık yoktur. SUDEP insidansı 45 yaş altında
0,05-0,1/1000 iken 45 yaş üstünde 3/1000 olarak belirlenmiştir. Ancak
nöbetleri erken yaşta başlayanlarda, dirençli epilepsisi olan,
politerapi alanlarda, IQ <70 olanlarda, epilepsi cerrahisi
yapılmış ve başarısız olmuş kişilerde
ve beraberinde başka hastalıkları olanlarda risk
artmaktadır. En yüksek risk ilaç uyum sorunu olan, sık jeneralize
tonik-klonik nöbet geçiren kişilerdir. Nedeni bilinmemekle birlikte
pulmoner, kardiyak, otonomik mekanizmaların biri veya birden
fazlasının etkili olduğu ileri sürülmektedir. Nöbet ile birlikte
santral ve obstrüktif apne, aşırı bronşiyal ve oral
sekresyon, pulmoner ödem ve hipoksi gibi nedenler solunumda değişikliklere
neden olmaktadır. Pulmoner ödem çoğu olguda görülmektedir. Diğer
bir olasılık da kardiyak otonomik kontrolun bozulması sonucu
kardiyak aritmilerin izlenmesidir. Etyopatogenezi bilinmediği için
önleyici tedavi de verilememektedir.
Epilepsi ve
Komorbidite
Epilepsi hastaların
komorbiditeleri fiziksel, psikiyatrik veya nörolojik durumlar ile
ilişkilidir. Fiziksel komorbid durumlar nöbetle ilişkili
yaralanmaları içerir. Bunlardan korunmak için epilepsi
hastalarının yatak çevresinde keskin objelerin kaldırılması,
banyo yaparken iyi olduklarının kontrol edilmesi, yüksekte ve
ağır makine başında çalışmamalar önerilir.
Epilepsi hastalarının IQ’ları çoğunlukla normal popülasyona
benzerdir. Ancak epilepsi sendromu, yaş, nöbet
sıklığı, nöbet süresi, kullanılan AEİler ve
epilepsi cerrahi komplikasyonları gibi bazı etkenler hastaların
kognitif durumlarını etkileyebilmektedir. Jeneralize epilepsili ve
nöbetleri kontrol altına alınamamış çocuklarda kognitif
etkilenme daha belirgin olmaktadır.
Depresyon başta olmak üzere
anksiyete, obsesif kompulsif bozukluk, panik bozukluğu, psikoz gibi
psikiyatrik hastalıklar epilepsi hastalarında sık görülmektedir
(normal popülasyona göre 2 kat). Depresyon, nöbetleri kontrol altına
alınmış hastaların %10-20’sinde görülürken nöbetleri
kontrol altına alınamamış hastalarda %20-60
oranlarında görülmektedir. Bunun yanında epilepsi hastalarında
intihar riski 2 kat artmaktadır. Epilepsi ve depresyon
birlikteliğinde, valproat, lamotirijin ve karbamazepin gibi AEİ’lerin
duygudurum düzenleyici etkisi ile depresyon tedavi edilebilmektedir. Ancak bu
ilaçların kullanılmasına rağmen depresif şikayetler
devam ediyorsa tedaviye bir antidepresan ilaç eklenebilir. Antidepresan ilaç
seçiminde selektif serotonin gerialım inhibitörleri nöbet
sıklığında değişikliğe yol açmamaları
nedeniyle tercih edilebilir. Psikoz epilepsi hastalarında nadiren ancak
genel popülasyona göre sık olarak gözükür. Epilepsi hastasında psikoz
varlığında postiktal psikozun ayrılması gerekir.
Bazı hastalarda nöbetlerin sıklaşması ve psikoz
atakları birbirini takip edebilir. Tedavide antipsikotik ilaçlar ve
elektrokonvulzif tedavi kullanılabilir (Ayrıca bakınız: Nörolojik
Hastalıkların Psikiyatrik Yansımaları).
Yapılan çalışmalar
epilepsi hastalarında demans ve Parkinson hastalığı gibi
nörodejeneratif hastalıklar, serebrovasküler hastalıkları ve
başağrısının sık olduğunu ortaya koymuştur.
Başağrısı tiplerinden özellikle migren epilepsi ile
sık birliktelik göstermektedir ve AEİ seçimini etkiler. Epilepsi
hastalarında uyku bozuklukları özellikle insomni, obstrüktif uyku
apne sendromu sık gözükür. Epilepsili hastaların polisomnografik
kayıtlarında REM uykusunda azalma, gece uyanıklık süresinde
uzama, REM latansında uzama görülmüştür.
KAYNAKLAR:
1)
Abou-Khalil BW. Antiepileptic
Drugs. Continuum (Minneap Minn) 2016;22(1 Epilepsy):132-156.
2)
Baykal B ve Altındağ E.
Nonkonvülzif Status Epileptikus. İstanbul: Korteks İletişim
Hizmetleri, 2018.
3)
Beghi E, Carpio A, Forsgren
L, ve ark. Recommendation for a definition of acute symptomatic seizure.
Epilepsia 2010;51(4):671-675.
4)
Capovilla G, Mastrangelo M,
Romeo A, Vigevano F. Recommendations for the management of "febrile
seizures": Ad Hoc Task Force of LICE Guidelines Commission. Epilepsia
2009;50 Suppl 1:2-6.
5)
Devinsky O, Hesdorffer DC, Thurman DJ, Lhatoo
S, Richerson G. Sudden unexpected death in epilepsy: epidemiology, mechanisms,
and prevention. Lancet Neurol 2016;15(10):1075-1088.
6)
Duncan JS, Winston GP, Koepp
MJ, Ourselin S. Brain imaging in the assessment for epilepsy surgery. Lancet
Neurol 2016;15(4):420-433.
7)
Fisher RS, Acevedo C,
Arzimanoglou A, ve ark. ILAE official report: a practical clinical definition
of epilepsy. Epilepsia 2014;55(4):475-482.
8)
Fisher RS, Cross JH, D'Souza
C, ve ark. Instruction manual for the ILAE 2017 operational classification of
seizure types. Epilepsia 2017;58(4):531-542.
9)
Glauser
T, Ben-Menachem E, Bourgeois B, ve ark. ILAE
treatment guidelines: evidence-based analysis of antiepileptic drug efficacy
and effectiveness as initial monotherapy for epileptic seizures and syndromes.
Epilepsia 2006;47(7):1094-1120.
10)
Koepp MJ, Caciagli L,
Pressler RM, Lehnertz K, Beniczky S. Reflex seizures, traits, and epilepsies:
from physiology to pathology. Lancet Neurol 2016;15(1):92-105.
11)
Krumholz A, Wiebe S, Gronseth GS, ve ark.
Evidence-based guideline: Management of an unprovoked first seizure in adults:
Report of the Guideline Development Subcommittee of the American Academy of
Neurology and the American Epilepsy Society. Neurology 2015;84(16):1705-1713.
12)
Kwan P, Arzimanoglou A, Berg AT, ve ark.
Definition of drug resistant epilepsy: consensus proposal by the ad hoc Task
Force of the ILAE Commission on Therapeutic Strategies. Epilepsia
2010;51(6):1069-1077.
13)
Kwon CS, Ripa V, Al-Awar O, Panov F, Ghatan S,
Jette N. Epilepsy and Neuromodulation-Randomized Controlled Trials. Brain Sci
2018;8(4).
14)
LaFrance WC, Jr., Baker GA, Duncan R,
Goldstein LH, Reuber M. Minimum requirements for the diagnosis of psychogenic
nonepileptic seizures: a staged approach: a report from the International
League Against Epilepsy Nonepileptic Seizures Task Force. Epilepsia
2013;54(11):2005-2018.
15)
Lapalme-Remis S, Cascino GD. Imaging for
Adults With Seizures and Epilepsy. Continuum (Minneap Minn) 2016;22(5,
Neuroimaging):1451-1479.
16)
Luciano D. Partial seizures of frontal and
temporal origin. Neurol Clin 1993; 11: 805-822.
17)
Panayiotopoulos, CP. A clinical guide to
epileptic syndromes and their treatment. 2nd edition, New York: Springer, 2010.
18)
Papetti L, Parisi P, Leuzzi V, ve
ark. Metabolic epilepsy: An update. Brain
& Development 2013;35:827–841.
19)
Ottman R, Hirose S, Jain S,
ve ark. Genetic testing in the epilepsies--report of the ILAE Genetics
Commission. Epilepsia 2010;51(4):655-670.
20)
Scheffer IE, Berkovic S,
Capovilla G, ve ark. ILAE classification of the epilepsies: Position paper of
the ILAE Commission for Classification and Terminology. Epilepsia
2017;58(4):512-521.
21)
Shih JJ, Fountain NB, Herman ST, ve ark.
Indications and methodology for video-electroencephalographic studies in the
epilepsy monitoring unit. Epilepsia 2018;59(1):27-36.
22)
Shorvon SD. The causes of
epilepsy: changing concepts of etiology of epilepsy over the past 150 years.
Epilepsia 2011;52(6):1033-1044.
23)
Tatum
WO, Rubboli G, Kaplan PW, ve ark. Clinical utility of
EEG in diagnosing and monitoring epilepsy in adults. Clin Neurophysiol
2018;129(5):1056-1082.
24)
Westbrook GL, Seizures and Epilepsy. In:
Kandel ER, Schwartz JH, Jessell TM (eds). Principles of neural science.
McGraw-Hill Co. 2000: 910-934.
25)
Wheless JW, Gienapp AJ,
Ryvlin P. Vagus nerve stimulation (VNS) therapy update. Epilepsy Behav
2018;88S:2-10.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
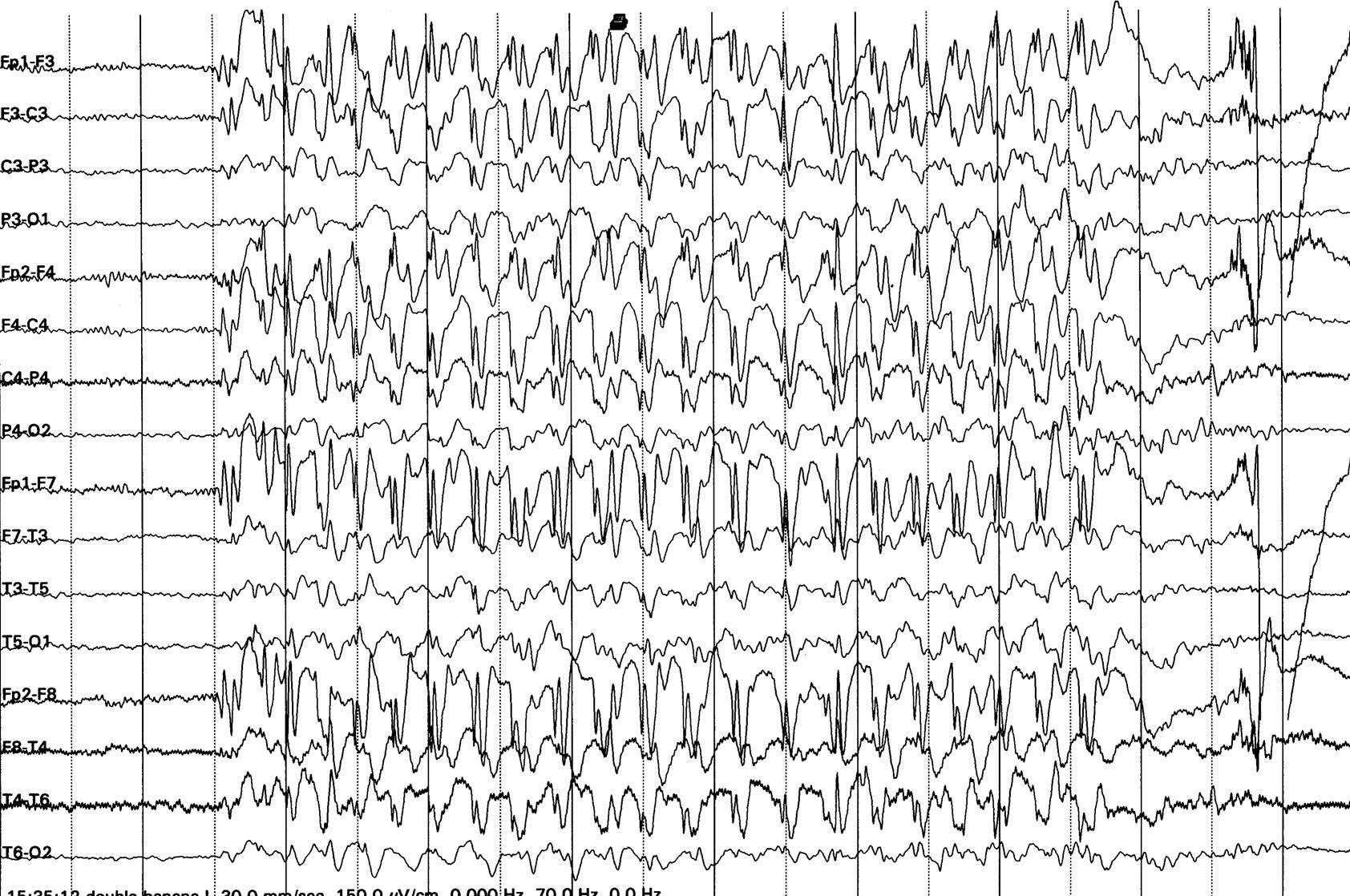{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
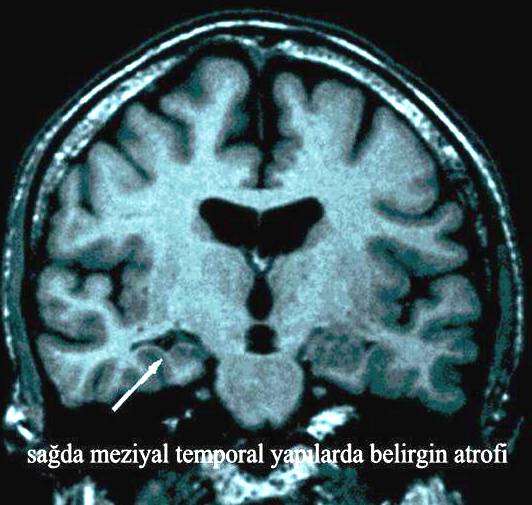{kind=link}
{kind=link}
{kind=link}
{kind=link}
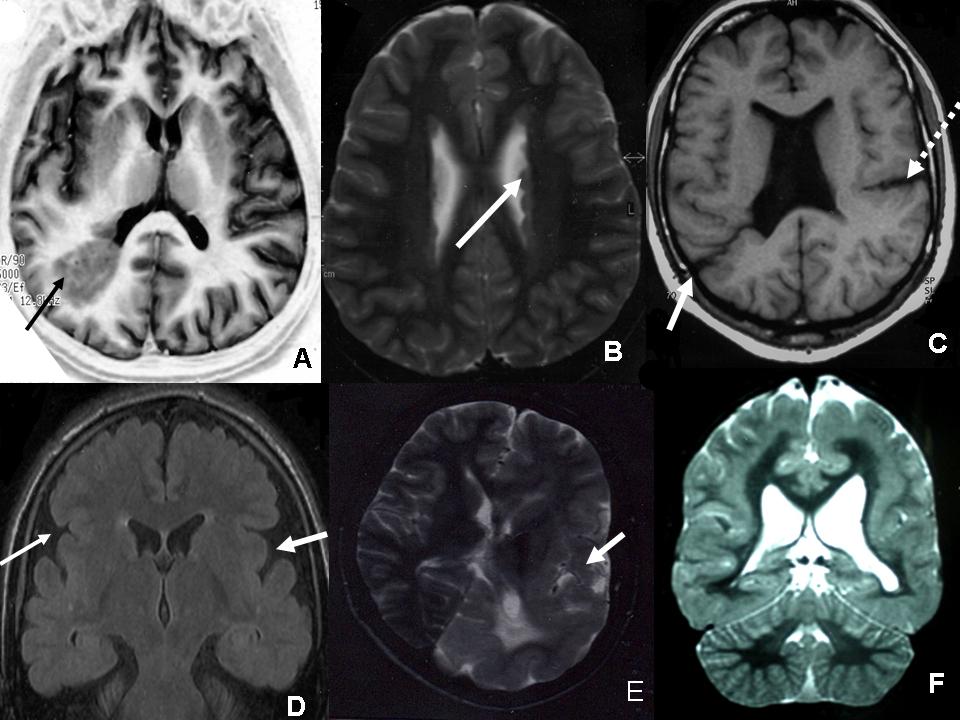{kind=link}
{kind=link}
{kind=link}
{kind=link}