PERİFERİK SİNİRLERİN YAYGIN VE ÇOK ODAKLI HASTALIKLARI
Yazanlar: Ayşe Nur Özdağ Acarlı, Arman Çakar, Yeşim Gülşen Parman, A. Emre Öge
Son güncelleştirme tarihi: 04.09.2020
Periferik nöropatilerin toplumdaki prevalansı %2 ile 3 arasındadır. İlerleyen yaşla birlikte bu oran artar ve 55 yaşın üzerinde %8’e, 65’in üzerinde ise %24’e kadar yükselir. Polinöropati gibi yaygın periferik nöropatilerin ortaya koydukları klinik bulguların çeşitliliği genellikle oldukça kısıtlıdır. Buna karşılık, bu tabloların nedenleri çok büyük çeşitlilik gösterir (Tablo 1). Bu nedenle, hastanın yaygın periferik nöropatisinin var olduğunu saptamak genellikle oldukça kolay, nedenini belirlemek ise daha zordur (eğer hastanın süregelen ağır diyabeti veya almakta olduğu nörotoksik ilaç gibi açıkça ortada bulunan bir neden yoksa). Klinik tablodaki küçük farklılıklar yaygın periferik nöropatide ayırıcı tanıya götüren ilk özelliklerdir.
Polinöropati, periferik sinirlerin aynı nedene ve fizyopatolojik süreçlere bağlı olarak hep birlikte, yaygın şekilde hastalanması ile ortaya çıkan bir klinik tablodur. Polinöropatileri oluşturan hastalık süreçleri ön planda hücre gövdesini etkiliyorsa bir nöronopati, başlıca akson hasarına neden oluyorsa bir aksonopati ve sinir liflerinin miyelin kılıfı primer olarak hasara uğruyorsa bir miyelinopati söz konusudur. En iyi nöronopati örneklerini spinal ön boynuz hücrelerini etkileyen ve sadece motor belirti ve bulgularla seyreden motor nöron hastalığı ile spinal arka kök gangliyonlarını haraplayarak başlıca duyusal belirtilere neden olan inflamatuvar duyusal poligangliyonopatiler (arka kök gangliyoniti) oluşturur. Birer polinöropati tablosu olmamakla birlikte, ilgili virüsün primer olarak etkilediği yerler, sırasıyla, spinal ön boynuz ve arka kök gangliyonu olan poliomiyelit ve varisella-zoster virüs infeksiyonları da nöronopati kavramı içinde ele alınabilir. Miyelin kılıfının ön planda hasara uğradığı başlıca polinöropatiler akut ve kronik inflamatuvar demiyelinizan polinöropatiler, herediter motor ve duyusal nöropatilerin bazı formları ve difteriye bağlı polinöropatidir. Primer aksonal hasarla seyreden polinöropatiler geniş bir liste oluşturur. Toksik, metabolik ya da nutrisyonel yetersizliğe bağlı polinöropatiler genellikle bu grupta yer alırlar.
Mononöropati multiplekste ise aynı hastalık süreci periferik sinirleri genellikle birbirinden ayrı zamanlarda yerleşen multipl odaklar halinde etkiler. Hastanın öyküsünden klinik tablonun zaman içinde farklı sinirlerin tutulmasıyla geliştiği anlaşılabilir, muayenede asimetrik multifokal periferik sinir harabiyetine ait bulgular saptanabilir. Buna karşılık, bazen multifokal lezyon odakları çok hızlı bir yayılma gösterir veya klinik belirti ve bulgular birbirleri ile birleşerek (konflüans) polinöropatiden ayrılamaz hale gelir. Mononöropati multipleks için en tipik örnekleri poliarteritis nodoza (PAN) ve eozinofilik granülomatöz polianjitis gibi vaskülitlere bağlı periferik nöropatiler oluşturur.
Polinöropatilerde klinik tanı, bir hastada polinöropati tablosunun varlığının gösterilmesi ve daha sonra bunun hangi nedene bağlı olduğunun ortaya konmasından ibarettir. Tablo 1’de görüldüğü gibi polinöropatiler çok değişik nedenlere bağlı olarak ortaya çıkabilir. Bir hastada temel olarak amaçlanan, tedavisi olanaklı bir polinöropatinin belirlenmesidir. Bu nedenle hastada polinöropati varlığı saptandıktan sonra klinik ve laboratuvar verilerine dayanılarak nedenin araştırılmasına yönelik bir analiz yapılması gerekir. Buradaki yöntem, klinik bulgular ve basit laboratuvar bulgularının ortaya koyduğu ipuçlarının yardımı ile geniş ayırıcı tanı listelerini daraltmak, sonra da bu daraltılmış ayırıcı tanı olasılıkları üzerinde daha derinlemesine laboratuvar araştırmaları yapmak şeklindedir. Nedeni bilinmeyen periferik nöropati ön tanısı ile özelleşmiş tıp merkezlerine gönderilen hastaların %76-87’sinde nedeni ortaya koyan bir tanıya varılabilmektedir. En sık rastlanan tanı grupları herediter, inflamatuvar demiyelinizan ve diğer hastalıklara (diyabet ve diğer metabolik hastalıklar, nutrisyonel yetersizlik, toksinler ve kanser) bağlı polinöropatilerdir.
Tablo 1. Başlıca periferik sinir hastalıkları (RJ Barohn ve AA Amato, değiştirilerek)
I. EDİNSEL
Metabolik bozukluklar
Diabetes mellitus
Böbrek hastalığına bağlı nöropatiler
Vitamin yetersizlikleri
İmmün bozukluğa bağlı
Guillain-Barré sendromu
Kronik inflamatuvar demiyelinizan polinöropati ve varyantları
Anti-MAG distal edinsel demiyelinizan simetrik nöropati
Multifokal motor nöropati
Radikülopleksus nöropatisi: servikal, torakal, lumbosakral
Vaskülit
Sarkoidoz
İnfeksiyona bağlı
Herpes zoster
Lepra, Lyme, HIV, CMV ve Epstein-Barr ile ilişkili
Kanser ve lenfoproliferatif hastalıklarla ilişkili
Lenfoma, miyeloma ve kanserle ilişkili
Paraneoplastik subakut duyusal nöronopati
Primer amiloidoz
İlaçlar veya toksinler
Kemoterapiye bağlı
Diğer ilaçlar
Ağır metaller ve endüstriyel toksinler
Mekanik/kompresif
Radikülopati
Mononöropati
Etyolojisi bilinmeyen
Kriptojenik duyusal ve duyusal-motor nöropati
Amiyotrofik lateral skleroz
II. HEREDİTER
Charcot-Marie-Tooth hastalığı ve ilişkili hastalıklar
Herediter duyusal-otonom nöropati
Ailesel brakiyal pleksopati
Ailesel amiloidoz
Porfiri
Diğer nadir periferik nöropatiler (Fabry hastalığı, metakromatik lökodistrofi, adrenolökodistrofi, Refsum hastalığı vb.)
Motor nöron hastalığı
Spinal müsküler atrofi
Ailesel amiyotrofik lateral skleroz
X-e bağlı bulbospinal müsküler atrofi
Herediter motor nöropati
Herediter spastik parapleji
Bu tabloda polinöropatilerin yanı sıra mononöropati, mononöropati multipleks ve spinal ön boynuz hastalıkları da yer almaktadır. Klinik uygulamada zaman zaman bu hastalık tablolarının birbirinden ayrılmasında güçlükle karşılaşılabilir.
MAG: miyeline bağlı glikoprotein, CMV: sitomegalovirüs, HIV: ”human immunodeficiency virus”.
POLİNÖROPATİLİ HASTADA TANI
Klinik Belirti ve Bulgular
Polinöropatilerde genellikle simetrik klinik bulgular ortaya çıkar. Polinöropatiyi ortaya çıkaran patolojik süreç yaygın aksonal dejenerasyon şeklinde de olsa, Guillain-Barré sendromunda (GBS) olduğu gibi multifokal segmental demiyelinizasyonla da seyretse polinöropatinin motor ve/veya duyusal belirti ve bulguları sıklıkla simetrik olarak yerleşir. Birçok polinöropati tablosunun altında yatan aksonal dejenerasyon süreci, periferik sinir aksonlarının distalden başlayarak proksimale doğru ilerleyen hasarı ile seyreder (“dying-back” nöropati veya distal aksonopati). Bunun nedeni hücre gövdesinin akson distali için gerekli proteinleri ya da enzimleri üretememesi veya aksonal transport sistemindeki bozukluk olabilir. Bu durumda periferik nöropati bulguları en uzun aksonların ulaştığı ekstremite distallerinden başlayarak ilerler. Polinöropatilerin bu tipik klinik görünümünde, duyusal belirtiler alt ekstremite distallerinden başlayan parestezi ve ağrılardan oluşur (eğer hastalık bu “pozitif” duyusal belirtilerle seyrediyorsa). Hastalık ilerledikçe yakınmalar alt ekstremitelerde proksimale doğru yayılırken –genellikle diz düzeyine kadar ulaştığında- üst ekstremite distallerinde de ortaya çıkar ve zaman içinde burada da proksimale doğru yayılır. Nörolojik muayenede ekstremite distallerinde eldiven ve çorap tarzında duyu kusuru saptanır. Kas kuvvetsizliği varsa öncelikle alt, daha sonra üst ekstremite distallerindedir. Özellikle ayak ekstensor kaslarındaki zaaf, yürürken ayağın yere takılması ya da topuklar üzerinde yürüme güçlüğü şeklinde ortaya çıkar. Tendon reflekslerinin azalması veya kaybolması da ekstremite distallerinden başlar. Aşil reflekslerinin kaybını zaman içinde diğer tendon reflekslerinin azalması izler. Klinik muayenede duyusal, motor ve refleks bozukluklarının birlikte, distal ve simetrik dağılımda olduğu bu tipik klinik görünüme birçok duyusal-motor polinöropati tablosunda rastlanır (diyabetik distal simetrik polinöropatide olduğu gibi) (Tablo 2).
Tablo 2. Distal simetrik polinöropatilerin sık rastlanan nedenleri (BC Callaghan ve ark., değiştirilerek)
|
Hastalık |
Yorum |
|
|
Metabolik |
|
|
|
|
Diyabet |
En sık neden, olguların %32-53’ü |
|
|
Prediyabet |
En duyarlı test glikoz tolerans testidir |
|
Kronik böbrek hastalığı |
Nedeni diyabetse nöropati özellikle şiddetlidir |
|
|
Kronik karaciğer hastalığı |
Nöropati genellikle hafiftir |
|
|
İdyopatik |
Tüm olguların %24-37’sidir |
|
|
Toksin (Alkol) |
İkinci en sık neden. Derinlemesine soruşturma gerektirir |
|
|
Kalıtsal |
|
|
|
|
CMT1 |
Kalıtsal demiyelinizan duyusal-motor polinöropati |
|
|
CMT2 |
Kalıtsal aksonal duyusal-motor polinöropati |
|
|
Ailesel amiloidoz |
Transtiretin mutasyonu en sıktır |
|
Nutrisyonel |
|
|
|
|
Vitamin B12 eksikliği |
200-500pg/mL arasındaysa metilmalonik asit düzeyi önemlidir |
|
|
Vitamin E eksikliği |
Serebellar ataksi nedeni olabilir |
|
|
Vitamin B6 eksikliği |
Düzeyi çok yüksek veya düşük olduğunda nöropatiye neden olabilir |
|
|
Tiamin eksikliği |
Ataksi, oftalmoparezi ve konfüzyonla ortaya çıkabilir |
|
|
Bakır yetersizliği |
Genellikle miyelonöropati şeklinde ortaya çıkar |
|
|
Gastrik “by-pass” cerrahisi |
Hangi faktörün sorumlu olduğunu belirlemek sıklıkla zordur |
|
|
Malabsorbsiyon sendromları |
Hangi faktörün sorumlu olduğunu belirlemek sıklıkla zordur |
|
İlaçlar |
|
|
|
|
Kemoterapi (vinkristin, sisplatin, taksol, bortezomib) |
|
|
|
Amiodaron |
Demiyelinizan nöropati yapabilir |
|
|
Fenitoin |
Genellikle yıllar boyu kullanımdan sonra |
|
|
Nükleosidler |
Nöropatinin nedenini ayırt etmek zor olabilir (HIV mi, tedavi mi?) |
|
|
Nitrofurantoin |
Böbrek yetersizliği varsa daha kötüdür |
|
|
Metronidazol |
Genellikle uzun süre, yüksek doz IV kullanımda |
|
|
Hidralazin |
Birlikte B6 kullanılarak sakınılır |
|
|
İzoniazid |
Birlikte B6 kullanılarak sakınılır |
|
|
Kolşisin |
Miyopatiye de neden olabilir |
|
Otoimmün |
|
|
|
|
Romatoid artrit |
Mononöropati multiplekse de sebep olabilir |
|
|
Lupus |
Mononöropati multiplekse ve CIDP benzeri polinöropatiye sebep olabilir |
|
|
Sjögren sendromu |
Duyusal nöronopati veya mononöropati multiplekse sebep olabilir |
|
|
Sarkoidoz |
Birçok nörolojik belirtiyle ortaya çıkabilir |
|
|
Sekonder amiloidoz |
Abdominal yağ ya da sural sinir biyopsisi tanıya yardımcı olur |
|
İnfeksiyonlar |
|
|
|
|
HIV |
Tedavisinde kullanılan ilaçlar da nöropatiye neden olabilir |
|
|
Hepatit B/C |
PAN ve kriyoglobulinemiyle birlikte mononöropati multiplekse neden olabilir |
|
Neoplastik |
|
|
|
|
MGUS |
İmmünofiksasyon paraproteini belirleme duyarlılığını arttırır |
|
|
Multipl miyelom |
IgG veya IgA paraproteinemisiyle birlikte olur |
|
|
Primer amiloidoz |
Yağ yastığı ya da sural sinir biyopsisi tanıya yardımcı olur |
|
CIDP: kronik inflamatuvar demiyelinizan polinöropati, CMT: Charcot-Marie-Tooth; HIV:”human immunodeficiency virus”, IgA: immünglobulin A, IgG: immünglobulin G, IV: intravenöz, MGUS: anlamı belirsiz monoklonal gamopati, PAN: poliarteritis nodoza. |
||
Bununla birlikte, hastanın öyküsü ve muayene bulgularında saptanan bazı ayrıntılar ya da kural dışılıklar nedene yönelik tanıda yardımcı olacak ipuçları sağlayabilir:
Motor, duyusal ve otonom sinir liflerinin tutulması: Bazı nöropatilerde motor, duyusal veya otonom fonksiyonlardan biri yalnız başına ya da diğerlerine oranla çok baskın şekilde bozulmuş olabilir. Sadece motor belirti ve bulgularla (kas kuvvetsizliği, atrofi, fasikülasyonlar) seyreden, duyusal bulguları olmayan bir periferik sinir sistemi hastalığı tablosunda motor nöron hastalığı ya da daha nadiren multifokal motor nöropati (MMN) en kuvvetli tanı olasılıklarıdır. Bazı periferik nöropatilerde ise motor belirti ve bulgular baskın olmakla birlikte muayenede genellikle daha hafif düzeyde duyusal bulgular da saptanır (Tablo 2).
Tablo 3. Ön planda motor belirtilerle seyreden nöronopati ve nöropatiler (RJ Barohn ve AA Amato, EP Bosch ve BE Smith, değiştirilerek)
Motor nöron hastalığı*
Multifokal motor nöropati*
Guillain-Barré Sendromu**
Akut motor aksonal nöropati*
Kronik inflamatuvar demiyelinizan polinöropati**
Osteosklerotik miyeloma**
Porfiriye bağlı nöropati**
Monomelik amiyotrofi (Hirayama sendromu)*
Diyabetik lumbosakral radikülopleksopati**
Kurşun intoksikasyonu**
Herediter motor-duyusal nöropati (Charcot-Marie-Tooth hastalığı)**
Herediter Motor Nöropati*
* Duyusal sinir iletimlerinin de normal ya da normale yakın olduğu saf motor sendromlar
**Muayenede genellikle duyusal bulgular da saptanır.
Bazı polinöropatilerde otonom sinir sistemi tutulmasına ilişkin bulgular belirgindir (Tablo 4). Ortostatik başdönmesi ve baygınlık, azalmış veya artmış terleme, sıcak intoleransı, mesane, barsak ve seks fonksiyonu bozuklukları hasta öyküsünde dikkatle araştırılmalı, gereğinde ortostatik hipotansiyon başta olmak üzere yatak başı otonom sinir sistemi muayenesi yöntemleri klinik muayeneye eklenmelidir. Diyabeti olmayan bir hastada otonom polinöropati belirti ve bulgularının olması ilk olarak amiloid nöropatisini ya da (çocuk ve genç yaştaki hastada) herediter duyusal-otonom nöropatileri (HSAN) akla getirmelidir. Nadiren idyopatik pandizotonomi sendromu, motor ve duyusal belirtileri olmayan bir periferik nöropatinin tek gösterisi olabilir.
Tablo 4. Otonom sinir sistemi tutulması ile seyreden polinöropatiler (BE Smith, değiştirilerek)
Akut
Akut pandizotonomik nöropati (idyopatik, otoimmün, paraneoplastik)
Guillain-Barré sendromu
Porfiri
Toksik: vinkristin, vacor
Kronik
Diabetes mellitus
Amiloid nöropatisi (ailesel ve edinsel)
Paraneoplastik duyusal nöronopati (malin inflamatuvar duyusal poligangliyonopati)
HIV’le ilişkili otonom nöropati
Herediter duyusal-otonom nöropati
HIV:”human immunodeficiency virus”.
Birçok duyusal veya duyusal-motor polinöropatide uyuşukluk, parestezi ve ağrı gibi yakınmalar ve muayenede ekstremite distallerinde belirgin duyu kusuru bulunur. Bunlar arasında diyabet, kanser, Sjögren sendromu, disproteinemiler, edinsel immün yetmezlik sendromu (acquired immunodeficiency syndrome=AIDS), B12 vitamini yetersizliği, sisplatin ve pridoksin intoksikasyonuna bağlı polinöropatiler ile, herediter ve idyopatik duyusal polinöropatiler sayılabilir. Bu derecede yaygın rastlanır olmaları, duyusal belirti ve bulgulardan ayırıcı tanıda yararlanmamızı güçleştirmektedir. Bununla birlikte, bazı duyusal özellikler ayırıcı tanı listesini daraltmakta işe yarayabilir. Bunlardan ilki polinöropatinin ağrılı olmasıdır. Ağrı, polinöropatili bir hastanın başlıca yakınmasını oluşturuyorsa Tablo 5’de görülen hastalıkları ön planda göz önünde bulundurmak gerekir.
Tablo 5. Ağrılı polinöropatiler (RJ Barohn, değiştirilerek)
Kriptojenik duyusal veya duyusal-motor polinöropati
Diabetes mellitus
Vaskülitler
Guillain-Barré sendromu
Amiloidoz
Toksik (arsenik, talyum)
HIV’le ilişkili distal simetrik polinöropati
Fabry hastalığı
HIV: ”human immunodeficiency virus”.
Kriptojenik duyusal polinöropati ve diyabete bağlı distal simetrik polinöropati, ağrılı nöropatiler arasında en sık rastlananlardır. Hastalarda alt ekstremite distallerinde (tabanlar ve ayak parmakları) baskın, istirahatte ve geceleri belirginleşen ağrılar vardır. Diyabette görülen bir diğer ağrılı nöropati şekli diyabetik lumbosakral radikülopleksopatidir (diyabetik amiyotrofi, proksimal asimetrik nöropati). Diyabetik mikroanjiyopati nedeniyle ortaya çıktığı düşünülen bu tabloda, genellikle bir alt ekstremitenin proksimalinde belirgin akut başlangıçlı ağrıyı günler sonra aynı ekstremitede kuvvetsizlik ve atrofi izler. Vaskülitlere bağlı nöropatilerde periferik sinirlerin tutulduğu ekstremitelerde daha çok distal ve asimetrik dağılımlı bir ağrı vardır. GBS’de ekstremitelerdeki simetrik uyuşma ve parestezilere ek olarak şiddetli sırt ve bel ağrısı olabilir.
Ayırıcı tanı listesini daraltabilecek diğer bir duyusal bozukluk, şiddetli propriyoseptif duyu kaybıdır. Bu durumda hastanın anamnezinde özellikle karanlıkta belirginleşen ağır denge bozukluğu ve ekstremitelerde inkoordinasyon, nörolojik muayenede ise belirgin vibrasyon ve pozisyon duyusu kaybı (ve bazen buna bağlı olarak ellerde psödoatetoz) vardır. Diğer duyu modalitelerinde daha geri planda bir azalma olabilir. Bu klinik tablo ön planda arka kök gangliyonlarını etkileyen bir duyusal nöronopatiyi düşündürmelidir (Tablo 6).
Derin duyu bozukluğu, subakut kombine dejenerasyonda olduğu gibi, medulla spinalis arka kordonlarının tutulmasını da gösteren bir bulgudur. Ancak, arka kordon tutulmasındaki derin duyu bozuklukları genellikle duyusal gangliyonopatilerde görülenlere oranla daha az belirgindir. Duyusal nöronopatilerde klinik bulgular asimetrik olma eğiliminde iken arka kordon tutulmasında daha simetriktir. Arka kordon lezyonlarında sık olarak piramidal traktus tutulmasına bağlı bulgular da görülürken, duyusal nöronopatilerde bu bulgulara genellikle rastlanmaz.
Tablo 6. Duyusal-ataksik nöropatiler ve nöronopatiler (BE Smith ve A Pestronk, değiştirilerek)
|
Duyusal nöronopatiler (poligangliyonopatiler) |
||
|
|
Paraneoplastik duyusal nöronopati |
|
|
|
Anti-Hu sendromu |
|
|
|
Sjögren sendromu |
|
|
|
İdyopatik |
|
|
Toksik polinöropatiler |
||
|
|
Sisplatin ve analogları |
|
|
|
Taksol ve diğer taksanlar |
|
|
|
Bortezomib |
|
|
|
Vitamin B6 megadozu |
|
|
|
Semisentetik penisilinler |
|
|
Yetersizlik/malabsorbsiyon |
||
|
|
Vitamin B12 eksikliği |
|
|
|
Vitamin E eksikliği |
|
|
İnflamatuvar demiyelinizan |
||
|
|
Guillain-Barré sendromu (Miller Fisher varyantı) |
|
|
|
IgM monoklonal gamopatisi |
|
|
|
Anti-MAG sendromu |
|
|
|
Anti-GD1b sendromu |
|
|
|
Kronik immün duyusal poliradikülopati |
|
|
İnfeksiyon |
||
|
|
Tabes dorsalis |
|
|
|
HIV’le ilişkili duyusal nöronopati |
|
|
Herediter |
||
|
|
Resesif sendromlar |
|
|
|
|
a-β-lipoproteinemi (Bassen-Kornzweig) |
|
|
|
Ataksi-Telanjiektazi |
|
|
|
Friedreich ataksisi |
|
|
|
|
|
|
|
ARSACS sendromu |
|
|
Dominant sendromlar |
|
|
|
|
Duyusal ataksik nöropati |
|
|
|
Duyusal ataksik nöropati2: 16q22 |
|
|
|
Duyusal ataksi (SNAX1) |
|
|
|
|
|
|
|
|
|
|
|
|
|
ARSACS: Charlevoix-Saguenay otozomal resesif spastik ataksi, HIV: ”human immunodeficiency virüs”, IgM: immünglobulin M, MAG: miyeline bağlı glikoprotein, SCA: spinoserebellar ataksi, SNAX1: “sensory ataxia 1”. |
||
Daha genel bir yaklaşımla, belirli duyu modalitelerinin ön planda tutulmasına dayanarak polinöropatileri kalın lif ve ince lif nöropatileri şeklinde ayırıp tanı listesi daraltılabilir. Periferik sinirlerde vibrasyon, pozisyon duyusu gibi derin duyular ve tendon refleks yayının aferent bölümü kalın miyelinli liflerle, buna karşılık ağrı ve ısı duyusu miyelinsiz ve ince miyelinli liflerle taşınır. İnce dokunma duyusu ise kalın ve ince miyelinli liflerce merkez sinir sistemine götürülür. Kalın miyelinli liflerin seçici olarak tutulduğu klinik tablolarda yaygın arefleksi, duyusal ataksi, psödoatetoz, vibrasyon ve pozisyon duyusu kaybı olur. Bu hastalarda duyusal ataksinin en göze çarpan klinik gösterisi Romberg belirtisidir. Hemen dikkati çekeceği gibi, sayılan klinik özellikler duyusal ataksik nöropatileri işaret etmekte ve duyusal gangliyonopatiler ön planda olmak üzere Tablo 6’da gösterilen hastalıkların düşünülmesini gerektirmektedir. İnce sinir liflerinin ön planda tutulduğu nöropatilerde ise (Tablo 7) altta belirgin olmak üzere ekstremite uçlarında ağrı ve ısı duyusu kaybı, ağrılı paresteziler ve otonom işlev bozuklukları görülür. Özellikle hastalığın erken dönemlerinde diğer duyu modaliteleri, motor işlevler ve denge korunmuştur; tendon refleksleri de korunmuş olabilir
Periferik nöropatilerin gösterilmesi ve ayırıcı tanısında yaygın olarak kullanılan sinir iletim incelemeleri, ön planda kalın miyelinli sinirlerin işlevine dayandığından ince lif nöropatilerinde sıklıkla normal bulunur. Bu nedenle, ince sinir liflerinin seçici olarak tutulduğu nöropatilerde objektif verilerin elde edilmesi güçtür. Gereğinde kantitatif duyusal test, otonom sinir sistemine yönelik yatak başı testleri ve elektrofizyolojik incelemelerden, sinir ve deri biyopsilerinden yararlanılabilir.
İnce sinir liflerinin seçici olarak tutulduğu polinöropatilerin başlıcaları amiloid nöropatisi, HSAN, kriptojenik duyusal polinöropati ve Fabry hastalığıdır. Bazı erken dönem diyabetik distal simetrik nöropati olgularında benzer özelliklere rastlanabilir. Polinöropatileri kalın ve ince lif nöropatisi tarzında ayırmak bir ölçüde yapaylık taşır. Birçok polinöropatide, özellikle hastalık progresyon gösterdikçe ilk tutulan duyu modalitesine diğerlerinin katıldığı görülür. Örneğin diyabetik distal simetrik polinöropatisi olan bir hastada dikkatli bir klinik muayene ile ağrı ve ısı duyusu kaybına dokunma ve derin duyu bozukluğunun eklendiği ortaya konabilir.
Tablo 7. İnce lif nöropatileri (BE Smith ve A Pestronk, değiştirilerek)
İdyopatik
Yanan ağız sendromu
Bozulmuş glikoz toleransı nöropatisi
Diabetes mellitus (nadir)
Amiloid nöropatisi (ailesel ve primer)
Lepra
Herediter duyusal-otonom nöropatiler
Fabry hastalığı
Tangier hastalığı
Ciguatera intoksikasyonu
Belirti ve bulguların dağılımı: Periferik nöropatilerde ortaya çıkan belirti ve bulguların vücut üzerindeki dağılım şekli ayırıcı tanıya yardımcı olabilir. Polinöropatilerde motor ve duyusal belirtiler genellikle ekstremite distallerinde belirgin olan simetrik bir yerleşim göstermekle birlikte, bu kuralın dışında kalan ve bu özellikleri ile tanıya yardımcı olan durumlar vardır. Kas kuvvetsizliği ekstremite proksimal kaslarında hakimse ya da distal kaslara ek olarak bu kas gruplarını da tutuyorsa ön planda inflamatuvar demiyelinizan polinöropatileri [GBS ve kronik inflamatuvar demiyelinizan polinöropati (chronic inflammatory demyelinating polyneuropathy=CIDP)] düşünmek gerekir.
Distal simetrik polinöropatilerin uzunluğa bağımlı özelliği nedeniyle, belirti ve bulgular alt ekstremite distallerinde daha önce başlar ve daha baskındır. Bu kuralın dışına çıkarak üst ekstremitelerden başlayan nöropatiler arasında kurşun nöropatisi (‘düşük el’ tablosu ile başlangıç), MMN, GBS’nin ve Charcot-Marie-Tooth hastalığının bazı varyantları, familyal amiloid nöropati tip 2, nadir olarak porfiri ve erişkin başlangıçlı Tangier hastalığı sayılabilir.
Belirti ve bulguların asimetrik veya fokal dağılımı da ayırıcı tanı listesini daraltmada yardımcı olur. Kas kuvvetsizliği ve atrofinin duyusal belirtiler olmaksızın bir ekstremitede baskın bir asimetriyle ortaya çıkması motor nöron hastalığını ya da MMN’yi akla getirir. Buna karşılık, duyusal ve motor belirtilerin multifokal ve asimetrik şekilde yerleştiği bir klinik tablo, vaskülitler başta olmak üzere mononöropati multipleks nedenlerinin araştırılmasını gerektirir. Tablo 8’de fokal ve asimetrik nörolojik bulgulara neden olan nöropatiler görülmektedir. Genel ayırıcı tanı yaklaşımını göstermek amacıyla bu tabloya asimetrik nörolojik tutuluma neden olan tüm periferik sinir hastalıkları (polinöropati, mononöropati multipleks ve mononöropatiler) alınmıştır.
Polinöropati tablosuna eklenen kranial sinir tutulmaları da dikkatle değerlendirilmelidir. Tablo 9’da kranial sinir tutulması görülen nöropatiler verilmiştir.
Tablo 8. Fokal/asimetrik bulgulara yol açan nöropatiler (RJ Barohn ve AA Amato, değiştirilerek)
Motor nöron hastalığı
Amiyotrofik lateral skleroz
Radikülopati- servikal veya lumbosakral
Radiks kompresyonu-disk fıtıklanması veya osteoartropatiye bağlı
Herpes zoster infeksiyonu (zona)
Meningeal karsinomatoz ve lenfomatoz
Sarkoidoz
Amiloid
Kronik immün duyusal poliradikülopati
Pleksopati – servikal, torakal, lumbosakral
İmmün kökenli/idyopatik
Neoplastik infiltrasyon
Diyabetik radikülopleksopati (Başlıca lumbosakral)
Ailesel brakiyal pleksopati
Herediter basınca duyarlılık nöropatisi
Mononöropati multipleks
Vaskülit
MMN
MADSAM
MAMA
Lyme hastalığı
Sarkoidoz
Lepra
HIV infeksiyonu
Hepatit B ve C
Amiloidoz
Herediter basınca duyarlılık nöropatisi
Kriyoglobulinemi
Kompresyon/tuzak nöropatileri
HIV: ”human immunodeficiency virüs”, MADSAM: multifokal edinsel demiyelinizan duyusal ve motor nöropati, MAMA: multifokal edinsel motor aksonopati, MMN: multifokal motor nöropati
|
Tablo 9. Kranial sinir tutulması görülen nöropatiler (A Pestronk, sadeleştirilerek) |
|||
|
II. Sinir |
|||
|
|
|||
|
|
Cıva |
||
|
|
Mitokondriyal: NARP sendromu |
||
|
|
|||
|
III, IV, VI Sinir |
|||
|
|
Botulizm (önce pupillalar tutulur) |
||
|
|
GD1b antikoru ile birlikte olan duyusal ataksik nöropati (CANOMAD) |
||
|
|
Diyabet (pupillanın kurtulduğu III. sinir felci) |
||
|
|
Miller Fisher sendromu |
||
|
V. Sinir |
|||
|
|
|||
|
|
Trigeminal duyusal nöropati |
||
|
|
|||
|
|
Sjögren sendromu |
||
|
|
|||
|
VII. Sinir |
|||
|
|
Bell felci |
||
|
|
Bell felci |
||
|
|
Poliradikülopatiler |
||
|
|
|
Sarkoidoz |
|
|
|
|
Akut immün nöropatiler |
|
|
|
|
|
Guillain Barré sendromu |
|
|
|
|
Fasiyal dipleji |
|
|
|
Lyme hastalığı |
|
|
|
|
Amiloid: Gelsolin |
|
|
|
|
||
|
|
|
Lepra |
|
|
|
Motor nöron sendromları |
||
|
|
|
||
|
|
|
Bulbospinal müsküler atrofi (Kennedy Hastalığı) |
|
|
VIII. Sinir |
|||
|
|
Sarkoidoz |
||
|
|
CMT |
||
|
IX. ve X. Sinir |
|||
|
|
Difteri |
||
|
|
Sarkoidoz |
||
|
|
Poliomiyelit |
||
|
|
Motor nöron hastalıkları |
||
|
|
Glossofaringeal nevralji |
||
|
XI. Sinir |
|||
|
|
Radyasyon |
||
|
|
Poliomiyelit |
||
|
|
Motor nöron hastalıkları |
||
|
XII. Sinir |
|||
|
|
Motor nöron hastalıkları |
||
|
|
|||
|
|
CMT 4D: Dil atrofisi |
||
ALS: amiyotrofik lateral skleroz, CANOMAD: kronik ataksik nöropati, oftalmopleji, M-protein, soğuk aglütininler ve anti-disialosil antikorlar, CMT: Charcot-Marie-Tooth, FOSMN: yüzden başlayan duyusal ve motor nöronopati, NARP: Nöropati, Ataksi, Retinitis Pigmentoza.
Üst motor nöron tutulması bulguları: Duyusal belirti ve bulguları olmayan bir hastada zaaf, atrofi ve fasikülasyon gibi alt motor nöron tutulması bulgularına üst motor nöron bulgularının eklenmesi kuvvetle motor nöron hastalığını düşündürür (Bakınız: Motor Nöron Hastalıkları).
Üst ekstremitelerde (genellikle asimetrik) alt motor nöron tutulması bulguları ile birlikte alt ekstremitelerde belirgin piramidal bulgular, spondilotik miyelopati başta olmak üzere servikal spinal lezyonlarda da görülür. Bu hastalarda genellikle servikal radikülopatileri işaret eden duyusal yakınma ve bulgular vardır. Ancak bazı hastalarda klinik verilerle motor nöron hastalığı ve servikal spondilotik miyelopatiyi ayırt etmek mümkün olmaz. Böyle olgularda servikal spinal görüntüleme [tercihen manyetik rezonans görüntüleme (MRG)] ve elektromiyografi bulguları birlikte değerlendirilerek ayırıcı tanı yapılmaya çalışılır. (Bakınız: Omurga ve Omurilik Hastalıkları).
Üst motor nöron tutulması bulguları duyusal belirtileri ön planda olan bir distal simetrik polinöropatiye ekleniyorsa, polinöropati ile birlikte olan bir kombine sistem hastalığını düşünmek gerekir (Bakınız: Omurga ve Omurilik Hastalıkları). Bunun en önde gelen nedeni B12 vitamini yetersizliğidir. Benzer bir tabloya bakır eksikliği de neden olduğundan, özellikle bariatrik cerrahi geçirenler, çoklu beslenme yetersizliği olanlar ve yüksek doz çinkoya maruz kalmış kişilerde (beslenme takviyesi veya bazı diş yapıştırıcıları yoluyla) serum bakır ve çinko düzeyinin araştırılması gerekir. Gereğinde aynı klinik tabloya neden olan “human immunodeficiency virüs” (HIV) infeksiyonu, karaciğer hastalığı, adrenomiyelonöropati gibi diğer nadir nedenler de araştırılır.
Klinik tablonun yerleşim şekli: Belirti ve bulguların başlangıç şekli, gelişme hızı ve seyri ile ilgili özellikler ayırıcı tanı listesini daraltmakta çok yardımcı olur. Nörolojik tablonun akut (4 haftadan az), subakut (4-8 hafta) ya da kronik şekildeki (2 aydan uzun) yerleşimi; monofazik, progresif veya tekrarlayıcı seyri hakkında bilgi edinilmeye çalışılmalıdır. GBS, porfiriye bağlı nöropati, vaskülitler, bazı toksik polinöropatiler, diyabetik lumbosakral radikülopleksopatiler akut-subakut seyir gösterirler. Tekrarlayıcı klinik seyir CIDP, akut porfiri, Refsum hastalığı, herediter basınca duyarlılık nöropatisi (HNPP), ailesel brakiyal pleksus nöropatisi ve bazı toksinlere aralıklı maruz kalma hallerinde görülür. Yavaş kronik progresyonla seyir, herediter nöropatilerde ve birçok metabolik, toksik ya da nutrisyonel yetersizliğe bağlı polinöropatide görülebilir.
Herediter nöropatiyi düşündüren veriler: Herediter nöropatiler, polinöropatiler içinde en sık rastlanan gruplardan birini oluşturur. Yıllar içinde çok yavaş bir seyirle ilerlemeleri, batma, iğnelenme, karıncalanma gibi pozitif duyusal yakınmaların çoğu kez bulunmaması nedeni ile hastalar ve özellikle onların hafif belirtili akrabaları tarafından uzun süre fark edilmezler. Bu nedenle hastaların aile fertleri ve akrabalarında benzer bir hastalığın bulunmadığına ilişkin ifadeleri yanıltıcı olabilir. Hastanın yanı sıra ulaşılabilen akrabalarının nörolojik muayenesi, bu kişilerde çukur ayak, çekiç parmak deformitelerinin araştırılması ve sinir iletim incelemelerinin yapılması bir herediter nöropatinin tanınmasını sağlayabilir. Tekrarlayan kompresyon mononöropatileri olan hastalarda HNPP akla getirilmelidir.
Öykü ve muayeneye ilişkin diğer özellikler: Birçok polinöropati sistemik hastalıklara bağlı olarak ortaya çıkar. Hastanın öyküsündeki halsizlik, iştahsızlık, kilo kaybı gibi özellikler böyle bir hastalığa dikkati çekebilir. Hastanın özgeçmişinde diyabet, hipotiroidizm, kronik böbrek yetersizliği, karaciğer hastalığı, intestinal malabsorbsiyon, habis hastalıklar, konnektif doku hastalıkları, HIV seropozitifliği, ilaç kullanımı, alkol ve beslenme alışkanlıkları, toksik maddelere maruz kalma, geçirilmiş infeksiyonlar gibi özellikler üzerinde dikkatle durulmalıdır.
Nörolojik muayenenin yanı sıra yapılacak dikkatli bir sistemik muayene ile polinöropatiye yol açan hastalık hakkında ipuçları elde edilebilir. Sinirlerin palpe edilmesi bazen tanıya yararlı veriler sağlayabilir. Tek bir sinirin hipertrofisi neoplastik bir oluşumu (nörofibrom, malign sinir kılıfı tümörü vb.) veya lokalize hipertrofik nöropatiyi düşündürür. Genel veya multifokal sinir hipertrofisi lepra, Tip I ve III nörofibromatoz, Charcot-Marie-Tooth hastalığı (CMT), akromegali, Refsum hastalığı ve kronik seyirli edinsel demiyelinizan polinöropatilerde [CIDP ve multifokal edinsel demiyelinizan duyusal ve motor nöropati (multifocal acquired demyelinating sensory and motor=MADSAM)] görülebilir. Polinöropatili bir hastada görülebilen ayak yaraları ve trofik deri değişikliklerinin yanısıra bulunabilecek bazı deri belirtileri tanı koydurucu olabilir (Tablo 8). Talyum zehirlenmesinde ortaya çıkan alopesi, arsenik ve talyum zehirlenmesinde görülen transvers tırnak çizgileri (Mees çizgileri), Fabry hastalığında abdomen ve kalçalarda görülen telanjiektaziler, kriyoglobulinemilerdeki purpurik deri döküntüleri, Polinöropati, Organomegali, Endokrinopati, M proteini, Deri (Skin) değişiklikleri (POEMS) sendromundaki deri hiperpigmentasyonu ve hipertrikoz bunlara örnektir (Tablo 10).
Tablo 10. Deri, tırnak ve saç belirtileri olan nöropatiler (BE Smith, değiştirilerek)
|
Hastalık |
Belirtiler |
|
Vaskülit |
Purpura, livedo retikülaris |
|
Kriyoglobulinemi |
Purpura |
|
Fabry hastalığı |
Anjiokeratoz |
|
Lepra |
Deride hipopigmentasyon |
|
Osteosklerotik miyelom (POEMS sendromu) |
Deride hiperpigmentasyon |
|
Porphyria variegata |
Büllü lezyonlar |
|
Refsum hastalığı |
İktiyoz |
|
Arsenik ve talyum zehirlenmesi |
Mees çizgileri |
|
Talyum zehirlenmesi |
Kellik |
|
Dev aksonal nöropati |
Sık, kıvırcık saç |
POEMS: Polinöropati, Organomegali, Endokrinopati, M proteini, Deri (Skin) değişiklikleri.
Yukarıda söz edilen klinik özelliklere dayanılarak periferik nöropatilerin başlıca klinik sunum şekilleri Tablo 11’de özetlenmiştir. Çok ve farklı nedenlere bağlı olarak ortaya çıktığını bildiğimiz nöropatilerde klinik gösterilerin birkaç maddede ele alınması aşırı bir basitleştirme gibi görünse de, böyle bir tablo ayırıcı tanıya yaklaşım için yararlı olacaktır.
Tablo 11. Nöropatilerde 10 tutulma şekli (RJ Barohn, değiştirilerek)
Şekil 1: Duyu kaybı ile birlikte simetrik proksimal ve distal kuvvetsizlik
Düşünülmesi gereken: İnflamatuvar demiyelinizan polinöropati [Guillain-Barré sendromu (GBS) ve kronik inflamatuvar demiyelinizan polinöropati (CIDP)]
Şekil 2: Kuvvetsizliğin bulunduğu veya bulunmadığı simetrik distal duyu kaybı
Düşünülmesi gereken: Kriptojenik duyusal polinöropati, metabolik hastalıklar, ilaçlar, toksinler, herediter [Charcot-Marie-Tooth (CMT), amiloidoz vb.]. Bakınız: Tablo 2
Şekil 3: Duyu kaybı ile birlikte asimetrik distal kuvvetsizlik
Multipl sinir, düşünülmesi gereken: Vaskülit, herediter basınca duyarlılık nöropatisi (HNPP), multifokal edinsel demiyelinizan duyusal ve motor nöropati (MADSAM), infeksiyöz [lepra, lyme, sarkoidoz, “human immunodeficiency virus” (HIV)]
Tek sinir veya tek bölge, düşünülmesi gereken: sinir kompresyonuna bağlı mononöropati veya radikülopati
Şekil 4: Duyu kaybı ile birlikte asimetrik proksimal ve distal kuvvetsizlik
Düşünülmesi gereken: Karsinomatozis veya lenfomatozise bağlı, idyopatik veya herediter [herediter basınca duyarlılık nöropatisi (HNPP), familyal] poliradikülopati veya pleksopati.
Şekil 5: Duyu kaybı olmadan asimetrik distal kuvvetsizlik.
Düşünülmesi gereken:
Üst motor nöron bulguları varsa: motor nöron hastalığı
Üst motor nöron bulguları yoksa:
1. İlerleyici müsküler atrofi (PMA)
a. Brakiyal amiyotrofik dipleji
b. Bacak amiyotrofik diplejisi
2. Multifokal motor nöropati (MMN)
3. Multifokal edinsel motor aksonopati (MAMA)
4. Juvenil motor amiyotrofi
Şekil 6: Üst motor nöron tutulması bulguları ile birlikte simetrik distal duyu kaybı ve arefleksi
Düşünülmesi gereken:
B12 yetersizliği ve diğer kombine sistem dejenerasyonu nedenleri.
Herediter hastalıklar [amiyotrofik lateral skleroz (ALS), metakromatik lökodistrofi (MLD), Friedreich]
Şekil 7: Duyu kaybı olmaksızın simetrik kuvvetsizlik
Düşünülmesi gereken:
A. Proksimal ve distal kuvvetsizlik: Spinal müsküler atrofi
B. Distal kuvvetsizlik: Herediter motor nöropati
Şekil 8. Orta hatta fokal proksimal simetrik kuvvetsizlik
Düşünülmesi gereken:
Boyun ekstensor kaslarında kuvvetsizlik: amiyotrofik lateral skleroz (ALS), primer lateral skleroz (PLS)
Bulber zaaf: ALS
Şekil 9: Kuvvetsizlik olmaksızın asimetrik propriyoseptif duyu kaybı
Düşünülmesi gereken: Duyusal nöronopati (gangliyonopatiler), kronik immün duyusal poliradikülopati.
Şekil 10: Otonom belirti ve bulgular
Düşünülmesi gereken: Otonom fonksiyon bozukluğu ile birlikte olan nöropatiler
Polinöropatilerde Laboratuvar İncelemeleri
Elektrofizyolojik testler
Sinir iletim incelemeleri ve iğne elektromiyografisi polinöropatilerin tanısında çoğu kere vazgeçilemeyecek bir yere sahiptir. Günümüzde sağlık kuruluşlarında yaygın olarak bulunan cihazlarla yapılan bu testler, hafif bir polinöropatinin objektif olarak ortaya konmasını ya da bir hastada ortaya çıkan kas zaafının, polinöropatilerin de aralarında bulunduğu hangi nöromüsküler hastalık grubuna bağlı olduğunun belirlenmesini sağlarlar. Bu testler yardımı ile aksonal ve demiyelinizan nöropatiler arasında ayrım yapmak ve böylece ayırıcı tanı listesini önemli ölçüde daraltmak mümkün olur (Bakınız: Sinir iletim incelemeleri ve elektromiyografi, Periferik sinirlerin anatomi, fizyoloji ve patolojik süreçleri).
Aksonal polinöropatilerde sinir iletim hızları normal veya hafif yavaş, duyusal ve bileşik kas aksiyon potansiyeli amplitüdleri düşüktür. Demiyelinizan polinöropatilerde ise (Tablo 12) sinir iletim hızları belirgin derecede yavaşlamıştır (normal alt sınırının %70’inden daha yavaş). Sinir iletim incelemeleri herediter ve edinsel demiyelinizan polinöropatilerin birbirinden ayrılmasında da yardımcı olur. Herediter polinöropatilerde sinir iletimlerinde homojen bir yavaşlama izlenirken, edinsel demiyelinizan polinöropatilerde (GBS, CIDP gibi) sinir iletim hızları aynı ekstremite segmentindeki farklı sinirler, aynı sinirin farklı segmentleri ve karşılıklı iki ekstremitenin aynı isimli sinirleri arasında belirgin değişiklik gösterir. Edinsel demiyelinizan nöropatilerde sinirler üzerinde iletim blokları gösterilebilir ya da GBS’nin erken döneminde olduğu gibi F yanıtlarının kaybolması, latanslarının uzayıp persistanslarının azalması başlıca elektrofizyolojik bulguyu oluşturabilir. İyi planlanarak yapılan elektrofizyolojik testler, klinik muayene ile saptanamayan asimetrik ve fokal sinir lezyonlarının ortaya konmasına (ve Tablo 7 de yer alan nöropatilerin belirlenmesine) yardımcı olabilir.
Tablo 12. Başlıca demiyelinizan polinöropatiler (A Pestronk, CH Chalk, RAC Hughes, değiştirilerek)
|
İmmün Kökenli |
|
Akut |
|
GBS ve varyantları |
|
Kronik |
|
CIDP |
|
Simetrik |
|
Multifokal (MADSAM) |
|
Multifokal motor nöropati |
|
Paraproteinemik demiyelinizan polinöropatiler |
|
DADS (M proteini ile birlikte olan ve olmayan) |
|
POEMS sendromu |
|
Herediter |
|
Charcot-Marie-Tooth hastalığı, Tip 1 ve diğer demiyelinizan formlar |
|
Herediter basınca duyarlılık nöropatisi |
|
Diğer genetik nedenler (Refsum hastalığı, metakromatik lökodistrofi gibi) |
|
İnfeksiyon |
|
Difteri |
|
HIV: Birlikte olan CIDP |
|
Lepra: (Lepromatöz: karışık aksonal, demiyelinizan) |
|
Toksinler |
|
Perheksilen toksisitesi |
|
Heksakarbonlar (Sinir iletim incelemelerinde demiyelinizan özellikler) |
|
Akdiken (Buckthorn) meyvesi zehirlenmesi |
|
Heksaklorofen |
|
Sodyum siyanat |
|
İlaçlar |
|
Ön planda demiyelinizan |
|
Karışık (aksonal ve demiyelinizan) |
|
Eozinofili-miyalji sendromu |
|
Altın |
|
Suramin |
|
Taksol |
|
MNGIE Sendromu |
CIDP: kronik inflamatuvar demiyelinizan poliradikülonöropati, DADS: distal edinsel demiyelinizan simetrik nöropati, GBS: Guillain-Barré sendromu, HIV: “human immunodeficiency virus”, MADSAM: multifokal edinsel demiyelinizan duyusal ve motor nöropati, MNGIE Sendromu: Miyopati ve eksternal oftalmopleji, Nöropati:Gastroİntestinal, Ensefalopati, NARP: Nöropati, Ataksi, Retinitis Pigmentoza, POEMS: Polinöropati, Organomegali, Endokrinopati, M proteini, Deri (Skin) değişiklikleri.
Sinir biyopsisi
Periferik sinir biyopsileri oldukça az sayıdaki nöropati nedeninin tanısı için gereklidir. Sinir biyopsisinden bilgi verici sonuçlar alınabilmesi için gönderilen materyalin bu konuda uzmanlaşmış, standart parafin ve dondurulmuş kesitlerin yanında ışık ve elektron mikroskopisi için preparatlar hazırlayabilen ve sinir lifi ayırma (“teasing”) yöntemini kullanabilen laboratuvarlarda incelenmesi gerekir. Bu nedenle, bazen komplikasyonlara (yara kapanma güçlükleri, biyopsi yerinde infeksiyon, sinir güdüğü nöromaları, biyopsi yapılan sinirin innervasyon alanında huzursuz edici duyusal bozukluklar gibi) yol açabilen sinir biyopsilerinin polinöropati düşünülen her hastaya yapılmaması, klinik bulgular ve diğer laboratuvar incelemeleri ile tanı konamayan ciddi ve ilerleyici nöropatiler için düşünülmesi gerekir. Sinir biyopsisinin tanısal değer taşıdığı durumların başında vaskülite bağlı nöropatiler ve amiloid nöropatileri gelir. Biyopsi ile yararlı bilgiler elde edilebilecek diğer periferik nöropatiler Tablo 13’da özetlenmiştir. Bunların birçoğunda da tanı için sinir biyopsisi kesin bir gereklilik taşımaz. Örneğin CMT 1A, HNPP, ailesel amiloidoz gibi durumlarda moleküler genetik tanı testleri biyopsiyi gereksiz kılabilir. Leprada aside dirençli mikroorganizmayı deri biyopsisinde göstermek sinir biyopsisi yapmaya oranla daha pratik olabilir.
Tablo 13. Sinir biyopsisi indikasyonları (EP Bosch ve BE Smith, değiştirilerek)
Sinir biyopsisinde tanı koydurucu bulgu görülenler:
Vaskülit*
Amiloidoz*
Sarkoidoz*
Lepra
Herediter basınca duyarlılık nöropatisi (tomaküler nöropati)**
Paraproteinemik nöropati (Anti-MAG antikorla birlikte IgM monoklonal gamopatisi)
Metakromatik lökodistrofi
Fabry hastalığı
Krabbe hastalığı
Dev aksonal nöropati
Poliglukozan cisimciği hastalığı
Sinirde tümör infiltrasyonu
Sinir biyopsisinde tanıyı düşündürücü bulgu görülenler:
Charcot-Marie-Tooth hastalığı tip 1
Refsum hastalığı
Kronik inflamatuvar demiyelinizan poliradikülonöropati
Amiodaron toksisitesi
Klorokin toksisitesi
Perheksilen toksisitesi
İnce lif nöropatileri***
*: Kombine sinir ve kas biyopsisi yapılması yararlı olur.
**: Günümüzde genetik test öncelik taşımakta ve genellikle yeterli olmaktadır.
***: Elektron mikroskopisinde
MAG: miyeline bağlı glikoprotein.
Periferik sinir biyopsilerinde distal sural sinir, alışılmış ve sık kullanılan bir örnekleme yeridir. Alternatif olarak süperfisiyel peroneal sinirin kullanılması, aynı insizyon ile peroneus brevis kasına ulaşılarak kas biyopsisi yapılmasına olanak sağlar. Bazı durumlarda radyal sinirin el sırtındaki yüzeyel deri dalı da biyopsi için kullanılabilir. Periferik sinirle birlikte alınan kas örneği özellikle vaskülitlerin ortaya konmasında yararlı olur.
İnce Lif Nöropatileri için Kullanılan İncelemeler:
Elektrofizyolojik İncelemeler: Periferik nöropatilerin belirlenmesi ve ayrımlanmasında en çok kullanılan laboratuvar yöntemleri sinir iletim incelemeleridir. Bunlar ön planda kalın miyelinli sinir liflerinin işlevlerini yansıttığından, başlıca ince sinir liflerini tutan nöropatilerde, en azından belirli bir süre için, tamamen normal kalabilirler. İnce lif nöropatilerinin bir kısmında otonom sinirler de tutulduğundan, elektromiyografi laboratuvarlarında yapılan otonom testler (sempatik deri yanıtları, R-R interval değişkenliği gibi) tanıya yardımcı olabilir. Ancak bu testlerin tanısal duyarlılığı yüksek değildir.
Ad ve C liflerini selektif olarak uyararak yapılan nosiseptif uyandırılmış potansiyel incelemeleri (lazer uyandırılmış potansiyeller ve ısı temaslı uyandırılmış potansiyeller) ince lif nöropatilerini göstermekte duyarlı elektrofizyolojik yöntemlerdir, ancak sinir sistemi hasarının lokalize edilmesinde başarılı değillerdir ve uygulanmaları standard elektromiyografi cihazlarına özel uyarıcıların eklenmesini gerektirir.
İnce lif nöropatisi düşünülen hastalarda deri biyopsisi ve kantitatif duyusal test gibi ek tanı yöntemleri kullanılabilir.
Deri biyopsisi: İntraepidermal sinir liflerini incelemek amacıyla uygulanan bir tanı yöntemidir. Bu yöntemde alınan yaklaşık 3 mm çapındaki “punch” biyopsiler basit ve ağrısız olduğundan aynı anda birçok farklı anatomik bölgeden örneklenebilir ve kolaylıkla tekrarlanabilir. Biyopsi materyalinde epidermis içindeki miyelinsiz aksonlar “protein gene product” (PGP) 9.5 gibi nöropeptidlerle boyanarak ışık mikroskopu ya da konfokal mikroskop yardımıyla incelenir ve sayılır (Şekil 1). Yaygın bulguları olan hastalarda biyopsi ekstremite distalinden ve proksimalinden alınarak mevcut ince lif nöropatisinin uzunluğa bağımlı bir niteliği olup olmadığına karar verilebilir; uzunluğa bağımlı olmayan ince lif nöropatileri daha çok immün kökenlidir. İnce lif nöropatilerinin tanısında %90 duyarlılığa ve %95-97 özgüllüğe sahip olduğu ifade edilen bu yöntem elektrofizyolojik incelemelerin çoğu kez normal kaldığı ağrılı distal polinöropatilerin incelenmesi ve takibinde altın standart tanı yöntemi olarak kullanılır hale gelmiştir. Bilimsel çalışmalarda kullanımı gittikçe yaygınlaşmakta olan deri biyopsisiyle dermal miyelinli lifler, ter bezi innervasyonu, otonom sistem etkilenimi veya amiloidoz varlığı araştırılabilmektedir.
Konfokal mikroskopi ile hastaların korneasında ince sinir lifi hasarını gösteren ve invazif olmayan bir yöntem de aynı amaçla kullanılabilir hale gelmiştir.
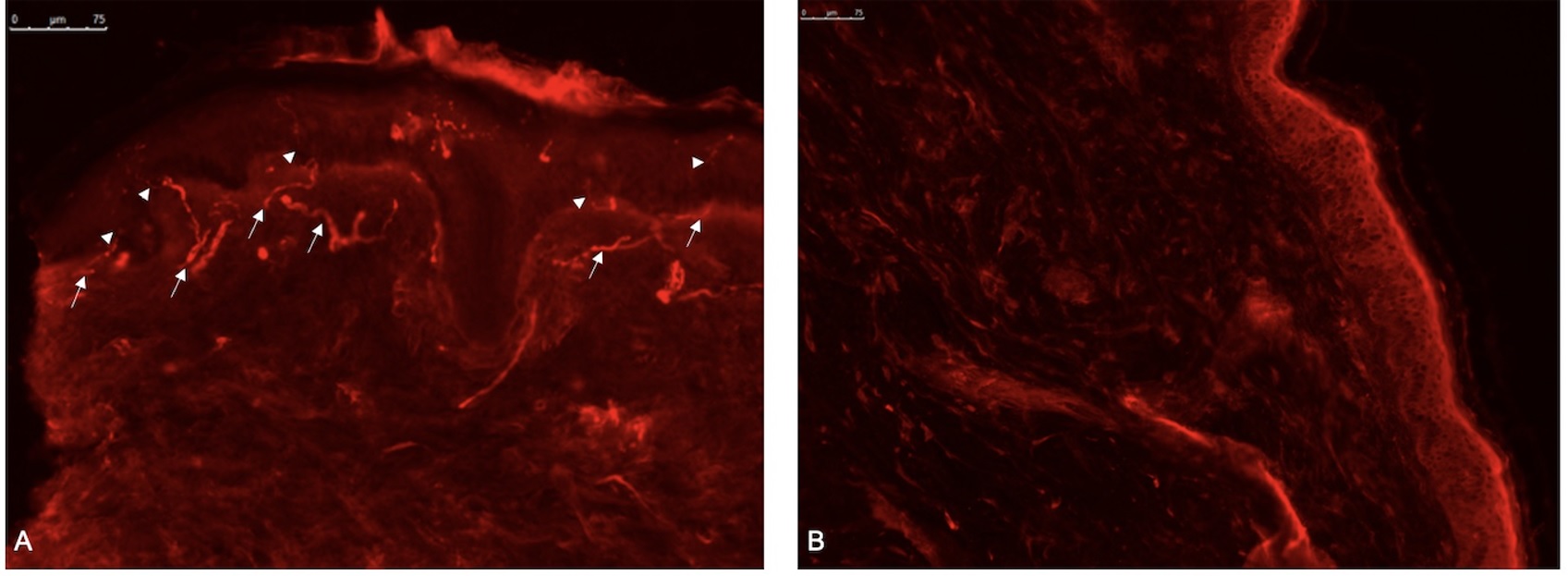
Şekil 1. ‘Protein gene product 9.5’ (PGP 9.5)’a karşı antiserum ile immün boyanan deri kesitleri (50 µm). A) Sağlıklı bireyin alt bacak bölgesine ait deri dokusunda subepidermal sinir pleksuslarından (oklar) doğan epidermal sinir lifleri (ok başları) görülmekte (X10). B) Bir kronik inflamatuvar demiyelinizan polinöropati (CIDP) hastasında epidermal sinir lifleri ve subepidermal sinir pleksusları görülemiyor (X5) (İstanbul Tıp Fakültesi Nöroloji Anabilim Dalı, Nöromüsküler Hastalıklar İnceleme Laboratuvarı arşivinden).
Kantitatif Duyusal Test (QST): Bir cihaz yardımı ile hastaya sunulan vibrasyon, sıcak ve soğuk uyaranlarına ilişkin duyum eşiklerinin çeşitli bilgisayar algoritmaları kullanılarak belirlenmesi ve sayısallaştırılmasını amaçlayan bir yöntemdir. Kalın miyelinli liflerin fonksiyonları vibrasyon tanıma eşiği ile incelenirken, ince sinir lifleri soğuk, sıcak, soğuk ağrısı ve sıcak ağrısı tanıma eşikleri ile araştırılır. Sıcak ağrısı eşiği ince lif nöropatilerini tanımada iyi bir duyarlılığa sahiptir. Özellikle objektif klinik ve elektrofizyolojik bulguları olmayan ağrılı ince lif nöropatisi olgularında C ve Ad liflerinin fonksiyon bozukluklarının ortaya konmasında önem taşır. Tanıda olduğu kadar tedavinin etkilerini takipte de yararlı olabilir. Bu yöntemle ilgili sorunlar, özel cihaz, tecrübe, iyi hasta kooperasyonu gerektirmesi ve zaman alıcı olmasıdır. Mevcut yazılımlarla hastanın temaruz ya da dikkatsizliğine bağlı tutarsız sonuçları tanımak ve düzeltmek de mümkündür. Şimdilik klinik tanı amacı ile kullanımı yaygın hale gelmemiş olan bu yöntem epidemiyoloji ve tedavi çalışmalarında kullanılacak düzeye erişmiştir.
İnce lif nöropatisi tanısında terleme yanıtının ölçülmesine dayanan yöntemler de kullanılabilir. Bunlardan termoregülatuvar terleme testi zaman alıcı, özel donanım ve tecrübe gerektiren bir yöntemdir. Kantitatif sudomotor akson refleks testinde (QSART) postgangliyonik sempatik liflerin bütünlüğünü araştırmak amacıyla lokal bir alanda cilde asetilkolin iyontoforezi yapılır ve ortaya çıkan terleme miktarı ölçülür. Genellikle bu konuda uzmanlaşmış laboratuvarlarda QST ile birlikte kullanılır. Yakın zamanda kullanımı daha kolay olan ve terleme ile derinin elektriksel direncinin değişmesine dayanan başka testler de kullanılmaya başlanmıştır.
Lomber ponksiyon
İnflamatuvar demiyelinizan poliradikülonöropatiler (GBS ve CIDP) ve sinir köklerinde tutulmanın olduğu diğer bazı periferik nöropatilerde beyin omurilik sıvısı (BOS) proteini artar*. GBS’de protein artışı genellikle 48 saatten (daha belirgin olarak ilk haftadan) sonra ortaya çıkar ve 3.-4. haftada en üst düzeye ulaşır. Meningeal karsinomatozis ve lenfomatozise bağlı poliradikülopati ile seyreden olgularda BOS’ta habis hücrelere rastlanabilir.

*Tarihsel değeri nedeniyle ‘albuminositolojik disosiasyon’ olarak bilinen bu durum, ilk olarak 1912’de Sicard ve Foix tarafından omurilik basılarında BOS’ta ‘beklenmeyen şekilde’ hücre artışı olmaksızın protein artışı varlığına işaret etmek için tanımlanmıştır. Guillain, Barré ve Strohl 1916’da Guillain-Barré sendromunu tanımlarken aynı özelliğin üzerinde durmuşlar ve ‘albuminositolojik disosiasyon’ bu hastalığın tanısı için temel laboratuvar bulgularından birisi olarak kabul edilmiştir (“Sur un syndrome de radiculonévrite avec hyperalbuminose du liquide céphalorachidien sans reaction cellulaire. Remarques sur le caractères cliniques et graphiques des réflexes tendineux” Bulletins et Mémoires de la Société Médicale des Hôpitaux de Paris, 1916)
Görüntüleme İncelemeleri
Akut ve kronik seyirli birçok radikülopatide (inflamasyon, infeksiyon veya karsinomatoza bağlı) spinal MRG incelemelerinde sinir köklerinin kalınlaşıp kontrast tuttuğu görülebilir (Şekil 2). Erişkin ve çocuk GBS olgularında da kauda ekuinayı oluşturan sinir köklerinin spinal MRG incelemelerinde bu özellikler gösterilmiştir. Bu bulgunun duyarlılığı elektrofizyolojik incelemeler ve BOS bulguları ile karşılaştırılabilecek derecede yüksek olmakla birlikte, özgül nitelikte değildir. Bu nedenle, şimdiki aşamada GBS tanısı için elektrofizyolojik incelemeler ve BOS incelemesinin yerini alacak durumda değildir.
CIDP ve immün kökenli kronik seyirli poliradikülonöropatilerin bir kısmında MRG incelemelerinde lumbosakral sinir köklerinin ve sinir pleksuslarının kalınlaşıp kontrast madde tuttuğu gösterilebilir. Bu bulgular da tanıya özgül nitelikte değildir, ancak kronik immün duyusal poliradikülopati gibi nadir durumlarda tanıya götürücü az sayıdaki laboratuvar bulgusundan birisi olabilir.
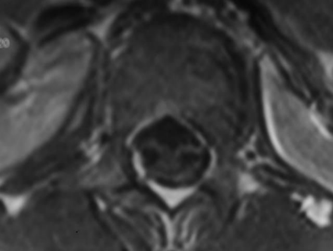
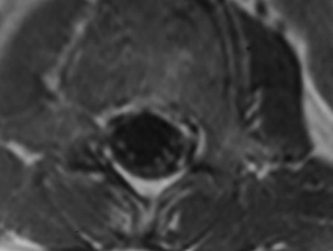
Şekil 2. Konus medullaris alt kesimi (solda) ve kauda ekuina düzeylerinden (sağda) geçen transvers T1 ağırlıklı kontrastlı MRI kesitlerinde kontrast tutan sinir kökleri görülüyor.
Polinöropatilerde Tanıya Gidiş
Hastanın ayrıntılı öyküsü ve nörolojik muayenesi mevcut polinöropatinin hangi ana grupta ele alınabileceğini büyük oranda belirler. Bundan sonra o gruba ve en olası tanılara uygun olan laboratuvar incelemelerinden kolay ve ucuz olanlar ile başlanıp gereğinde daha güç ve pahalı yöntemlere doğru ilerlenir. Ayrıntılı bir klinik muayene ve elektrofizyolojik testler, incelemede kullanılacak diğer laboratuvar testlerinin seçiminde yardımcı olur.
Klinisyenin karşılaştığı polinöropati tablolarının büyük bölümünü kronik, distal simetrik, uzunluğa bağımlı, duyusal-motor polinöropatiler oluşturur. Bu tablo içinde başvuran hastalarda etyolojiyi araştırmak için başvurulabilecek laboratuvar araştırmaları Tablo 14’de özetlenmiştir. Bunlar arasında nedeni ortaya koymada en verimli olan laboratuvar araştırmaları diyabet araştırmak için kullanılan testler, vitamin B12 düzeyi ve metabolitleri ile serum immün fiksasyon elektroforezidir. Herediter nöropatiler için yapılacak genetik testler genellikle klinik fenotip, geçiş paterni ve elektrofizyolojik özelliklere (aksonal ya da demiyelinizan tipte olmasına) dayanılarak istenir. En yüksek oranda anormallik bulunan testler PMP22 genindeki CMT 1A duplikasyonu ve HNPP delesyonudur. CMT2 için yapılan testlerde verimlilik oranı düşüktür. Yeni nesil gen dizileme tekniklerinin yerleşmesiyle daha fazla genetik kökenli nöropatide tanıya varılır hale gelinmektedir.
Tablo 14. Kronik uzunluğa bağımlı nöropatide önerilen araştırmalara (JC Watson ve PJB Dyck, değiştirilerek)
Tam kan sayımı
Böbrek fonksiyonları
Karaciğer fonksiyonları
Eritrosit sedimentasyon hızı (kuru göz/ağız ve duyusal nöronopati varsa ENA)
Açlık kan şekeri* (%11), hemoglobin A1C* (%26)
2 saatlik oral glikoz tolerans testi**
Tiroid stimulan hormon
Monoklonal protein* (serum protein immün fiksasyon elektroforezi) (%10)
Vitamin B12 (%2)*** (metilmalonik asit ile birlikte %9)*
İnfeksiyonlar (risk varsa ya da endemik bölge ise): Lyme, HIV
Aile öyküsü, çukur ayak, çekiç parmak*
*En yüksek oranda anormal bulunanlar (bulunma oranları ile)
**: Diyabet ve bozulmuş glikoz toleransı tanısı için. Bu ikinci durumun polinöropati ile nedensel ilişkisi olduğu konusu bir derece tartışmalıdır.
***Normal B12 serum düzeyi bu vitaminin eksikliğini dışlatmaz. Sınır değerlerde (200-500pg/mL) serum düzeyi olan hastaların %5-10’unda metilmalonik asit düzeyi yüksektir. Bu nedenle metilmalonik asit düzeyi (homosistein düzeyi ile birlikte olabilir) incelemeye eklenmelidir.
a. Muayene ve araştırmalar sonucunda olguların %74-82’sinde neden belirlenebilir. Nedenin ortaya konamadığı %20-25 hastada genellikle hızlı progresyon ve belirgin sakatlık olmaz (Kronik kriptojenik (idyopatik) duyusal polinöropati)
ENA: “extractable” nükleer antijen, HIV: ”human immunodeficiency virüs”.
Kronik distal simetrik duyusal-motor nöropatilerin dışında kalan polinöropatiler, önceki bölümlerde üzerinde durulan özelliklerine göre farklılaşan tanı olasılıklarına göre araştırılmalıdır (Tablo 15). Bu hastalıkların ortak özellikleri ciddi ağırlıkta ve progresyon gösterme eğiliminde olmaları ve birçoğunun özgül tedavisinin mümkün olmasıdır. Bu nedenle, bu hastalıkların hepsinde gerekli elektrofizyolojik incelemeler yapılarak tutulum paterni ayrıntılı şekilde belirlenmeli ve mümkünse nöromüsküler hastalıklar alanında tecrübeli bir meslektaşa danışılarak inceleme ve tedaviler planlanmalıdır.
Tablo 15. Polinöropatinin klinik şekline göre tanı testleri (TD Levine ve DS Sapesrtein, KG Gwathmey ve N Jovanovich, değiştirilerek)
|
|
Olası Tanı |
Testler |
|
İnce lif nöropatisi |
Diyabet |
Açlık şekeri, HbA1C, OGTT |
|
Bozulmuş Glikoz Toleransı |
OGTT, HbA1C |
|
|
Sjögren sendromu |
SS-A, SS-B, Schirmer testi, tükrük bezi biyopsisi |
|
|
|
Sarkoidoz |
Serum ACE düzeyi |
|
|
Primer sistemik amiloidoz |
Serum immünofiksasyon |
|
|
|
Kantitatif immünglobulin düzeyleri |
|
|
|
Serum serbest hafif zincirleri |
|
|
|
Doku biyopsisi (yağ, rektal, deri, diğer) |
|
|
Familyal amiloidoz |
Genetik testler: transtiretin, gelsolin, APOA1, Biyopsi (dudak) |
|
|
SLE |
ANA, anti-dsDNA, anti-Smith kompleman, antifosfolipid, sedimentasyon, CRP |
|
|
Fabry hastalığı |
Serum α-galaktozidaz düzeyi |
|
|
Sodyum kanalopatileri Nav 1.7, Nav 1.8) |
Genetik testler: SCN9A, SCN10A |
|
|
Herediter duyusal-otonom nöropatiler |
Genetik testler (birçok mutasyon mevcut) |
|
|
HIV/AIDS |
HIV virüs yükü, CD4 sayısı |
|
|
Kriyoglobulinemi |
Hepatit C antikor ve PCR, kriyoglobulinler |
|
Duyusal ataksi |
B 12 eksikliği |
Vitamin B12, metilmalonik asit + homosistein |
|
|
E vitamini eksikliği |
Serum E vitamini düzeyi |
|
|
Sjögren sendromu |
SS-A, SS-B, tükrük bezi biyopsisi |
|
|
HIV |
HIV serolojisi |
|
|
Paraneoplastik |
Anti-Hu, anti-CV2/CRMP5, toraks grafi/BT, PET |
|
|
Vitamin E eksikliği |
Vitamin E düzeyi |
|
|
Tabes dorsalis |
Sifiliz serolojisi |
|
|
Vitamin B6 toksisitesi |
Vitamin B6 düzeyi |
|
|
Platinum türevleri toksisitesi |
Klinik öykü |
|
|
Miller Fisher sendromu |
EMG, anti-GQ1b |
|
|
Akut duyusal ataksik nöropati |
EMG, anti-GD1b |
|
|
Anti-MAG antikor sendromu |
EMG, immün fiksasyon, anti-MAG |
|
|
CISP |
SEP, kontrastlı spinal MRG, BOS |
|
Saf motor nöropati |
AMAN |
EMG, BOS, anti-GM1, GD1a antikorları |
|
MMN |
EMG, Anti-GM1, serum immünofiksasyon, kantitatif immünglobulin düzeyleri |
|
|
|
ALS |
EMG, görüntüleme |
|
|
Kennedy hastalığı |
Androjen reseptör geni |
|
|
Batı Nil virüsü |
Seroloji, BOS’ta PCR |
|
Akut motor- duyusal nöropatiler |
GBS |
EMG, BOS |
|
AMSAN |
EMG, BOS |
|
|
Mononöropati müultipleks |
Sedimentasyon, ANA, ANCA, Hepatit B ve C serolojisi, kriyoglobulinler, HIV, sinir biyopsisi |
|
|
|
Diyabetik lumbosakral radikülopleksopati |
Açlık kan şekeri, HbA1C |
|
Subakut motor- duyusal nöropati |
CIDP |
EMG, BOS, bazı olgularda sinir biyopsisi |
|
MADSAM |
EMG, BOS, bazı olgularda sinir biyopsisi |
|
|
DADS |
EMG, BOS, serum immün fiksasyonu, kantitatif immünglobülin düzeyleri, serum serbest hafif zincirleri |
|
|
|
Anti-MAG sendromu |
Anti-MAG titresi |
|
|
Herediter basınca duyarlılık nöropatisi |
PMP 22 delesyonu |
|
|
Sarkoidoz |
Serum ACE düzeyi, BOS |
|
|
Batı Nil virüsü |
Seroloji, BOS |
|
|
Lenfomatoz/Karsinomatoz menenjit |
BOS (sitoloji ile-tekrarlanması gerekebilir) |
|
|
Negatif sonuçlar-ilerleyici seyir |
Sinir biyopsisi |
|
Kronik motor- duyusal nöropatiler |
Diyabet |
Açlık kan şekeri, HbA1C |
|
Bozulmuş glikoz toleransı |
OGTT |
|
|
Vitamin B12 eksikliği |
Vit B12 düzeyi, metilmalonik asit + homosistein |
|
|
|
Paraproteinemi |
Serum immünofiksasyonu, kantitatif immünglobülin düzeyleri, serum serbest hafif zincirleri, anti-MAG titresi |
|
|
CMT1 |
PMP duplikasyon, PMP delesyon, Cx32, MPZ, MFN2 |
|
|
CMT2 |
EGR2, FIG4, GARS, GDAP1, HSPB1, LMNA, MFN2, MPZ, PRX, RAB7 |
|
|
Sjögren sendromu |
SS-A, SS-B, tükrük bezi biyopsisi |
|
|
Negatif sonuçlar-ilerleyici seyir |
BOS, sinir biyopsisi |
|
Miyelonöropati |
Miyelopati-nöropati birlikteliği |
Spinal MRG |
|
|
Vitamin B12 eksikliği |
Vit B12 düzeyi, metilmalonik asit + homosistein |
|
|
Nitröz oksit maruziyeti |
B12 eksikliğine bağlı miyelonöropati gelişir |
|
|
Bakır eksikliği |
Serum bakır ve çinko düzeyi |
|
|
HSP |
Genetik test |
|
Akut ve kronik otonom nöropatiler |
Akut otonom gangliyonopati |
Gangliyonik asetilkolin reseptör antikorları, voltaj kapılı potasyum kanal antikorları, GAD-65 antikorları, anti-Hu |
|
GBS |
EMG, BOS |
|
|
Diyabet |
Açlık kan şekeri, HbA1C |
|
|
Primer sistemik amiloidoz |
Serum immünofiksasyonu,
kantitatif immünglobulin düzeyleri, serum
serbest hafif zincirleri |
|
|
|
Sjögren sendromu |
SS-A, SS-B, tükrük bezi biyopsisi |
|
|
Ailesel amiloidoz |
Transtiretin genetik incelemesi |
ACE: “angiotensin converting enzyme”, AIDS: “acquired immunodeficiency syndrome”, ALS: amiyotrofik lateral skleroz, AMAN: akut motor aksonal nöropati, AMSAN: akut motor duyusal aksonal nöropati, ANA: anti-nükleer antikor, ANCA: anti-nötrofil sitoplazmik antikor, BOS: beyin omurilik sıvısı, CIDP: kronik inflamatuvar demiyelinizan polinöropati, CISP: kronik immün duyusal poliradikülopati, CMT: Charcot-Marie-Tooth, CRP: C-reaktif protein, Cx32: konneksin 32, dsDNA: “double stranded” deoksi nükleik asit, EGR2: “Early Growth Response 2”, EMG: elektromiyografi, GAD: glutamik asit dekarboksilaz, GARS: glisil tRNA sentetaz, GBS: Guillain-Barré sendromu, GDAP1: “Gangliozide-induced differentiation-associated protein 1”, HbA1C: hemoglobin A1C, HIV: “human immunodeficiency virus”, HSP: herediter spastik paraparezi, HSPB1: “Heat-shock” protein 1, LMNA: lamin, MADSAM: multifokal edinsel demiyelinizan duyusal ve motor nöropati, MAG: miyeline bağlı glikoprotein, MFN2: mitofusin-2, MMN: multifokal motor nöropati, MPZ: “Myelin Protein Zero”, MRG: manyetik rezonans görüntüleme, OGTT: oral glikoz tolerans testi, PCR: polimeraz zincir reaksiyonu, PMP 22: “Peripheral Myelin Protein 22”, PRX: periaksin, RAB7: “Ras-related in brain 7”, SEP: somatosensoriyel uyandırılmış potansiyeller, SLE: sistemik lupus eritematozus, SS-A: Sjögren sendromu ilişkili antijen A, SS-B: Sjögren sendromu ilişkili antijen B.
HEREDİTER POLİNÖROPATİLER
Herediter nöropatiler, genetik bir bozukluk sonucu ortaya çıkan hastalıklardır. Bu hastalıkların bazılarına merkez sinir sistemi tutulumu da eşlik edebilir. Fenotipik özellikler, aileler arasında veya aynı aile içinde değişkenlikler gösterebilir. Son yıllarda moleküler biyoloji alanındaki önemli gelişmeler herediter nöropatilerin önemli bir bölümüne tanı konmasını kolaylaştırmıştır. Böylelikle hem bu nöropatilerin tanısı için başvurulabilen ve invazif bir işlem olan sinir biyopsisine gerek kalmamakta, hem de daha önceleri klinik, elektrofizyolojik ve histopatolojik özelliklere dayandırılan sınıflamaların yerini artık genetik sınıflamalar almaktadır. Gün geçtikçe mutasyon sayısı da artmaktadır. Saptanan genetik defektler ayrıca hücre biyolojisinde rol oynayan proteinlerin görevleri konusunda ipuçları sağlamaktadır. Bu proteinlerin hücre içinde üstlendikleri fonksiyonlar ve birbirlerini nasıl etkiledikleri konusunda ilerleyen dönemde daha çok bilgi edinilecektir. Edinilen bilgiler ile de yeni tedavi perspektifleri oluşması yönündeki çalışmalar ivme kazanacaktır. Bu bölümde herediter nöropatiler içinde en sık görülen CMT’den söz edilecektir (Tablo 17). Bu Tablo incelendiğinde hem demiyelinizan hem de aksonal özellikteki nöropatilere aynı genlerin mutasyonunun yol açabildiği de görülmektedir. Buradan mutasyonların tek başına belirleyiciliğinden çok, proteinler arası etkileşimin nihai fenotipe yol açtığı anlaşılabilir. Benzer şekilde aynı genler, farklı kalıtım paterni ile de hastalığa neden olabilmektedir. Tablo 16’te ise diğer herediter nöropatiler ve nöropatilerin eşlik ettiği hastalıklar yer almaktadır.
Tablo 16. Herediter polinöropatiler (Washington University Home Page, https://neuromuscular.wustl.edu/time/hmsn.html‘den uyarlanmıştır)
|
1) CMT (Ayrıca bakınız: Tablo 17) 2) Herediter motor sendromlar a) Herediter ALS -dominant, resesif, X’e bağlı, çocukluk çağında başlayanlar b) Bulber sendromlar c) SMA d) Distal herediter motor nöropati (dHMN). Dominant, resesif, X’e bağlı 3) HSAN a) HSAN1 b) HSAN2 c) HSAN3 d) HSAN4 e) HSAN5 f) HSAN7 g) HSAN ve demans h) Spastik paraparezi ile herediter duyusal nöropati i) Konjenital ağrı duyarsızlığı j) Paroksismal ciddi ağrı sendromu k) Primer eritromelalji l) İnce lif nöropatisi 4) Metabolik defektlere bağlı nöropatiler a) Lipid b) Lipoprotein c) Fitanik asit d) Lizozomal depo hastalıkları (Fabry hastalığı, Krabbe hastalığı, Metakromatik lökodistrofi, Niemann-Pick hastalığı) e) Amiloidoz f) Porfiri 5) Merkez sinir sistemi tutulumuyla birlikte giden herediter nöropatiler a) Dev aksonal nöropatiler b) Friedreich hastalığı c) Nöroakantositoz d) Herediter spastik paraparezi (bazı tipleri) e) Spinoserebellar ataksi (bazı tipleri) |
CMT: Charcot-Marie-Tooth, SMA: spinal müsküler atrofi, HSAN: herediter duyusal-otonom nöropatiler
Charcot-Marie-Tooth Hastalığı (CMT)
Ondokuzuncu yüzyılın sonlarında Charcot, Marie ve Tooth tarafından tanımlanan bu hastalık herediter nöropatiler içinde en sık rastlanılanıdır (prevalansı 1/2500). Hastalık, genellikle ilk iki onyılda bulgu vermeye başlar ve yavaş progresyon gösterir. Klinik tablonun ağırlığı, aynı aile içinde hasta bireyler arasında değişkenlik gösterebilir. Nadiren ciddi fonksiyon kaybına yol açar. Hastalık, 1968’de Dyck ve Lambert tarafından klinik, elektrofizyolojik ve histopatolojik verilere göre sınıflanarak, doksanlı yılların başına kadar Herediter Motor Duyusal Nöropati (HMSN) adıyla anılmıştır. Moleküler biyoloji alanındaki gelişmelerle birlikte bu gruptaki nöropatilere, ilk tanımlayanlara atfen yeniden Charcot-Marie-Tooth hastalığı (CMT) adı verilmiştir.
Bazı değişiklikler gündeme gelmiş olsa da, hastalık halen, elektrofizyolojik bulgular ve kalıtım tipini temel alacak şekilde sınıflandırılmaktadır. Buna göre medyan sinir motor ileti hızı 38 m/sn’den düşük olan ve otozomal dominant olarak kalıtılan grup CMT1 olarak adlandırılırken, medyan motor ileti hızı 38’den yüksek, aksonal tip olan, CMT2’yi oluşturmaktadır. Otozomal resesif olarak kalıtılan demiyelinizan formlar CMT4, aksonal formlar ise AR-CMT2’yi oluşturmaktadır. Hastalığın X kromozomuna bağlı olarak kalıtılan tipi ise CMTX olarak adlandırılmaktadır. Erken başlangıçlı, motor gelişme geriliği ile seyreden formlar için kullanılan CMT3 veya “Dejerine-Sottas Hastalığı” terimi artık kullanılmamaktadır. Ancak, mevcut sınıflandırmanın yetersiz kaldığı durumlar mevcuttur. Bunlara örnek olarak, aynı aile üyelerinde medyan motor ileti hızının aksonal veya demiyelinizan sınırlar içerisinde değişkenlik gösterdiği otozomal dominant olarak kalıtılan CMT formları görülebilir. Bu grup dominant intermediate CMT (DI-CMT) olarak adlandırılmaktadır. [Harding ve Thomas’ın geniş bir hasta serisi üzerinde yaptıkları araştırmaya dayanarak, elektrofizyolojik incelemede median sinirin motor ileti hızının 38 m/s’den düşük saptandığı hastalarda demiyelinizasyonun, yüksek olanlar da ise ön planda akson kaybının olduğunu kabul etmek gelenek olmuştur.]
Bu sınıflandırma sonrasında, kesin tanı için kaçınılmaz olan son nokta genetik testtir. Tüm CMT olgularının yaklaşık %80 kadarını oluşturan CMT1’in en sık sebebi olan, “Peripheral Myelin Protein 22” (PMP22) duplikasyonu ilk yapılması gereken genetik testtir. Daha sonra ise, ülkemiz gibi genetik olarak heterojen bir altyapıya sahip ülkelerde, yeni nesil dizileme yöntemlerinin kullanılması uygun olacaktır. Birden fazla genin aynı anda gözden geçirilebilmesini mümkün kılan bu incelemeden elde edilen büyük verilerin doğru analizi önem taşır. Özellikle hastalık oluşturmayan polimorfizmlerin ayrımının yapılması tanı kesinliğini arttırmaktadır.
Tablo 17. Herediter Motor Duyusal Nöropatiler (Washington University Home Page, https://neuromuscular.wustl.edu/time/hmsn.html‘den uyarlanmıştır)
|
CMT1
CMT 1B: P0 protein (MPZ); 1q23 CMT 1C: LITAF; 16p13 CMT 1D: EGR2; 10q21 CMT 1E: PMP-22 (nokta mutasyonu): 17p12 CMT 1F: NEFL; 8p21 CMT 1G: PMP2; 8q21
CMT4 CMT 4B1: MTMR2; 11q22 CMT 4B2: SBF2; 11p15 CMT 4B3: SBF1; 22q13 CMT 4C: SH3TC2 (KIAA1985); 5q32 CMT 4D (Lom): NDRG1; 8q24 CMT 4E: EGR2; 10q21 CMT 4F: Periaxin; 19q13 HMSN-Russe (4G): HK1; 10q22 CMT 4H: FGD4; 12q12 CMT 4J: FIG4; 6q21 CMT 4K: SURF1; 9q34
AR-CMT2 A (B1): Lamin A/C; 1q22 B (B2): PNKP; 19q13 PNKP: 19q13 F/Distal HMN: HSPB1; 7q11 H/Piramidal bulgular: 8q21 K/Hoarseness: GDAP1; 8q21 P: LRSAM1; 9q33 R: TRIM2; 4q31 S: IGHMBP2; 11q13 T: MME; 3q25 X: SPG11; 15q21 A2B: MFN2; 1p36 EE: MPV17; 2p23 AHNAK2: 14q32 EGR2; 10q21 HSJ1/DNAJB2; 2q35 MCM3AP (GANP): 21q22 PRPH: 12q13 SACS: 13q12 CMT+Nöromiyotoni: HINT1; 5q31 |
CMT2 CMT 2A1: KIF1B; 1p36 CMT 2B: RAB7; 3q21 CMT 2C: TRPV4; 12q24 CMT 2D: GARS; 7p14 CMT 2E: NEFL; 8p21 CMT 2F/ Distal HMN: HSPB1; 7q11 CMT 2I: P0; 1q22 CMT 2J: P0; 1q22 CMT 2K: GDAP1; 8q21 CMT 2L: HSPB8; 12q24 CMT 2M: DNM2; 19p13 CMT 2N: AARS; 16q22 CMT 2O: DYNC1H1; 14q32 CMT 2P: LRSAM1; 9q33 CMT 2Q: DHTKD1; 10p14 CMT 2U: MARS; 12q13 CMT 2V: NAGLU; 17q21 CMT 2W: HARS; 5q31 CMT 2Y: VCP; 9p13 CMT 2Z: MORC2; 22q12 CMT 2CC: NEFH; 22q12 CMT 2DD: ATP1A1; 1p13 CMT 2: TFG; 3q12 CMT 2: DGAT2; 11q13 CMT 2: MME; 3q25 CMT 2: JAG1; 20p12
X’e bağlı 1: GJB1 (CX32); Xq13 2: Xp22.2 3: Xq27 4 (Cowchock): AIFM1; Xq26 5: PRPS1; Xq22 6: PDK3; Xp22
|
Genlerle ilgili kısaltmalar için ilgili bölümlere bakınız.
CMT1
Otozomal dominant olarak kalıtılan bu hastalık, genellikle ilk ya da ikinci onyılda başlar. Ayaklarda pes kavus ve çekiç parmak deformitesi görülebilir (Şekil 3a). Alt ekstremitelerde belirgin distal zaaf ve atrofi klinik tabloya hakimdir. Atrofi daha çok peroneal bölgedeki kasları tutar. Bu nedenle ince bilekli ve yukarıda adı geçen ayak deformiteleri olan hastalarda ilk tanı olasılığı CMT hastalığıdır. Subjektif duyusal yakınmalar daha geri plandadır. Ancak muayenede daha çok uçları tutan hipoestezi bulunabilir. Vibrasyon duyusu genellikle bozuktur. Derin tendon refleksleri hastalığın hafif formlarında ya da başlangıcında alınabilir fakat hastaların yarısından fazlasında tamamıyla kayıptır. Elektrofizyolojik ve histopatolojik (Şekil 3b) incelemeler demiyelinizan özellikler gösterir, buna ikincil akson kaybı da eşlik eder.

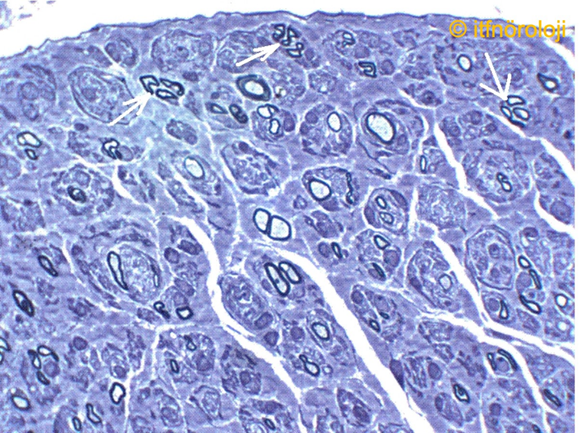
Şekil 3a. Charcot-Marie-Tooth (CMT) hastalığında bacak distallerinde atrofi, pes kavus ve çekiç parmak deformitesi.
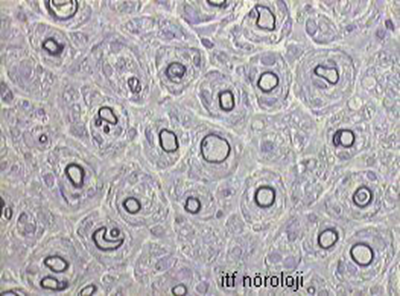
Şekil 3b. Charcot-Marie-Tooth Tip 1A (CMT 1A). Yüzeyel peroneal sinir biyopsisinde miyelinli aksonların kısmen yok olduğu, tüm alana Schwann hücre proliferasyonuna bağlı olarak soğan zarı görünümünün hakim olduğu izlenmektedir (Thioninle boyanmış enine yarı ince kesit. X40).
Günümüzde, genetik incelemelerdeki gelişmeler sonucunda, tanımlanan tiplerin
sayısında artış olmuştur. Diğerlerine kıyasla daha sık rastlanan birkaç alt
tipin klinik özellikleri aşağıda anlatılmıştır:
CMT 1A: Tüm CMT1 olgularının %70-90’ını oluşturur. On yedinci kromozomda, kompakt miyelin proteinlerinden PMP22 ile ilgili olan bölgenin (17p11.2) duplikasyonu sonucu ortaya çıkar. Genellikle klasik CMT fenotipine neden olur. Çok daha az sayıda hastada, PMP22 genine ait nokta mutasyonları CMT 1A’ya kıyasla daha ağır bir fenotip olan CMT 1E alt tipine sebep olmaktadır. Bu tipteki mutasyonlara sıklıkla sağırlık da eşlik eder.
CMT 1B: Birinci kromozomda yine bir kompakt miyelin proteini olan “Protein Zero” veya MPZ geninin nokta mutasyonu sonucu oluşur (1q22-23). Ülkeler arası farklılıklar izlense de tüm CMT1 olgularının yaklaşık %5-10’u bu mutasyona bağlıdır.
CMT 1C: Bu form CMT 1A ve CMT 1B’den çok daha az sayıda hastada, 16. kromozomda (16p13.3-p12) yer alan, protein degradasyonunda rol oynadığı düşünülen “lipopolysaccharide-induced tumor necrosis factor” (LITAF) genindeki mutasyonlar sonucunda ortaya çıkar.
CMT 1D: Bu grupta, DNA’ya bağlanarak gen ekspresyonunda düzenleyici rol üstlenen ve Schwann hücre gelişimini etkileyen “Early Growth Response 2” (EGR2) proteinine ait mutasyonu olan hastalar yer almaktadır.
CMT4
Otozomal resesif (OR) geçen demiyelinizan CMT hastalığına CMT4 adı verilir. Hastalığın başlangıç yaşı, otozomal dominant tiplere kıyasla genellikle daha küçüktür ve klinik bulgular daha ağır seyreder. Ülkemizde, akraba evliliklerinin sıklığından dolayı, literatüre göre daha yüksek oranda otozomal resesif geçiş olduğunu bilmekteyiz. En sık görülen tipler ve bunların fenotipik özellikleri aşağıda gösterilmiştir:
CMT 4A: “Gangliozide-induced differentiation-associated protein” (GDAP1-kromozom 8q21.1). Ağır bir fenotipe neden olur. Kranial sinir tutulumu eşlik edebilir. Otozomal dominant olarak da kalıtılabilir. Aksonal tipte polinöropatiye de neden olabilir (bkz: CMT2K).
CMT 4B1: “Myotubularin-related protein-2” (MTMR2-kromozom 11q22.1). Motor gelişim basamaklarının geri olduğu ağır bir fenotipe neden olur.
CMT 4B2: “Myotubularin-related protein-13” (MTMR13 (SBF2)-kromozom 11q22.1). Motor gelişim basamaklarının geri olduğu ağır bir fenotipe neden olur.
CMT 4C: “SH3 Domain And Tetratricopeptide Repeats 2” (SH3TC2-kromozom-5q23-q33), bu mutasyonda erken başlayan kifo-skolyoz sıklıkla izlenir.
CMT 4D Lom: Roman’larda görülür, “N-myc Downstream-Regulated Gene 1” (NDRG1-kromozom 8q24)
CMT 4F: “Periaxin” (PRX-kromozom 19q13). Duyusal ataksinin belirgin olarak izlendiği, erken başlangıçlı ağır bir fenotipe neden olur.
CMTX
Çok büyük bir kısmı X kromozomunun uzun kolundaki (Xq13-22) “Gap junction beta 1” (GJB1) geninin mutasyonuna bağlı gelişir. Bu gen miyelinin kompakt olmadığı, Ranvier boğumuna yakın bölgelerdeki iyon alış-verişini düzenleyen proteinleri (gap-junction proteinleri) kodlar. Dünyada, CMT 1A dan sonra en sık görülen genotiptir ve tüm CMT olgularının yaklaşık %20-25’ini oluşturmaktadır. Ülkemizde ise bu oran diğer ülkelere göre oldukça düşüktür. Erkeklerde klinik tablo daha ağırdır. Demiyelinizasyonla birlikte aksonal tutulum da izlenebilmektedir. Aksonal tutulum genellikle kadınlarda görülür. Ayrıca CMT’nin bu tipinde diğerlerinden farklı olarak asimetrik özellikler ve merkez sinir sistemi tutulumuna ait bulgular da görülebilmektedir. Bu hastaların kranial MRG incelemelerinde ak maddeyi tutan, multipl skleroz hastalığındaki lezyonlara benzer lezyonlar görülebileceği bildirilmiştir.
CMT2
CMT1’e göre daha nadir görülür. Otozomal dominant olarak kalıtılır. Klinik özellikler CMT1’e benzer. Daha önce de belirtildiği gibi akson kaybının elektrofizyolojik ve histopatolojik özelliklerini taşır (Şekil 4). Moleküler biyoloji alanındaki gelişmeler sonucunda birçok yeni gen defekti tanımlanmıştır. Bunlardan bazılarının özellikleri aşağıda özetlenmiştir:
CMT 2A2A: En sık olarak izlenen CMT-2A2A’ya, Mitofusin-2 (MFN2) genindeki mutasyonlar neden olur. Merkez sinir sistemi tutulumu, optik atrofi gibi değişik özellikler taşıyan klinik tablolar izlenebilir. Aynı gendeki homozigot mutasyonlar ise bir AR-CMT2 alt tipi olan CMT 2A2B’ye neden olmaktadır.
CMT 2B: GTP bağlayan ve veziküler transportta rol üstlenen RAB7 mutasyonu sonucunda ortaya çıkar. Ağır duyu kaybına bağlı olarak ayaklarda ülsere lezyonlar izlenebilmektedir.
CMT 2C: TRPV4 genindeki mutasyonlara bağlı olarak gelişir. Bu proteindeki bozukluklar kalsiyum transportunda bozukluğa neden olur.
CMT 2D: Glisil tRNA sentetaz (GARS) mutasyonu sonucunda ortaya çıkar. Motor tutulumun daha baskın olduğu bir fenotipe neden olur. Herediter distal motor nöropati (dHMN) tip 5 ile alleliktir.
CMT 2E: Bu hastalardaki mutasyon, akson transportunda rol alan nörofilamanlarla ilgili “neurofilament light chain” (NFL) genindedir. Hastaların %30’unda işitme kaybı görülebilir. CMT 2F: “Heat-shock” proteinlerinden olan HSPB1’in mutasyonu bu hastalığa yol açar. dHMN tip 2B ile alleliktir.
AR-CMT2
Otozomal resesif geçişli aksonal tipte polinöropati nedeni olan AR-CMT2’de tanımlanan genlerin sayısı da son yıllarda artış göstermiştir. CMT4’e benzer olarak bu grup da ülkemizde diğer ülkelere kıyasla daha sık olarak görülmektedir. Sebep olan genlerin birçoğunda allelik hastalıklar izlenmektedir. Örneğin AR-CMT2A nedeni olan Lamin A/C genindeki mutasyonlar aynı zamanda Emery-Dreifuss müsküler distrofi ve kavşak tipi müsküler distrofiye de neden olabilmektedir. Bu grupta, daha önce de söz edildiği üzere CMT 4A’ya da neden olan GDAP1 ve CMT 2A2A nedeni olan MFN2 de bulunmaktadır. Ayrıca nöromiyotoni ile birlikte izlenen “histidine triad nucleotide-binding protein 1” (HINT1) ve daha çok ileri yaşta görülen “membrane metalloendopeptidase” (MME) de bu grubun içindedir.
Şekil 4. Otozomal resesif Charcot-Marie-Tooth Tip 2 (CMT2; GDAP1 mutasyonu). Sural sinir biyopsisinde miyelinli akson yoğunluğunda önemli ölçüde azalmayla birlikte, geriye kalan aksonların miyelin kılıflarının genellikle ince olduğu görülmektedir. Ayrıca rejenerasyon odaklarının (oklar) varlığı gözlenmektedir. (Thioninle boyanmış enine yarı ince kesit. X40).
Herediter basınca duyarlılık nöropatisi (HNPP)
Otozomal dominant geçen bu hastalıkta, gelip geçici, ağrısız duyusal-motor defisitler izlenmektedir. Elektrofizyolojik bulgular, özellikle periferik sinirlerin sıklıkla tuzaklandığı yerlerde iletim bloklarının hakim olduğu demiyelinizan bir nöropatiyi telkin eder. En çok etkilenen sinirler, peroneal, ulnar, radyal sinirlerdir. Bazı hastalarda tekrarlayıcı defisitler kronik ve ilerleyici bir seyre de yol açabilir. Sinir biyopsisindeki tipik görünüm fokal miyelin kalınlaşmasıdır, buna Latince’de sosis anlamına gelen “tomaküla” adı verilir (Şekil 5a ve Şekil 5b). Hastalık, 17. kromozomda, PMP22’yi kodlayan bölgenin (17p11.2) delesyonu sonucu ortaya çıkmaktadır. Yukarda da söz edildiği gibi aynı gendeki duplikasyonlar CMT 1A, nokta mutasyonları ise CMT 1E fenotipine neden olmaktadır. HNPP’de kimi zaman brakiyal pleksus da tutulabilir. Ancak bu tablo, brakiyal pleksusun tutulduğu ağrılı ataklar ile seyreden ailevi nöraljik amiyotrofiden ayırt edilmelidir. SEPT9 gen mutasyonu sonucunda gelişen ve otozomal dominant olarak kalıtılan bu hastalıkta, belirgin hipotelorizm nedeniyle tipik bir yüz görünümü izlenir (Modigliani tipi yüz görünümü).
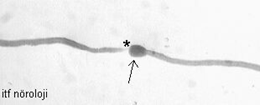
Şekil 5a. Ailesel basınca duyarlılık nöropatisi. Tek tek sinir liflerini izole etme yöntemi uygulamasında (teasing) paranodal tomaküla (ok) ve komşuluğunda demiyelinizasyon izlenmektedir (Osmium tetroksit, X40).

Şekil 5b. Ailesel basınca duyarlılık nöropatisi. Thioninle boyanmış enine yarı ince kesitte hipermiyelinize lif izlenmektedir (tomaküla, ok) (X40).
Tedavi
Hastalıkta halen kanıtlanmış bir tedavi seçeneği bulunmamaktadır. Bunun en büyük sebebi de hastalığın son derece yavaş ilerlemesi nedeniyle klinik çalışma oluşturmanın zorluğudur. Ayrıca hastalığın genetik altyapısının heterojen olması da kısıtlılıklara neden olmaktadır. Günümüze dek birçok tedavi seçeneği denenmiştir. Bunlardan biri olan askorbik asidin farklı dozları istenen etkiyi gösterememiştir. Benzer şekilde progesteron antagonizmasını hedef alan tedavi çalışmalarında da olumlu etki gösterilememiştir. Son dönemde CMT 1A için GABAB reseptör agonisti olan baklofen, opioid reseptör antagonisti olan naltrekson ve hücre içi enerji yolaklarının bir metaboliti olan D-Sorbitol moleküllerinin kombinasyonunu içeren PXT3003 molekülü, “transforming growth factor beta” (TGF-β) sinyal mekanizmalarını inhibe eden ACE-083 ve Nörotrofin-3 ilişkili tedavi seçenekleri umut vaad etmektedir. Hayvan modellerinde, aksonal rejenerasyon üzerinden etkisini gösteren Nörotrofin-3 ile olumlu sonuçlar elde edilmiştir. Bunun üzerine adeno-ilişkili virüs (scAAV1.tMCK.NTF3) aracılığıyla Nörotrofin-3 gen transferinin etkilerini inceleyen Faz 1/2a çalışması planlanmıştır ve sonuçları merakla beklenmektedir (NCT03520751). Ayrıca, MTMR ailesindeki mutasyonlara bağlı olarak gelişen CMT 4B1 ve CMT 4B2’de, Niacin/Niaspan molekülünün, miyelin yapısını düzelterek etkili olabileceği preklinik çalışmalarda gösterilmiştir. Bu molekülün de yakın bir tarihte klinik çalışmasının başlayacağı düşünülmektedir.
Bundan sonraki bölümde CMT hastalığına kıyasla çok daha nadir görülen bazı herediter nöropatiler anlatılacaktır.
Herediter Duyusal Otonom Nöropati (HSAN)
HSAN, heterojen genetik altyapıya sahip bir grup hastalıktan oluşmaktadır. Miyelinsiz ve ince miyelinli duyusal sinir lifleri, kalın miyelinli liflere göre daha belirgin olarak etkilenmektedir. En sık görülen alt tiplerin özellikleri aşağıda belirtilmiştir:
HSAN1: En sık rastlanan alt gruptur. Otozomal dominant olarak kalıtılır. En sık alt tipi olan HSAN 1A, “serine palmitoyltransferase, long chain base subunit 1” (SPTLC1) mutasyonuna bağlı olarak gelişir. Yakınmalar sıklıkla yirmili yaşlarda başlar. Ağrı ve ısıya duyarsızlık ve buna eklenen ani saplanıcı batıcı ağrılar mevcuttur. Ağrıya duyarsızlık nedeniyle yanıklar, kırıklar, göz ve kulaklarda sonucu körlük ve sağırlığa kadar varabilen travmalar izlenebilir. Genetik alanındaki gelişmeler sonucunda SPTLC1 haricinde SPTLC2, “atlastin 3” (ATL3), ATL1, DNA metiltransferaz 1 (DNMT1) genlerinin de HSAN1 fenotipine neden olduğu gösterilmiştir.
HSAN2: Bu gruptaki ilk tanımlanan “with no K (lysine) protein kinase 1” (WNK1) geni HSAN 2A’dan sorumludur. Hastaların yakınmaları, erken çocukluk döneminde başlar. Otozomal resesif kalıtım tipi izlenir. Dokunma, propriyosepsiyon ve vibrasyon hissi kayıptır. Derin tendon refleksleri alınamaz. Epizodik hiperhidrozis izlenebilir. Ayıca mental retardasyon da kliniğe eşlik edebilir. HSAN2 fenotipine ayrıca FAM134B, “axonal transporter of synaptic vesicles” (ATSV) ve “sodium channel protein type 9 subunit alpha” (SCN9A) da neden olabilir. SCN9A mutasyonları, konjenital ağrı duyarsızlığı (congenital insensitivity to pain=CIP-1) nedenidir.
HSAN3: “Inhibitor of kappa light polypeptide gene enhancer in B-cells, kinase complex-associated protein” (IKBKAP) genindeki mutasyonlar sonucunda gelişir. Otozomal resesif kalıtım paterni gösterir. Riley-Day Sendromu olarak da anılan bu hastalıkta disotonomi bulguları klinikteki ana bileşendir.
HSAN4: Konjenital ağrı duyarsızlığı ve anhidrozis (CIPA) olarak da adlandırılır. En geç erken çocukluk dönemine kadar bulgu verir. Ağrı duyarsızlığı ve kendine zarar verme davranışı baskın klinik özelliklerdir. Nörotrofik tirozin kinaz reseptör 1 (NTRK1) gen defektine bağlı gelişir ve otozomal resesif olarak kalıtılır. Anhidrozise bağlı hiperpireksi atakları izlenebilir.
HSAN5: “Nerve growth factor beta” (NGFβ) mutasyonuna bağlı olarak gelişir ve otozomal resesif kalıtım paterni izlenir. Anhidrozis tabloya eşlik etmez. Bunun haricinde HSAN-4 benzeri bir klinik tabloya neden olur.
HSAN6: Distonin (DST) geninin mutasyonlarına ikincil olarak gelişir ve otozomal resesif kalıtım paterni izlenir. Erken çocukluk çağında başlar ve disotonomi bulgularının eşlik ettiği ağrıya duyarsızlık sendromuna neden olur.
HSAN7: HSAN-1 gibi otozomal dominant olarak kalıtılır. Şikayetler doğumla birlikte başlar. Mutasyon, voltaj kapılı sodyum kanalı 1.9’u (NaV1.9) kodlayan SCN11A genindedir. Konjenital ağrı duyarsızlığı (CIP-2) nedenidir.
HSAN8: “PR domain zinc finger protein 12” (PRDM12) mutasyonuna bağlıdır ve otozomal resesif olarak kalıtılır. Konjenital ağrı duyarsızlığı (CIP-3) nedenidir.
Bu hastalıkların hiçbiri için hastalığın seyrini değiştirici bir tedavi seçeneği mevcut değildir. Özellikle yara bakımı ve sekonder infeksiyonların engellenmesi, ampütasyonların ve sepsis gibi komplikasyonların önlenmesi açısından büyük önem taşımaktadır. Ayrıca otonom bulguların tedavisi de kritik önem taşımaktadır. HSAN 1A’da nörotoksik sfingolipid metabolitlerinin birikimini engelleme amacıyla L-Serin molekülü denenmektedir.
Ailevi Transtiretin Amiloidoz
Otozomal dominant geçişli olan bu nöropati, 18. kromozomdaki (18q11.2-12.1), “transtiretin” proteinini kodlayan TTR genindeki mutasyonlar sonucunda gelişmektedir. TTR genindeki mutasyonlar, transtiretin proteinini destabilize ederek doğal olan tetramer formun amiloidojenik monomer forma dönüşmesine ve periferik sinirler başta olmak üzere dokularda birikmesine neden olurlar. Hastalık Portekiz, Japonya ve İsveç’te endemiktir. Ülkemizde de artan sıklıkta vakalar bildirilmektedir. Hastalık esas olarak otonom tutulumun da eşlik ettiği, duyusal ve motor polinöropati tablosuna neden olur. Polinöropatiye, karpal tünel sendromu gibi tuzak nöropatisi tabloları da eşlik edebilir. Periferik sinir sistemi dışında, kalp böbrek, göz, gastrointestinal sistem ve merkez sinir sistemi de başlıca etkilenen bölgelerdir. Klinik tablo mutasyon tipine göre belirgin farklılıklar gösterebilir. Örneğin, tüm dünyada en sık olarak izlenen Val30Met mutasyonu klasik bir fenotipe neden olur. Balkan ülkelerinde daha sık olarak izlenen Glu89Gln mutasyonunda ise kalp tutulumu ve karpal tünel sendromu çok daha erken izlenebilmektedir. Hastalığın tanısı TTR genetik incelemesi ile konulur. Ancak asemptomatik taşıyıcıları, hastalardan ayırmada dokulardaki amiloid birikimini göstermek de önemlidir (Şekil 6). Bu amaçla dudak, abdominal cilt altı yağ dokusu biyopsisi ulaşım kolaylığı açısından sinir biyopsisine kıyasla tercih edilmektedir. Erken tedavi, hastaların fonksiyonel durumu açısından önem taşımaktadır. Bu nedenle asemptomatik hastaların belli aralıklarla takip edilmesi çok önemlidir. Bundan 10 yıl önceye kadar, karaciğer transplantasyonu, hastalıkta bilinen tek tedavi seçeneği iken, son yıllarda yeni ilaç alternatifleri ortaya çıkmıştır. Bu ilaçlar esas olarak iki yolla etkisini göstermektedir. Tafamidis meglumine ve diflunisal gibi tetramer stabilizatörleri esas olarak transtiretinin tetramerik yapısını koruyarak, monomerlerine ayrışmasını önlemeyi hedeflemektedir. TTR gen susturucu tedaviler ise (patisiran, inotersen) karaciğerden transtiretin yapımını baskılayarak etkilerini göstermektedir. Çok yakında, bu mekanizmalar ile etkilerini gösteren yeni ilaçların da eklenmesi beklenmektedir. Ayrıca biriken amiloidi hedef alan monoklonal antikorların da yakın gelecekte kullanılması beklenmektedir.
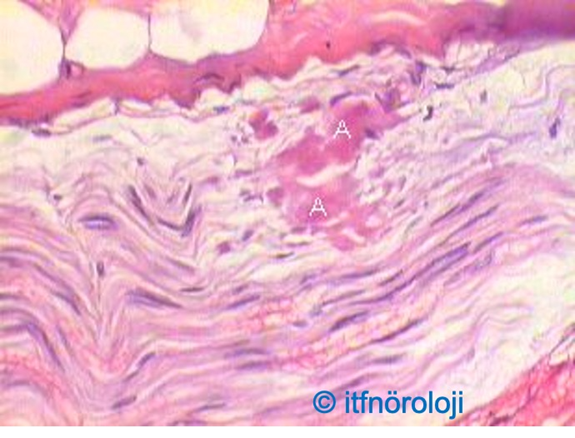
Şekil 6. Ailevi transtiretin amiloidoz. Endonöriumda lifler arasını infiltre etmiş amiloid (A) deposu (Hematoksilen eosinle boyanmış boyuna parafin kesit X10.)
Porfiri
“Heme” sentezindeki defekt sonucu gelişir. Süksinil CoA ve glisinden “heme” oluşana kadar geçen aşamalardaki enzim defektleri hastalığın alt tiplerine neden olmaktadır. Otozomal dominant geçişlidir. Nöropati, karın ağrısı atakları ve psikiyatrik bozuklukları takiben 2-3 gün içinde ortaya çıkar. Akut başlangıçlı yaygın kas kuvvetsizliği ile seyrettiğinden GBS’ye benzer. En sık olarak akut intermittan porfiri alt tipi, söz konusu nöropati tablosuna neden olmaktadır. Tablonun başlangıcında proksimal olan zaaf genellikle asimetriktir. Solunum sıkıntısı ve otonom belirtiler eşlik edebilir. Kranial sinir tutulumu hastaların yaklaşık %75’ine eşlik eder. Elektrofizyolojik olarak aksonal tutulumun ön planda olması GBS’nin en sık görülen demiyelinizan tipinden [akut inflamatuvar demiyelinizan poliradikülonöropati (AİDP)] ayırır. Nöropatinin düzelmesi akson hasarının derecesine bağlıdır. Sulfonamid, griseofulvin, barbitürat, fenitoin gibi ilaçlar porfiri ataklarının ortaya çıkmasına neden olur. Akut hepatik porfiride kullanımı onaylanan, delta amino levulinik asit sentaz 1 (ALAS1) genine yönelik bir “small interfering RNA” (siRNA) molekülü olan givosiran, akut intermittan porfiride de özellikle sık atak geçiren hastalarda kullanılmaya başlanmıştır.
Dev Aksonal Nöropati
Çocukluk
çağında başlayan motor ağırlıklı nöropatiye neden olan bir hastalıktır. Otozomal
resesif geçişlidir. Gigaxonin (GAN) genindeki mutasyonlar sonucunda
ortaya çıkar. İskelet deformitelerinin de eşlik ettiği tabloda giderek bozulan
yürümenin yanında, merkez sinir sistemi tutulumuna ait bulgular da bulunur.
Bunlar başlıca, piramidal belirtiler, ataksi ve mental geriliktir. Bu hastalar
20 yaşına gelmeden kaybedilirler. Fenotipik özelliklerinden biri sert, kıvırcık
saçlardır (“kinky hair”). Histopatolojik olarak nodal veya internodal yerleşimli,
segmenter olarak çap genişlemesi gösteren aksonların varlığı tanı koydurtucudur
(Şekil 7).
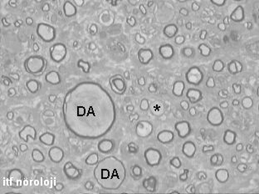
Şekil 7. Dev aksonal nöropati. Sural sinir biyopsisinde genel olarak akson çapına göre ince miyelin kılıfına sahip olan ya da demiyelinizasyona uğramış (*) lifler ve dev aksonlar (DA) görülmekte (Thioninle boyanmış enine yarı ince kesit. X40).
Friedreich Hastalığı
Otozomal resesif geçişlidir. Çalışmalar 9q13 lokusundaki frataksin (FXN) geninde GAA trinükleotid tekrar artışına bağlı olduğunu göstermiştir. Hastaların az bir kısmında trinükleotid tekrar artışı ve nokta mutasyonu, birleşik heterozigot durumda, atipik bir fenotipe neden olabilir. Her iki allelde nokta mutasyonu bulunmasının yaşamla bağdaşmadığı düşünülse de, kliniğimiz tarafından bu durumun CMT benzeri bir fenotipe yol açtığı gösterilmiştir. Hastalık tipik olarak ilk ya da 2. onyılda başlar. Refleks kaybı ve propriyoseptif defisitle karakterize, ilerleyici nitelikteki duyusal nöropatiye serebellar ataksi ve dizartri eşlik eder. Babinski delili bulunur. Ayrıca, kardiyomiyopati, endokrin bozukluklar, katarakt gibi multi-sistemik tutulum bulguları da görülür. Prognoz kötüdür, hastalar genellikle 35 yaş civarında kaybedilir. Sentetik bir koenzim Q10 analoğu olan, mitokondri membranında elektron taşınmasında düzenleyici rolü olan ve oksidatif stres üzerinden etkisini gösteren idebenonun Friedreich ataksisi tedavisinde bazı çalışmalarda olumlu sonuçlar verdiği bildirilmiştir. Oksidatif stres dışında, frataksin proteini aracılı metabolik yolakları hedef alan MIN-102 (NCT03917225), hücre içi frataksin düzeyini arttırarak etkisini gösteren ve HIV tedavisinde de kullanılan etravirine (NCT04273165), FXN gen ekspresyonunu arttırmayı hedefleyen nikotinamid (NCT03761511) ile ilişkili çalışmalar halen devam etmektedir. Ayrıca adeno-ilişkili virüs ile gen transferi ile ilgili preklinik çalışmalar hız kazanmıştır. (Ayrıca bakınız: Hareket Bozuklukları /Ataksiler).
BAĞIŞIKLIK BOZUKLUĞUNA BAĞLI POLİNÖROPATİLER
İNFLAMATUVAR DEMİYELİNİZAN POLİNÖROPATİLER
Guillain-Barré Sendromu (GBS)
Poliomiyelitin toplumdaki sıklığının azalmasından sonra GBS, akut yaygın gevşek felce neden olan hastalıkların en sık görüleni halini almıştır (insidens: 1-2/100.000). Çocukluktan ileri yaşlara kadar her yaş grubunda rastlanan otoimmün kökenli bir hastalıktır. Erkeklerde kadınlara göre biraz daha sık görülmektedir. Genellikle başka bir hastalığı olmayan kişilerde ortaya çıkar. Başka bir sistemik veya otoimmün hastalıkla birlikte olması sık karşılaşılan bir durum değildir. Birbirlerinden bazen klinik yönleriyle, bazen de daha çok laboratuvar özellikleri ile ayrılan farklı alt grupları vardır (Tablo 18).
Tablo 18. Guillain-Barré sendromlarının sınıflanması (EP Bosch ve BE Smith, değiştirilerek)
Akut inflamatuvar demiyelinizan poliradikülonöropati (AİDP)
Akut motor aksonal nöropati (AMAN)
Akut motor-duyusal aksonal nöropati (AMSAN)
Miller Fisher sendromu (MFS) ve diğer bölgesel varyantlar
Faringeal-servikal-brakiyal
Paraparetik
Yüz felçleri
Saf okülomotor
GBS’nin fonksiyonel varyantları
Akut pandizotonomi
Saf duyusal GBS
Ataksik GBS
Akut İnflamatuvar Demiyelinizan Poliradikülonöropati (AİDP)
GBS terimi, pratikte hastalığın bu en sık rastlanan klasik inflamatuvar demiyelinizan formu için kullanılır. Çünkü bu form Batı dünyasında GBS olgularının yaklaşık olarak %85-90’ında görülür.
Hastaların büyük kısmında GBS’ye ait yakınmaların başlangıcından önceki 1-4 haftalık süre içerisinde geçirilen bir üst solunum yolu ya da gastrointestinal traktus infeksiyonu, cerrahi girişim ya da aşılanma gibi bir olay vardır. AİDP’de immün saldırının hedefi periferik sinirin miyelin kılıfıdır. Hastalığın en sık yerleşme şekli, simetrik olarak bacaklardan başlayıp kollara, daha sonra yüze, orofaringeal kaslara ve ağır olgularda solunum kaslarına doğru yükselen kas kuvvetsizliği ile olur. Daha seyrek olarak, yaklaşık %10, kollarda veya kranial alan kaslarında güçsüzlükle de başlayabilir. Birçok kere polinöropati sendromu için tanımlanan genel dağılım şeklinin dışına çıkan ve ekstremite proksimallerinde de belirgin olan bir kas kuvvetsizliği görülür. Hastaların yaklaşık olarak %80’inde eşlik etmekte olan duyusal yakınmalar genel olarak daha geri plandadır ve ekstremite uçlarında parestezi ya da nadir olmayarak sırt, bel ve bacak ağrıları şeklindedir. Sinir kökü inflamasyonuna bağlı olarak ortaya çıkan ağrı yakınması hastalığın ilk semptomu olabilir ve 1/3 olguda kas güçsüzlüğünden önce başlayabilir. Nörolojik muayenede, tanımlanan alanlardaki kas kuvvetsizliğine ek olarak, hemen daima, tendon reflekslerinin yaygın şekilde kaybolduğu ya da azaldığı görülür. Ancak nadir olarak hastalığın başlangıcında ve özellikle sadece motor ve aksonal formlarında tendon refleksleri normal bulunabilir. Objektif duyusal bulgular daha çok ekstremite uçlarındaki hafif yüzeyel hipoestezi veya azalmış vibrasyon ve pozisyon duyusu ile sınırlıdır.
Olguların %10-30 kadarında mekanik ventilasyonu gerektiren solunum yetersizliği gelişir. Hastaların yarısında, en sık fasiyal paralizi olmak üzere kranial sinir felçleri görülür. Orofaringeal kaslarda kuvvetsizlik olguların yarısında, göz hareketi felçleri %10-20 kadarında gelişir. Nadir olarak papilla ödemi görülebilir ve çok artmış BOS proteini ile birlikte olur (>200 mg/dL). Otonom sinir sistemi bozuklukları hastaların yarısı kadarında görülür ve şiddetli motor zaafı, solunum yetersizliği olan olgularda daha sıktır. Otonom belirti ve bulgular arasında EKG değişiklikleri (T dalgası anormallikleri, ST segment depresyonu, QRS genişlemesi, QT uzaması ve çeşitli kalp blokları), kalp aritmileri (sinüzal taşikardi, bradikardi, ventriküler taşikardi, atrial flutter, atrial fibrilasyon ve asistoli), ortostatik hipotansiyon ve hipertansiyon krizleri, daha nadiren geçici idrar retansiyonu, terleme bozuklukları ve paralitik ileus sayılabilir. Ciddi ve kalıcı nitelikte sfinkter kusuru GBS tanısından uzaklaştırıcı bir ölçüt olarak ele alınmalıdır. Tablo 19’da GBS tanısını destekleyen ve bu tanıdan uzaklaştıran faktörler ele alınmıştır. Otonom tutuluma bağlı olarak meydana gelebileceği düşünülen uygunsuz antidiüretik hormon (ADH) sendromu da GBS’nin bir diğer komplikasyonudur. Ek olarak, otonom sistem bozukluğuna bağlı akut hipertansiyonla ilişkili posterior reverzibl lökoensefalopati sendromu da erişkin ve çocuk GBS olgularında bildirilmiştir.
Tablo 19. Tipik bir GBS tablosu için tanıyı kuvvetlendiren ve bu tanıdan uzaklaştıran faktörler (PA van Doorn ve ark., değiştirilerek)
Tanı için gerekli
Kol ve bacaklarda ilerleyici kuvvetsizlik (sadece bacaklardan başlayabilir)
Azalmış, kayıp tendon refleksleri
Tanıyı kuvvetle destekler
Semptomların günler boyunca-4 haftaya kadar ilerleyici seyri
Hafif duyusal belirti ve bulgular
Kranial sinir tutulması (özellikle bilateral fasiyal kas kuvvetsizliği)
Otonom fonksiyon bozukluğu
Ağrı (sıklıkla vardır)
Yüksek BOS proteini
Tipik elektrofizyolojik bulgular
Tanıdan şüphe ettirir
Başlangıçta şiddetli pulmoner fonksiyon bozukluğu ve ekstremitelerde sınırlı kuvvetsizlik
Başlangıçta şiddetli duyusal bulgular ve sınırlı kuvvetsizlik
Başlangıçta mesane-barsak fonksiyon bozukluğu
Başlangıçta ateş
Keskin duyu seviyesi
Sınırlı kuvvetsizlik, solunum bozukluğu yok, yavaş ilerleme (CIDP akla gelmeli)
Kuvvetsizlikte belirgin ve ısrarlı asimetri
Kalıcı mesane ve barsak fonksiyon bozukluğu
BOS’ta artmış mononükleer hücre (>50/mm3)
BOS’ta polimorfonükleer hücre varlığı
BOS: beyin omurilik sıvısı, CIDP: kronik inflamatuvar demiyelinizan polinöropati.
Kas kuvvetsizliği, şiddet ve dağılım alanı olarak progresyonunu genellikle 1-3 hafta içerisinde tamamlar (olguların %50’si 1, %80’i 3 ve %90’ı 4 haftada maksimum kas zaafına ulaşır). İki aydan daha uzun süre progresyon gösteren edinsel demiyelinizan polinöropati olgularının CIDP olarak değerlendirilmesi gerekir. Progresyon aşamasında fulminan bir seyir izleyerek 24-48 saat içinde solunum yetersizliğine kadar ilerleyen hastalar da görülebilir. Progresyonun durmasından sonra hastalık 2-4 haftalık bir plato ve bunu izleyen yavaş bir iyileşme dönemine girer. Hastaların yaklaşık dörtte birinde intravenöz immünglobulin (IVIg) ya da plazmaferez tedavileri sırasında veya tedaviden kısa süre sonra kötüleşme görülebilir.
GBS seyrinde ortaya çıkan kas kuvvetsizliğinin şiddeti büyük değişkenlik gösterir. Hastaların bir kısmı ekstremite ve yüz kaslarında hafif kuvvetsizlikle ambulatuvar kalabilirken, bir kısmında kuadripleji ve mekanik ventilasyon gerektiren solunum yetersizliği gelişir. Çağdaş yoğun bakım koşullarına rağmen olguların %2-12’sinde hastalık solunum yetersizliğine bağlı sorunlar, pulmoner embolizm ya da otonom bozukluklara bağlı kardiyovasküler olaylar nedeni ile ölümle sonlanır. Diğer hastalar 6-12 ay süren (ağır seyirli olgularda 1,5-2 yıla kadar uzayan) bir iyileşme dönemi geçirirler. Hastaların %75 kadarı yeterli bir iyileşme göstererek işine dönebilir hale gelir. Yaklaşık % 5-10’unda hareketi engelleyecek şiddette motor defisitler kalır. Olguların %2-5’inde GBS nüksü gözlenir.
Akut Motor Aksonal Nöropati (AMAN)
Kuzey Çin’de yaz aylarında görülen ve epidemiler oluşturan bir GBS formu olarak tanımlanmıştır. Diğer ülkelerde de sporadik olarak görülen bu form Batı ülkelerinde rastlanan GBS’lerin %5’ten azını oluşturur. Klinik tablo başlıca motor belirti ve bulgularla seyreder, duyusal bulgular görülmez. Kranial sinir tutulması nadirdir. Periferik sinirlerde inflamasyonun geri planda olduğu ve demiyelinizasyonun bulunmadığı bir aksonal hasar söz konusudur. Daha hızlı bir progresyon görülmekle birlikte, prognoz genellikle AİDP’de karşılaşılandan farklı değildir. Elektromiyografide başlangıçta düşük amplitüdle kaydedilen distal motor yanıtlar iyileşme ile birlikte hızla yükselebilir. Bu bulgular olasılıkla motor sinir terminallerinde ya da Ranvier nodunda reverzibl bir iletim bloğuna işaret etmektedir. İletim hızlarında belirgin derecede yavaşlama ya da distal latanslarda kayda değer bir uzama yoktur. Olguların bir kısmında akson hasarına bağlı yavaş ve yetersiz iyileşme görülürken, çoğu hızlı şekilde iyileşir. Akson hasarını izleyen sinir rejenerasyonunun beklenen yavaşlığı göz önüne alındığında bu hızlı iyileşme paterni şaşırtıcı görünmektedir (aşağıya bakınız).
Akut Motor-Duyusal Aksonal Nöropati (AMSAN)
Motor ve duyusal belirtilerle seyreden ve periferik sinirlerde inflamasyon-demiyelinizasyon yerine aksonal hasarın söz konusu olduğu bir GBS formudur (Feasby ve ark., 1986). Kas zaafının progresyonu daha hızlı, şiddeti daha fazladır. Akson hasarını izleyen yavaş sinir rejenerasyonu nedeni ile iyileşme yavaş ve çoğu kere yetersizdir. Akut motor aksonal nöropatinin (AMAN) daha ağır ve yaygın seyreden, motor ve duyusal sinir liflerini birlikte tutan bir şekli olduğu düşünülmektedir.
Miller Fisher Sendromu (MFS)
GBS’nin ekstremite ataksisi, arefleksi ve eksternal oftalmopleji ile seyreden bir diğer alt tipidir. Hastalar oftalmoplejiye bağlı çift görme, dengesizlik, hareketlerinde beceriksizlik ve bazen ekstremitelerde parestezilerden yakınır. Pupillalar genellikle normaldir. Ptoz, orofaringeal kaslar ve yüz kaslarında kuvvetsizlik olabilir. Olguların yaklaşık 1/3 ünde ekstremite ve solunum kaslarında da kuvvetsizlik gelişir. Kısmi formlarında ataksinin eşlik etmediği oftalmopleji ya da oftalmoplejinin olmadığı ataksik nöropati görülebilir. Bazı MFS hastalarında fiks dilate pupiller gelişebilir. Çok az sayıda patolojik incelemenin olduğu MFS’de ekstra-aksiyal okülomotor sinirlerde demiyelinizasyon varlığı gösterilmiştir. Bickerstaff beyinsapı ensefaliti ile MFS arasında bir ilişki olduğu, benzer otoimmün süreçlerin periferik ve merkez sinir sistemi yapılarını etkileyerek bu iki tabloyu ve bazen her ikisinin özelliklerini bir arada barındıran olguları ortaya çıkarabildiği ileri sürülmüştür (Tablo 20).
Faringeal-Servikal-Brakiyal Felç
Faringeal-servikal-brakiyal felç GBS’nin orofaringeal, boyun ve omuz kaslarının akut olarak gelişen güçsüzlüğü ve yutma bozukluğu ile seyreden bir alt tipidir. Fasiyal kaslarda da zaaf bulunabilir. Alt ekstremite kas gücü ve tendon refleksleri korunmuştur. Bazı uzmanlar MFS, Bickerstaff ensefaliti ve faringeal-servikal-brakiyal felç tablolarını birbiri ile örtüşen özellikler gösterebilen anti-GQ1b antikor sendromu olarak ele almaktadır.
Paraparetik Form
Kas güçsüzlüğünün hastalık başlangıcında alt ekstremitelerde sınırlı kaldığı, görece hafif bir GBS varyantıdır. Takipleri sırasında bazı olgularda üst ekstremite güçsüzlüğü eklenebilir. Bununla birlikte, bu alt tipteki çoğu hastanın üst ekstremite derin tendon refleksleri azalmış ya da kayıptır ve yaklaşık %90 olgunun üst ekstremite elektrofizyolojik incelemelerinde anormallik saptanır.
Diğer Varyantlar
Akut pandizotonomide diyare, kusma, başdönmesi, karın ağrısı, ileus, ortostatik hipotansiyon, üriner retansiyon, pupiller anomali, değişmeyen kalp hızı, terleme, tükrük ve gözyaşı salgısında azalma gibi semptomlar izlenir. IVIg’e yanıt verir.
Saf duyusal GBS’de kalın miyelinli duyusal lifler etkilenir ve ağır duyusal ataksi ile seyreder. Derin tendon refleksleri kayıptır ve hafif motor tutulum görülebilir. GD1b antikorları ile ilişkili olabileceği bulunmuştur.
Hastalık öncesi olaylar
Yukarıda belirtildiği gibi GBS’li olguların %90 kadarında hastalığın 1-3 hafta öncesinde geçirilmiş bir infeksiyon söz konusudur (üst solunum yolu infeksiyonu %60, gastrointestinal infeksiyon %30). En sık olarak belirlenen infeksiyon ajanı Campylobacter jejuni’dir (Tablo 21). AMAN ve akut motor duyusal aksonal nöropati (acute motor sensory axonal neuropathy=AMSAN) olgularında %60-70, AİDP olgularında ise %30’a varan oranlarda C. jejuni infeksiyonu bildirilmektedir. Epstein-Barr virüsü, sitomegalovirüs (CMV), Haemophilus influenzae ve Mycoplasma pneumoniae GBS ile ilişkisi bilinen infeksiyon ajanlarıdır. Hepatit A, B, E ve C, varisella-zoster virüs (VZV), HIV ve herpesvirüs infeksiyonlarının da GBS ile ilişkisi bildirilmiştir. C. jejuni infeksiyonunu izleyen GBS’ler daha çok motor belirtilerle ve aksonal tutulma ile seyretmektedir. Ayrıca bu olgular daha yavaş iyileşme göstermektedir ve artmış özürlülük oranları ile daha kötü prognoza sahiptir. CMV infeksiyonundan sonra gelişen GBS olgularının ise daha genç, duyusal belirti ve bulguları belirgin, daha sık kranial sinir tutulması ve solunum yetersizliği olan hastalar olduğu bildirilmiştir. Koronavirus hastalığı 2019 (COVID-19) ve zika virüs infeksiyonlarının eşlik ettiği GBS olguları bildirilmekle birlikte, aralarında doğrudan nedensel bir ilişkinin varlığı henüz aydınlatılamamıştır. GBS’ye öncelik ettiği bildirilen diğer olaylar, aşılanmalar (influenza, kuduz, tetanoz ve difteri toksoidleri, oral poliomiyelit aşısı), cerrahi girişimler ve bazı ilaçların kullanımıdır (streptokinaz, suramin, gangliozidler ve eroin). GBS, sistemik lupus eritematozus (SLE) gibi hastalıkların seyrinde ya da Hodgkin hastalığı, erken HIV infeksiyonu, kemik iliği veya organ transplantasyonu nedeniyle yapılan farmakolojik immünosupresyon gibi immün yetersizlik durumlarında da ortaya çıkabilir. GBS ile aşılanmalar arasında bir ilişki bulunsa bile bu zayıf ve düşük olasılıklı bir ilişkidir. Örneğin, influenza aşısı ile GBS riskinde küçük bir artış görülebileceği bildirilse de, bunun hastalığın kendisi ile gelişebilecek olan tüm sağlık risklerine oranla çok daha az bir öneme sahip olduğu düşünülmektedir. Ayrıca influenza hastalığı ile GBS gelişme riski, aşısına oranla birkaç kat daha fazladır. Bununla birlikte, GBS geçiren olgularda relaps riskini düşürmek için influenza aşılamasını 1 yıl kadar geciktirmek yerinde bir davranıştır. Herhangi bir aşılamanın ardından GBS geliştiğinden kuşkulanılan bir hastada aynı aşının yeniden uygulanmasından kaçınmak gerekir. Son olarak, COVID-19’a karşı halihazırda geliştirilmekte olan aşıların toplum içerisinde yaygın bir şekilde uygulanmaya başlamasının ardından GBS sıklığında bir artış olabileceği ihtimali üzerinde de durulmaktadır.
Patoloji ve patogenez
Patolojik değişiklikler GBS alt tipine göre değişiklik gösterir. AİDP ve MFS’de fokal inflamatuvar yanıt periferik miyeline ya da miyelini yapan Schwann hücrelerine karşı oluşur. AİDP formunda başlıca histopatolojik bulgular endonöriumda mononükleer inflamatuvar infiltrasyon ve sinir liflerinde segmental demiyelinizasyondur. Sinir köklerinden distal intramüsküler sinir dallarına kadar periferik sinirlerin her tarafında bu lezyonlar görülmekle birlikte, daha çok motor kökler ve proksimal pleksus segmentleri tutulur (Şekil 8). AMAN ve AMSAN formlarında ise iltihabi infiltrasyonun görülmediği bir akson hasarı söz konusudur.
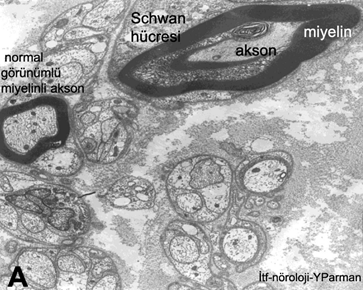
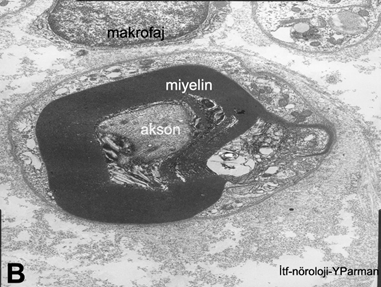
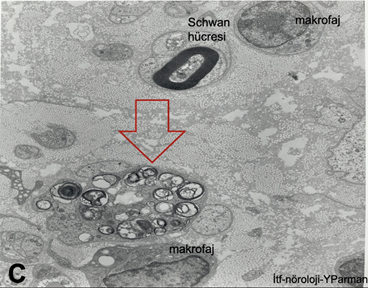
Şekil 8. Akut inflamatuvar demiyelinizan polinöropatide (AİDP) elektron mikroskopisi bulguları. A ve B: GBS’de makrofaj aracılı «myelin stripping» (uranyl asetat), C: Makrofaj aracılı demiyelinizasyonu izleyen miyelin parçalanması ve akson dejeneresansı (ok) (A: X4150, B: X6450, C:X2250).
GBS’ye yol açan patolojik süreçleri periferik sinirlerin yapısında yer alan antijenlere karşı oluşan otoantikorların başlattığı kabul edilmektedir. GBS’li hastaların kanlarında oldukça yüksek oranda (yaklaşık %60) antigangliozid antikorlar gösterilmiştir. Gangliozidler periferik sinir membranlarında “lipid salları” denen katmanlarda bulunur ve membran bütünlüğünün korunmasında görev alırlar. IgG anti GM1, GM1b, GD1a ve GalNAc-GD1a antikorlar motor ağırlıklı olgular ya da AMAN ve AMSAN’lı hasta gruplarında daha yüksek oranda görülürler. GD3, GT1a, ve GQ1b’ye karşı antikorlar ise oftalmopleji ve MFS ile ilişkili görünmektedir. Antigangliozid antikorlar içerisinde belirli bir GBS alt grubu ile en anlamlı ilişkiyi gösteren IgG anti-GQ1b antikorudur. Bu antikor MFS olgularının yaklaşık %90’ında akut fazda yüksek titrelerde saptanır ve klinik düzelmeyle birlikte kaybolur (Tablo 20 ve 21). Bazı antikorlar ise tek tek gangliozidlere değil, farklı gangliozid komplekslerinin oluşturduğu yeni konformasyonel epitoplara bağlanırlar. Yakın zamanda GD1a/GD1b ve GD1b/GT1b komplekslerine karşı antikorların daha ağır seyirli GBS formlarından sorumlu olduğu gösterilmiştir.
Tablo 20. Anti-glikolipid antikorlar ve ilişkili oldukları otoimmün polinöropati sendromları (HJ Willison ve N Yuki, değiştirilerek).
|
Klinik Sendrom |
Antikorun karşı olduğu glikolipid |
Antikor izotipi |
|
Kronik duyusal-motor demiyelinizan nöropati |
SGPG, SGLPG |
IgM (monoklonal) |
|
Kronik ataksik nöropati |
GD1b, GD2, GD3, GT1b, GQ1b |
IgM (monoklonal) |
|
Multifokal motor nöropati |
GM1, GD1b, asialo-GM1 |
IgM (poliklonal veya monoklonal) |
|
Akut motor aksonal nöropati |
GM1, GM1b, GD1a, GalNac-GD1a
|
IgG |
|
Miller Fisher sendromu |
GQ1b, GT1a |
IgG |
|
Bickerstaff beyinsapı ensefaliti Akut oftalmoparezi Ataksik Guillain-Barré sendromu Faringeal-servikal-brakiyal felç |
GT1a (GQ1b) |
IgG |
SGLPG: sülfat-3-glukronil laktozaminil paraglobozid, SGPG: sülfat-3-glukronil paraglobozid
Tablo 21. GBS’de antigangliozid antikorlar, gangliozid lokalizasyonları ve birlikte olan klinik tablolar (S Kusunoki ve ark., değiştirilerek)
|
Hedef antijen
|
Sınıf |
Antijenin bulunduğu yer |
Klinik özellikler |
|
GQ1b |
IgG |
III, IV, VI. kranial sinirlerin paranodal miyelini, Bazı arka kök gangliyon nöronları |
MFS Oftalmoplejili GBS Bickerstaff beyinsapı ensefaliti |
|
GD1 monospesifik |
IgG |
Paranodal miyelin Arka kök gangliyonu büyük nöronları
|
AİDP Ataksik GBS |
|
GalNAc-GD1a
|
IgG |
Periaksonal membran |
AMAN Saf motor GBS |
|
LM1 |
IgG |
Miyelin |
AİDP |
|
GM1 |
IgG |
Belirlenmemiş |
AMAN Saf motor GBS |
|
GD1a |
IgG |
Belirlenmemiş |
AMAN |
AİDP: akut inflamatuvar demiyelinizan poliradikülonöropati, AMAN: akut motor aksonal nöropati, GBS: Guillain-Barré sendromu, MFS: Miller Fisher sendromu.
Tablo 22. Nöropati ile ilişkisi gösterilmiş mikroorganizmaların taklit ettikleri glikolipidler (HJ Willison ve N Yuki, değiştirilerek)
|
Mikroorganizma |
Taklit edilen glikolipid |
|
Campylobacter jejuni |
GM1, GM1b, GD1a, GalNac-GD1a, GD3, GT1a, GQ1b |
|
Haemophilus influenzae |
GM1, GT1a |
|
Mycoplasma pneumoniae |
Galaktoserebrosid |
|
Sitomegalovirüs |
GM2 |
AİDP formunda Schwann hücresi yüzeyindeki epitoplara bağlanan antikorların kompleman aktivasyonuna yol açtığı, bunun da miyelinin vakuolizasyonu, yıkımı ve makrofajlarla fagositozuna varan bir süreci başlattığı öne sürülmüştür. Süreç içerisinde hem hücresel hem de hümoral immün yanıtın rol oynadığı düşünülmektedir. Aktive olmuş T hücrelerinin invazyonunu makrofaj aracılı demiyelinizasyon takip etmekte, Schwann hücreleri ile miyelinde kompleman ve immünglobulin birikimi görülmektedir. Henüz AİDP ile ilişkili olabilecek spesifik bir miyelin antijeni/leri saptanmamış olmakla birlikte Ranvier nodunda yer olan gliomedin, kontaktin, “transiently expressed axonal surface glycoprotein-1” (TAG-1), moesin ve nörofasin gibi özel protein yapılarına karşı antikorların varlığı bildirilmiştir. Henüz üzerinde daha fazla araştırmaya ihtiyaç duyulsa da, nodal kompleksleri hedef alan immün yanıtın GBS’nin patolojik kaskadında önemli bir yer tuttuğu düşünülmekte ve bu durum nodo-paranodopati terimiyle ifade edilmektedir (Şekil 9). Nod bölgesi protein ve glikolipid yapıdaki olası potansiyel antijenler açısından oldukça zengindir. Bu bölge ayrıca fonksiyonel olarak da antikor birikimi, kompleman aktivasyonu ve makrofaj toplanması gibi patolojik saldırılara karşı hassas bir yapıdadır. AMAN ve AMSAN’da ise otoimmün olayların ilk hedefinin Ranvier nodlarında aksonal membranda yer alan gangliozid yapısındaki antijenler olduğu, buralara bağlanan antikorların bir yandan muhtemelen iyon kanalı disfonksiyonuna yol açan olayları başlattığı, diğer yandan kompleman aktivasyonunu izleyerek Ranvier nodlarına makrofajların çekilmesi, periaksonal mesafenin açılması, buraya makrofajların migrasyonu ve sonuçta akson dejenerasyonu ile giden bir sürece yol açtığı düşünülmektedir. AMAN’lı olguların bir kısmında gelişen aksonal hasarla uyumlu şekilde ağır kas zaafı, yavaş ve yetersiz iyileşme ile belirlenen bir klinik seyir görülmektedir. Bir kısım olgularda ise, iyi seyirli AİDP olgularında görülenden farksız hızlı ve tam bir klinik düzelme dikkati çekmektedir. Aksonal dejenerasyonu izleyen rejenerasyon için gereken uzun zaman göz önüne alındığında bu durum şaşırtıcı görünmektedir. Bu olgularda aksonal membrana bağlanan antikorların iyon akımlarını etkileyerek iletim bloklarına yol açtığı; sonuçtaki hızlı iyileşmenin ise iletim bloklarının düzelmesi ile sağlandığı düşünülmektedir. Patolojik süreç bu geri dönüşümlü iletim bloğu aşamasından iyileşmeye doğru gitmez, akson hasarı oluşturacak şekilde ilerlerse AMAN/AMSAN olgusunun ağır ve güç iyileşen bir klinik seyri olacaktır. Hafif kas kuvvetsizliği ve hızlı iyileşme ile seyreden AMAN olgularında şaşırtıcı bir diğer bulgu olarak tendon reflekslerinin korunmuş olduğu da görülebilir.
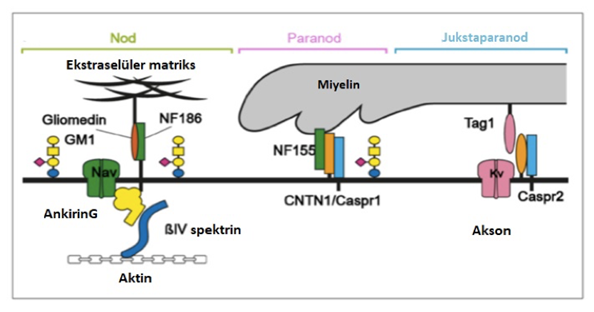
Şekil 9. Nod, paranod ve jukstaparanod bölgelerinin sadeleştirilmiş organizasyonu (A Uncini, değiştirilerek)
GBS’ye yol açan otoantikorların ortaya çıkış mekanizmaları ile ilgili bilimsel veriler, bunların bir infeksiyon ajanına karşı oluşturulduğunu ve periferik sinirlere yöneldiğini düşündürmektedir. GBS ile ve özellikle onun aksonal formları ile ilişkisi en sık gösterilen bakteriyel organizma olan C. jejuni, dünya çapında yaygın bir bakteriyel enterit nedenidir. Çeşitli olgu serilerinde, yakında geçirilmiş bir C. jejuni infeksiyonu ile başlıca motor bulgular ve akson hasarı ile seyreden GBS ve yüksek anti GM1, anti GD1a veya anti GQ1b antikor titreleri arasında ilişki olduğu görülmüştür. MFS’li olgulardan izole edilen C. jejuni örneklerinde GQ1b benzeri epitoplar da bulunmuştur. Bu bulgular, yüzeyindeki antijenik yapıları gangliozidler gibi sinir dokusu yapı maddelerini taklit eden mikroorganizmalara karşı konağın oluşturduğu antikorların sinir dokusuna yönelerek hastalık sürecini başlatabileceğini düşündürmektedir (moleküler taklit mekanizması, Tablo 22). C. jejuni enteriti olmasına karşın GBS gelişmeyen hastalarda ise anti-gangliozid antikorları üretilmemektedir. GBS’li olgulardan izole edilen C. jejuni suşlarının gangliozidlerin karbonhidrat epitoplarını taklid eden lipo-oligosakkaridler (LOS) oluşturdukları gösterilmiş, LOS tiplerinin oluşumundan sorumlu olan bakteri gen kümeleri belirlenmiştir. Bu kümelerdeki belirli gen varyantları gangliozid benzeri LOS’ların ekspresyonundan sorumlu görünmektedir. Bu şekilde belirli bir gangliozidin moleküler yapısının LOS tarafından taklit edilmesi antigangliozid antikorların özgüllüğünü ve buna bağlı olarak, söz konusu gangliozidlerin sinir sisteminde baskın olduğu bölgelere göre ortaya çıkacak GBS varyantlarını belirler görünmektedir (Tablo 21,22). Saf motor/aksonal nöropatisi olan hastalardan elde edilen C. jejuni suşları yüzeylerinde GM1 ve GD1a benzeri LOS’lar sunarken, oftalmopleji ile birlikte olan GBS olguları ve MFS’li olgulardan elde edilenler GD3, GT1a, GD1c benzeri LOS’lar sunmaktadırlar. Bir tavşan modelinde GM1 benzeri LOS ile immünizasyon yoluyla anti-GM1 antikorlar ve klinik planda aksonal polinöropati oluşturulmuştur. Tavşan modeliyle yapılan ileri incelemelerde anti-GM1 antikorlarının Ranvier nodunda voltaj kapılı sodyum kanallarına bağlandığı görülmüştür. Benzer şekilde, bir MFS fare modelinde anti-GQ1b antikorlarının presinaptik sinir terminalinde yerleşen sodyum kanallarına bağlandığı gösterilmiştir. Bu süreç sodyum kanal blokajına yol açarak reverzibl ileti bozukluğuna neden olabilir. Antikor bağlanması kompleman aracılı olmaktadır ve kompleman inhibitörleri ile iyileşme gözlenebilmektedir. Moleküler taklit mekanizması H. influenzae gibi bazı başka infeksiyon ajanları için de gösterilmiştir. Diğer yandan, C. jejuni infeksiyonu geçiren kişilerin 1/1000’inden azında GBS gelişmesi, gangliozidleri taklid eden C. jejuni varyantları ile infekte olan ailelerde bile salgınlar görülmemesi gibi özellikler, GBS’nin ortaya çıkışında konağa ait faktörlerin de çok önemli olduğunu göstermektedir.
Akut-subakut seyirli oldukça saf bir otonom nöropati olarak tanımlanan akut pandizotonomi (otoimmün otonomik gangliopati=AAG) başlıca ortostatik hipotansiyon, yetersiz terleme, gözyaşı ve salya salgılanmasında azalma, gastrointestinal motilite bozuklukları, atonik mesane, impotans ve sabit kalp atım hızı ile seyreder. Bu hastaların bir kısmında da hastalığa öncelik eden infeksiyon ya da aşılanma gibi bir olay bulunur. AAG olgularının önemli bölümünde gangliyonik tipte (a-3 tipi) nikotinik asetilkolin reseptörlerine karşı antikorlar bulunur.
Laboratuvar Bulguları
GBS’de tanıya en çok yardımcı olan laboratuvar yöntemleri elektrofizyolojik incelemeler (elektromiyografi ve sinir iletim incelemeleri) ile BOS incelemesidir.
Rutin laboratuvar incelemeleri: Eritrosit sedimentasyon hızı, lökosit sayısı ve CK düzeyinde hafif artış olabilir. Uygunsuz ADH salımına bağlı hiponatremi görülebilir.
Beyin omurilik sıvısı (BOS): Protein düzeyi artmıştır. Genel olarak hücre artışı görülmez (albuminositolojik disosiyasyon). Kan-BOS bariyerinin bozulmasına bağlı olan protein artışı genellikle 48 saatten (daha belirgin olarak ilk haftadan) sonra ortaya çıkar ve 3.-4. haftada en üst düzeye ulaşır. Birinci haftanın sonunda olguların %66’sında BOS protein yüksekliği bulunurken, %10 kadarında BOS protein düzeyi hiçbir bir zaman yükselmez. GBS’li olguların BOS’larında oligoklonal bandların varlığı gösterilebilir. BOS’taki hücre sayısı genel olarak mm3’te 10 mononükleer hücreyi geçmez (nadiren 50/mm3 mononükleer hücre görülebilir). BOS pleositozu ile seyreden bir GBS olgusunda leptomeningeal malign hastalıklar, Lyme hastalığı, HIV infeksiyonu ya da poliomiyelit gibi diğer tanı olasılıkları düşünülmelidir.
Elektrofizyolojik incelemeler: AİDP de ilk anormal elektrofizyolojik bulgu genellikle uzun latanslı, düşük persistanslı ya da kaydedilemeyen F yanıtlarıdır. Bu bulgu sinir kökleri ya da pleksuslar gibi proksimal sinir segmentlerindeki demiyelinizasyonu yansıtır. F yanıtlarının araştırılması sırasında gözlenen tek (normal bireylerde görülebildiği tibial sinir F incelemesi hariç) ve özellikle multipl A dalgaları da GBS’nin erken bir elektrofizyolojik tanı bulgusu olarak ele alınır. İlerleyen haftalar içinde multifokal demiyelinizasyonla seyreden polinöropatiyi gösteren elektrofizyolojik veriler yerleşir. Motor iletim incelemelerinde sinir iletim hızları belirgin derecede ve multifokal nitelikte bir yavaşlama gösterir, sinir üzerinde uyarım noktası proksimale doğru kaydıkça iletim blokları ve kas yanıtlarında dispersiyonla karşılaşılır. Polinöropatinin şiddetine bağlı olarak duyusal aksiyon potansiyelleri geç latanslı ve düşük amplitüdlü olarak elde edilir ya da kaydedilemez. Üst ekstremite distallerinde median ve ulnar sinir duyusal aksiyon potansiyelleri kaydedilemez ya da çok düşük amplitüdlü bulunurken sural sinir duyusal aksiyon potansiyelinin elde edilebilir olması inflamatuvar demiyelinizan polinöropatiler için oldukça kuvvetli bir tanı kriteridir ve GBS’de erken dönemden itibaren saptanabilir (sural kurtulma paterni). İğne elektromiyografisinde erken dönemde sadece motor ünite potansiyeli seyrelmesi izlenir. İlerleyen haftalarda az ya da çok miktarda sekonder aksonal hasarın gelişmesiyle fibrilasyon ve pozitif diken potansiyelleri ile parsiyel denervasyon-reinnervasyon sürecinin diğer bulguları görülmeye başlanır. Göz kırpma refleksi ve fasiyal sinir iletim incelemeleri hastalık tanısında yardımcı incelemeler olarak kullanılabilmektedir.
AMAN olgularının motor iletim incelemelerinde bileşik kas aksiyon potansiyeli (BKAP) amplitüdlerinin belirgin derecede düşük olduğu görülür. Bu olguların iğne elektromiyografisinde yoğun denervasyonu yansıtan fibrilasyon ve pozitif diken potansiyelleri ile seyrelmiş motor ünite potansiyeli aktivitesi dikkati çeker. MFS olgularının elektrofizyolojik incelemelerinde ise başlıca duyusal bir aksonopatiyi yansıtan bulgular elde edilir. Duyusal aksiyon potansiyelleri kaydedilemez ya da düşük amplitüdlü bulunurken, genellikle kayda değer düzeyde motor sinir iletim bozukluğu saptanmaz.
Motor iletim incelemelerinde ekstremite distalinden yapılan elektriksel uyarımla elde edilen BKAP amplitüdlerinin çok düşük olması ağır motor akson hasarını gösterebileceğinden, yeterli iyileşme açısından kötü prognozu işaret eden bir faktör olarak ele alınır (Ayrıca bakınız: Nörolojide Laboratuvar İncelemeleri, Elektromiyografi). Diğer yandan, demiyelinizasyon bulguları olan GBS olgularının daha yüksek oranda solunum yardımına gereksinimi olduğu da bilinmektedir.
Antigangliozid antikorlar: Patogenez bölümünde söz edildiği gibi bazı antigangliozid antikorlarla bazı GBS formları arasında ilişki vardır. Ancak bunlardan sadece IgG anti-GQ1b ile MFS arasındaki ilişki tanıya yardımcı olabilecek kadar yüksektir. MFS varlığı düşünülen hastalar dışındaki GBS olgularında antigangliozid antikor araştırmasının belirgin bir tanısal yardımı olmaz. Bununla birlikte, IgG anti-GM1, GD1a gibi antikorların yüksek titrede varlığı AMAN tanısını destekleyici bir bulgu olarak ele alınabilir.
Sinir biyopsisi: Klinik veriler, elektrofizyolojik incelemeler ve BOS bulguları tanı için hemen daima yeterli olduğundan GBS olgularında sinir biyopsisine gerek duyulmaz.
Sinir ultrasonu ve manyetik rezonans görüntüleme (MRG): Özellikle çocuk yaş grubunda daha az invazif bir alternatif olarak tanıya katkı sağlayabilmektedir. Semptom başlangıcından 1-3 gün sonra sinirlerde genişleme görülebileceği, fakat bu genişlemenin hafif olduğu ve segmentli olarak dağıldığı bildirilmektedir. GBS’de en fazla spinal sinir kökleri ve proksimal sinir segmentleri tutulmakla birlikte, etkilenen bölgelerin dağılımı açısından hastalık alt tipleri arasında farklılıklar görülebilir. MRG incelemesi ise ayırıcı tanıda yer alan patolojilerin dışlanması amacıyla kullanılabileceği gibi, sinir köklerinde genişleme ve kontrastlanma varlığının gösterilmesi ile GBS tanısına katkı sağlayabileceği düşünülmektedir.
Ayırıcı Tanı
Tablo 23’de hızlı gelişen genel kas zaafı ile seyreden ve GBS ile ayırıcı tanısı gereken hastalık tabloları verilmiştir.
Tablo 23. GBS’yi taklit edebilecek durumlar (IR Bella, değiştirilerek)
|
Hastalık |
Başlıca ayırt edici özellikler |
|
Myasthenia gravis |
Tendon refleksleri korunur Oküler kaslardaki kuvvetsizlik belirgin Antikolinesteraz ilaçlara yanıtlı EMG: ardısıra uyarımla motor yanıtta dekrement, tek lif EMG’sinde artmış jitter ve blok |
|
Botulizm |
Belirgin bulber tutulma olur Otonomik tutulma (pupillalar) EMG: normal sinir iletim hızları, düşük amplitüdlü kas yanıtları, yüksek frekanslı ardısıra uyarımla ya da kısa süreli maksimal kası ile kas yanıtlarında anlamlı amplitüd artışı |
|
Kene paralizisi |
1-2 günde hızlı progresyon |
|
Toksik nöropatiler |
EMG: genellikle aksonal polinöropati ile uyumlu bulgular |
|
Organofosfor toksisitesi |
Akut kolinerjik reaksiyon görülür |
|
Porfirik nöropati |
Mental bozukluk Karın ağrısı EMG: Başlıca motor aksonal bir polinöropatiyi gösteren bulgular |
|
Difterik nöropati |
Öncelik eden farenjit Yavaş gelişim Palatal felç, akomodasyon felci Miyokardit |
|
Poliomiyelit |
Kaslarda kuvvetsizlik, ağrı ve duyarlılık Duyu korunmuştur BOS: Protein ve hücre artışı |
|
Periyodik paralizi |
Refleksler normaldir Kranial sinirler ve solunum korunur Serum K+ düzeyi anormal bulunabilir |
|
Kritik hastalık polinöropatisi |
Sepsis ve multiorgan yetersizliği (2 haftadan uzun) EMG: Aksonal polinöropati ile uyumlu |
|
Akut yoğun bakım miyopatisi |
Kuadriparezi ve arefleksi vardır Nöromüsküler bloker ilaç ve kortikosteroid kullanımını izler Travma, status astmatikus, organ transplantasyonu vb. ile ilgili yoğun bakım tedavisini izler EMG ve diğer laboratuvar bulguları miyopati ile uyumludur |
|
Vaskülitlere bağlı nöropati |
Sistemik belirti ve bulgular Duyusal ve motor bulgularda asimetri vardır Refleksler kas zaafı ile orantılı olarak azalır |
|
Kuduz |
Ateş Duyusal ve motor bulgular asimetriktir Kas spazmları, ajitasyon, anormal mental durum |
BOS: beyin omurilik sıvısı, EMG: elektromiyografi.
Tedavi
Destekleyici tedavi: Yukarıda anlatıldığı gibi, GBS’nin seyri sırasında solunum yetersizliği başta olmak üzere hastanın yaşamını tehlikeye sokan birçok sorunla karşılaşılabilir. Ağır felç ve çok sayıda tıbbi komplikasyonun görülebildiği akut dönemin geçmesinden sonra ise hastaların çoğu için yeniden normal, üretken bir yaşama başlama olasılığı vardır. Bu nedenle GBS’nin akut döneminde dikkatli ve özenli bir tıbbi bakımın uygulanması çok önemlidir.
Hastalığın erken döneminde gelişebilecek solunum yetersizliği açısından dikkatli bir gözlem yapılması gerekir. Kas kuvvetsizliğinin progresyon gösterdiğinden kaygı duyulan her hasta hastaneye yatırılarak izlenmelidir. Kuvvetsizliği belirginleşen hastaların olanaklar elverdiğince yoğun bakım ünitelerine alınması gerekir. Bir hastanın mekanik ventilasyona gereksinimi olacağı konusunda en dikkat çekici parametreler, hastalık başlangıcından hospitalizasyona kadar olan sürenin 7 günden kısa olması, yatakta başını kaldırmada güçlük, bulber fonksiyon kusuru varlığı ve hastanın vital kapasitesinin %60’ın altına düşmesidir. Zorlu vital kapasite ve maksimum inspirasyon basıncı sık olarak (2 saatte bir) izlenmelidir. Yeterli oksijenizasyon ve gelişmesi muhtemel hiperkapninin arteriyel kan gazları ile izlenmesi yararlı olur. Vital kapasitenin hızlı düşüş göstermesi ya da 15 ml/kg’ın altına inmesi halinde solunum yardımı uygulanmalıdır.
Hastada solunum yetersizliğini gösteren klinik belirtilerin de dikkatle izlenmesi gerekir. Bunlar huzursuzluk ya da uyuklama hali, artan solunum sayısı, tek nefeste sesli olarak 20’ye kadar sayamama, aksesuar solunum kaslarının kullanılması, inspiryumda karın duvarının paradoksal içe hareketi ve öksürme kuvvetinin azalmasıdır. Solunum cihazları ile yardım için başlangıçta trakeal intübasyon yeterli olur. Çünkü solunum yardımı uygulanan hastaların yaklaşık üçte birinde birkaç gün içinde ekstübe edilmelerini sağlayacak kadar hızlı bir düzelme olur. Solunum yetersizliği bir haftadan daha fazla uzayan olgularda trakeostomiye başvurulması gerekir.
Otonom sinir sistemi bozuklukları, özellikle kan basıncı, kalp ritmi ve sıvı dengesi açısından izlenmelidir. Hipotansiyonla mücadele için sıvı infüzyonları yapılabilir. Hipertansiyon ancak uzun süreli ve şiddetli olması halinde tedavi edilmelidir; otonom disfonksiyon nedeniyle antihipertansiflerle aşırı hipotansiyon oluşturma riski mevcuttur. Bu amaçla kısa etkili alfa-adrenerjik blokerler kullanılabilir.
Hastanın beslenmesi diğer önemli bir genel bakım konusudur. Birkaç günlük bir intravenöz sıvı tedavisinin ardından besin yetersizliğine bağlı doku değişiklikleri ortaya çıkmaya başlayacağından, yutamayan hastaları ilk hafta içinde nazogastrik tüp veya gastrostomi ile beslemeye başlamalı ya da parenteral nütrisyon uygulanmalıdır. Göğüs fizyoterapisi egzersizleri ile pulmoner hijyen korunmaya çalışılmalıdır. GBS’nin sık görülen bir komplikasyonu olan bronkopnömoni önlenmeye çalışılmalı, gelişebilecek solunum yolu infeksiyonları enerjik şekilde tedavi edilmelidir. Hastaların yatağa bağımlı olduğu dönemde derin ven trombozu ve pulmoner embolizme karşı profilaktik subkutan heparin tedavisi uygulaması gerekir.
Ağır kas kuvvetsizliği olan hastaların yatak içindeki pozisyonlarının çok sık olarak değiştirilmesi, hem yatak yaralarını ve kompresyon nöropatilerini önlemek, hem de ekstremitelerindeki ağrı ve huzursuzluk hissini azaltmak açısından çok önemlidir. Hastanın yatırıldığı ilk günlerden itibaren hafif fizyoterapi egzersizlerine başlanmalı ve ekstremitelerde kontraktürlerin gelişmesi önlenmeye çalışılmalıdır. GBS seyrinde sık görülen ekstremite ve sırt ağrılarına karşı hafif analjeziklerin kullanılması tercih edilmeli, narkotiklerden sakınılmaya çalışılmalıdır (konstipasyon oluşturma riski).
İmmünoterapi: GBS’nin immünolojik tedavisinde plazmaferez ya da IVIg kullanılır. Çok merkezli kontrollü çalışmalar, bu tedavilerin hastalık başlangıcından itibaren ilk 3-4 hafta içerisinde uygulanması halinde tedavi verilmeyenlere oranla belirgin derecede daha hızlı bir iyileşme sağladığını ve bu iki tedavi seçeneğinin etkinlikleri arasında kayda değer bir farklılık olmadığını göstermiştir. Tedaviye daha erken başlanması (ilk 2 hafta) daha yüksek etki sağlar.
Plazmaferez genellikle 2 haftalık bir süre içerisinde uygulanacak 5 seansta toplam 5 plazma hacmi kadar değişim hedeflenerek yapılır. Hafif olgularda 2 seans plazmaferez yeterli olurken, orta ve ağır şiddetteki hastalara 4 plazmaferez seansının gerekli olduğu, ağır hastaların ek olarak uygulanan 2 seanslık plazmaferezden (toplam 6) ek bir yarar sağlamadığı gösterilmiştir. Plazmaferez iyi bir venöz ulaşım yolu (santral venöz kateter gibi) gerektirir ve ancak büyük tıp merkezlerinde uygulanabilen bir tedavi yöntemidir. Kardiyovasküler instabilitesi ve otonom disfonksiyonu olan hastalar tarafından tolere edilmesi, çocuklara uygulanması çok güçtür. Sepsis gibi ağır yan etkileri daha sıktır. Bu nedenlerle uygulanması daha kolay olan (plazmaferez ile hemen hemen aynı olan yüksek maliyeti dışında) IVIg yaygın kullanım alanı bulmaktadır. IVIg genel olarak 2 g/kg olan toplam dozun 5 gün içinde 0,4 g/kg lik günlük dozlar halinde verilmesiyle uygulanır. Hastaların %10’dan azında gribal semptomlar, başağrısı, kimyasal menenjit, bulantı, yorgunluk hali gibi hafif yan etkiler görülür. Bunlar tedavi öncesinde verilecek difenhidramin gibi antialerjikler, parasetamol ve düşük doz kortikosteroid uygulamasıyla önlenebilir. Ağır yan etkiler çok nadirdir. Bunlar arasında anafilaktik reaksiyon (başlıca IgA yetersizliği olan hastalarda görülür, bu nedenle tedavi öncesinde immünglobulin A düzeylerine bakılması gerekir), böbrek yetersizliği (daha çok renal hastalığı olan hastalarda görülür, tedavi öncesinde böbrek fonksiyonları yönünden bir değerlendirme yapılması yerinde olur) ve inmeye neden olabilen tromboembolik komplikasyonlar sayılabilir. 2018’de “International Guillain-Barré Syndrome Outcome Study (IGOS)” veri bankasının geriye dönük olarak tarandığı gözlemsel bir çalışmada, kötü prognoza sahip GBS olgularına 2. kez IVIg kürü uygulanmasının ek bir fayda sağlamadığı saptanmıştır. Plazmaferez ve IVIg tedavilerinin kombine edilmesinin tek tek uygulanmalarına bir üstünlüğü olmadığı gösterilmiştir.
Plazmaferez ve IVIg uygulanan hastaların %3-10’unda tedavi sonrasındaki ilk bir iki hafta içerisinde GBS nüksü görülebilir (Tedavi ile ilişkili dalgalanma). Bu durumda genellikle ikinci bir IVIg kürü uygulanır (2-5 günde 2g/kg). Bu tablonun görüldüğü olguların akut başlangıçlı CIDP’lerden ayırt edilip uygun tedavi verilmesinde güçlükle karşılaşılabilir. CIDP hastalarının yaklaşık olarak %5-16 kadarında hastalık akut olarak başlar ve bu hastaların hemen hemen hepsi başlangıçta GBS tanısı alır. Üçten fazla yeni kötüleşme epizodu görülen ya da hastalık başlangıcından itibaren 9. haftadan sonra kötüleşen olgularda akut başlangıçlı CIDP’yi düşünmek gerekir. GBS’yi düşündüren akut bir başlangıcı olduğu halde IVIg tedavisine yanıt vermeyen olgularda da ağır seyirli, tedaviye kötü yanıtlı GBS’nin yanısıra akut başlangıçlı CIDP olasılığını akla getirmek gerekir.
GBS’de kortikosteroidler genellikle etkisizdir. IVIg ile kortikosteroidlerin kombine kullanımı da yalnız başına IVIg uygulamasına oranla daha yararlı olmamıştır. Tedaviye eklenen mikofenolat mofetilin de yararlı bir katkısı gösterilememiştir. GBS tedavisinde henüz IVIg ve plazmaferez dışında etkili bulunan bir farmakolojik ajan bulunmamaktadır. Bir insan monoklonal antikoru olan ve kompleman faktör C5’e yüksek afinitiyle bağlanarak aktif formu olan C5a’ya dönüşümünü engelleyen ekulizumab tedavisi ile ilgili çalışmalar ise devam etmektedir.
GBS’de prognozu ağırlaştıran başlıca faktörler, ileri yaş, başka bir ciddi tıbbi hastalıkla birlikte olma, GBS’nin ağır ve hızlı seyretmesi, hastalığın ilk haftasında entübasyon ve ventilatör desteğine ihtiyaç duyulması, ilk elektrofizyolojik incelemelerde motor kas yanıtlarının ileri derecede düşük amplitüdlü olması ve hastalığa öncelik eden C. jejuni infeksiyonunun varlığıdır. Buna karşılık, bu ağır prognoz parametrelerinin tümünü taşıyan bir hastanın bile 6 ay sonra yardımsız yürüyebiliyor olma şansı, plazmaferez ya da IVIg uygulanması halinde, tedavi görmeyenlere oranla iki kat fazladır (%20 ye karşı % 40).
AAG olgularında yukarıda anlatılan immünolojik tedavilerin yanısıra antikolinesteraz ilaçların uygulanması ile özellikle ortostatik hipotansiyon tedavisinde başarının arttığı bildirilmiştir.
GBS’li olguların izlenmesi ve tedavisi ile ilgili prensiplerin bir özeti Tablo 24’te verilmiştir.
Tablo 24. GBS tedavisi (IR Bella, değiştirilerek)
|
Klinik durum |
İzleme ve tedavi |
|
Çok hafif GBS |
Hastaneye yatırılır. |
|
Yardımsız yürüyor, solunum sorunu yok |
Gözlenir. Plazmaferez veya IVIg (uygulanması düşünülebilir). |
|
Yardımla yürüyor, solunum sorunu yok |
Olanak varsa yoğun bakım ünitesine alınır, yoksa VK nin sık olarak ölçülebileceği, yoğum bakım imkanlarına ulaşımın nispeten kolay olduğu bir yerde tutulur. AKG izlenir. IVIg veya plazmaferez (4 değişim) uygulanır. |
|
Yürüyemiyor, hafif solunum yetersizliği var |
Yoğun bakım ünitesine alınır. VK sık olarak izlenir. AKG izlenir. Şu hallerde entübasyon yapılır: VK <15 ml/kg, ya da 4-6 saat içinde düşme eğilimi gösteriyor, Orofaringeal parezi ve aspirasyon Solunum yorgunluğu bulguları ve VK 15 ml/kg. IVIg veya plazmaferez (4 değişim) uygulanır.
|
|
Yürüyemiyor, solunum yardımına gereksinimi var |
Yoğun bakım ünitesine alınır. Otonom sinir sistemi disfonksiyonu izlenir Hipotansiyona karşı sıvı infüzyonu ile mücadele edilir. Sürekli hipertansiyon varsa kısa etkili a-adrenerjik blokerler uygulanabilir. Yatak yaralarına ve kompresyon nöropatilerine karşı sık olarak yatağında çevrilir, pozisyonu değiştirilir. Fizik tedaviye başlanır. IVIg veya plazmaferez (4 değişim) uygulanır. |
AKG: arteriyel kan gazları, IVIg: intravenöz immünglobulin, VK: vital kapasite.
Kronik Seyirli İnflamatuvar Demiyelinizan Polinöropatiler
Kronik İnflamatuvar Demiyelinizan Polinöropati (CIDP)
GBS’de olduğu gibi otoimmün mekanizma ile gelişen ve periferik sinirlerde multifokal demiyelinizasyonla seyreden bir hastalıktır. Prevalansı 100.000’de 1-9 arasındadır. Prevalans değerlerindeki coğrafi bölgeler arası bu değişkenliğe çalışmalarda farklı tanı kriterlerinin kullanılmış olması ve hastalığın klinik açıdan çeşitlilik göstermesinin neden olabileceği düşünülmektedir. CIDP hemen hemen her yaşta görülebilmekle birlikte genellikle en çok 5. onyılda görülür ve erkeklerde daha sıktır. Genel olarak, ekstremitelerin distal ve proksimallerini oldukça simetrik şekilde tutan kas kuvvetsizliği söz konusudur. Hastalığın erken evrelerinde kuvvetsizlik distal kaslara sınırlı bulunabilir. GBS’den farklı olarak, kas kuvvetsizliği aylar içerisinde yavaş ve düzenli, basamaklı ya da remisyon ve alevlenmelerle giden bir progresyon gösterir. Hastalık 2-4. onyıllarda daha çok ataklı bir seyir gösterirken 5-7. onyıllarda daha çok progresif seyir izlenir. GBS olgularının hemen tümü hastalık başlangıcından sonraki ilk ay içinde progresyonunu tamamladığı halde, CIDP tanısı için 2 aydan daha uzun bir progresyon öyküsüne gereksinim vardır. Hastaların büyük kısmı el ve ayaklarda uyuşmalardan, daha azı parestezilerden yakınır, ağrı nadirdir. Duyu kusuru kas kuvvetsizliğine göre daha geri planda olmakla birlikte, ekstremite distallerinde özellikle vibrasyon ve pozisyon duyusu gibi kalın miyelinli sinir liflerini işaret eden duyuların azaldığı görülür. Derin duyu kaybına bağlı duyusal ataksi ve pozitif Romberg belirtisi izlenebilir. Tendon refleksleri oldukça simetrik olarak azalmış veya kaybolmuştur. Hastaların %10 kadarında kalınlaşmış hipertrofik sinirler palpe edilebilir (dirsekte ulnar, bilekte radial sinir, peroneal sinir, posterior auriküler sinir gibi). GBS’den farklı olarak kranial ve otonom sinir tutulmaları seyrek ve hafiftir. Solunum yetersizliğine neden olan interkostal kas ve diyafram tutulmasına da nadir olarak rastlanır. Semptom ve bulguların 4 hafta içinde en ağır düzeye ulaştığı akut başlangıçlı CIDP olguları da (%13) bildirilmiştir. Akut başlangıçlı CIDP olgularını GBS’den ayırt etmek güç olsa da CIDP olgularında duyusal semptomların daha baskın olması, 8 haftadan sonra kötüleşmenin hala devam etmesi ya da bu süre içinde en az 3 atakla seyretmesi, otonom tutulumun eşlik etmemesi, fasiyal zaaf, öncesinde infeksiyon öyküsü ya da mekanik ventilasyon ihtiyacının daha nadir olması GBS ile ayrım açısından akılda tutulmalıdır.
CIDP’de en duyarlı laboratuvar yöntemi BOS incelemesidir. Olguların %90’ında (GBS’de olduğu gibi) BOS’ta hücre artışı olmaksızın protein yükselmesi (albuminositolojik disosiyasyon) mevcuttur. BOS hücre sayısı 10/mm3’ün altındadır (daha yüksek hücre değerleri HIV infeksiyonu, Lyme hastalığı, sarkoidoz ya da sinir köklerinin lösemik, lenfomatoz infiltrasyonunun araştırılmasını gerektirir). Oligoklonal band CIDP hastalarında %65’e varan oranlarda pozitif saptanabilir. Elektrofizyolojik testlerde, GBS’de olduğu gibi, periferik sinirlerde multifokal demiyelinizasyonu yansıtan bulgular saptanır. Özellikle motor iletim incelemelerinde farklı periferik sinirler ve bu sinirlerin değişik segmentleri arasında farklılık gösteren (üniform olmayan) iletim yavaşlamaları, distal motor latanslarda uzama, gecikmiş F yanıtları ve kısmi iletim blokları (proksimal amplitüdlerde %30-50 azalma) ya da anormal temporal dispersiyon (proksimal motor yanıt süresinde %30 ve üzeri uzama) görülür. Bununla birlikte, hastaların seyrek rastlanan bir bölümünün multisegmental demiyelinizasyonun bu tipik özelliklerini göstermediğini, bunların klinik özellikleri ve diğer laboratuvar bulguları ile tanınması gerektiğini hatırlamak gerekir.
Otopsi, MRG ve ultrason incelemeleri ile CIDP’de inflamatuvar lezyonların daha çok spinal kök, periferik sinirlerin proksimal segmentleri ve pleksuslarda yerleştiği, fakat periferik sinir sistemi boyunca yaygın bir şekilde de olabileceği gösterilmiştir. Bununla birlikte proksimal sinirler ve sinir köklerine erişmek güç olduğundan çoğu biyopsi sural sinirden yapılır. Sinir biyopsilerinde kronik segmental demiyelinizasyon-remiyelinizasyona bağlı değişiklikler, miyelinli lif kaybı, endonöral ve perinöral ödem ve hafif derecede perivasküler iltihabi hücre infiltrasyonu görülür. Demiyelinizan değişikliklerin gösterilmesinde en hassas yöntem liflerin “teasing” metoduyla ayrılarak incelenmesidir ve %50-80 duyarlılığa sahiptir (Şekil 10). Olası komplikasyonları, elde edilen histopatolojik bulguların birçok kere CIDP için özgün olmaması ve hastaların çoğuna klinik ve diğer laboratuvar bulguları ile tanı konabilmesi nedeniyle sinir biyopsisine genellikle başvurulmaz. MRG incelemelerinde yer yer genişlemiş, intensitesi artmış ve kontrast tutan periferik sinirler ve sinir kökleri görülmesi de tanıya yardımcı olabilir (Şekil 11).
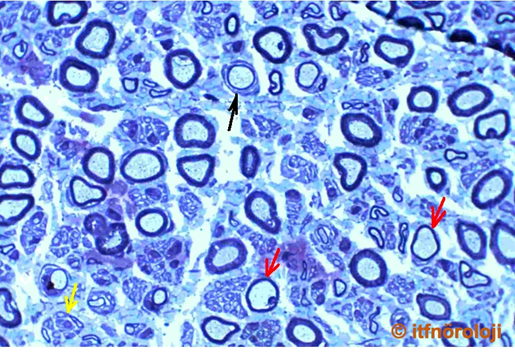
Şekil 10a. Kronik inflamatuvar demiyelinizan polinöropati (CIDP) olgusunun sural sinir biyopsisinde miyelinli akson yoğunluğunda azalmanın yanında çapına oranla ince miyelin kılıfı olan (kırmızı oklar) aksonlar (remiyelinizasyon) ve rejenerasyon odağı (sarı ok) ve Schwann hücre proliferasyonu (siyah ok) görülmekte. (Thioninle boyanmış enine yarı ince kesit. X40).

Şekil 10b. Bir kronik inflamatuvar demiyelinizan polinöropati (CIDP) olgusunda sinir lifi ayırma yöntemiyle segmental demiyelinizasyon odakları (oklar) (Osmiyum tetroksit, X10)
CIDP patogenezinde hem hücre aracılı hem de hümoral mekanizmaların birlikte rol oynayarak periferik sinirlerde hasara yol açtığı düşünülmektedir. GBS ve ‘relapsing-remitting’ deneysel allerjik nörit ile olan immünopatolojik benzerlikler CIDP’nin aktif T lenfosit, makrofaj, kompleman ve otoantikorların birlikte rol aldığı, henüz net olarak bilinmeyen periferik sinir antijenlerine karşı immün bir saldırı ile meydana geldiğini düşündürmektedir. Ayrıca CIDP’de IVIg, plazmaferez, kortikosteroid gibi immün sistemi hedef alan tedavilerin etkili olması da etyolojide otoimmünitenin rol oynadığının kanıtıdır.
Bazı CIDP hastalarının sinir biyopsileri incelendiğinde nodal ve paranodal bölgelerde ayrılma olduğu, ayrıca nod/paranod bölgelerinde yapısal bütünlüğün sürdürülmesi açısından gerekli olan proteinlerin anormal şekilde var olduğu ve dağılım gösterdiği görülmüştür. Son dönemde CIDP vakalarının bir kısmında nodal antijenlerin hastalıkta önemli rol oynadığına dair bulgular saptanmıştır. Yapılan çalışmalar ile CIDP hastalarının %4 ile 18’inde, Ranvier nodunda ve paranodal bölgelerde yer alan nörofasin (NF) 155, NF-186, kontaktin-1 (CNTN1) ve kontaktin-ilişkili protein 1 (Caspr1) gibi hücresel adezyon moleküllerine karşı otoantikorların varlığı gösterilmiştir. Seropozitif hastalar ayrıntılı olarak incelendiğinde antikor negatif CIDP olgularından farklı bir takım klinik özellikler gösterdikleri; hastalığın subakut ve ağır klinik özelliklerle başladığı, önemli ölçüde engelliliğe neden olacak şekilde ağır tremor, duyusal ve serebellar ataksi, distal baskın zaaf varlığı ve IVIg’e yetersiz yanıt verdiği dikkati çekmiştir.
Nadiren CIDP semptomlarının çok hafif olduğu ve hayat kalitesinin etkilenmediği vakalarda tedavi gereksinimi olmasa da, CIDP hastalarının büyük kısmında hastalığa bağlı belirgin kötüleşme görülmekte ve tedaviye ihtiyaç duyulmaktadır. CIDP’de etkili olduğu kanıtlanmış birinci basamak tedaviler kortikosteroidler, IVIg ve plazmaferezdir. Bu tedaviler klinik tablo ve hastanın kişisel yanıtına göre değişkenlik gösterse de birbirleriyle eşit etkinliğe sahiptir ve bu tedavilerden her biri ile hastaların %70-80’inde klinik düzelme elde edilebilir. Biri ile yanıt alınamayan ya da başlangıçta alındığı halde daha sonra olumlu etkinin kaybolduğu hastalarda bir diğeri ile başarılı tedavi sağlanabilir. Olguların yaklaşık %90’ında ilk aylar içinde tedaviye olumlu yanıt alınır. Ancak hastalığın tekrarlama eğilimi yüksektir ve hastaların ancak üçte biri kadarında bir dönemde tedaviyi tamamen kesmek mümkün olabilir. Bu nedenle, genellikle ilk tedavi ile yeterli bir iyilik elde edildikten sonra relapsları önleyecek uygun bir idame tedavisini yürütmek gerekir.
CIDP’de birinci basamak tedavinin seçiminde göz önünde bulundurulması gereken faktörler hastalığın ciddiyeti, eşlik eden diğer hastalıklar (diyabet ve gastrointestinal problemleri olanlar için steroidlerin, böbrek yetersizliği olanlar için IVIg’in riskli olması gibi), tedavilerin yan etki profilleri (steroidlerde en yüksek), maliyeti (plazmaferez ve IVIg pahalıdır) ve tedavinin ulaşılabilirliğidir (plazmaferez ancak büyük tıp merkezlerinde uygulanabilmektedir). Hastalığın ağır seyrettiği CIDP hastalarında hızlı immünomodulasyon sağlayan IVIg, plazmaferez veya pulse uygulama ile yüksek doz kortikosteroidlerin tercih edilmesi önerilmektedir. Hastalığın sinsi ilerlediği CIDP olgularında ise amaç remisyonun sağlanması olduğundan başlangıç tedavisi olarak steroid tedavisi tercih edilebileceği, glukokortikoidlerin kontrendike olduğu durumlarda ise 2. basamak tedavi olarak metotreksat, siklosporin, mikofenolat mofetil ve azatioprin gibi immünsupresif ajanların uygulanabileceği önerilmektedir. Bununla birlikte, bu tedavilerin CIDP’de etkinlikleri henüz net bilinmemektedir. İlk uygulanan birinci basamak tedaviye yanıt vermeyen olgularda diğer birinci basamak tedavilere geçilmesi ya da iki tedavinin birlikte uygulanması önerilmektedir.
CIDP tedavisinin en ucuz seçeneği olan kortikosteroid uygulamasına 1-1,5 mg/kg’lık prednizolon (erişkinde 100 mg/gün’e kadar) dozu ile başlanır. Kuvvetsizliğin düzelmeye başlamasıyla birlikte (bu genellikle 6-8 haftada sağlanır), doz azaltılarak günaşırı tedaviye geçilebilir. Kas kuvvetinde oldukça sabit bir iyilik elde edildikten sonra ise doz yavaşça azaltılır (birkaç haftada bir 5 mg gibi). Günaşırı 20 mg gibi düşük dozlara varıldıktan sonra doz azaltımının çok daha yavaş yapılması, tüm tedavi sırasında kronik steroid kullanımına ait komplikasyonların yakından gözlenmesi ve gerekirse tedavi edilmesi gerekir. Ağır seyirli ve tedaviye dirençli olgularda ya da uzun süreli steroid kullanımının yan etkilerinden kaçınmak amacıyla pulse kortikosteroid uygulamaları da yapılabilmektedir.
IVIg, CIDP’nin bir diğer standart tedavi yöntemi haline gelmiş ve etkinliği son 15 yılda tamamlanan birçok randomize kontrollü çalışma ile gösterilmiştir. Genellikle GBS’de olduğu gibi 5 (veya 2) günde tamamlanan toplam 2g/kg’lik ilk kürle başlanır. Sonraki tedavi süreci aynı dozun 1-1,5 ayda bir tekrarlanması ya da daha düşük dozların (0,4 g/kg gibi) daha sık aralıklarla verilmesi (haftada 1-2 kez) ile sürdürülür. Zaman geçtikçe daha düşük dozların daha geniş aralıklarla verilmesi amaçlanır. Haftada bir gibi çok sık aralıklı tedaviye bağımlı hale gelen ya da aylar ve yıllar geçtikçe IVIg tedavisine refrakter hale gelen olgular vardır. Bu ikinci grup hastalarda arada uygulanacak plazmaferez tedavisi hastayı tekrar IVIg’e yanıt verir hale getirebilir. Plazmaferez aynı zamanda hızla kötüleşen ve çabuk yanıt alınması istenilen durumlarda yararlı bir tedavi seçeneğidir. CIDP’nin kronik seyri nedeniyle GBS’ye göre daha seyrek değişimler şeklinde uygulanır. Yeni geliştirilen subkutan Ig (SCIg) tedavisi ile tedavinin kolaylaştırılması ve intravenöz uygulama sırasında görülen IgG düzeyinde dalgalanmaların engellenmesi amaçlanmıştır. Olgu serileri ve retrospektif çalışmalar hasta uyumunun yüksek olması gibi yararları olduğunu da söylerken, 2018’de yayınlanan bir çalışmada SCIg’in CIDP’de etkili ve iyi tolere edilebilen bir tedavi şekli olduğu gösterilmiştir.
Kortikosteroid ve IVIg’e cevap alınamayan ya da yan etkiler nedeniyle bu tedavilerin uygulanamadığı olgularda tedaviye sitostatik ilaçlar eklenebilir (azatiopirin, mikofenolat mofetil, metotreksat, siklofosfamid, siklosporin). Bugüne kadar immünsupresif ajanların CIDP’de yararlı olduğuna dair gözlemsel çalışmalar bildirilmiş olmakla birlikte, bunların hiçbiri bu tedavilerin etkinliğini ispatlayacak yeterli kanıt düzeyine sahip randomize kontrollü çalışmalar değildir. İnterferon-beta ve fingolimod ile yapılan kontrollü çalışmalarda bu tedavilerin etkili olmadığı bulunmuştur. CIDP’de kullanılmakta olan 2. basamak tedaviler Tablo 25’de izlenmektedir. Monoklonal antikor tedavilerine bakıldığında rituksimabın CIDP’de, özellikle anti-CNTN1 ve anti- NF155 pozitif olgularda etkili bir tedavi şekli olduğuna dair az sayıda olgu serisi bulunmaktadır. Gelecekte yapılacak kontrollü çalışmalarda rituksimab ve benzeri ilaçlarla başarılı sonuçlar alınacağı umulmaktadır. Az sayıda olguda takrolimus, etanersept gibi ilaçlar denenmiştir. Otolog kök hücre transplantasyonu uygulanarak başarılı şekilde tedavi edilmiş olgular bildirilmiş olmakla birlikte, mortalitesi yüksek olan bu tedavinin etkisi ve uygulanabileceği özel gruplarının ileri çalışmalarla belirlenmesi gerekmektedir.
Tablo 25. CIDP’de kullanılmakta olan 2. basamak tedavi seçenekleri ve önerilen dozları* (D Cocito, değiştirilerek)
|
ilaç |
Doz |
|
Azatiopirin |
100-200 mg/gün |
|
Rituksimab |
375mg/m² /hf/4 hf |
|
Siklofosfamid |
1 gr/m²/ay veya 2mg/kg |
|
Mikofenolat Mofetil |
1-2 gr/gün |
|
Siklosporin A |
100-300mg/gün |
|
Metotreksat |
7.5-15mg/hf |
|
İnterferon alfa |
800bin-3 milyonU 1-3 hf |
|
İnterferonβ1a |
6 milyonU |
*Tabloda 2. basamak tedavide en çok yararlılığı bildirilen ilaçlar sırasıyla yer almaktadır.
CIDP ile birlikte olan hastalıklar
CIDP’nin klinik ve elektrofizyolojik özelliklerini gösteren polinöropati tabloları bazı sistemik hastalıklar ve diğer tıbbi durumlarla birlikte olabilir. Bunların başlıcaları, HIV infeksiyonu, iltihabi barsak hastalığı, SLE, diabetes mellitus (DM), kemik iliği ve organ transplantasyonu (doku reddi ve “graft versus host disease” durumlarında) ve lenfomalardır. Bu hastalık durumları ile birlikte olan ve olmayan CIDP formları arasında oluşum mekanizmaları ve seyir özellikleri açısından temel farklılıklar olup olmadığı iyi bilinmemektedir. Örneğin diyabetik hastalarda gelişebilen ve klinik, elektrofizyolojik özellikler açısından CIDP’ye çok benzeyen polinöropati tablosunun CIDP mi yoksa bir diyabetik polinöropati (DPN) formu mu olduğu konusu iyi bilinmemektedir. Bazı yazarlar diyabette CIDP sıklığının arttığını ve hastaların %9-26’sında CIDP bulunduğunu bildirmiştir. Diğer taraftan, iyi dokümante edilmiş epidemiyolojik çalışmalarda diyabetiklerde tipik CIDP prevalansında artış bulunmamıştır.
CIDP’li olguların bir kısmının kanında bir monoklonal protein (M-proteini) saptanır, tüm CIDP olgularında immünofiksasyon elektroforezi ile monoklonal protein araştırılmalıdır. CIDP’nin tipik klinik ve laboratuvar bulguları ile seyreden olgularda bu M-proteini genellikle IgG veya IgA grubundandır. Monoklonal protein saptanan olguların bir kısmında plazmositoma ya da lenfoma gibi lenfoproliferatif bir hastalık bulunur. Yapılacak araştırmalarla bu hastalıkların saptandığı durumlarda her biri için uygun olan tedavi uygulanır. Bu hastalıkların bulunmadığı M-proteini pozitif durumlar anlamı belirsiz monoklonal gamopati (MGUS; monoclonal gammopathy of undetermined significance) olarak isimlendirilir (aşağıya bakınız). MGUS’la birlikte olan CIDP, M-proteini negatif CIDP’lerden klinik seyir ve tedaviye yanıt açısından belirgin bir farklılık göstermez. MGUS’la birlikte olsun olmasın, CIDP olgularının uzun süreli takipleri sırasında lenfoproliferatif bir hastalık gelişebilir. Bu nedenle, özellikle başlangıçta tedaviye yanıt verip sonradan vermez olan hastalar, tedaviye yanıtsız kronik progresif seyir izleyenler ve MGUS’la birlikte olan tüm CIDP olgularında immünolojik durum aralıklı olarak yeniden değerlendirilmeli ve kemik grafileri ve gerekirse kemik iliği biyopsisi yapılmalıdır.
Lenfoproliferatif hastalıkların yanısıra küçük hücreli akciğer karsinomu, pankreas, kolon kanserleri, kolanjiokarsinom ve melanomun paraneoplastik komplikasyonu olarak gelişen CIDP olguları yayınlanmıştır. Prokainamid, siklosporin ve takrolimus gibi ilaçların toksik etkisi ile gelişen CIDP’ye benzer nöropatiler de bildirilmiştir.
CIDP’nin Klinik Olarak Sınıflanması
CIDP’de klinik spektrum distal ya da proksimal baskın zaaf, sadece motor ya da duyusal form, asimetrik ya da fokal tutulum olacak şekilde çok geniştir (Şekil 12). “European Federation of Neurological Societies and the Peripheral Nerve Society (EFNS/PNS)” CIDP kılavuzunda hastalık ‘tipik CIDP’ ve ‘atipik CIDP’ olarak sınıflandırılmıştır (Tablo 26). Buna göre tipik CIDP tanımı, bu hastalığın simetrik proksimal ve distal kas kuvvetsizliği ile seyreden klasik formu için kullanılmalıdır. CIDP grubuna giren hastalar immünmodülatuvar tedavilere, eşlik eden bir monoklonal gamopati olsun veya olmasın iyi yanıt verirler. Tedavi yanıtının iyi olması CIDP tanısı için destekleyici kriterler arasında yer almaktadır (Tablo 27).
Atipik CIDP grubunda ön planda distal yerleşimli, duyusal baskın belirti ve bulgularla seyreden hastalar yer alır. Klinik planda aksonal tipte uzunluğa bağımlı bir nöropatiyi düşündüren bu bulgulara karşın elektrofizyolojik incelemeler edinsel demiyelinizan bir polinöropati ile uyumludur. Bu grup distal edinsel demiyelinizan simetrik nöropati (distal acquired demyelinating symmetric=DADS) olarak isimlendirilir Hastaların başlıca yakınması ekstremite distallerinde duyu bozukluğu, dengesizlik ve ellerdeki tremordur (Tablo 28). Vibrasyon duyusu ağrı ve ısı duyularına oranla daha baskın şekilde bozulmuştur. Hastaların başlıca duyusal yakınmaları olmasına karşın, motor sinir iletim incelemelerinde genellikle yaygın ve oldukça homojen bir iletim yavaşlaması bulunur, iletim blokları nadirdir. Buna karşılık motor distal latanslar daha proksimal segmetlerdeki yavaşlamayı da aşan oranda, belirgin derecede uzundur (Terminal latans indeksi <0,25). Sinir biyopsisine nadiren başvurulmakla birlikte, histopatolojik olarak elektron mikroskopisinde miyelin intraperiod çizgilerinde genişleme gösterilmesi bu hastalık için oldukça tipiktir. Olguların immünmodülatuvar tedaviden yararlanma oranları çok düşüktür. IVIg ve plazmaferez, kortikosteroidler ve sitostatik ilaçlar, a-interferon ve rituksimabdan bir derece yararlanan hasta grupları bildirilmiştir. Bununla birlikte hastalığın seyri çok yavaştır ve genellikle belirgin bir sakatlığa yol açmaz. İyi gidişli olguları immünmodülatuvar tedavi vermeden, sadece yakınmalarına yönelik semptomatik tedaviler uygulayarak izlemenin yararı kanıtlanmamış ve yan etkisi yüksek ilaç uygulamalarına oranla daha uygun olduğu düşünülmektedir
DADS fenotipli hastaların yaklaşık 2/3’ünde, hemen daima kappa hafif zinciri olan bir IgM paraprotein vardır. DADS-M adı verilen bu grubun %80’den fazlasını 6. onyılın üzerindeki erkek hastalar oluşturur. Akademik merkezlerde DADS-M olgularının sıklığı CIDP olgularınınki kadardır. Olguların yaklaşık yarısından fazlasında anti-MAG antikorlar bulunur. Bazı olgularda sulfatidler, sadece SGPG veya GD1b’ye karşı antikorlar da saptanır. Nöropatinin bu antikorların miyeline bağlanması nedeniyle ortaya çıktığı düşünülmektedir.
DADS fenotipli olguların IgM paraproteinemisi olmayanları DADS-I (idyopatik) ismi altında gruplandırılır. Bu grup oldukça heterojendir ve olguların bir kısmı immünmodülatuvar tedavilere oldukça iyi yanıt verir. Bu grup “duyusal CIDP” olarak adlandırılmış olan edinsel demiyelinizan polinöropatiye benzemekle birlikte, DADS fenotipi için uzunluğa bağımlı paternde distal simetrik duyusal belirti ve bulgular aranırken, sadece duyusal CIDP olgularında genellikle daha geniş-yaygın duyu kaybı bulunur. Sadece duyusal CIDP’li hastaların elektrofizyolojik incelemelerinde motor sinirlerde de iletim yavaşlamaları ve iletim blokları saptanır ve tedaviye daha iyi yanıt alınır.
Sadece duyusal CIDP’nin farklı bir formu olarak kronik immün duyusal poliradikülopati de bildirilmiştir. İlk kez 2014’de Sinnreich ve arkadaşları tarafından bildirilen 15 vakanın birkaç yıl boyunca sadece duyusal semptomlarının olduğu, bu esnada yapılan iletim incelemelerinin normal bulunduğu, duyusal uyandırılmış potansiyellerinde (SEP) gecikme, BOS protein düzeylerinde artış ve lomber MRG görüntülerinde lomber köklerde genişleme olduğu izlenmiştir. Kök biyopsisi ile sürecin inflamatuvar olduğu doğrulanmış ve hastaların hepsi kortikosteroid ya da IVIg ile iyileşme göstermiştir.
Atipik kronik inflamatuvar demiyelinizan polinöropatilerden bir diğeri fokal-multifokal klinik belirti ve bulgularla seyreden fokal CIDP ve MADSAM (Lewis-Sumner sendromu) nöropatidir (Tablo 27). Fokal CIDP’de brakiyal ya da lumbosakral pleksus ya da bir veya daha fazla sayıda üst/alt periferik sinir etkilenimi olur. Hastalar kortikosteroid ya da IVIg tedavilerinden fayda görür. MADSAM ise asimetrik, iki veya daha çok periferik sinirin dağılımına uyan multifokal ve yavaş progresif bir seyir izler (Şekil 11). Motor belirtilere duyusal belirti ve bulgular eşlik eder ve genellikle BOS proteini yüksektir. Ayrıca duyusal sinir iletim incelemelerinde patolojik bulgular elde edilir. Kortikosteroid ve IVIg tedavisine yanıt yaklaşık aynı düzeyde ve oldukça iyidir (%70 kadar).
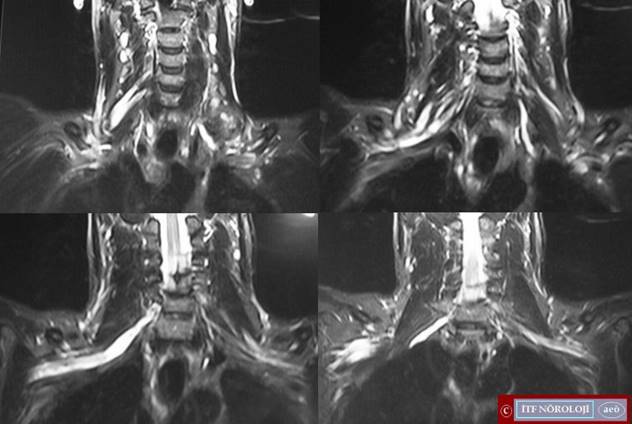
Şekil 11. MADSAM olgusunun koronal planda T2 ağırlıklı manyetik rezonans görüntüleme (MRG) kesitlerinde iki yanlı kalınlaşmış ve hiperintens görünümlü servikal sinir kökleri ve brakiyal pleksusları (Dr. Fikret Aysal’ın izniyle).
Takipleri süresince semptomların sadece motor tutulma ile sınırlı kaldığı, duyusal semptom ya da bulguların eşlik etmediği vakalar atipik CIDP’ler içinde yer alır ve motor CIDP olarak adlandırılmıştır. Duyusal iletim incelemeleri ve sural sinir biyopsisi normaldir. EFNS/PNS’nin 2010 kılavuzunda da yer aldığı gibi, hastalarda sıklıkla steroid tedavisi ile iyileşme gözlenmez hatta steroidle kötüleşme de olabilir. Sadece duyusal CIDP’de olduğu gibi, motor CIDP tanısı yalnızca klinik bulgulara mı dayanmalı yoksa tanı için normal duyusal iletim çalışması koşulu aranmalı mı konusu da henüz netleşmemiştir.
|
Tablo 26. EFNS/PNS CIDP klinik tanı kriterleri (EFNS/PNS Birleşik Görev Grubu CIDP tanı kılavuzu, 2010) |
|
|
(1) İçerme kriterleri |
|
|
(a) Tipik CIDP |
|
|
Tüm ekstremitelerde en az 2 ay içerisinde gelişmiş kronik progresif, hecmelerle seyreden ya da tekrarlayan simetrik proksimal ve distal zaaf ve duyusal disfonksiyon; |
|
|
Tüm ekstremitelerde derin tendon reflekslerinde azalma veya kayıp |
|
|
Kranial sinirler nadiren etkilenebilir |
|
|
(b) Atipik CIDP (hala CIDP düşündürür fakat farklı özelliklere sahiptir) |
|
|
Aşağıdakilerden biri, aksi halde (a) geçerlidir (etkilenmemiş ekstremitelerde tendon refleksleri normal olabilir) |
|
|
Ağırlıklı olarak distal (DADS) |
|
|
Asimetrik (MADSAM, Lewis-Sumner sendromu) |
|
|
Fokal (Alt veya üst ekstremitelerde brakiyal ve lumbosakral pleksusun bir veya daha fazla motor periferik sinir tutulumu) |
|
|
Sadece motor ya da |
|
|
Sadece duyusal (primer duyusal nöronun santral uzantısının etkilendiği kronik immün duyusal poliradikülopatiyi de kapsar) |
|
|
(2) Dışlama kriterleri |
|
|
Lyme hastalığı, difteri, polinöropatiye yol açabilen ilaç ve toksin maruziyeti |
|
|
Herediter demiyelinizan nöropati (CMT ve diğer) |
|
|
Belirgin sfinkter kusuru |
|
|
Multifokal motor nöropati tanısı |
|
|
Miyelin ilişkili glikoproteine karşı antikor titrelerinin yüksek olduğu IgM monoklonal gamopatisi |
|
|
Demiyelinizan nöropatiye yol açan diğer nedenler: POEMS sendromu, osteosklerotik miyelom, diyabetik ve diyabetik olmayan lumbosakral radikülopleksus nöropatisi. Periferik sinir sistemi lenfoması ve amiloidozu nadiren demiyelinizan özellikler taşır |
|
CIDP: kronik inflamatuvar demiyelinizan polinöropati, CMT: Charcot-Marie-Tooth hastalığı, DADS: distal edinsel demiyelinizan simetrik nöropati, IgM: immünglobulin M, MADSAM: multifokal edinsel demiyelinizan duyusal ve motor nöropati, POEMS: Polinöropati, Organomegali, Endokrinopati, M proteini, Deri (Skin) değişiklikleri.
|
Tablo 27. CIDP tanısı için destekleyici kriterler (EFNS/PNS Birleşik Görev Grubu CIDP tanı kılavuzu, 2010, değiştirilerek) |
|
1. BOS protein artışı (lökosit sayısı <10/mm3) |
|
2. MRG incelemesinde kauda ekuina, lumbosakral veya servikal sinir köklerinde ya da brakiyal veya lumbosakral pleksuslarda hipertrofi ya da kontrast madde tutulumu olması |
|
3. En az bir duyusal sinirde anormal elektrofizyolojik bulgular: |
|
a. Normal sural ile anormal medyan sinir (karpal tünel sendromu olmaksızın) ya da radyal sinir duyusal aksiyon potansiyeli amplitüdleri; veya |
|
b. İleti hızının normalin alt sınırının %80’inden yavaş olması (eğer duyusal aksiyon potansiyeli amplitüdü normalin alt sınırının %80’inden düşük ise normal alt sınırının %70’inden yavaş olması); veya |
|
c. Merkez sinir sistemi hastalığı olmaksızın gecikmiş duyusal uyandırılmış potansiyeller |
|
4. İmmünmodülatuvar tedaviyi takiben objektif klinik iyileşme |
|
5. Sinir biyopsisinin elektron mikroskopisi ya da “teasing” metodu ile ayrılmış liflerinde demiyelinizasyon ve/veya remiyelinizasyon varlığının açıkça gösterilmesi |
BOS: beyin omurilik sıvısı, MRG: manyetik rezonans görüntüleme.

Şekil 12. Kronik seyirli edinsel demiyelinizan nöropatiler (Saperstein, değiştirilerek)
CIDP: kronik inflamatuvar demiyelinizan polinöropati, CMT: Charcot-Marie-Tooth hastalığı, DADS: distal edinsel demiyelinizan simetrik nöropati, DADS-I: idyopatik DADS, DADS-M: IgM ilişkili DADS, IgM: immünglobulin M, MADSAM: multifokal edinsel demiyelinizan duyusal ve motor nöropati, POEMS: Polinöropati, Organomegali, Endokrinopati, M proteini, Deri (Skin) değişiklikleri.
Tablo 28. Kronik seyirli edinsel demiyelinizan nöropatiler (JS Katz, değiştirilerek)
|
|
Tipik CIDP |
DADS-M |
DADS-I |
MADSAM |
MMN |
|
Klinik Özellikler |
|||||
|
Kuvvetsizlik |
Simetrik proksimal+distal |
Distal veya yok |
Distal veya yok |
Asimetrik Distal>proksimal Üst >alt ekstremite |
Asimetrik Distal>proksimal Üst >alt ekstremite |
|
Duyu kaybı |
Simetrik |
Simetrik |
Simetrik |
Multifokal |
Yok |
|
Refleks kaybı |
Simetrik |
Simetrik |
Simetrik |
Multifokal veya yaygın |
Multifokal veya yaygın |
|
Elektrofizyoloji |
|||||
|
Anormal BKAP |
|
|
|
|
|
|
Yavaşlama |
Simetrik |
Simetrik |
Simetrik |
Multifokal |
Multifokal |
|
Uzun distal latans |
Bazen |
Daima |
Bazen |
Bazen |
Bazen |
|
İletim bloğu |
Sık |
Mutad |
Nadir |
Sık |
Sık |
|
Anormal DAP |
Genellikle simetrik |
Daima simetrik |
Genellikle simetrik |
Sık, multifokal |
Normal |
|
Laboratuvar |
|||||
|
Artmış BOS Proteini |
Genellikle |
Genellikle |
Bazen |
Genellikle |
Normal |
|
Monoklonal protein |
%20, genellikle IgG veya IgA LPH açısından değerlendirilmeli |
IgM-k Tanım gereği |
%20, IgG veya IgA LPH açısından değerlendirilmeli |
Nadir |
Nadir |
|
Anti-MAG antikor |
Yok |
>%70 |
Yok |
Yok |
Yok |
|
Anti-GM1 antikor |
Nadir |
Yok |
Yok |
Nadir |
%40-50 |
|
Anti-sulfatid antikor |
Yok |
Mutad değil veya MAG’la kombine |
Yok |
Yok |
Yok |
|
Duyusal sinir biyopsisi: Demiyelinizasyon/ remiyelinizasyon |
Sık |
Sık |
Sık |
Sık |
Minimal bulgular |
|
Tedaviye yanıt |
|||||
|
Prednizon |
Evet |
Zayıf |
Muhtemel |
Evet |
Hayır |
|
Plazmaferez |
Evet |
Zayıf |
Muhtemel |
Mümkün |
Hayır |
|
IVIg |
Evet |
Zayıf |
Muhtemel |
Evet |
Evet |
|
Siklofosfamid |
Evet |
Zayıf |
Muhtemel |
Mümkün |
Evet |
BKAP: Bileşik kas aksiyon potansiyeli, BOS: beyin omurilik sıvısı, DAP: Duyusal aksiyon potansiyeli, IVIg: intravenöz immünglobulin, LPH: Lenfoproliferatif hastalık, MAG: miyeline bağlı glikoprotein.
Multifokal Motor Nöropati (MMN)
Ekstremitelerde asimetrik kuvvetsizliğe neden olan otoimmün kökenli bir polinöropatidir. CIDP’nin bir formu olup olmadığı konusu uzun süre tartışılmış olan bu nöropati, artık daha çok ayrı bir hastalık olarak ele alınmaktadır. Sıklıkla üst, nadiren alt ekstremite distalinde asimetrik kuvvetsizlikle başlar. Hasta hekime el kaslarında kuvvetsizlik, düşük el veya düşük ayak gibi nedenlerle başvurur. Kas kuvvetsizliği, belirli bir veya birkaç periferik sinirin innervasyon alanına lokalize edilebilen bir dağılım gösterir. Kuvvetsizliğin varlığına karşın atrofi yoktur ya da ilerlemiş olgularda görülür. Kaslarda kramp ve fasikülasyonlar görülebilir. Bunların yoğun olduğu bölgelerde nörojenik kas hipertrofisi gözlenebilir (Şekil 13). Duyu kusuru yoktur ya da çok hafif derecededir. Tendon refleksleri korunmuştur, fakat zayıf ve atrofik ekstremitelerde azalmış ya da kayıp olabilir. Hastalık aylar ve yıllar içinde yavaş bir progresyonla aynı ekstremitedeki diğer sinirlere ya da diğer ekstremitelere doğru yayılır. Yavaş ve sinsi progresyonu yüzünden başlangıcından tanıya kadar geçen süre genellikle uzundur.
Şekil 13. Sol el başparmağının adduksiyon-opozisyonu şeklinde kasılmalardan yakınan multifokal motor nöropati (MMN) olgusunda sol tenar kasların hipertrofisi.
BOS genellikle normaldir. Elektrofizyolojik incelemelerde motor sinirler üzerinde iletim bloklarının ve multifokal demiyelinizasyonu gösteren diğer bulguların saptanması tanıya çok yardımcı olur. Bununla birlikte MMN’nin tipik klinik özelliklerini gösteren fakat iletim bloğu saptanamayan olgu bildirimleri de bulunmaktadır. Bu durum iletim bloğunun rutin elektrofizyoloji incelemeleri ile saptanamayacak kadar proksimalde ya da distalde yerleşmesi ile açıklanabilir. Fakat klinik olarak etkilenmeyen ekstremitede, kas gücü tam bulunan sinirlerde de iletim bloğu saptanabileceği akılda tutulmalıdır. Fokal blok saptanan ya da saptanmayan hastaların klinik özellikleri ile tedavi yanıtlarının farklılık göstermediği bildirilmektedir. Hastaların %40-80’inin serumlarında IgM grubundan anti-GM1, asialo GM1 veya GD1a gangliozid antikorları saptanır. Bu antikorların yüksek titrede pozitif olması MMN için oldukça spesifiktir. Fakat bu yöntemin duyarlılığı azdır ve düşük titrede anti-GM1 antikorlar diğer bazı otoimmün kökenli nöropatilerde de görülebilir. Bu nedenle, antigangliozid antikorların aranmasının tanıya elektrofizyolojik test verilerine ek bir katkı sağlama olasılığı düşüktür.
Sınırlı sayıda MMN hastasının, yapılan fasiküler sinir biyopsi incelemelerinde demiyelinizasyon bulgularına rastlanmaması ve elektrofizyolojik incelemelerde geçici iletim bloklarının görülebilmesi hastalığın etyopatogenezinde Ranvier nodunda fonksiyonel bir bozukluğun rol oynayabileceğini de düşündürmektedir. Yapılan bir çalışmada, 11 MMN hastasının %55’inde anti-GM1 pozitif bulunmasına rağmen, yalnızca bir tanesinde deney hayvanlarının “teasing” metodu ile ayrılan sinir liflerinde indirekt immünfloresan yöntemiyle paranod bölgesinde boyanma saptanmıştır. Aksine, aynı çalışmada iletim bloğu olan 11 CIDP hastasının 9’unda nod, paranod ve/veya miyelin bölgesinde boyanma olduğu gösterilmiştir. Bu bulgular MMN ve CIDP’nin farklı hastalık mekanizmalarına ve hedef antijenlere sahip olduğu görüşünü desteklemektedir.
MMN’nin ayırıcı tanı açısından önemli bir özelliği amiyotrofik lateral sklerozla (ALS) benzerlik göstermesidir (Bakınız: Motor Nöron Hastalıkları). Günümüzde birisi tedavi edilebilen diğeri edilemeyen bu iki hastalığın birbirinden ayırd edilmesi, MMN’nin ALS’ye oranla çok daha seyrek oranda görülmesine karşın önemlidir (büyük tıp merkezlerinde ALS/MMN rastlanma sıklığı 50/1). MMN’de sıklıkla üst ekstremitelerde asimetrik başlayan kas kuvvetsizliği ve fasikülasyonlar görülmesi, duyu kusurunun olmaması ALS ile benzerlik gösterir. Kas kuvvetsizliğinin ALS’deki gibi ön boynuz ya da radiks segmentlerine değil, periferik sinirlere uygun bir dağılım göstermesi, kas atrofisi bulunmaması ya da ALS’ye oranla hafif olması MMN tanısı yönündeki klinik bulgulardır. Tendon reflekslerinde belirgin canlılık, klonus ve patolojik reflekslerin varlığı, bulber kasların tutulması ALS’yi düşündürür. Yine de MMN ve ALS’nin progresif alt motor nöron tutulması ile giden formunun klinik planda ayrılması bazen çok güç olabilir. Bu durumda motor iletim incelemelerinde sinir trunkusları üzerinde iletim bloklarının gösterilmesi MMN tanısına ulaşılmasını sağlar. Ancak, ALS’li hastalarda da nadir olarak yalancı iletim bloklarına rastlanabileceği hatırlanmalı ve elektrofizyolojik inceleme tekniği açısından dikkatli olunmalıdır.
Elektrofizyolojik incelemelerle iletim bloğunun gösterilemediği, klinik bulguların MMN telkin ettiği olgularda yardımcı tanı yöntemi olarak brakiyal pleksus MRG ve/veya sinir ultrasonografisinden de yararlanılabilmektedir. MRG ile MMN olgularının %35 ile %50’sinde, servikal sinir kökleri ve brakiyal pleksus gibi olası proksimal iletim bloğu bölgeleri görüntülenerek, T2 hiperintensitesi ve sinirlerde ödem varlığı gösterilebilmektedir. Ultrason incelemeleri de MMN’nin ALS’den ayırt edilmesinde umut vaad eden bir yöntem olabilir. Motor lif içeren miks tipteki periferik sinirlerin çaplarının MMN hastalarında ALS’ye oranla genişlemiş olduğunu bildiren yayınlar bulunmaktadır.
CIDP’den faklı olarak, MMN kortikosteroid ve PE tedavisine yanıt vermez. Günümüzde MMN tedavisi için ilk tercih edilen IVIg’dir. MMN’de etkililiği gösterilmiş ilk ilaç ise intravenöz siklofosfamiddir. İlk klinik serilerde 8 gün içerisinde verilen 3 g/m2 lik ilk doz ve onu izleyen aylık 0,5-1 g/m2 intravenöz (veya 2 mg/kg oral) kürlerle %70 olguda klinik iyilik sağlanmıştır. Bu dozlarda yan etkilerin çok fazla olması nedeniyle halen IVIg’e yanıt vermeyen hastalar için 0,5-1 g/m2 lik başlangıç dozu önerilmekte, idame dozunun yan etkilere ve hastanın toleransına göre ayarlanması tavsiye edilmektedir. Alopesi, bulantı, kusma, hemorajik sistit, infertilite, fırsatçı infeksiyonlar ve şiddetli kemik iliği depresyonu tedbir alınması gereken başlıca yan etkilerdir. Siklofosfamid, sayılan yan etkilerine ek olarak uzun vadede tümör gelişimi ile ilişkili bulunduğundan, çoğu oldukça genç yaşta olan ve uzun süre tedavi edilmesi gereken MMN’li hastalarda -IVIg’e yanıtsız olanlar dışında- günümüzde kullanılmamaktadır.
CIDP de anlatıldığı şekilde uygulanan IVIg tedavisine hastaların çoğunda (%80) günler ya da haftalar içinde iyi yanıt alınır. Yaşlı ve belirgin atrofisi olan hastaların tedaviye yanıtları daha kötüdür. Tedaviye yanıt vermeyen olguların yanısıra, sürdürülen IVIg uygulamasına bir derece yanıt veren ancak yavaş şekilde progresyon gösteren olgular da vardır (iletim bloklarının ya da akson hasarının süregelmesi nedeniyle). Genellikle hastalığın tekrarlamasını engellemek için aralıklı IVIg uygulamaları gerekir (CIDP konusuna bakınız). MMN olgularının IVIg ile devam tedavilerinde zamanla doz artışı yapılmasına ihtiyaç duyulabilmektedir. IVIg tedavisi MMN’de etkili bulunmakla birlikte hastalığın ilerleyişini kısmen kontrol altına alabilmekte ve akson kaybını ancak yavaşlatabilmektedir. Bir çalışmada yüksek doz IVIg ile devam tedavisi uygulanan olgularda aksonal dejenerasyonun belirgin derecede azaldığı ve hatta reinnervasyonun sağlandığı bildirilmiştir. CIDP’de olduğu gibi SCIg tedavisinin IVIg ile benzer tedavi etkinliğine sahip olduğu bildirilmektedir. Rituksimab ve ekulizumab tedavileri ile olumlu sonuç bildiren çalışmalar da yayınlanmıştır. Siklosporin, metotreksat, azatiopirin ve interferon beta-1a ile klinik durumu stabilize olan az sayıda olgu da bildirilmiştir.
Multifokal Edinsel Motor Aksonopati
Klinik özellikleri MMN ile uyumlu olan, buna karşılık elektrofizyolojik incelemelerinde segmental demiyelinizasyon özelliklerinin saptanmayıp sadece aksonal tutulma bulgularının görüldüğü, bir kısmı immünmodülatuvar tedaviye yanıt veren nadir olgular da bildirilmiştir (multifokal edinsel motor aksonopati, MAMA). MMN’de nadir olmayarak elektrofizyolojik incelemelerle iletim bloğu varlığının gösterilemeyebileceği bilinmektedir. Bu durum göz önüne alındığında MAMA hastalarının esasında sekonder aksonal dejenerasyon bulgularının hakim olduğu MMN olguları olabileceğine dair soru işaretleri akla gelmektedir. Böyle özellikleri olan hastaların bilinmesi, tedaviye yanıtlı olabilecek az sayıdaki olgunun tanınmasını sağlamakla birlikte, yanıt vermeyecek birçok hastada gereksiz, pahalı ve bazen zararlı olabilecek tedavi girişimlerine yol açma ihtimalini ortaya çıkarmaktadır. Bu nedenle, immün kökenli olduğundan kuşkulanılan fakat tanısal sınıflama açısından tereddüde düşülen olgular için polinöropati tanı ve tedavisinde tecrübe kazanmış merkezlere danışmaya çalışılmalıdır.
Paraproteinemik Nöropatiler
Nedeni bilinmeyen kronik seyirli bir polinöropatisi olan hastalarda monoklonal proteinlerin (M proteini) varlığı araştırılmalıdır. İdyopatik polinöropatisi olan hastaların yaklaşık %10’unda bir monoklonal gamopati vardır. Bir hastada M proteinlerinin varlığının gösterilmesi, primer amiloidoz, multipl miyelom, lenfoma, osteosklerotik miyelom, Waldenström makroglobulinemisi, kriyoglobulinemi ve diğer lenfoproliferatif hastalıkların araştırılmasını gerektirir. Ancak M proteini saptanan hastaların 2/3 kadarında altta yatan bir hastalık bulunamaz ve bu tablo anlamı belirsiz monoklonal gamoapati (MGUS; monoclonal gammopathy of undetermined significance) olarak isimlendirilir. Bu ismin verilmesinin sebebi, bir kısım MGUS’lu hastanın uzun süreli takiplerinde multipl miyelom başta olmak üzere habis plazma hücresi diskrazilerinin ortaya çıkmasıdır (10 yıl sonra yaklaşık %20, 20 yıl sonra 1/3 ünde). M proteini bulunan hastalarda polinöropatinin nasıl oluştuğu kesin olarak anlaşılamamakla birlikte, bazı M proteinlerinin miyelin ve aksolemmanın bazı komponentlerine karşı antikor özelliği gösterdiği bilinmektedir.
Rutin serum protein elektroforezi küçük miktardaki M proteinlerini belirlemede genellikle yetersiz kalır. İmmün elektroforez veya tercihen immünofiksasyonla M proteinleri saptanır, ağır ve hafif zincir tipleri belirlenir. İdrar protein elektroforezi multipl miyelom ve primer amiloidozla birlikte olan hafif zincir gamopatilerinin gösterilmesini sağlayabilir. M proteini ve nöropatisi olan tüm hastalarda Bence-Jones proteinürisi araştırılmalı, immünglobulinler kantititif olarak incelenmeli, litik ya da sklerotik kemik lezyonlarını araştırmak amacıyla radyografiler yapılmalıdır. Habis plazma hücresi diskrazileri ile MGUS’un ayırt edilmesi için kemik iliği aspirasyon ya da biyopsisi gerekebilir.
Anlamı Belirsiz Monoklonal Gamoapati (MGUS)
İleri yaştaki kişiler arasında MGUS prevalansı oldukça yüksektir (50 yaşın üstünde %1, 70 yaşın üstünde %3). MGUS saptanan hastaların %30-70’inde polinöropati bulunur. Genel popülasyonda en sık görülen M-proteini IgG iken, paraproteinemik polinöropati hastalarında en sık olarak (yaklaşık %50’sinde) IgM görülür. MGUS nöropatisi genellikle orta ve ileri yaştaki hastalarda görülen, sinsi başlangıçlı, aylar ve yıllar içinde yavaş progresyon gösteren bir polinöropatidir. Klinik tablo sıklıkla distal ve simetrik, duyusal-motor bir polinöropati şeklindedir. Daha nadir olarak klinik ve laboratuvar bulguları CIDP’ye benzeyen bir poliradikülonöropati görülür. Elektrofizyolojik incelemelerde demiyelinizan ya da karışık aksonal ve demiyelinizan polinöropatiyle uyumlu bulgular elde edilir. Paraproteini IgM grubundan olan hastalarda daha belirgin duyu kaybı ve duyusal ataksi ile elektrofizyolojik incelemelerde motor distal latanslarda çok belirgin uzama gözlenebilir. IgM paraproteinemisine özgü bu nöropati, daha önce CIDP’nin bir varyantı olabileceği de düşünülen ve yukarıda sözü edilen DADS-M olarak adlandırılmaktadır. Buna karşılık, IgG/IgA gibi monoklonal proteinlere ise sıklıkla uzunluğa bağımlı duyusal-motor aksonal nöropati eşlik eder ve bunlar yavaş bir seyir gösterdiğinden genellikle tedavi gerektirmez. Fakat IgM dışı paraproteinemilerin tipik CIDP de dahil olmak üzere her çeşit periferik nöropati tablosu ile birlikte de görülebileceği akılda tutulmalıdır.
Nörolojik bulguları hafif ve hastalık progresyonu yavaş olan olgular tedavi verilmeden periyodik kontrollerle izlenebilir. Progresif nörolojik defisiti olan hastalardan CIDP’ye benzer klinik ve elektrofizyolojik bulguları olanlar immünmodülatuvar tedavilere iyi yanıt verirler. Bu hastalarda plazmaferez, IVIg veya prednizolon tedavileri (bazen bir sitostatik ilaçla birlikte olmak üzere) uygulanır. IgM MGUS olgularında çeşitli immünoterapi yöntemlerine genel olarak IgG ve IgA MGUS’lu hastalara oranla daha güç yanıt alınır.
IgM MGUS’u olan DADS-M hastalarının en az yarısında monoklonal protein, bazı periferik sinir glikoproteinlerinin (miyeline bağlı glikoprotein=MAG, P0) bir karbonhidrat epitopu ile reaksiyon gösterir. Bu antikor benzeri aktivite, paraproteinemik nöropatilerin otoimmün hastalıklarda görülen mekanizmalarla ortaya çıktığını düşündürmektedir. MAG’a karşı antikor aktivitesi ELISA veya Western blot yöntemi ile gösterilebilir ya da sinir dokusunda immünohistokimyasal yöntemlerle ortaya konabilir. Elektron mikroskopisinde miyelin lamellerinin aralarının genişlemiş görünümü (myelin splitting) anti-MAG nöropatileri için karakteristiktir.
Tedaviye dirençli olduğu bilinen anti-MAG pozitif DADS-M’de rituksimabın denendiği iki plasebo kontrollü çalışmadan ilkinde tedaviyle yürümede düzelme ve antikor titresinde azalma sağlandığı gösterilmiştir. Diğerinde ise özürlülük skorlarında anlamlı bir değişiklik saptanmamıştır. Her ne kadar rituksimab tedavisinin kullanımı için yeterli kanıt düzeyi bulunmasa da, etkili olduğu bildirilen olgu serileri de göz önüne alındığında, genç erişkinlerde ya da motor zaaf veya yürüme güçlüğü gibi ileri derecede özürlülüğe sahip hastalarda kullanılabileceği düşünülmektedir.
Daha az invazif bir inceleme olan cilt biyopsisi ile miyelinli sinir liflerinde IgM birikimi olduğu ve ayrıca anti-MAG antikorlarının enjeksiyonu ile deney hayvanlarında kompleman aracılı periferik sinir demiyelinizasyonunun geliştiği gösterilmiştir. Bununla birlikte yakın zamanda anti-insan doğal öldürücü hücre 1 (HNK1) antikorlarının da anti-MAG nöropatinin tanısında yüksek oranda duyarlılığa ve özgüllüğe sahip olduğu bildirilmiştir. Her ne kadar başka çalışmalar ile desteklenmesi gerekse de anti-HNK1 antikor düzeylerinin tedavi ile azalması ve anti-MAG’ın aksine hastalığın ağırlığı ile bağıntı göstermesi dikkat çekicidir.
CANOMAD (kronik ataksik nöropati, oftalmopleji, M-protein, soğuk aglütininler ve anti-disialosil antikorlar) IgM MGUS ile ilişkili nadir bir hastalıktır. Klinik tablo Miller Fisher sendromuna benzerdir, fakat kronik ilerleyicidir, bazen de ataklarla seyreder. Tanı klinik bulgulara eşlik eden GD1b, GD3, GQ1b ve GT1b gibi disialosillenmiş gangliozid antikorlarından oluşan IgM paraproteinemisinin saptanması ile konulmaktadır. IgM antikorların yaklaşık yarısı soğuk aglütininlerdir. IVIg ve rituksimab ile klinik iyileşmenin olduğu bildirilmekle birlikte randomize kontrollü çalışma bulunmamaktadır.
Primer Sistemik Amiloidoz
Primer amiloidozda amiloid hafif zincirleri (amyloid light chain: AL) dokularda birikir. AL amiloidozu tek başına görülebileceği gibi multipl miyelom ve Waldenström makroglobulinemisinde de görülür. Yaşlılarda ve erkeklerde daha sık rastlanan primer sistemik amiloidozda, hastaların yaklaşık 1/5’inde uzunluğa bağımlı distal aksonal dejenerasyonla seyreden bir polinöropati gelişir. Sinir dokusunda amiloid başlıca endonöral küçük damarların etrafında bir kılıf halinde birikir. Bu görünüm amiloidozdaki sinir hasarının hipoksiye bağlı olarak geliştiğini düşündürür. Amiloid birikimlerinin sinir liflerine bası yaparak ya da doğrudan toksik hasara yol açabileceği de düşünülmektedir. Sinsi başlangıçlı ve yavaş progresif seyirli polinöropatide klinik tabloya ekstremite distallerinde ağrılar, ağrı ve ısı duyusunda baskın hipoestezi ve otonom belirtiler hakimdir. Hastaların yaklaşık %25 inde fleksör retinakulumda amiloid birikimine bağlı karpal tünel sendromu vardır.
Olguların %90 kadarının serum veya idrar immünofiksasyon elektroforezinde monoklonal protein veya hafif zincirler (sıklıkla l) saptanır. Tanı, dokularda amiloid birikiminin histopatolojik olarak gösterilmesi ile konur. Bu amaçla yapılan abdominal yağ aspirasyonu, rektal biyopsi ya da kemik iliği aspirasyonu olguların %80’inde pozitiftir. Kombine sinir ve kas biyopsisinde hastaların %90’ında amiloid birikimi saptanır. Sinir dokusunda amiloid birikimi bulunan fakat plazma hücre diskrazisini gösterecek veri elde edilemeyen hastalar familyal amiloid nöropatiler yönünden de araştırılmalıdır (Bakınız: Herediter Polinöropatiler). Kalp ve böbrek tutulumu olan hastalarda prognoz kötüdür (ortalama sağkalım süresi 6-7 ay). Organ tutulmaları olmayan amiloid nöropatisi olgularında daha iyi prognoz (ortalama sağkalım süresi 40 ay) vardır. Uygun hastalarda kök hücre nakli ile 10 yıllık sağ kalım oranının tam yanıt veren olgularda %53, tüm hastalara bakıldığında ise %43 olduğu bildirilmektedir. Kök hücre nakline uygun olmayan %75-80 oranındaki primer amiloidoz hastasında sıklıkla aralıklı oral melfalan ve prednizon tedavisi uygulanmaktadır. Bu tedavi renal ve kardiyak amiloidozun progresyonunu yavaşlatır, fakat nöropatinin seyrini etkilemez. Daha az sıklıkta uygulanmakta olan tedaviler ise talidomid, lenalidomid, pomalidomid, daratumumab ve bortezomibtir.
Multipl Miyelom
Multipl miyelomda vertebra miyelomuna bağlı omurilik ya da sinir kökü basıları sıktır. Henüz tedavi uygulanmamış multipl miyelom olgularında başlangıçta polinöropati görülme oranı %5 ile %20 olarak bildirilmektedir. Hastalık seyri sırasında gerek hastalığın kendisi gerekse tedavi için uygulanan kemoterapötik ajanların etkisiyle hastaların yaklaşık %75’inde periferik nöropati gelişebilmektedir. Ayrıca multipl miyelom hastalarının %30-40’ında primer (AL) amiloidoz eşlik edebileceğinden, nöropati saptanan multipl miyelom hastalarında sinir biyopsisi ile amiloid birikimi varlığının araştırılabileceği akılda tutulmalıdır.
Osteosklerotik Miyelom: Miyelomlar arasında %2 oranında görülen osteosklerotik miyelomlarda periferik nöropati gelişme oranı %85 tir. Bu hastalıkta plazma hücresi proliferasyonu, sklerotik kemik lezyonları şeklinde kendini gösteren tek veya multipl plazmositomlar şeklindedir. Multipl miyelomdan farklı olarak hastalar daha genç yaştadır ve çoğu erkektir. Polinöropati klinik ve laboratuvar özellikleriyle CIDP’ye benzer (CIDP tablosu nedeniyle incelenen tüm erişkin hastalarda M proteini ve kemik lezyonları araştırılmalıdır). Motor belirtileri baskın, simetrik proksimal ve distal kas zaafıyla seyreden edinsel demiyelinizan bir polinöropati tablosu görülür. BOS’ta protein miktarı yüksektir (genellikle >100 mg/dl). Olguların %90’ında M proteini bulunur (genellikle l hafif zinciri ve IgG, IgA ağır zincirlerinden oluşur). Hastaların çoğunda POEMS (Crow-Fukase) sendromunun multisistem belirtilerinden bir veya birkaçı vardır [POEMS= Polinöropati, Organomegali, Endokrinopati, M proteini, Deri (Skin) değişiklikleri]. Başlıca deride hiperpigmentasyon (Şekil 14a ve 14b), hepatomegali, jinekomasti, testiküler atrofi ve parmaklarda çomaklaşma ile diyabet ve hipotiroidizm görülür. POEMS sendromunun belirtilerinin neoplastik plazma hücrelerinin vasküler endotelyal büyüme faktörü (VEGF) isimli sitokini aşırı miktarda üretmelerinden kaynaklandığı düşünülmektedir. Hastaların çoğunun serumunda çok yüksek düzeyde VEGF bulunur ve hastalık aktivitesi ile korelasyon gösteren bu belirteç POEMS için majör tanı kriterlerinden biri olarak kabul edilmektedir.
Radyografilerde özellikle vertebra, pelvis ve kaburga kemiklerinde sklerotik veya karışık litik-sklerotik lezyonlar görülür. Bu lezyonların ortaya konabilmesi için bazen direkt radyografiler yeterli olmaz, şüphe edilen bölgelerde kemik dokusunun bilgisayarlı tomografi gibi daha ileri bir görüntüleme yöntemiyle incelenmesi gerekebilir (Şekil 14c). İzole plazmositomun varlığını kanıtlamak için genellikle açık biyopsi yapılması gerekir. Tek lezyonu olan hastalarda bu lezyonun total olarak çıkarılması ya da tümör irradyasyonu hastaların klinik bulgularında dramatik düzelme sağlayabilir. Multipl lezyonu olan hastalara kombine melfalan ve prednizolon tedavisi uygulanır. Yeni bir çalışmada otolog periferik kan kök hücresi transplantasyonundan belirgin şekilde yararlanan olgular bildirilmiştir ve <65 yaş hastalarda bir tedavi seçeneği olabileceği düşünülmektedir. Yapısal olarak talidomide benzeyen fakat daha az nörotoksik olduğu bilinen lenalidomid ile kombine deksametazon tedavisine iyi yanıt alınabildiğine dair olgu bildirileri bulunmaktadır. Bortezomibin nöropati bulgularını kötüleştirebileceğine dair endişeler olmakla birlikte POEMS sendromunda tek başına ya da kombine (steroid ya da steroid ile birlikte siklofosfamid şeklinde) tedavi olarak etkili olduğu ve sistemik bulguları da iyileştirebildiği bildirilmiştir.
Şekil 14a. POEMS sendromlu olgunun avuç içinde hiperpigmente lekeler.
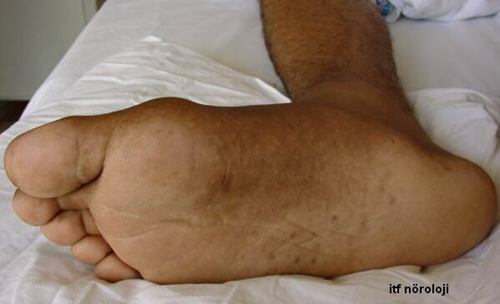
Şekil 14b. Benzer lekelerin ayak tabanındaki görünümü.
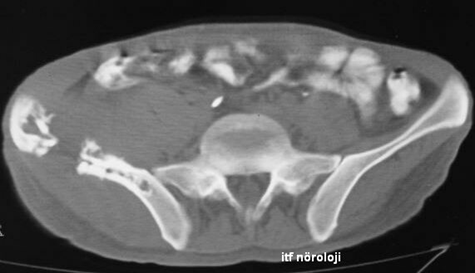
Şekil 14c. Aynı hastanın pelvis bilgisayarlı tomografisinde sağ ilyak kanatta litik-sklerotik lezyon görülmekte.
Perİferİk Sİnİr VaskülİtleRİ
Nöropatinin eşlik ettiği vaskülitler, primer sistemik vaskülitler, sekonder sistemik vaskülitler ve sistemik olmayan/lokalize vaskülitler olarak üç ana grup ve bunların alt grupları şeklinde değerlendirilir:
Tablo 29. Nöropatinin eşlik ettiği vaskülitlerin sınıflandırılması (JBP Dyck, değiştirilerek).
1- Primer sistemik vaskülitler
a. Küçük çaplı damar vaskülitleri (ANCA+ vaskülitler)
i. Mikroskopik polianjitis
ii. Eozinofilik granülomatöz polianjitis (Churg-Strauss sendromu)
iii. Granülomatöz polianjitis (Wegener granülomatozu)
iv. Esansiyel miks kriyoglobulinemi (Çok daha nadir olarak vaskülitik nöropatiye neden olur)
v. IgA ilişkili vaskülit (Çok daha nadir olarak vaskülitik nöropatiye neden olur)
b. Orta çaplı damar vaskülitleri
i. PAN
c. Büyük çaplı damar vaskülitleri
i. Dev hücreli arterit
2- Sekonder sistemik vaskülitler
a. Bağ dokusu hastalıkları (Romatoid artrit, Sjögren sendromu, sistemik lupus eritematozus, sistemik skleroz, dermatomiyozit, miks bağ dokusu hastalığı)
b. Sarkoidoz
c. Behçet hastalığı
d. İnfeksiyon (Hepatit B, Hepatit C, HIV, lepra, Lyme Hastalığı, HTVL-1 gibi)
e. Bazı ilaçlar
f. Malignite
g. İnflamatuvar barsak hastalığı
h. Hipokomplamanlı ürtikerli vaskülit sendromu
3- Sistemik olmayan lokalize vaskülitler
a. Sistemik olmayan vaskülitik nöropati
b. Diyabetik/non-diyabetik radikülopleksus nöropatileri
c. Lokalize kutanöz/vaskülitik vaskülit.
ANCA: anti-nötrofil sitoplazmik antikor, HIV: “human immunodeficiency syndrome, HTLV: “human T-cell lymphotropic virüs”, PAN: poliarteritis nodosa.
Primer sistemik vaskülitler, orta ve küçük çaplı damarların cidarında iltihap ve nekroz oluşturarak bu damarların oklüzyonuna ve sulama alanlarında iskemiye neden olurlar. Sinir sistemi dahil olmak üzere birçok organı etkileyen bu hastalıklarda yaşamı tehdit eden ve tedavisi mümkün olan organ yetersizlikleri ortaya çıktığından erken tanı önemlidir. Primer sistemik vaskülitlerin seyrinde periferik sinir sistemi tutulması sıktır (Tablo 30). Primer sistemik vaskülitlerden, küçük çaplı damar vaskülitleri alt grubundaki mikroskopik polianjitiste arteriol, kapiller ve venüller etkilenir. Glomerülonefrit ve pulmoner hemorajiler sıktır, periferik sinir tutulması hastaların %35-75’inde izlenir. Aynı gruptaki Churg-Strauss sendromunda da birçok organ etkilenmekle birlikte akciğerler ön planda tutulur. Pulmoner infiltrasyonlar, tekrarlayan astım atakları, ateş ve eozinofili sıktır. Hastaların yaklaşık yarısında periferik nöropati görülür. Wegener granülomatozu üst ve alt solunum yolları ile böbrekleri etkiler. Solunum yollarında granülomlar ve nekrotizan glomerülonefrit sıktır. Astma ve eozinofili yoktur. Olguların 1/5 kadarında nöropati görülür. Orbita ve kavernöz sinüslerdeki granülomlara bağlı kranial sinir tutulmaları gelişebilir. Orta çaplı damar vaskülitlerinden olan PAN, küçük ve orta boy arterleri tutarak böbrekler, karaciğer, barsaklar, deri ve kaslar ile merkez ve periferik sinir sistemini tutar. Olguların yaklaşık yarısında nöropati gelişir, 1/3-1/2 kadarında hepatit B yüzey antijeni (HbsAg) pozitiftir. Hepatit B ile birlikte olan olguların daha seyrettiği bildirilmiştir.
Tablo 30. Periferik nöropatiye sıklıkla neden olan sistemik vaskülitlerin klinik özellikleri ve tedavileri (KC Gorson, değiştirilerek)
|
Periferik sinir % |
Üst solunum % |
Alt solunum % |
Renal % |
Gastro-intestinal % |
Artralji Artrit % |
Kalp
% |
Deri
% |
MSS
% |
cANCA/ PR3 % |
pANCA/ MPO |
Damar boyutu |
Diğer |
Tedavi |
Viral |
|
|
Primer sistemik vaskülitler |
|||||||||||||||
|
Wegener granülomatozu1 (granülomatöz polianjitis) |
40-50 |
95 |
70-85 |
70-80 |
<5 |
60-70 |
10-25 |
40-50 |
5-10 |
75-90 |
5-20 |
Küçük-orta |
Hayır |
Steroid+ Sitotoksik/ rituksimab |
Hayır |
|
Churg Strauss sendromu1 (eozinofilik granülomatöz polianjitis) |
60-80 |
50-60 |
40-70 |
10-40 |
30-50 |
40-50 |
10-40 |
50-55 |
5-30 |
3-35 |
2-50 |
Küçük-orta |
Ateş, eozinofili |
Steroid |
Hayır |
|
Mikroskopik polianjitis1 |
60-70 |
Hayır |
15-70 |
75-90 |
30 |
40-60 |
10-15 |
50-65 |
10-15 |
10-50 |
50-80 |
Küçük |
Ateş |
Steroid+ Sitotoksik/ rituksimab |
Hayır |
|
Poliarteritis nodosa2 |
35-75 |
Hayır |
Hayır |
Hayır |
15-55 |
50-75 |
5-30 |
25-60 |
3-17 |
Nadir |
Nadir |
Orta |
Ateş, hipertansiyon |
Steroid |
Hepatit B %33-50 |
|
Sekonder sistemik vaskülitler |
|||||||||||||||
|
Romatoid vaskülit |
50 |
Hayır |
5-30 |
10-25 |
10-30 |
90-100 |
10-30 |
30-90 |
5-15 |
Hayır |
Hayır |
Orta |
Ateş, kilo kaybı, nodüller |
Steroid |
Hayır |
cANCA=Sitoplazmik immünfloresan paterni nötrofil serin proteinaz 3’e (PR3) yönelik anti-nötrofil sitoplazmik antikor (ANCA); pANCA= Perinükleer immünfloresans paterni miyeloperoksidaza (MPO) yönelik ANCA. 1=Küçük çaplı damar vaskülitleri; 2= Orta çalı damar vaskülitleri
Bağ dokusu hastalıklarının seyrinde vaskülit geliştiğinde ortaya çıkan patolojik ve klinik tablo PAN’dakine benzer. Romatoid artrit toplumda sık görülen bir hastalık olduğundan, romatoid vaskülit en sık karşılaşılan vaskülitik nöropati nedenlerindendir. Ancak, modern antiromatizmal tedaviler ile insidansı azalmaktadır. PAN’daki gibi küçük ve orta boy arterleri tutar. Romatoid vaskülit genellikle yerleşmiş romatoid artriti olan hastalarda görülür. Erkekler ve ileri yaş hastalarda risk biraz daha fazladır. Bu nedenle, yerleşmiş romatoid artrit tanısı bulunmayan, ancak romatoid faktörü pozitif olan hastalarda diğer vaskülit nedenleri araştırılmalıdır. Sjögren sendromu ve SLE seyrinde vaskülitik tutulum romatoid artrite kıyasla daha nadirdir. SLE daha çok kronik inflamatuvar demiyelinizan nöropati benzeri bir tabloya neden olurken, Sjögren sendromunda ince lif nöropatisi, distal simetrik nöropati ve kranial nöropatiler vaskülitik nöropatiye göre daha sık olarak karşımıza çıkar. Hepatit C virüsü ile ilişkili kriyoglobulinemik vaskülitte ise vaskülitik nöropati hastaların yaklaşık %65’inde görülür. Glomerülonefrit, palpabl purpura, ülserasyonlar gibi sistemik bulgular da eşlik edebilir. HIV ile ilişkili vaskülitik nöropati, hastalığın neden olduğu bir diğer nöropati tipi olan HIV-ilişkili distal simetrik nöropatiye kıyasla oldukça nadirdir. İlaçlarla ilişkili vaskülitik nöropati, diğer vaskülit nedenleri dışlandıktan sonra düşünülmesi gereken bir tablodur. Ancak nadiren de olsa minosiklin, tümör nekroz faktör alfa (TNF-α) inhibitörleri, son dönemde kanser tedavisinde yaygın olarak kullanılan immün kontrol noktası inhibitörleri ile ilişkili vaskülitik nöropati olguları bildirilmiştir. İlaçlar dışında, kötüye kullanılan kokain gibi maddeler ile de vaskülitik nöropati görülebilir. Vaskülitik nöropatisi olan hastaların yaklaşık %10’una malign hastalıklar eşlik eder.
Biyopsi ile kanıtlanmış vasküliti olan hastaların 1/3 kadarında sistemik bir hastalığı gösteren klinik ve laboratuvar bulguları yoktur (sistemik olmayan/nonsistemik vaskülitik nöropati). Histopatolojik bulguların sistemik nekrotizan vaskülitlerin aynısı olmakla birlikte, sadece periferik sinir ve kas dokusuna sınırlı olduğu bu tablo en sık karşılaşılan vaskülitik nöropatidir. Hastalarda sistemik tutulmayı düşündürecek klinik ve serolojik bulgular genellikle yoktur. Progresyonu yavaş, atakları daha seyrek olan bu hastalıkta yaşamı tehdit eden komplikasyonlar genellikle ortaya çıkmaz. Yine de, başlangıçta sistemik bulguları olmayan bu hastaların bir kısmında deri tutulması görüldüğünü, %10’dan azında zaman içinde büyük organ etkilenmesine giden bir sistemik yayılma geliştiğini hatırlamak gerekir. Tutulan sinirlere ait belirti ve bulgular yavaş fakat iyi bir düzelme gösterdiğinden, nörolojik tablonun prognozu da oldukça iyidir.
Vaskülitik nöropatilerde çapları 50-300 mm arasında değişen arteriol, kapiller, venül, küçük ve orta boy arterlerde yoğun tıkanmalar gelişerek sinir dokusu iskemisine neden olur. İskemi sinir liflerinde aksonal dejenerasyona yol açar. Vasküler inflamasyonun mediyatörü olduğu düşünülen başlıca faktörler, dolaşan immün kompleksler, anti-endotelyal ve antisitoplazmik antikorlar, sitotoksik T lenfositleri ve gecikmiş tip aşırı duyarlılıktır. İlaçlar, HIV, Hepatit B ve C virüsleri gibi infeksiyon ajanları bağışıklık yanıtının tetikleyicileri olarak düşünülmekle birlikte, olguların çoğunda etken belirlenememiştir.
Vaskülit sürecinin dokuları multifokal ve rasgele bir şekilde tutmasına bağlı olarak sinirler asimetrik ve yamalı şekilde lezyona uğrar. Bu nedenle vaskülitler multifokal/asimetrik periferik sinir tutulmasına yol açan nedenlerin prototipini oluşturur (Tablo 8). Vaskülitlere bağlı periferik nöropatiler farklı klinik görünümlerde karşımıza çıkabilir. Olguların çoğu mononöropati multipleks tablosu ile hekimin karşısına gelir. Bir kısmında ise birden çok sinire ait tutulma bulguları oldukça hızlı şekilde birbirinin üzerine eklenerek bir veya birden çok ekstremitede oldukça yaygın fakat asimetrik kas zaafı ve duyu kusuruna neden olan birleşik (konflüan) bir görünüm alır (asimetrik polinöropati görünümü). Olguların %30 kadarında yoğun ve yaygın vaskülitin neden olduğu subakut seyirli, simetrik distal duyusal-motor bir polinöropati görünümü vardır. Bazı hastalarda öykünün dikkatli alınmasıyla nörolojik belirtilerin fokal ve asimetrik başlayıp basamaklı bir ilerleme gösterdiği ortaya çıkarılabilir. Mononöropati multipleks şeklinde klinik seyir gösterenlerde genellikle akut ve ağrılı başlangıç ve hızlı bir progresyon izlenir. Ancak, kronik ve yavaş bir progresyonun söz konusu olduğu hastalara da rastlanır. Yukarıda belirtilen nörolojik bulguların ateş, kırıklık, kilo kaybı, kas ve eklem ağrıları, solunum sistemi belirtileri, hematüri, karın ağrısı, cilt döküntüsü ve gece terlemeleri gibi sistemik belirtilerle ya da açıklanamayan diğer organ tutulmaları ile birlikte olması sistemik nekrotizan vaskülitlerin varlığını düşündürmelidir.
Tanıya yardımcı olacak başlıca laboratuvar incelemeleri eritrosit sedimentasyon hızı, tam kan sayımı ve lökosit formülü, idrar incelemesi ve böbrek fonksiyonlarına yönelik diğer incelemeler, karaciğer enzimleri, romatoid faktör, anti-siklik sitrüllenmiş peptid (anti-CCP), antinükleer antikor, ekstrakte edilebilir nükleer antijenler, serum komplemanları (C3, C4, CH 50), anti-nötrofil sitoplazmik antikorlar, kriyoglobulinler, hepatit B antijen ve antikoru, hepatit C antikoru ve serum immünofiksasyon elektroforezidir.
Vaskülitik nöropatisi olan tüm hastalarda anti-nötrofil sitoplazmik antikorlar (ANCA) bakılmalıdır. Sitoplazmik ANCA (c-ANCA), sıklıkla granülomatöz polianjitis ile birliktelik gösterir. Perinükleer ANCA (p-ANCA) ise daha sık olarak mikroskobik polianjitis ve eozinofilik granülomatöz polianjitis hastalarında pozitif saptanır (Tablo 30). Miyeloperoksidaza karşı gelişen ANCA, MPO-ANCA olarak adlandırılır. Bu tipteki antikorlar genellikle p-ANCA tarzında boyanma tarzı gösterirler. Proteinaz 3’e karşı gelişen antikorlar ise (PR3-ANCA) sıklıkla c-ANCA şeklinde boyanırlar. Son yıllarda nörofilaman hafif zincir düzeyinin sistemik vaskülitlerde periferik sinir tutulumu için bir biyobelirteç görevi gördüğü öne sürülmüştür. Ayrıca damar çeperlerinden hipoksiye yanıt olarak sentezlenen VEGF düzeyinin de vaskülitik nöropatide arttığı izlenmiştir.
Elektrofizyolojik incelemeler periferik sinirlerde akson hasarı ile seyreden tutulmayı gösterir. Nadir olarak, vaskülitik nöropati atağının erken dönemlerinde lezyona uğrayan sinir segmentlerinde elektrofizyolojik olarak tam veya parsiyel iletim bloğu bulguları saptanabilir (‘yalancı iletim bloğu’, Wallerian dejenerasyonun tamamlanmasından önce geçici olarak). Bu bulgu vaskülitik nöropatinin kompresyon ve tuzaklanmaya bağlı sinir lezyonları ile ve inflamatuvar demiyelinizan nöropatilerle karıştırılmasına neden olabilir. Sinir iletim incelemeleri ve iğne elektromiyografisi, klinik olarak gözden kaçan fokal ve asimetrik sinir tutulması bulgularını ortaya koyarak tanıya yardımcı olabilir. BOS genellikle normaldir.
Vaskülitik nöropatilerin kesin tanısı histopatolojik olarak damar duvarlarında destrüksiyonla birlikte transmural mononükleer hücre infiltrasyonu ve nekrozun gösterilmesi ile konur (Şekil 15). Deri lezyonları olan hastalarda lezyon yerinden alınacak deri biyopsisi tanıyı sağlar. Birçok kere vasküliti tespit etme olasılığını arttırmak amacıyla kombine sinir ve kas biyopsilerinin alınması tavsiye edilir. Vaskülitik nöropati düşünülen hastalarda uzun, komplikasyon riski yüksek tedavilerin uygulanması söz konusu olduğundan ve tedaviye başlandıktan sonra histopatolojik incelemeler güçleşeceğinden doku biyopsisine bir an önce başvurulması yerinde olur.

Şekil 15a. Damarın tüm katmanlarını tutan mononükleer hücre infiltrasyonu (non-nekrotizan vaskülit) (Yüzeyel peroneal sinir biyopsisi, HE, X20)
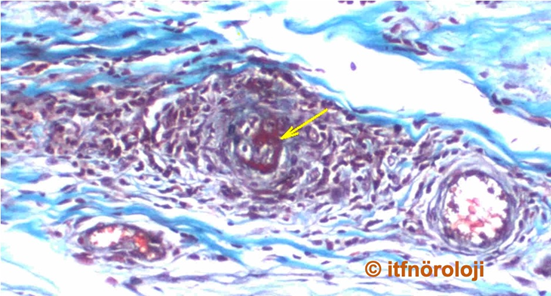
Şekil 15b. Nekrotizan vaskülit. Yüzeyel peroneal sinir biyopsisinde tümüyle tıkalı bir damarın cidarında fibrinoid nekroz (ok) (Modifiye gomori trikrom (MGT), X20)
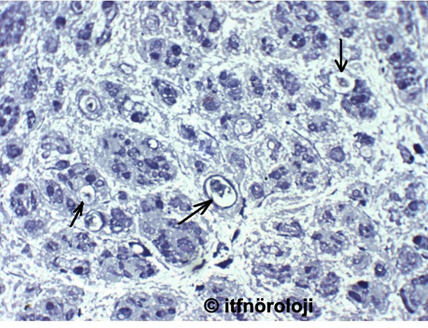
Şekil 15c. Miyelinli aksonların hemen tümüyle dejenere olduğu ileri aşamada vaskülit olgusu. Waller dejenerasyonunun çeşitli evrelerine ait bulguları olan lifler (Oklar) (Thionin, X40)
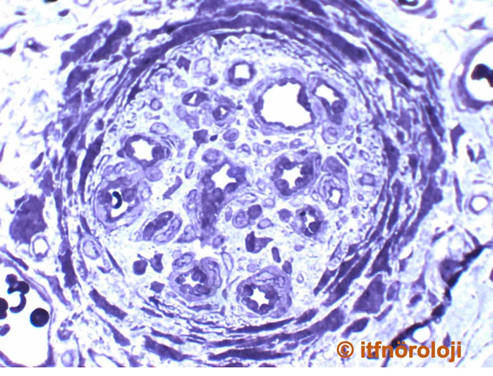
Şekil 15d. Vaskülitik nöropati olgusunda tam tıkalı epinöral damarda endotelle döşeli revaskülarizasyon alanları (Thionin, X40).
Vaskülitlerin önde gelen tedavi ajanı kortikosteroidlerdir. Sistemik nekrotizan vaskülitlerin tedavisinde, ilerlemekte olan multipl organ ve sinir hasarını durdurmak amacıyla genellikle agresif bir yaklaşım benimsenir. Steroidler yüksek dozla oral başlanır (prednizolon, 1-2 mg/kg/gün) veya fulminan olgularda başlangıçta 3-5 gün yüksek doz intravenöz (1000 mg/gün) verildikten sonra oral tedaviye geçilir. Wegener granülomatozu ve mikroskopik polianjitis olguları, multipl organ tutulması olan ve kortikosteroid tedavisine rağmen ilerleyen olgularda kortikosteroid ve sitostatik ajan tedavisine kombine olarak başlanır. Remisyon oluşturmada en etkili olan sitotoksik ilaç siklofosfamiddir. Genellikle oral yolla 2 mg/kg/gün dozda verilir.
İntravenöz siklofosfamid uygulamasının daha az yan etkisi olduğu, ancak nüks riskini yükselttiğini düşündüren bulgular mevcuttur. Siklofosfamid uygulanan hastalarda ilk ayda haftada bir, daha sonra ayda bir tam kan sayımı yapılır. Total lökosit sayısının 3500/ml’nin, nötrofil sayısının 1500/ml’nin altına düşmesi halinde dozun titre edilmesi veya ilacın kesilmesi gerekir. Hemorajik sistit gelişimini engelleyici tedbirler alınır. Siklofosfamidle remisyon elde edildikten sonra metotreksat veya azatiopirin ile idame tedavisine geçilebilir. Aynı amaçla mikofenolat mofetil ve leflonomid de kullanılmıştır. Belirgin bir klinik iyileşme ortaya çıkıncaya kadar iki ilaçla birlikte tedavi yapılır. Klinik remisyon görüldükten sonra prednizolon tedavisi yavaş olarak günaşırı uygulamaya dönüştürülür ve daha sonra azaltılır (ayda bir 5-10 mg, doz azaldıkça daha yavaş). İdame tedavisi tüm hastalık aktivitesi kaybolduktan sonra 1 yıl daha sürdürülür. Kortikosteroid ve sitostatik kombine tedavisi sistemik nekrotizan vaskülitlerde oldukça iyi bir remisyon oranı sağladığı gibi, bu hastalıklara bağlı ölümleri de belirgin derecede azaltmıştır. Nonsistemik vaskülitik nöropati ve dev hücreli arteritlerde organ hasarına bağlı yaşamsal tehlike olasılığı düşük olduğundan, tedaviye sadece kortikosteroidle başlanabilir. Nonsistemik vaskülit nöropatide spontan remisyonlar da görülebildiğinden, tedavi edilecek hastaları seçmek güçtür ve klinik tablonun ağırlığına, seyir özelliklerine dayanılır. Bu olgularda kortikosteroid ve sitostatiklerin kombine edilmesinin düzelme oranını arttırdığı, ancak daha yüksek oranda ilaca bağlı yan etkilere sebep olduğu gösterilmiştir. Bu tedavilere yanıt vermeyen refrakter hastalarda ise IVIg, rituksimab ve plazmaferez denenebilir. Özellikle rituksimabın son yıllarda mikroskobik polianjitis ve Wegener granülamatozunun indüksiyon tedavisinde ilk seçenek olarak siklofosfamide alternatif olabileceği düşünülmektedir. Refrakter PAN tedavisinde ise infliksimab ile olumlu sonuçlar bildirilmiştir.
Hepatit B, C ve HIV infeksiyonu ile birlikte olan vaskülitlerde kronik immünosupresyon viremiyi arttırabileceği için genel olarak kontrendikedir. Ancak hepatit B ile birlikte olan PAN’da kısa süreli olarak 1-2 haftalık steroid uygulamasını izleyen antiviral tedavi, interferon alfa-2b veya lamivudin verilmektedir). Hepatit C’nin tedavisindeki gelişmeler ise son yıllarda önemli ölçüde hız kazanmıştır. Eskiden daha sıklıkla kullanılan interferon tedavilerinin yerini doğrudan etkili antiviral ajanlar almıştır. Özellikle zarf proteinine ve diğer yapısal proteinlere karşı geliştirilen antiviral tedaviler ve proteaz inhibitörleri Hepatit C virüsünün tedavisinde oldukça etkilidir. Ancak bu etki Hepatit C virüsü ile ilişkili kriyoglobulinemik vaskülitte daha az olarak izlenir. Bu olgularda serumda dolaşan kriyoglobulinlerin plazmaferez ile hızlı şekilde ortadan kaldırılması ile iyi sonuçlar alınabilmektedir. Ayrıca rituksimabın polinöropati bulgularına yararlı etkisi olduğunu bildiren açık çalışmalar mevcuttur.
METABOLİK BOZUKLUKLARA BAĞLI POLİNÖROPATİLER
Diyabetik Nöropati
DM gelişmiş batı ülkelerinde periferik nöropatilerin en sık nedenidir. Diyabetle birlikte yaşama süresi uzadıkça nöropati prevalansı artar. Yirmi beş yıllık diyabeti olan hastaların yaklaşık yarısında diyabetik nöropati gelişir.
Diyabetik nöropatilerin farklı klinik görünümleri Tablo 31’de verilmiştir.
Tablo 31. Diyabetik nöropatiler (R Pop-Busui ve ark., değiştirilerek)
________________________________________
· Diffüz nöropatiler
§ Distal simetrik polinöropatiler
· İnce lif baskın polinöropatiler
· Kalın lif baskın polinöropatiler
· Miks tipte polinöropatiler (En sık)
§ Diyabetik otonom nöropati
§ Diyabetik nöropatik kaşeksi
· Mononöropatiler
§ İzole kranial veya periferik nöropatiler (okülomotor nöropati, ulnar nöropati, femoral nöropati gibi)
§ Mononöritis multipleks
· Radikülopati ve/veya pleksopatiler
§ Radikülopleksus nöropatisi (lumbosakral poliradikülopati, proksimal motor amiyotrofi)
§ Torakal radikülopati
*Diyabette sıklığı artan nöropatiler
§ Tuzak nöropatileri
§ Kronik inflamatuvar demiyelinizan polinöropati
§ Radikülopleksus nöropatisi
§ Tedavi ile ilişkili akut ağrılı ince lif nöropatisi
Hiperglisemik Nöropati
Yeni tanı konmuş veya iyi kontrol altında olmayan diyabetik nöropatilerde görülür. Alt ekstremite distallerinde rahatsızlık verici dizesteziler ve ağrılarla seyreder. Gliseminin kontrol altına alınması ile hızlı şekilde düzelir. Elektrofizyolojik incelemelerde yavaş bulunan sinir iletim hızları da hipergliseminin düzelmesini izleyerek hızla normale döner.
Diyabetik Polinöropati (DPN, Distal Simetrik Polinöropati)
En sık görülen diyabetik nöropati şeklidir. Genellikle uzun süre hiperglisemiye maruz kalmış, retinopati ve nefropati gibi diğer komplikasyonları da ortaya çıkmış olan tip I ve tip II DM’li hastalarda görülür. Birçok kere tip II DM’nin ortaya çıkış zamanı kesin olarak bilinemediğinden hastalığın ilk klinik bulgusu olarak da karşımıza çıkabilir. Ekstremite distallerinde yanma, batma ya da sızlama şeklinde ağrılar, paresteziler, hipoesteziden oluşan duyusal belirti ve bulgulara otonom sinir sistemi tutulmasına ilişkin bulgular ve hafif derecede distal motor zaaf ve atrofi eklenir. Yakınmalar alt ekstremite distallerinden başlayarak sinsi şekilde proksimale doğru yayılır. Üst ekstremitelere ilişkin yakınmalar genellikle distal simetrik polinöropatiye değil, karpal tünel sendromu gibi bir mononöropatiye bağlıdır (aşağıya bakınız). Şiddetli ağrı, genellikle sanılanın aksine, DPN’de sık bir belirti değildir. Şiddetli ağrı daha çok kilo kaybı ile birlikte olan, subakut başlangıçlı diyabetik nöropatik kaşekside ya da radikülopleksus nöropatisinde görülür.
Hafif otonom tutulma hem tip I hem tip II DM olgularında görülür. Şiddetli otonom nöropatiye ise daha çok tip I DM’de rastlanır. Başlıca belirtiler cinsel fonksiyon bozukluğu, gastroparezi, diyabetik diyare, mesane atonisi, ayaklarda terleme kaybı ve çeşitli kardiyovasküler bozukluklardır. Hastaların bir kısmında ayağa kalkınca baş dönmesi ya da senkop gibi postüral hipotansiyon belirtileri vardır. Anormal kalp ritmi değişkenliği ve buna bağlı kardiyak aritmilerin diyabetik hastaların uzun vadeli sağkalım oranlarını azaltan önemli faktörlerden olduğu düşünülür. DM’li olgulardaki ani kardiyak ölümlerden diyabetik otonom nöropati sorumlu tutulmuş olmakla birlikte, aterosklerotik kalp hastalığı ve nefropatinin bunlarla daha fazla korelasyon gösterdiğin ortaya konmuştur.
Ön planda miyelinsiz ve ince miyelinli sinir liflerini etkileyen DPN, süreç içinde yerleştiğinde büyük oranda geriye dönüşsüzdür. Klinik tabloya ayak ülserleri ve nöropatik artropati (Charcot eklemleri) eklenebilir. Elektrofizyolojik incelemelerde demiyelinizasyonun da eşlik edebildiği, genellikle aksonal hasarla seyreden duyusal-motor bir polinöropatiyi yansıtan bulgular saptanır. Ön planda ince sinir lifleri tutulan erken dönem hastalarında sinir iletim incelemeleri normal bulunabilir.
DPN tanısı, uzun süreli kötü glisemik kontrole maruz kalan ve genellikle diğer DM komplikasyonları olan hastalarda duyusal baskın distal simetrik aksonal polinöropatinin varlığında, benzer bir polinöropatiyi oluşturacak diğer nedenlerin de dışlanmasıyla konulur. Ayırıcı tanıda değerlendirilmesi gereken diğer polinöropatiler arasında amiloid nöropatisi, herediter nöropatiler, immün kökenli nöropatiler (Sjögren sendromu, paraproteinemi ile birlikte olanlar), B vitamini yetersizliklerine bağlı olanlar, hipotiroidi, üremik nöropati, vb. sayılabilir.
Akut Ağrılı Diyabetik Nöropati [“Diyabetik Nöropatik Kaşeksi” (Ellenberg, 1974)]
Bu nadir görülen sendrom diyabetik duyusal polinöropatinin farklı bir formunu oluşturur. Özellikle alt ekstremitelerde, çok nadir olarak üst ekstremiteler ve gövde üzerinde akut ya da subakut başlangıçlı, geceleri daha belirgin, yanıcı, sızlayıcı ağrı ile karakterizedir. Erkeklerde ve tip II diyabeti olan hastalarda daha sık olarak görülür. Buna deride yaygın kontakt hiperestezisi eşlik eder. DM’nin süre ve şiddeti ile bağıntısız olan, bazen sıkı glisemi kontrolünü izleyerek ortaya çıkan bu tabloya şiddetli ve hızlı kilo kaybı, depresyon ve impotans eşlik edebilir. Objektif nörolojik bulgu çok azdır. Tedavisi çok iyi glisemi kontrolü ile olur (tedavi ile presipite olan olgularda bile). Monofazik seyir gösterir ve genellikle 6-9 aylık bir süre içerisinde iyileşme görülür.
Mononöropatiler
Kranial nöropatiler: Akut başlangıçlı 3. ve 6. sinir felçleri genellikle ileri yaşta, glisemi kontrolü kötü olan hastalarda ortaya çıkar. Olguların yarısı kadarında göz çevresi ve ardında şiddetli ağrı ile başlar. Aylar içinde spontan ve tam düzelme gösterir. Diyabetik 3. sinir felcine genellikle pupilla katılmaz. Bunun nedeni, sinirin orta kesimindeki, muhtemelen iskemik kökenli olan lezyondan sinirin periferinde seyreden otonom liflerin etkilenmemesidir. Bazı diyabetik olguların akut başlangıçlı izole kranial sinir felçlerinden beyinsapındaki küçük iskemik lezyonların sorumlu olduğu da gösterilmiştir.
Diyabetik hastalarda 7. sinir felcinin de daha sık görüldüğü iddia edilmiştir. Ancak diyabet ve Bell felci toplumda sık görülmektedir ve bu iki hastalık durumu arasında nedensel bir bağlantı olduğuna ilişkin kanıtlar yoktur. Diyabetik bir hastada gelişen periferik yüz felcinin bir diyabetik nöropati formu olarak değil, Bell felcinde (ya da bu sinirin diğer lezyonlarında) olduğu şekilde ele alınması uygun olur (Bakınız: Kranial Nöropatiler). Bazı yazarlar DM varlığının Bell felcinin iyileşmesi açısından kötü bir prognostik faktör olduğunu öne sürmüştür.
Diyabetik Radikülopleksus Nöropatisi (DRPN, Proksimal Diyabetik Nöropati, Diyabetik Amiyotrofi, Bruns-Garland Sendromu)
Distal simetrik polinöropatiye oranla daha az görülen (Tip II diyabette %1,1, tip I de % 0,3) bu diyabetik nöropati formu, akut veya subakut, gürültülü ve sıklıkla asimetrik başlangıcı ile dikkati çeker. Daha çok 50 yaşın üstündeki erkek hastalarda ve kilo kaybı ile birlikte görülür. Bu hastalarda uzun süreli ve kötü kontrol edilen diyabet öyküsü, ya da DM’nin retinopati, nefropati gibi komplikasyonları genellikle yoktur. En sık görülen formu olan lumbosakral radikülopleksus nöropatisi (DLRPN), genellikle bir alt ekstremitede, başlıca kalça ve uyluk üzerinde şiddetli ağrı ile başlar. Sıklıkla femoral sinir alanındaki ağrı ile başlaması nedeniyle diyabetik femoral nöropati adı ile anılmıştır. Ancak bu isim tüm klinik tabloyu tanımlayamadığından DRPN tercih edilmektedir. Klinik tabloya günler içinde aynı ekstremitenin kök kaslarında belirgin olan kuvvetsizlik ve giderek atrofi eklenir. Nörolojik muayenede tutulan tarafta femoral sinir ya da üst lumbosakral pleksusu ilgilendiren alanda hipoestezi, kök ve uyluk kaslarında zaaf ve atrofi, patella refleksinin kaybı dikkati çeker. Olguların birçoğunda ağrı, duyu kusuru, kas kuvvetsizliği ve atrofi aynı ekstremitede dizaltında, siyatik sinirin motor ve duyusal dallarını ilgilendiren alana da uzanır. Akut ağrıyı izleyen proksimal ve distal zaafla giden klinik tablo birçok kere kompresif lumbosakral radikülopati ile karıştırılır. Bilgisayarlı tomografi veya spinal MRG incelemelerinde hafif diskopati bulguları olan hastalar, bu görünüm genel popülasyonda çok sık rastlanan bir görüntüleme bulgusu olduğundan ilgili disklere yönelik gereksiz cerrahi girişimlere maruz kalabilirler. Aynı atak sırasında, izleyen ataklarla ya da bazen yavaş progresif bir seyirle karşı ekstremitede de benzer belirti ve bulgular gelişebilir.
Torakal radikülopleksus nöropatileri gövde üzerinde band tarzında ağrı ve nadir olarak karın kaslarında segmenter atoni ile seyreder. Bu olgulardaki ağrılar viseral kökenli ağrılar ve herpes zoster infeksiyonu ile karıştırılabilir. Üst ekstremiteleri ilgilendiren ve nöraljik amiyotrofiye (idyopatik akut brakiyal pleksusu nöropatisi) benzeyen bir servikal-brakiyal radikülopleksopati de görülebilir (Bakınız: Spinal Sinirlerin Hastalıkları). Proksimal diyabetik nöropatiye genellikle diyabetik distal simetrik nöropatiye ilişkin bulgular da katılabilir.
“Diyabetik amiyotrofi” adı altında tanımlanan klinik tabloyu da bu grupta ele almak gerekir. Alt ekstremite kök kaslarında simetrik ağrısız kuvvetsizlik ve atrofi ile giden olgular bildirilmiş olmakla birlikte, bunların yukarıda tanımlanan asimetrik başlangıçlı şekilden farklılığı şüphelidir.
Proksimal diyabetik nöropatinin oluşumundan, spinal sinir köklerini ve pleksusları asimetrik şekilde tutan ve bazen otoimmün kökenli vaskülit şeklinde seyreden diyabetik mikroanjiopati sorumlu tutulur (aşağıya bakınız). Genellikle aylar içinde spontan düzelme görülmekle birlikte, olguların bir kısmında iyileşme yetersiz olur. Hastaların %20 kadarında tekrarlama bildirilmiştir.
Sinir iletim incelemeleri genellikle distal simetrik polinöropatiyi yansıtan bulgular verir. İğne elektromiyografisinde semptomatik ekstremitenin lezyona uğrayan radiks-pleksus segmentlerinden innervasyon alan kaslarında ve paraspinal kaslarda yoğun parsiyel denervasyon-reinnervasyonu gösteren bulgular saptanır. BOS proteini genellikle artmıştır. Nadiren eritrosit sedimentasyon hızı ve romatoid faktör yüksekliğine, antinükleer antikor ve immün aracılı hastalıkların diğer laboratuvar belirteçlerinin yüksekliğine rastlanır.
DLRPN’nin ayırıcı tanısında spinal köklerin disk fıtıklanmalarına veya diğer dejeneratif spinal süreçlere bağlı basıları, intraabdominal tümöral veya iltihabi kitlelere bağlı sinir kökü-pleksus bası ve infiltrasyonları, alt ekstremitenin arteriyel ve venöz dolaşım bozuklukları ile kalça ekleminin travmatik ve dejeneratif ağrılı süreçleri akla getirilmelidir.
Tuzak Nöropatileri
DPN, tuzak nöropatilerinin oluşumunu kolaylaştırabilir. Subklinik DPN’si olan bir hasta, en sık görülen tuzak nöropatisi olan karpal tünel sendromuna ait klinik yakınmalarla hekimin karşısına çıkabilir. Hastanın yakınma ve bulgularının şiddeti tuzak nöropatisine yönelik özgün tedavileri (cerrahi girişim gibi) gerektirecek düzeye varabilir. Bazı hastalarda ise tuzak nöropatisinin belirti ve bulguları olmaksızın elektrofizyolojik incelemelerde sinirlerin mutad tuzaklanma yerlerinde, en sık olarak bilekte median, dirsekte ulnar sinir olmak üzere iletim yavaşlamaları saptanır (subklinik tuzak nöropatisi).
Diyabette CIDP
Bazı yazarlar diyabetlilerde CIDP sıklığının genel topluma oranla yüksek olduğunu öne sürmüşlerdir. Bununla birlikte, özellikle DM’nin toplumda sık rastlanan bir hastalık olması nedeniyle, bu iki hastalık arasında tesadüfün ötesinde bir ilişki bulunduğu kanıtlanamamıştır. Yine de, bazı diyabetik nöropati formları ile CIDP arasında, BOS’ta protein artışı, sinir iletimlerinde demiyelinizan özellikler ve immünmodülatuvar tedaviden yararlanma gibi ortak özellikler bulunması ilgi çekicidir. DM’li bir hastada ortaya çıkan polinöropatinin hızlı ve motor belirtileri baskın ağır bir seyir göstermesi, muayenede tendon reflekslerinde genel azalma bulunması, sinir iletim incelemelerinde multifokal demiyelinizasyonun açık bulgularının saptanması ve BOS proteininin çok yüksek olması halinde CIDP varlığı düşünülerek tedavinin ona uygun şekilde planlanması yerinde olur.
Tedavi ile İlişkili Akut Ağrılı İnce Lif Nöropatisi (İnsülin Nevriti)
İnsülin tedavisine yeni başlanan hastalarda alt ekstremite distallerinde ortaya çıkan duyusal belirtilerle karakterize, Caravati tarafından 1933 yılında tanımlanmış bir tablodur. Sıklıkla otonomik yakınmalar da eşlik eder. Retinopati ve nefropati bulgularının da belirginleşmesi ile birliktelik gösterebilir. Nörolojik muayene ve elektrofizyolojik testlerde çok az objektif bulgu mevcuttur. Sıkı glisemik kontrolün yol açtığı tekrarlayıcı hipogliseminin mi, yoksa insülinin tetiklediği başka mekanizmaların mı bu tabloya yol açtığı iyi bilinmemektedir.
Glikoz Tolerans Bozukluğunda Polinöropati
Kontrolsüz çalışmalar glikoz tolerans bozukluğu (IGT) olan kişilerde idyopatik aksonal polinöropati oranının genel toplumdan daha yüksek olduğunu göstermiş, fakat bu bulgu kontrollü çalışmalarla doğrulanamamıştır. Söz konusu polinöropatinin distal belirti ve bulguları baskın, uzunluğa bağımlı hafif bir aksonal polinöropati şeklinde seyrettiği, bu özellikleri ile DPN’nin hafif bir formunu teşkil ettiği düşünülmektedir.
Patogenez
Tablo 26’da görüldüğü gibi son yıllarda diyabetik nöropati formlarını genel olarak distal-simetrik (duyusal-otonom-motor) ve fokal-multifokal nöropatiler (mononöropatiler, mononöritis multipleks, kranial nöropatiler) ile radikülopati ve pleksopatiler (DLRPN) olarak üç gruba ayırma eğilimi vardır. Distal-simetrik polinöropatilerin oluşumuna daha çok diyabete bağlı sinir metabolizması bozukluğunun neden olduğu, proksimal-asimetrik formların ise diyabetik küçük damar hastalığına bağlı olarak ortaya çıktığı düşünülmektedir. Tüm diyabetik nöropati formlarını tek hipotezle açıklamaya çalışan görüşler de vardır. Örneğin, diyabetik küçük damar hastalığına bağlı multifokal çok sayıda lezyona ait belirtilerin bir araya toplanarak (konflüans göstererek) bir distal simetrik polinöropati görünümü ortaya çıkarabileceği de savunulmaktadır. Tablo 32, DM’li hastalarda görülen farklı nöropati tabloları için en olası patogenetik mekanizmaları özetlemektedir.
Tablo 32. Diyabetik nöropatilerin fizyopatolojik klasifikasyonu (PJB Dyck, değiştirilerek)
|
Fizyopatolojik mekanizma |
Nöropati alt-tipi
|
|
Metabolik-mikrovasküler-hipoksik |
Diyabetik polinöropati (DPN) |
|
|
Diyabetik otonom nöropati
|
|
İnflamatuvar-immün |
Radikülopleksus nöropatileri (DRPN) |
|
|
Kranial nöropatiler |
|
|
Ağrılı distal nöropati ve kilo kaybı (diyabetik kaşeksi)
|
|
|
Diyabette CIDP
|
|
Kompresyon ve tekrarlayıcı hasar |
Bilekte median nöropati |
|
|
Dirsekte ulnar nöropati |
|
|
Fibula başında peroneal nöropati
|
|
Diyabet komplikasyonları |
Ketoasidoz nöropatisi |
|
|
Kronik böbrek yetersizliği nöropatisi |
|
|
Büyük damar iskemisi ile birlikte nöropati
|
|
Tedavi ile ilişkili |
İnsülin nevriti |
|
|
Hiperinsülin nöropatisi |
Metabolik mekanizmalar: “Diabetes Control and Complications Trial" (DCCT) isimli klinik çalışma kan şekerini çok sıkı şekilde kontrol altına alan yoğun insülin tedavisinin diyabetik nöropati gelişme riskini 5 yılda %64 oranında azalttığını göstermiştir. Bu bulgu, yüksek kan glikozunun yol açtığı metabolik bozuklukların diyabetik nöropatiye yol açabileceğine ilişkin en kuvvetli kanıtı teşkil etmektedir. Kronik hiperglisemi lipid, alkol şekerleri ve miyoinozitol metabolizmasında bozukluklara yol açar. Üzerinde en çok durulan ve çalışılan metabolik hipotez, poliol yolu ile ilgili olandır. Glikoz, aldoz redüktaz enzimi tarafından sorbitole dönüştürülür. Hiperglisemi nedeniyle bu metabolik yolun aşırı çalışması hücre içinde sorbitol birikimine yol açar. Bu birikim hücre içinde miyoinozitol ve taurinin azalmasına, bu da hücre metabolizmasının bozulmasına neden olur. Hücre içinde miyoinozitol ve taurin azalmasının Na+-K+-ATPaz aktivitesi azalmasına ve sinir iletim hızının düşmesine neden olduğu gösterilmiştir. Aldoz redüktaz inhibitörleri uygulaması ve diyete miyoinozitol eklenmesi ile diyabetik nöropatiyi tedavi etmeyi amaçlayan çalışmalardan bugüne kadar yüz güldürücü ve kliniğe uygulanabilecek sonuç alınamamıştır. Bununla birlikte, farklı ilaçlarla yapılacak yeni çalışmalardan daha olumlu sonuçlar alınması beklenebilir.
Nörotrofik faktörlere ilişkin bozuklukların diyabetik nöropatiden sorumlu olabileceğine ilişkin bulgular da elde edilmiştir. Hayvan deneylerinde ve diyabetik hastalar üzerindeki çalışmalarda NGF ve beyin kaynaklı nörotrofik faktör düzeylerinin azaldığı ve hedef dokulardan sinir hücre gövdesine retrograd aksonal transportunun bozulduğu gösterilmiştir. Başka bir çalışmada diyabetik hastaların derisinde NGF ekspresyonunun azalması ile ince lif nöropatisinin başlangıç bulguları arasında bir bağlantı olduğu görülmüştür. Diğer bazı çalışmalarda nörotrofin-3 ve insüline benzer büyüme faktörü azalımı ile diyabetik nöropati arasında ilişki bulunmuştur. Nörotrofik faktörlerle nöropatilerin önlenmesi ve tedavisini amaçlayan preklinik araştırmalar olumlu sonuçlar vermekle birlikte, bugüne kadar yapılan klinik çalışmalarda birbiri ile uyumsuz sonuçlar elde edilmiştir.
Diyabetik nöropatinin patogenezinde rol aldığı düşünülen ve tedavisi konusundaki çalışmaları yönlendiren diğer metabolik hipotezler, oksidatif stres ve serbest radikal sentezindeki artış (alfa-lipoik asit uygulaması çalışmaları) ve kalıcı hipergliseminin yol açtığı proteinlerin non enzimatik glikasyonudur (yıkılamayan artmış glikasyon son ürünleri). Ortaya çıkan bu glikasyona uğramış proteinlerin “receptor for advanced glycation endproducts (RAGE)” reseptörleri üzerinden inflamasyon kaskadını başlattığı öne sürülmektedir. Ayrıca hiperglisemi sonucunda polyADP-riboz polimeraz (PARP) aktivasyonu sonucunda serbest radikal oluşumunun arttığı da saptanmıştır. PARP inhibisyonu ile deneysel diyabetik nöropati modellerinde olumlu sonuçlar elde edilmiştir. Yukarıda söz edildiği gibi, bu hipotezlere dayanılarak günümüze kadar gerçekleştirilen çalışmalarda diyabetik nöropatinin önlenmesi ve iyileştirilmesinde kullanılabilecek, etkisi kanıtlanmış klinik tedavi yöntemleri ortaya çıkmamıştır. Endonöral kan akımını arttırdığı öne sürülen prostaglandin analoglarının verilmesi, diyetin linolenik asitle zenginleştirmesi (evening primrose=akşam çuhaçiçeği yağı), antioksidan olarak E vitamini ve alfa lipoik asit uygulanmasının etkilerinin kanıtlanması gerekmektedir. Bazı ülkelerde ağrılı nöropati tedavisinde yoğun şekilde kullanılmakta olan alfa lipoik asit, bir çift-kör çalışmada etkili bulunmuştur.
Vasküler mekanizmalar: Özellikle fokal/asimetrik diyabetik nöropatilerin oluşumundan sorumlu tutulan vasküler mekanizmalara ilişkin en önemli kanıtlar klinik ve histopatolojik verilerden kaynaklanmaktadır. Gerçekten de, multifokal seyirli nöropatilerin en önemli nedenlerinden birisi sinirleri besleyen küçük damarların hastalığıdır (Bakınız: Vaskülitlere Bağlı Nöropatiler). Histopatolojik olarak diyabetik mikroanjiopatide damar duvarlarında kalınlaşma ve endonöral küçük damar lümenlerinde daralma görülür. Bu patolojik sürecin başlıca bileşenleri bazal lamina duplikasyonu, endotel ve perisit hücrelerinin dejenerasyonu, endotelyal fenestrasyon ve ayrışmadır. Diyabetik lumbosakral radikülopleksopatisi olan hastaların sinir biyopsilerinde mikroanjiopatiye sekonder iskemik değişikliklerin varlığı gösterilmiştir (aksonal dejenerasyon, multifokal sinir lifi kaybı, fokal perinöral fibroz ve kalınlaşma, hasar nöromaları, neovaskülarizasyon ve organellerin biriktiği şiş sinir liflerinin varlığı). Mikrovasküler hasarın sinir liflerinde aksonal hasarın yanısıra akson distrofisine bağlı sekonder segmental demiyelinizasyona da neden olduğu görülmüştür.
Vasküler hipotezle metabolik görüşleri ve yaygın polinöropati oluşum mekanizmasını bağdaştıran yorumlar da vardır. Endonöral hipoksinin sinir liflerinde aksonal transportu ve Na+-K+-ATPaz aktivitesini azalttığı düşünülür. Bu bozukluklar da aksonal dejeneresans ve sonuçta sinir iletim hızının yavaşlamasına neden olur. Ancak bu hipotez ile hastalığın neden duyusal ve otonom sinirleri daha çok seçtiğini açıklamak güçtür.
Diyabetik nöropatinin gelişiminde otoimmün mekanizmaların rolü konusundaki görüşler nöropatinin tedavisinde yeni yaklaşımların tartışılmaya başlamasına neden olmuştur. Özellikle diyabetik radikülopleksus nöropatisi olan hastaların sinir biyopsilerinde mikroskopik vaskülit varlığını (epinöral küçük damarların duvar ve çevrelerinde inflamasyon, damar duvarında nekroz ve eski kanama bulguları) gösteren çok sayıda çalışma vardır. Bu bulgular, özellikle fokal/asimetrik diyabetik nöropatilerin tedavisinde kortikosteroidler ve IVIg gibi immünmodülatuvar tedavilerin uygulanmasını gündeme getirmiştir. Kendiliğinden regresyon gösterme eğilimi olan bu nöropatilerde uygulanan immünmodülatuvar tedavilerin etkili olup olmadığı tartışılmakla birlikte, bu tedaviler birçok merkezde ağır seyirli ve yeterli düzelme göstermeyen olgular üzerinde denenmektedir. Çok merkezli, prospektif, kontrollü bir çalışma ile IV metilprednizolon uygulamasının ağrı ve diğer duyusal belirtiler gibi ikincil sonlanım kriterleri üzerine etkili olduğu, erken uygulanması halinde DLRPN olgularında yararlı olabileceği kanısına varılmıştır. Kortikosteroid uygulamasının, glisemi kontrolünü kötüleştirme potansiyeli göz önünde bulundurularak hastanın diyabet uzmanının yakın kontrolü altında yapılması gerekir. Yukarıdaki sözü edilen mekanizmalar sonucunda ortaya çıkan ve hastalığın en rahatsız edici bulgularından biri olan nöropatik ağrının tedavisi ayrıca ayrıntılı bir şekilde tartışılacaktır.
Tedavi
Bugün için diyabetik nöropatinin önlenmesi ve tedavisinde en etkin yöntem kan glikoz düzeyinin iyi kontrol altında tutulmasıdır. Diyabetli hastalarda nöropati oluşumuna neden olan metabolik bozuklukları düzeltmeye yönelik daha özgün tedavi yöntemlerinin etkisi şimdiye kadar kanıtlanamamıştır.
Diyabetik nöropatinin ağrı, otonom bozukluklar ve duyu kaybı gibi semptomlarına yönelik tedaviler özel bir önem taşır. Aşağıda söz edilecek semptomatik tedaviler benzer belirtilerle seyreden bütün polinöropatilerde uygulanabilir.
Nöropatik ağrının tedavisinde en çok farmakoterapiden yararlanılır. Bir hastada hangi ilacın yararlı olacağını önceden belirlemek çok zordur ve birçok kere ilaçların sıra ile “titre edilerek” denenmesi gerekir. Nöropatik ağrı farmakoterapisinin temel prensipleri her keresinde tek bir ilacın denenmesi, bir ilaca en düşük dozla başlanıp maksimal yarar sağlanıncaya ya da tolere edilemeyen yan etkiler ortaya çıkıncaya kadar dozun yavaşça arttırılması, ağrı azalması anlamlı, yan etkiler tolere edilebilir, hasta fonksiyon ve aktivitesi iyi olduğu sürece tedavinin sürdürülmesi olarak özetlenebilir. Hastanın tolere edebildiği bir ilacın etkisiz olduğuna karar verilip bir başkasına geçmeden önce ilk ilaçla 4-6 hafta tedavinin sürdürülmesi uygun olur (Bakınız: Nöropatik Ağrı).
Otonom bozukluklara ait belirtiler çeşitli tedavi girişimleri ile azaltılabilir. Semptomatik ortostatik hipotansiyonu olan hastaların uyudukları yatakların baş kısmını biraz yükseltmek, yemeklerinde 2 fincan koyu çay ya da kahve içmelerini, az öğünde çok yemek yerine küçük ve çok sayıda öğün halinde yemelerini önermek, günlük su ve tuz alımlarını arttırmak yararlı olabilir. Bacaklardaki venöz kapasitansı azaltmak için varis çorapları yararlı olmakla birlikte hastalar tarafından tolere edilmeleri güçtür. Plazma volümünü arttırmak için fludrokortizon (0,1-0,6 mg/gün) kullanılabilir. Alfa-agonist midodrin vazomotor ve venomotor tonusu arttırarak ayaktayken kan basıncını arttırır ve ortostatik hipotansiyon belirtilerini azaltır. Prostaglandin sentezini inhibe eden nonsteroid anti-inflamatuvar ilaçlardan ibuprofen ve indometazin ya da doğrudan etkili bir alfa-agonist olan fenilpropanolamin aynı amaçla kullanılabilir. Gecikmiş mide boşalmasına karşı sık ve küçük öğünler ve sıvı diyet önerilir. Yemeklerden yarım saat önce 10 mg metoklopramid uygulaması yararlı olur, yüksek dozlarında ekstrapiramidal semptomlar görülebilir. Bir diğer alternatif ilaç yemeklerden önce 10-20 mg şeklinde önerilen cisapriddir. Diyabetik diyarenin bakteriyel kökenli olduğu düşünülüyorsa kısa kürler halinde tetrasiklin veya eritromisin uygulanır. Anormal motiliteden kaynaklandığı kanısı oluşuyorsa loperamid verilebilir. Bazı hastaların rahatsız edici diyarelerinde klonidin uygulaması yararlı olur. Genitoüriner yakınmalar konusunda genellikle bir üroloğun işbirliğine gereksinim olur. Nörojenik mesanesi olan hastaların gün içinde sık idrara giderek rezidüel idrar miktarını azaltmaları önerilir. Daha şiddetli olgularda elle abdominal kompresyona ya da hastanın kendi başına uygulayacağı aralıklı kateterizasyona gereksinim doğar. Erektil impotans için ürolog kontrolü altında oral sildenafil, tadalafil, kavernöz cisimler içine vazodilatatör madde uygulaması yapılabilir ya da penil implant uygulamasına gidilir.
Nöropatiye bağlı duyu kusuru, ayak ülserlerine ve distal eklemlerde hasara (nöropatik artropati) neden olabilir. Diyabetik bir hastanın kronik ayak yaraları, çoğu kez farkedilmeyen ağrısız travmalar, vasküler yetersizlik ve sekonder infeksiyonların ortak sonucudur. Bunların önlenmesi tedavisinden kolay olduğundan, diyabetik nöropatisi olan hastanın ayakları kendisi tarafından her gün, doktoru tarafından sık sık muayene edilmelidir. Yara olması muhtemel travma yerlerinde kronik bası etkenlerinin ortadan kaldırılması ve gelişen yaraların derhal uygun şekilde tedavi edilmesi, gangren nedeniyle ekstremite kaybının ve sepsis gibi sistemik komplikasyonların önlenmesi açısından çok önemlidir.
Polinöropati semptomlarının tedavisi planlanırken uygulanacak ilaçların yan etki profili dikkatle gözden geçirilmeli, nöropatinin kendisinin ya da çoğu kez yaşlı bir diyabetik olan hastanın kardiyovasküler sorunlarının hastayı ilaç yan etkilerine daha açık bir hale getirdiği unutulmamalıdır. Örneğin, ağrı tedavisi için yüksek dozda başlanılan trisiklik antidepresan ilaçlar, o ana kadar belirti vermeyen otonom nöropatiye bağlı şiddetli ortostatik hipotansiyonun ortaya çıkmasına neden olabilir. Kardiyak otonom tutulması ya da primer kalp hastalığı olan hastalarda meksiletin gibi kalp ritmini etkileyen ilaçlar çok dikkatle kullanılmalıdır. Ortostatik hipotansiyona yönelik plazma volümünü arttırıcı tedaviler uygulanmadan önce konjestif kalp yetersizliği ya da iyi kontrol altına alınmamış hipertansiyon bulunmamasına dikkat edilmelidir.
Diyabetik radikülopleksus nöropatisi, şiddetli ağrının yanı sıra iyileşmesi bazen yavaş ve yetersiz olan kas kuvvetsizliğine de neden olur. Bu nedenle, ağrıya yönelik tedavilerin yanında dikkatli bir fizyoterapi ve psikolojik destek programının uygulanması önem taşır. Hasta mevcut tablonun genellikle monofazik özellikte olduğu ve mutlaka bir derece düzeleceği konusunda ikna edilmeye çalışılmalıdır. Özellikle çalışma çağında olan hastaların, ağrıları azalıp klinik tabloları düzelmeye başladığında, gereğinde ortotik cihazlardan da yardım alarak, mümkün olduğunca hızlı bir şekilde iş yaşamına döndürülmesine gayret edilmelidir.
Üremik Polinöropati
Kronik böbrek yetersizliğine bağlı polinöropatiler 1960’lı yıllardan itibaren, diyaliz ve transplantasyonun üremik hastalara daha uzun yaşama şansı sağlaması ile tanınmaya başlamıştır. Kronik böbrek yetersizliğinde nöropati prevalansı, böbrek yetersizliğinin şiddet ve süresine bağlı olarak %10 ile %80 arasında değişmektedir.
Üremik nöropati, muhtemelen böbrekle atılamayan sistemik toksinlerin birikimine bağlı olarak gelişir. Nöropatiye neden olan spesifik bir toksin belirlenememiş olmakla birlikte, guanidin bileşikleri, miyoinozitol, parathormon, orta moleküler ağırlıklı maddeler ve hiperkalemi potansiyel nörotoksik faktörler arasında sayılmaktadır. Bunun yanısıra, hastaların bir kısmında aynı zamanda hem böbrek hastalığı hem de nöropati gelişimine neden olabilecek diyabet gibi sistemik hastalıklar bulunduğundan, polinöropatinin gelişimi birkaç faktöre birden bağlı olabilmektedir. Aynı nedenle, bir hastada üremik nöropatiden söz edilebilmesi için aylardan beri devam eden kronik son aşama böbrek yetersizliğinin (kreatinin klirensi <10ml/dk) bulunması, periferik sinirleri etkileyecek ilaç toksisitesi ile diyabet, vaskülit, amiloidoz gibi sistemik hastalıkların ekarte edilmiş olması gerekir. Nitrofurantoin ve kolşisin böbrek yetersizliği olan hastalarda sık rastlanan toksik nöropati nedenleridir.
Üremik nöropati erkeklerde kadınlardan daha sık görülür. Klinik özellikler distal ve simetrik, duyusal belirtileri baskın duyusal-motor polinöropati şeklindedir. Başlıca semptomlarını huzursuz bacaklar, bacak krampları, distal parestezi ve uyuşukluklar ile parmaklarda yanmalar oluşturur. Önde gelen muayene bulguları distal duyu kaybı (özellikle vibrasyon duyusu), refleks azalması ve ayak parmak ekstansiyonunda simetrik kuvvetsizliktir. Nadiren, diyalizin ilk haftalarında GBS’yi taklit eden ani bir başlangıçla ortaya çıkabilir. Son aşama böbrek yetersizliği olan diyalizdeki hastalarda bazen subakut, motor bileşeni baskın bir polinöropati gelişebilir. Bu durumdaki bazı hastalar geleneksel hemodiyalizden yüksek-akım hemodiyalizine geçilmesi ile düzelirler.
Üremik hastalarda karpal tünel sendromu arteriovenöz fistüllere ya da transvers karpal ligamentte (b-2 mikroglobulinle birlikte olan) amiloid birikimine bağlı olarak ortaya çıkabilir. İskemik monomelik nöropati, arteriovenöz fistüller nedeniyle gelişen ve hemen daima birlikte aterosklerotik damar hastalığı olan diyabetik üremik kişilerde ortaya çıkan bir komplikasyondur. Ani ağrılı başlangıcı olan bu komplikasyonda kalıcı nörolojik defisitlerin önlenmesi için fistülün hızla cerrahi olarak kapatılması gerekir.
Üremik nöropati patolojik olarak, periferik sinir sisteminin en distal kesimlerinde belirgin olan aksonal dejenerasyonla seyreder. Segmental demiyelinizasyon, aksonal atrofiye sekonder olarak gelişir. Elektrofizyolojik incelemelerde sinir iletim hızlarında genel yavaşlama ve distal latanslarda uzama izlenir. Sinir iletim yavaşlamasının olmadığı erken aşamalarda geç yanıtların (H refleksi ve F yanıtları) latansı uzamış bulunur. BOS proteini sıklıkla artmış olmakla birlikte, üremik nöropatisi olan hastalarda bu incelemeye nadir olarak ihtiyaç duyulur.
Diyaliz periferik nöropatiyi önler, stabilize eder ya da iyileştirebilir. Hemodiyaliz sırasında nöropatinin kötüleşmesi diyalizin sıklaştırılması veya süresinin uzatılmasını gerektirebilir. Sinir fonksiyonlarına etki açısından hemodiyaliz ve periton diyalizi arasında belirgin fark yoktur. Böbrek transplantasyonunun nöropatiyi düzeltici etkisi daha belirgindir. Transplantasyonu izleyen aylarda şiddetli üremik nöropatilerde bile düzelme görülebilir. Son aşama böbrek yetersizliği olan hastalarda yüksek akımlı hemodiyaliz ve eritropoetin uygulaması B6 vitamin yetersizliğine neden olabilir. Böyle olgularda günlük rejime 60mg pridoksin eklenmesinin duyusal belirtileri azalttığı gösterilmiştir. Nöropatik ağrı tedavisinde trisiklik antidepresanlar doz ayarlamasına gerek olmadan kullanılabilir. Gabapentin için renal doz ayarlaması yapılması gerekir.
Karaciğer Yetersizliğine Bağlı Polinöropati
Karaciğer hastalığı olanların sinir biyopsileri ve sinir iletim incelemelerinde oldukça yüksek oranda anormallik bulunmakla birlikte, semptomatik nöropati nadir ve hafif şiddettedir. Prospektif çalışmalar, kronik karaciğer hastalığı olanlarda yüksek oranda otonom tutulma varlığını göstermiştir. Karaciğer transplantasyonu bekleyen hastalar arasında otonom nöropati bulgusu olanların ölüm oranları, olmayanlara oranla belirgin derecede daha yüksek bulunmuştur. Bu nedenle transplantasyon bekleyen ileri karaciğer hastalarının otonom fonksiyonlar açısından incelenmesi ve otonom tutulması olanların erken transplantasyon açısından değerlendirilmesi gerektiği savunulmaktadır. Başarılı ortotopik transplantasyondan sonra nöropatinin düzeldiğine dair gözlemler mevcuttur. Primer biliyer sirozla birlikte olan ağır ataksik duyusal bir nöronopati ile hiperlipidemi ve cilt ksantomlarıyla birlikte olan ağrılı dizestezik duyusal bir polinöropati bildirilmiştir. Tablo 33 karaciğeri etkileyen hastalıklarla birlikte olan ve farklı mekanizmalarla gelişen başlıca nöropatileri özetlemektedir.
Tablo 33. Karaciğer hastalıklarında görülen nöropatiler (EP Bosch, değiştirilerek).
|
Hastalık |
Nöropati |
Tedavi |
|
Viral hepatit A, B, C |
AİDP, CIDP |
IVIg, plazmaferez |
|
HCV, mikst kriyoglobulinemi ile birlikte veya değil |
Duyusal, duyusal-motor polinöropati, Mononöropati multipleks |
Plazmaferez, kortikosteroidler, siklofosfamid, interferon alfa, ribavirin |
|
Kronik karaciğer hastalığı |
Duyusal nöropati, kardiyovagal dizotonomi |
Karaciğer transplantasyonu |
|
Karaciğer transplantasyonu |
CIDP |
IVIg, plazmaferez |
|
Primer biliyer siroz |
Ksantomatöz nöropati, duyusal nöronopati |
? |
|
Kolestatik karaciğer hastalığında vitamin E eksikliği |
Duyusal nöropati veya spinoserebellar sendrom |
Vitamin E, TPGS |
AİDP: akut inflamatuvar demiyelinizan poliradikülonöropati, CIDP: kornik inflamatuvar demiyelinizan polinöropati, HCV: Hepatit C virüs, IVIg: intravenöz immünglobulin, TPGS: Tokoferol polietilen 1000-glikol süksinat.
İNFEKSİYONLARA BAĞLI NÖROPATİLER
Lepra
Eski çağlardan beri bilinen ve ortaçağda Avrupa’da endemik olan lepra, halen başlıca tropikal ve subtropikal bölgelerde rastlanan bir infeksiyondur. Dünya nüfusunun tümüne bakıldığında en sık periferik nöropati nedenlerinden birisidir. Ancak, sağlık koşullarındaki iyileşme ve çok droglu tedavi uygulaması ile dünyadaki prevalansı da düşmektedir. Kayıtlı olgu sayısı 1991’de 1980’lerin başındaki sayının yaklaşık yarısına (5,4 milyon) düşmüş, 1996’da 0,9 milyona inmiştir.
Lepra, periferik sinirlerin Mycobacterium leprae tarafından oluşturulan primer bir infeksiyon hastalığıdır. Bu aside dirençli, Gram pozitif basil zorunlu bir intrasellüler parazittir. Bütün bakteriler içinde en düşük çoğalma hızına sahip olandır; deneysel olarak infekte edilmiş farede bölünme süresi 13 gündür. İnsandan insana geçişi güçtür. Üst solunum yolu ile alındığı, nazal mukozadaki ilk kolonizasyondan sonra yavaşça vücuda dağıldığı düşünülür. Tahmin edilen inkübasyon dönemi 3-10 yıldır. Bakterinin çoğalabildiği dokular günlük ortalama sıcaklığı 27-300 C olan bölgelerde yer alır. Bu nedenle deri ve yüzeysel seyirli sinirler infeksiyondan etkilenen başlıca vücut yapılarıdır.
Hastalığın klinik belirtileri, konağın M. lepraeya karşı geliştirdiği bağışıklık yanıtının yoğunluğu tarafından belirlenir. Yoğun bir hücresel bağışıklık yanıtı bakteriyi sınırlarken hastanın dokularını tahrip eder (tüberküloid lepra). Buna karşılık, konağın bağışıklık yanıtı minimal olduğunda bakterinin çoğalmasına ve yayılmasına karşı koymaz, lepra antijenlerine karşı anerji vardır ve minimal inflamatuvar reaksiyonla birlikte yaygın deri ve sinir lezyonları oluşur (lepromatöz lepra). Hastalığın bu iki uç formu arasında 3 ara formu tanımlanmıştır.
Lepranın kardinal semptomu, çoğu kez ağrısız yaralanmalarla fark edilen duyu kaybıdır. Bakterinin sıcaklıkla ilgili gereksinimleri nedeniyle sıcaklığı düşük vücut bölgelerindeki deri sinirleri etkilenir. Bu nedenle, klinik tablo tam olarak geliştiğinde duyu kaybının polinöropatilerde görülenden farklı olarak kulak kepçesi, zigomatik kemiklerin üzeri ve eklemlerin ekstensor yüzlerini seçen bir dağılım gösterdiği izlenir. Büyük sinirler yüzeye yakın seyrettikleri bölgelerde tutulur. Bu sinirler sert ve şişkin şekilde palpe edilebilir (büyük auriküler sinir, ulnar sinir dirsek segmenti, bilek lateralinde radyal sinir deri dalı, peroneal ve sural sinirler). Özellikle tüberküloid formda deri sinirleri iyi sınırlanmış bir alanda tutulur. Buna dermis ve epidermis lezyonlarının da katılması ile tipik hipopigmente anestetik maküller oluşur. Duyu bozukluğunun ağır olduğu olgularda özellikle ağrı duyusu kaybı, tekrarlayan travmalar sonucu ekstremite deformitelerine ve otoampütasyonlara neden olur. Motor zaaf, duyu kaybı iyice yerleşmeden önce ortaya çıkmaz, geliştiğinde ise asimetrik bir dağılım gösterir, ayırıcı tanıda diğer mononöropati multipleks nedenlerini düşünmek gerekir. Fasiyal sinirin küçük dallarının selektif tutulması sonucunda frontal kaslar ve orbikülaris okuli kasında izole zaaf olabilir. Bazen nöropatik bulgular deri lezyonları olmadan ortaya çıkabilir (saf nöritik lepra). Polinöropatilerin çoğunluğunda azalan ya da kaybolan tendon reflekslerinin leprada genellikle korunmuş olması ayırıcı tanıda yardımcı olur.
Lepranın tanısı deri ve gereğinde sinir biyopsilerinde M. leprae’nin gösterilmesi ile konur. Deri biyopsilerinde bakteri yoğunluğuna bakılarak infeksiyonun tedaviye yanıtı da izlenebilir. Tedavi spesifik kemoterapiden ve hastalığın oluşturduğu deformitelerin önlenmesi ve düzeltilmesinden oluşur. Az basilli tüberküloid formların tedavisinde 3 antibiyotiğin tek doz verilmesi yeterli olur (rifampisin 600 mg, ofloksasin 400mg, minosiklin 100 mg). Çok basilli formlarda (lepromatöz ve “borderline”) yine üçlü antibiyotik tedavisi yapılır (dapson 100 mg/gün, rifampisin 600mg/gün, klofazimin 50 mg/gün), fakat tedavi 2 yıl sürdürülür (mümkünse deri smear testi negatif gelene kadar, sonra uygun lepra ilacı monoterapisi ile devam edilir).
HIV İnfeksiyonu ve Nöropati
Çok aktif antiretroviral tedavinin (HAART) 1990’ların ortalarından itibaren kullanıma girmesi ile HIV infeksiyonu olan hastaların yaşam süreleri uzamış; geç dönem nörolojik komplikasyonlarına açık olan bu hastalarda merkez sinir sistemi komplikasyonları azalırken periferik nöropatiler sık bir komplikasyon olma özelliğini sürdürmüştür (Bakınız: Sinir Sistemi İnfeksiyonları). HIV ile beklenen yaşam süresinin uzaması hem HIV’in kendisine hem de diğer infeksiyöz ajanlara ve nörotoksik tedavilere maruziyet zamanının uzamasına yol açmaktadır. HIV infeksiyonunun serokonversiyon aşamasından AIDS’in ileri devrelerine kadar farklı dönemlerinde birçok periferik nöropati tablosu ile karşılaşılır (Tablo 34). HIV-1 infeksiyonlu hastalarda klinik bulgularla tanı konan periferik nöropati prevalansı %15-30 kadardır. Elektrofizyolojik inceleme verileri kullanıldığında bu oran belirgin derecede artmaktadır. CD4 sayısı nöropati sıklığı ile ters ilişki gösterir. CD4 sayısı normal olan hastalarda daha çok inflamatuvar demiyelinizan polinöropatiler görülürken, düşük olanlarda distal simetrik polinöropatiler ve lumbosakral poliradikülopatilere rastlanır.
Tablo 34. HIV infeksiyonlu hastalarda periferik nöropatiler (CH Chalk, değiştirilerek)
Distal ağrılı nöropati
Nutrisyonel yetersizliğe bağlı nöropatiler
Antiretroviral ilaçlara bağlı nöropatiler
GBS
CIDP
Mononöropati multipleks
Akut lumbosakral poliradikülopati
CIDP: kronik inflamatuvar demiyelinizan polinöropati , GBS: Guillain-Barré sendromu.
Akut ve kronik inflamatuvar demiyelinizan polinöropatiler tipik olarak başka semptomu olmayan HIV infeksiyonlu hastalarda (bazen serokonversiyon döneminde) görülür. Normal popülasyona göre hastaların daha hızlı kötüleştiği bildirilmektedir. Bu olgularda AİDP atakları daha sık olarak tekrarlamakta ya da CIDP gelişebilmektedir. Normalde CIDP daha çok ataklı bir seyir gösterirken, HIV ile infekte CIDP olgularında genellikle yavaş progresif bir seyir izlenmektedir. Normal popülasyona göre CIDP başlangıç yaşı çok daha erkendir. Klinik tablo HIV infeksiyonu olmayan hastalardaki gibidir. BOS’ta lenfositik pleositoz (20-50/mm3) ve sinir biyopsilerinde belirgin iltihabi hücre infiltrasyonu görülür. Bu nedenle inflamatuvar demiyelinizan polinöropatisi olan hastalarda HIV-1 risk faktörlerinin bulunması, BOS’ta hücre görülmesi, pozitif hepatit B serolojisi veya poliklonal hipergamaglobulinemi varlığı HIV araştırılmasını gerektirir. Bu olgulardaki akut (GBS) ve kronik (CIDP) inflamatuvar demiyelinizan polinöropatiler de, -HIV infeksiyonu bulunmayan hastalarda olduğu gibi- plazmaferez ve IVIg tedavisine iyi yanıt verir. HIV pozitif CIDP hastalarında kısa süreli steroid tedavisinin güvenilir ve etkili olabileceği de bildirilmektedir.
Distal simetrik polinöropatiler HIV infeksiyonunun en sık görülen nörolojik komplikasyonudur ve prevalansı farklı serilerde %9 ile %63 arasında bildirilmektedir, geç evrelerde daha sık görülür. Başlıca belirtileri ayaklarda yanmalar, ağrılı dizesteziler ve distal duyu azalmasıdır. Distal kaslarda hafif kuvvetsizlik ve otonom fonksiyon bozukluğu olabilir. Belirgin derecede immünsupresyonun varlığında daha sık görüldüğü ve ağır seyrettiği bildirilmektedir. HIV’de viremi düzeyi ile distal simetrik polinöropati gelişme sıklığı ve şiddetinin korelasyon (bağıntı) gösterdiğini bildiren yayınlar bulunmakla birlikte, bu bulguların desteklenmediği çalışmalar da bildirilmiştir. Ayrıca birçok çalışmada güçlü antiretroviral tedavi (ART) kullanımı ile birlikte HIV ilişkili distal simetrik polinöropati insidansının azaldığı saptanmıştır. Hastaların bir kısmında polinöropatiye yol açabilecek B12 vitamini ve folat yetersizliği, ya da nörotoksik ilaç uygulaması [vinkristin, dapson, izoniazid, talidomid, nükleosid revers transkriptaz inhibitörleri (dideoksisitidin, dideoksiinosin, stavudin)] gibi nedenler bulunur. Özellikle tüberküloz tedavisi sırasında izoniazidin yeterli piridoksin verilmeksizin kullanıldığı HIV olguları, distal simetrik polinöropati gelişmesi açısından yüksek riske sahiptir. Bir kısım olgulardaki polinöropatinin nedeni ise belirlenememiştir. Araştırmacıların çoğuna göre nöropatinin doğrudan HIV infeksiyonuna bağlı olması ihtimali düşüktür.
Patogenezde HIV infeksiyonunun makrofaj hücrelerinde disregülasyona yol açarak, TNF-α, interferon alfa, interlökin 6 (IL-6) gibi proinflamatuvar sitokinlerin ve nitrik oksit ya da serbest radikaller gibi inflamatuvar mediatörlerin salınmasına neden olabileceği düşünülmektedir. Bu sürecin başlıca miyelinsiz ya da ince miyelinli olmak üzere sinir liflerine zarar verdiği varsayılmaktadır. Bir diğer olası mekanizma ise HIV’in yüzey proteini gp120 aracılığıyla Schwann hücre kemokin reseptörünü (CXCR4) aktive ettiği ve bunun hem nöronal apoptoza hem de TNF-α gibi proinflamatuvar sitokin salınımı ile sinir hücrelerinde doğrudan toksik hasara yol açtığı yönündedir.
Didanozin ve sitavudin gibi bazı antiretroviral ilaçların nörotoksik etkisi çok merkezli, randomize çalışmalar ile gösterilmiştir. Nöropati insidansı doz bağımlıdır ve ilaca maruziyet süresinin uzaması ile artış göstermektedir. Genellikle ilaca başladıktan 7 ile 9 hafta sonrasında semptomlar başlamaktadır. Ellerde daha fazla etkilenme olabileceği bildirilmekle birlikte elektrofizyolojik incelemelerle HIV ilişkili distal simetrik polinöropatiden ayrılması mümkün değildir. Her ne kadar bu ilaçlar artık HIV’in birinci basamak tedavisinde kullanılmasa da, viral yükü baskılama ve immün fonksiyonun geri kazanılmasındaki yararlarının potansiyel nörotoksik etkilerinin önüne geçtiği bilindiğinden, kullanımlarına dair herhangi bir sınırlama da getirilmemiştir.
Sinir biyopsilerinde miyelinli ve miyelinsiz lif kaybı görülür. Distal simetrik polinöropatisi olan HIV-1 pozitif hastaların bir kısmında nekrotizan vaskülit bulunmuştur. Özellikle elektrofizyolojik incelemelerin negatif bulunabildiği erken evrelerde olmak üzere altın standart tanı yöntemi cilt biyopsisidir. Fakat hastanın öykü ve kliniği distal simetrik polinöropati ile uyumlu ise cilt biyopsisi yapılmasının gerekmediği düşünülmektedir.
HIV ile ilişkili ve ilaca bağlı distal simetrik polinöropati için tedavi seçenekleri kısıtlıdır. Etkili ART ile polinöropati riskinin azaldığı gözükmektedir. Bununla birlikte polinöropati gelişmesi halinde eğer muhtemel nörotoksik bir ilaç -sitavudin, didanozin ya da talidomid gibi- kullanılıyorsa kesilmelidir. Nükleosid analoğu dozunun azaltılmasını takip eden ilk 1-6 haftada nöropati semptomlarında kötüleşme (“coasting” fenomeni), ardından 3-19 hafta boyunca kademeli olarak iyileşme görülür. Semptomatik tedavide kullanılabilen bazı antidepresan ve antiepileptik ilaçların ART ajanları ile etkileşebileceği akılda tutulmalıdır.
Mononöropati ve mononöropati multipleks: HIV ya da eş zamanlı hepatit B veya C infeksiyonlarından kaynaklanan immün komplekslerin perinöral damarlarda birikmesiyle meydana gelen vaskülit nedeniyle olabileceği de varsayılmaktadır. Tek veya iki yanlı fasiyal paraliziler HIV-1 serokonversiyon döneminde görülebilir. Birkaç kranial veya spinal sinirin tutulduğu, CD4 sayısı 200/ml den yüksek hastalarda nöropatilerin prognozu iyidir ve genellikle spontan düzelme gösterir. Buna karşılık, ağır defisite neden olan mononöropati multipleks tabloları genellikle CD4 sayısı 200/ml den az, immün yetersizliği olan hastalarda ortaya çıkar. Bu hastalarda CMV infeksiyonu araştırılmalıdır. CMV infeksiyonunun ortaya konması ya da bundan klinik olarak şüphe duyulması halinde hemen gansiklovir tedavisine başlanması gerekir. CMV infeksiyonunun bulunamaması halinde nöropatinin en kuvvetli nedeni periferik sinir vaskülitidir. Bu olgular kortikostreoidler başta olmak üzere immünoterapiden yararlanırlar.
Akut lumbosakral poliradikülopati (progresif poliradikülopati): Genellikle CMV infeksiyonunun neden olduğu en ağır AIDS komplikasyonlarından biridir. Lumbosakral sinir köklerinin CMV tarafından invazyonuna bağlı olarak hızla progresyon gösteren bir kauda ekuina sendromu (gevşek bir paraparezi, sfinkter fonksiyon bozukluğu, perinede duyu azalması ve alt ekstremitelerde tendon reflekslerinin kaybı) ile seyreder. İleri dönemlerinde üst ekstremiteler ve kranial alanda tutulmaya ait bulgular ortaya çıkar.
BOS’ta genellikle polimorfonükleer ağırlıklı (%40‘tan çok) olmak üzere 50/mm3 ten fazla hücre vardır. Protein düzeyinin artmış, glikozun azalmış olması beklenir. Ancak bu tipik BOS bulgularına olguların ancak yarısı kadarında rastlanır. Hastaların yine yarısı kadarında BOS CMV kültürü pozitiftir. CMV DNA’sının PCR ile gösterilmesi güvenilir ve duyarlı bir tanı testidir. MRG incelemesinde kauda ekuina liflerinin kontrast madde tuttuğu gösterilebilir. Mevcut klinik ve BOS bulguları ile –CMV infeksiyonu gösterilememiş olsa bile- hemen gansiklovir ve foskarnet tedavisi (birlikte veya monoterapi olarak) başlanmalıdır. CMV ensefalitinde etkili olduğu bilinen sidofovirin progresif radikülopatideki etkisi bildirilmemiştir. Tedavi edilmeyen olgular hızla progresyon göstererek 2-30 günde ölümle sonlanır. Tedavi ile nörolojik tablonun ilerleyişinin durdurulması ve hatta düzelmesi mümkün olabilir.
HIV pozitif hastalarda eş zamanlı olarak tüberküloz görülme sıklığı artmıştır. Tüberküloz menenjiti miyeloradikülopati ya da sadece kauda ekuina sendromu olarak da bulgu verebilmektedir. Erken tanı ve tedavi ile prognoz daha iyidir. Bu nedenle semptomlar yeni başladığı sırada tüberküloz menenjiti olabileceğini akla getirmek önemlidir.
Lumbosakral poliradikülopatiye yol açabilecek diğer nedenler arasında sifiliz, toksoplazmoz ve leptomeningeal lenfomatoz sayılabilir. HIV infeksiyonu ile birlikte olan bir diğer tablo, habis olmayan bir CD8 lenfositoz olan diffüz infiltratif lenfositoz sendromudur. Multipl iç organ ve periferik sinir tutulması görülen bu sendrom kortikosteroid veya antiretroviral tedavi uygulamasına cevap verebilir.
Oldukça yeni bir kavram olan immün rekonstitüsyon inflamatuvar sendromu’nda (IRIS), güçlü antiretroviral tedavi ile HIV-1 RNA düzeyleri azalıp CD4 hücre sayıları artan olgularda daha önce var olan infeksiyöz veya otoimmün hastalıkların şiddetlenmesi söz konusudur. Yakın zamanda IRIS’le ilişkilendirilen 2 GBS olgusunda immünosupresan tedavi altında CD4 sayısında azalmayla birlikte klinik düzelme gözlendiği bildirilmiştir.
TOKSİK POLİNÖROPATİLER
Nöropati oluşturan toksik maddeler üç grupta incelenebilir: ilaçlar, endüstriyel toksinler ve çevresel toksinler (Tablo 35). Bir kimyasal maddenin nörotoksik olduğunun kanıtlanması kolay değildir ve bunun için ideal olarak hastada diğer nöropati nedenlerinin dışlanmış olması, toksine maruz kalan çok sayıda kişide aynı nörolojik belirtilerin ortaya çıkması, toksinle temasın kesilmesini izleyerek nöropatinin gerilemesi, nörotoksisitenin hayvan deneyi ve doku kültürleriyle gösterilmiş olması gerekir.
İlaçlar
İlaçlara bağlı nöropatilere oldukça sık rastlanır (Tablo 35). Nöropatisi olan bir hastanın öyküsünde bu hastalığa neden olabilecek bir ilaç kullanımının araştırılması önem taşır. Çünkü, kullanılan ilaç ile polinöropati arasında mantıklı bir nedensellik ilişkisi kurulabilirse, mevcut polinöropatinin nedenini araştırmak konusunda uzun ve yorucu araştırmalar yapmaya gerek kalmaz.
İlaçlara bağlı polinöropatiler genellikle aksonal dejenerasyonla seyreder. Klinikte ekstremite distallerinde baskın simetrik duyusal ya da duyusal-motor bulgular vardır. Ağrı sık karşılaşılan bir yakınmadır. Toksik madde ile temasın kesilmesinden sonra aylar içinde iyileşme olur. Bazı hastalarda sorumlu ilacın kesilmesine rağmen klinik bulgular haftalar süren bir progresyon gösterebilir (“coasting”). Bazı ilaçlar ise distal aksonopati yerine arka kök gangliyonlarını etkileyerek ataksik duyusal bir nöronopatiye (sisplatin, piridoksin megadozu) ya da demiyelinizan bir polinöropatiye (perheksilen, amiodaron, suramin) yol açarlar. Son dönemde özellikle metastatik ve ileri evre kanser hastalarında kullanılmaya başlanan immün kontrol noktası [programlı ölüm 1 (PD-1) ve sitotoksik T lenfosit ile ilişkili antijen 4 (CTLA-4)] inhibitörleri ile de otoimmünite ile ilişkili yan etkiler bildirilmeye başlanmıştır. Bu ilaçların kullanımı ile periferik sinir sisteminde GBS ve CIDP tabloları görülebilmektedir. Tablo 36’de toksik nöropatiler en çok yol açtıkları belirtilere ve tutulan periferik sinir yapılarına göre gruplanmıştır.
Gelecekte nörotrofik faktörlerin kullanımının yaygınlaşması ve bu maddelerin kemoterapi ile birlikte uygulanmasının kanser ilaçlarının polinöropati yapıcı etkilerini engelleyeceği umulmaktadır.
|
Tablo 35. Nöropati nedeni olan başlıca toksik maddeler (RA Lewis, değiştirilerek) |
|||
|
Çevresel ve Endüstriyel Toksinler |
|
İlaçlar
|
|
|
Ağır Metaller |
Arsenik |
Antibiyotikler ve diğer Anti-infeksiyöz Maddeler |
Kloramfenikol |
|
|
Kurşun |
Klorokin |
|
|
|
Civa |
Dapson |
|
|
|
Platin |
İzoniazid |
|
|
|
Talyum |
|
Metronidazol |
|
|
|
|
Nitrofurantoin |
|
Heksakarbonlar |
N-heksan |
|
Zalsitabin (dideoksisitidin) ve |
|
|
Metil-n-bütil keton |
|
diğer nükleosid analogları (stavudin, didanosin) |
|
|
Metil-etil keton (N-heksanı potansiyelize edebilir) |
|
|
|
|
|
|
|
|
Akrilamid |
|
Kemoterapi ve Antikanser Maddeler |
Sisplatin |
|
Karbon disülfid |
|
Paklitaksel |
|
|
Etilen glikol |
|
Mizonidazol |
|
|
|
|
Suramin |
|
|
Organofosfatlar |
Triortokrezil fosfat |
|
Talidomid, Leflunomid |
|
|
|
|
Vinkristin |
|
Yenilerek Alınan |
Ciguatera - |
|
Bortezomib |
|
|
ciguatoxin ve maitotoxin |
|
Etoposid, teniposid |
|
|
Paralitik deniz kabuklusu |
|
|
|
|
Zehirlenmesi-Saksitoksin |
Antiromatizmal |
Klorokin |
|
|
Şişen balık (fugu)-Tetrodotoksin |
Ve |
Kolşisin |
|
|
|
İmmünsupresan |
Altın |
|
Böcek Sokmaları |
Kene paralizisi |
Maddeler |
FK 506 (Prograf) |
|
ve Hayvan |
Tropikal kurbağa derisi toksinleri |
|
|
|
Toksinleri |
|
Kardiyovasküler İlaçlar |
Amiodaron |
|
|
|
Hidralazin |
|
|
Nörotoksik |
Cassava (Menihot esculenta)- |
|
Perheksilin |
|
Bitkiler |
siyanid |
|
Propafenon |
|
|
Tırmanan zambak (Gloriosa |
|
Simvastatin ve diğer |
|
|
superba) |
|
statinler |
|
|
Mandrake (Podophyllium peltatum)- |
|
|
|
|
podofilotoksin |
Psikiyatrik ve Sedatif İlaçlar |
Disülfiram (Antabus) |
|
|
Cataranthus roseus-vinka alkaloidi |
Glutetimid |
|
|
|
Tullidora (Karwinskia humbodtiana) |
|
|
|
|
|
Diğer İlaçlar |
Pridoksin (Vitamin B6) |
|
|
|
|
Fenitoin (Difenil hidantoin) |
Tablo
36.
Klinik
ve patolojik özelliklerine göre toksik nöropatiler (A
Pestronk, değiştirilerek).
|
Aksonal |
Demiyelinizan |
Karışık |
||
|
Duyusal |
Duyusal ve Motor |
Motor |
“Buckthorn”
(Cehri) Klorokin Difteri |
Amiodaron |
|
Almitrin Brentuksimab Dioksin Fenitoin Flekanid Iksabepilon İnterferon-α Lenalidomid Levodopa |
Akrilamid Siyanid Disülfiram İpilimumab Podofilin Taksol Triortokrezil
fosfat Vakor (PNO) |
β-bungarotoksin Botulizm Gangliozidler Kurşun Cıva |
||
Endüstriyel ve Çevresel Toksinler
Endüstride kullanılan bir kimyasal maddenin nörotoksik etkisi, genellikle aynı işyerinde çalışan birçok işçinin aynı zamanda hastalanması ile ortaya çıkar. Toksik maddenin belirlenmesi ve işçilerin bu madde ile temasının kesilmesi ile de “salgın” kontrol altına alınır. Endüstriyel ortamda polinöropati yapmış olan başlıca maddeler akrilamid, alil klorid, karbondisülfid, dimetilaminopropionitril ve heksakarbon bileşikleridir (N-heksan ve metil N-butil keton). Sterilizasyonda kullanılan etilenokside maruz kalan hastane çalışanlarında polinöropatiler gösterilmiştir. Triortokrezilfosfat karışan gıda maddeleri ile toksik nöropati salgınları görülmüştür.
Heksakarbonlar ortak metabolitleri olan 2,5 heksandion aracılığı ile polinöropatiye neden olurlar. Bu nöropatide lokal nörofilament birikimine bağlı multifokal aksonal şişmeler (dev aksonal nöropati) ve sekonder segmental demiyelinizasyon olur. Toksik nöropatilerin çoğunda yaygın aksonal dejenerasyonu yansıtan elektrofizyolojik bulgular saptanırken, heksakarbon nöropatilerinde iletim yavaşlaması ve parsiyel iletim bloklarının saptanmasının nedeni budur. Klinik tablo ekstremite distallerinde parestezi ve duyu azalımı ile başlamakla birlikte, olguların çoğunda belirgin derecede kas kuvvetsizliği gelişir. Ağır olgularda kas zaafının oldukça hızlı yerleşmesi, ekstremite distallerinin yanısıra proksimal kas gruplarını da etkilemesi nedeniyle inflamatuvar demiyelinizan polinöropatilerle karıştırılabilir. Sinir iletim incelemelerinde demiyelinizasyon varlığını gösteren bulguların saptanması ayırıcı tanıyı daha da zorlaştırabilir. Heksakarbon nöropatilerinde BOS bulgularının normal olması, aynı işyerinde birçok işçinin aynı zamanda hastalanması ve gereğinde yapılacak sinir biyopsisinde tipik patolojik bulguların gösterilmesi tanı koydurur. Heksakarbonlar merkez sinir sisteminde özellikle uzun traktuslara hasar verebildiğinden, ağır seyirli olguların iyileşme aşamasında polinöropati bulguları azalırken piramidal iritasyon bulguları ortaya çıkabilir. Bazı yapıştırıcı solüsyonlarda çözücü olarak bulunan N-heksan, yurdumuzda zaman zaman ayakkabı ve deri eşya üreten atölyelerde çalışan işçiler arasında oldukça ağır polinöropatilere yol açmıştır. Benzer yapıştırıcıları inhale ederek uyuşturucu etkisinden yararlanmaya çalışan kişilerde de ileri kas zaafı ile seyreden polinöropatiler bildirilmiştir.
Metaller
Metaller genellikle element hallerinde değil, organik ve inorganik bileşenler halindeyken nörotoksiktir. Metal nöropatilerinin tanısına birlikte olan hematolojik ve diğer sistemik bulgular yardımcı olur.
Arsenik zehirlenmesinde polinöropatiye karın ağrısı, kusma ve pansitopeni eşlik eder. Akut başlangıçlı ve ağır seyirli olgular GBS ile karıştırılabilir. Avuç içleri ve ayak tabanlarında hiperkeratoz ve soyulma, tırnaklarda soluk transvers bandlar (Mees çizgileri) görülebilir. Kurşun nöropatisiyle birlikte karın ağrısı ve anemi görülür. Nöropati motor ağırlıklıdır ve klasik olarak radial siniri ön planda etkileyip düşük el tablosuna yol açtığı bilinir. Talyum zehirlenmesinde gastrointestinal semptomlar ve ağrılı, distal duyusal-motor polinöropati olur. En göze çarpan belirti, genellikle nöropatinin başlangıcından 2-3 hafta sonra ortaya çıkan alopesidir. Talyum zehirlenmesinde de Mees çizgileri görülebilir.
Metal nöropatilerinde tanı, hastalık öyküsünde intoksikasyon şüphesi bulunan, polinöropati bulgularına sistemik yakınma ve bulguların eklendiği olguların kan, idrar, saç ve tırnaklarda yüksek metal düzeylerinin gösterilmesi ile konur. Tedavi toksik madde temasının kesilmesiyle olur. Tedavide yaygın şekilde kullanılmış olan penisilamin, dimerkaprol (BAL), etilendiamin tetraasetik asit (EDTA) gibi şelatör maddelerin muhtemelen yararlı etkisi vardır. Ancak bunların toksik nöropatilerin tedavisindeki etkilerini araştıran kontrollü çalışmalar da mevcut değildir.
Organofosfat İntoksikasyonları
Organofosfat bileşikleri dünyada insektisit olarak yaygın şekilde kullanılmakta, intoksikasyonları, tarım ve bahçe bakımı amacıyla püskürtülürken inhalasyon veya deri yoluyla alınarak, intihar amacıyla ya da kaza eseri yutularak olmaktadır. Piyasalarda var olan binlerce organofosfat markasının gelişmekte olan ülkelerdeki kontrolü, muhtemelen Batı ülkelerine oranla daha zayıftır.
Organofosfat intoksikasyonlarının en sık görülen Tip I sendromu, asetikolinesteraz enziminin geri dönüşsüz inhibisyonuna bağlı yoğun muskarinik stimülasyon nedeniyle gelişir. Şiddeti alınan bileşiğin toksisitesine ve alındığı miktara göre değişir. Toksik maddenin alınmasından sonraki ilk gün içinde ortaya çıkan bulantı, kusma, diyare, terleme, hipersalivasyon, istemsiz idrar ve gaita yapma, taşikardi veya bradikardi görülür. İleri olgularda emosyonel labilite, yorgunluk, kognitif bozukluklar ve uyanıklık bozukluğu, konvülziyon ve koma gibi merkez sinir sistemi belirtileri görülebilir.
Ara Tip veya Tip 2 organofosfat sendromu iskelet kasındaki nikotinik asetilkolin reseptörlerinin aşırı uyarılmasına bağlıdır. Toksik maddeye maruz kalınmasından 12-96 saat sonra ortaya çıkar. Akut ve ara sendromlar arasında genellikle 1-4 günlük semptomsuz bir dönem vardır. Solunum yetersizliği genellikle ilk belirtidir. Bunu proksimal kaslar ve boyun fleksörlerinde kuvvetsizlik izler. Kranial alan kasları tutulabilir, fakat distal ekstremite kasları genellikle kurtulur. İyileşme 5-15 günde başlar. Atropin, muskarinik reseptörlere özgül olduğu için ara sendromun engellenmesi veya tedavisinde işe yaramaz.
Bazı organofosfat bileşikleriyle intoksikasyondan 7-21 gün sonra bir gecikmiş polinöropati tablosu ortaya çıkar. Bu nöropatinin gelişmesinin, hastanın daha önce Tip 1 ve Tip 2 sendromları geçirmiş olması ile bir ilişkisi yoktur. Bazı hastalarda toksik maddeye kronik maruz kalma sonucunda ortaya çıktığı düşünülmektedir. Sinir sisteminde nörotoksik esteraz veya nöropati target esteraz ismi verilen reseptör proteinin fosforilasyonu sonucu gelişir. Genellikle subakut başlangıçlı, en şiddetli haline yaklaşık 2 haftada ulaşan, duyusal –motor, periferik-santral bir aksonopati şeklindedir. Bacaklarda ağrılı paresteziler ve kramplar ortaya çıkar. Alt ekstremite distallerinden başlayan motor belirtiler düşük ayak, daha sonra el kaslarında kuvvetsizlik ve geç dönemde yaygın kas kuvvetsizliği görünümündedir. Uzun süreli intoksikasyona bağlı ALS’ye benzer tablolar bildirilmiştir. Bulanık görme ve ataksi olabilir. Medulla spinalisin uzun traktuslarının tutulmasına bağlı spastisite ve hiperrefleksi bulunabilir. Elektrofizyolojik incelemeler aksonal bir polinöropati ve sekonder demiyelinizasyonu yansıtır. Polinöropati aylar içerisinde yavaş bir düzelme gösterir, ancak omurilik tutulmasına ait belirtiler genellikle gerilemez.
Triortokrezil fosfat insektisit olmayan, sanayide çeşitli amaçlarla kullanılan bir organofosfattır. Birçok ülkede, gıdalara karışması ile ciddi aksonal polinöropati tablolarına yol açmıştır.
BESLENME YETERSİZLİĞİNE VE ALKOLİZME BAĞLI POLİNÖROPATİLER (Ayrıca bakınız: Sinir Sisteminin Nutrisyonel Hastalıkları)
Beslenme yetersizliklerine bağlı polinöropatiler genellikle şiddetli kıtlık çekmekte olan toplumlarda ya da esir ve konsantrasyon kamplarında kalanlarda görülmüştür. Starvasyona bağlı polinöropatilerde beslenmede eksik olan ve polinöropatiye yol açan asıl maddeyi belirlemek güç olmakla birlikte, B grubu vitaminlerinden bir veya birkaçının eksikliği söz konusudur. Bu nöropatilerde genellikle distal aksonal dejenerasyonla seyreden duyusal ya da duyusal-motor bir polinöropati ortaya çıkar. Olguların birçoğunda merkez sinir sistemi ve kalp, deri gibi diğer organların tutulmasına bağlı bulgular vardır. Vitamin yetersizliğine bağlı polinöropatilerde tedavinin ana kuralı hastaya vakit geçirmeden uygun vitaminlerin verilmesidir. Bununla birlikte, polinöropatisi olan bütün hastalara vitamin tedavisi uygulamanın hiçbir bilimsel dayanağı yoktur (Tablo 37).
Tablo 37. Vitamin eksiklikleri ile birlikte olan nöropatiler (KG Gwathmey ve N Jovanovich, değiştirilerek)
|
Vitamin |
Klinik |
Doğrulama testleri |
Tedavi önerileri |
|
B12 vitamini |
Miyelonöropati |
Serum B12 ve metilmalonik asit düzeyleri, Pernisiyöz anemi söz konusuysa gastrin ve intrensek faktör antikorları |
B12 1000 mg IM, 1 ay haftada bir, sonra ayda bir, Geri dönebilir bir neden yoksa devamlı |
|
Folik asit |
Duyusal nöropati |
Serum folik asit |
1mg/gün |
|
Bakır yetersizliği |
Miyelonöropati |
Serum bakır düzeyi, Çinko toksisitesi |
Element bakır, oral 1 hafta 8 mg/gün Sonra haftada bir 2 mg düşür, 2mg/gün ile idame, Geri dönebilir bir neden yoksa devamlı |
|
E vitamini |
Duyusal nöropati, belirgin ataksi ve serebellar bulgularla birlikte |
Serum E vitamini düzeyi |
50-200 IU/gün, oral (Geri dönebilir bir neden yoksa) |
|
B1 vitamini (tiamin) |
Duyusal-motor polinöropati (motor baskın) |
Serum B1 vitamini düzeyi |
100 mg IV veya IM, 3-5 gün süreyle, sonra 50mg/gün oral |
|
B6 vitamini (piridoksin) Eksikliği veya toksisitesi |
Eksiklik: Uzunluğa bağımlı nöropati Toksisite: Duyusal nöronopati |
Serum B6 vitamini düzeyi |
Eksiklik: İzoniazid ve uzun süreli hidralazin tedavisi alanlarda 50mg/gün oral Toksisite: B6 alımını durdur |
Nutrisyonel yetersizliğe bağlı polinöropatilerin prototipi olan Beriberi, B1 vitamini (tiamin) yetersizliğinde ortaya çıkar. Başlıca belirtileri kalp yetersizliği ve polinöropatidir. İlk olarak ekstremite distallerinde ağrılı duyusal yakınmalar ortaya çıkar. Hastalığın ilerlemesi ile distal kaslarda kuvvetsizlik gelişir. Tedavi oral (absorbsiyon bozukluğu varsa parenteral) tiamin uygulaması ve dengeli beslenmenin sağlanması ile olur.
Nikotinik asidin (niasin) diyetteki eksikliği pellegraya neden olur. Bu sendromda olguların yaklaşık yarısında bir duyusal-motor polinöropati gelişir. Hastalarda dermatit ve diyarenin görülmediği durumlarda klinik tablo tiamin yetersizliğine bağlı nöropatilerden ayrılamaz.
B6 vitamini (piridoksin) yetersizliği izoniazid (INH), hidralazin ve nadiren penisilamin tedavisi sırasında ortaya çıkar. Bu ilaçlar yapısal olarak B6 vitaminine benzer ve piridoksin koenzim aktivitesi ile çatışırlar. Genellikle sinsi başlangıçlı distal ve simetrik duyusal-motor bir polinöropati gelişir. INH tedavisine piridoksinin eklenmesiyle (100 mg/gün) polinöropati önlenir. Piridoksinin çok yüksek dozda alınması da (600-3000 mg/gün) duyusal bir polinöropatiye neden olur.
B12 vitamini yetersizliğinde subakut kombine dejenerasyon ortaya çıkar (Bakınız: Omurga ve Omurilik Hastalıkları). Bu tabloya hafif bir polinöropati eklenebilir.
Malabsorbsiyona bağlı E vitamini yetersizliği ekstremite ve yürüme ataksisi, bozulmuş vibrasyon ve pozisyon duyusu, arefleksi, oftalmopleji ve pigmenter retinopati ile seyreden bir spinoserebellar sendroma yol açar (Ayrıca bakınız: Dejeneratif Ataksiler). Klinik belirtiler kronik E vitamini eksikliğinin başlangıcından yıllar sonra ortaya çıkar ve yavaş ilerler. Arka kök gangliyonu hücrelerinin periferik sinirlerde ve medulla spinalis arka kordonlarında uzanan geniş çaplı aksonlarının dejenerasyonu ile giden bu tablodan, muhtemelen nöronların E vitamini eksikliğine bağlı antioksidan korunmalarının kaybolması sorumludur. Tanı, klinik bulgular, serumda çok düşük E vitamini düzeyleri (<5mg/ml) ve bozuk yağ absorbsiyonunun gösterilmesi ile konur. E vitamini uygulaması hastalığın progresyonunu durdurur.
Bariatrik Cerrahi
Obezitenin görülme sıklığındaki artış ile birlikte bariatrik cerrahi uygulanan hasta sayısı da gün geçtikçe çoğalmaktadır. Dolayısıyla bariatrik cerrahi sonrası gelişen nörolojik komplikasyonların insidansı da artmakta ve yılda yaklaşık %16 sıklığında olduğu tahmin edilmektedir. Bariatrik girişimler gıda alımının kısıtlanması ve/veya barsaktan emilimin azalması ile nutrisyonel yetersizliğe neden olmaktadır. Bunlardan gastrik bandlama ile vertikal bandlama prosedürleri gıda alımının kısıtlanmasına neden olurken, “Roux-en-Y” gastrik “by-pass” ve biliopankreatik diversiyon-duodenal “switch” operasyonları ise hem kısıtlayıcı hem de malabsorpsiyona yol açan girişimlerdir. Son iki girişim ince barsakta geniş kısmın devre dışı bırakılmasına neden olduğundan vitamin ve mikronutrisyon yetersizlikleri daha fazla görülür. Sıklıkla tiamin (B1), B12, folat, vitamin D, vitamin E ve bakır yetersizliği görülmektedir. Bu nedenle hastalar cerrahi sonrası yüksek protein ve düşük yağ oranına sahip gıdalar ile beslenmeli, vitamin takviyesi almalı ve hastaların düzenli aralıklarla kan incelemeleri yapılmalıdır.
Cerrahi sonrası ensefalopati, optik nöropati, miyelopati, poliradikülonöropati ve polinöropati gibi sinir sisteminin çeşitli bölgelerinin etkilendiği komplikasyonlar görülmekte ve nutrisyonel yetersizliğe bağlı olarak geliştikleri düşünülmektedir. Akut, subakut ve kronik polinöropatiler, fokal tuzak nöropatileri, nöropati ile ilişkili posterior lateral miyelopati, radikülopleksopati bildirilmiş olan periferik sinir sistemi komplikasyonlarıdır. Cerrahi öncesi diyet yapanlarda ve cerrahi sonrası hızlı kilo kaybı, diyare, “dumping” sendromu, bulantı ve kusma, iştah kaybı ve önerilen takviye edici gıdaların alınmaması durumlarında periferik nöropati daha sık olarak gelişmektedir. Bununla birlikte, çoğu bildiride hastaların vitamin ve mikronutrisyon değerleri ayrıntılı olarak araştırılmamış, bir kısmında ise ayrıntılı incelemelere rağmen eksiklik bulunduğuna dair kesin değerler saptanmamıştır.
Tiamin deposu yaklaşık olarak 20 günde tükenebildiğinden, cerrahi sonrası erken dönemde tiamin eksikliğine bağlı olarak ağrılı akut periferik nöropati ya da aksonal natürde fakat klinik olarak GBS’ye çok benzeyen bir akut poliradikülonöropati izlenebilir. Bu tablonun gelişiminde immünite aracılı mekanizmaların da etkili olduğu öne sürülmektedir. Bu hastaların tedavisinde vitamin ve eser elementlerin uygun bir şekilde replasmanı dışında, IVIg ve plazmaferez gibi immünolojik tedavilerin de uygulanması önerilmektedir. Bunun haricinde subakut kalın lif polinöropatisi ve yanan ayak sendromuna yol açan primer ince lif nöropatisi görülebilmektedir. Geç dönemde ise B12, bakır, folat ya da vitamin E yetersizliklerine bağlı olarak miyelopati gelişebilir ve bu bariatrik cerrahinin en fazla özürlülüğe neden olan komplikasyonudur. Bu tabloda ataksi, spastisite, derin tendon reflekslerinde artış, propriyosepsiyon ve vibrasyon duyusu ile yüzeyel ağrı ve ısı duyularında kayıp saptanır. Operasyondan uzun yıllar sonra ise yavaş ilerleyici, distal ağrılı paresteziler (yanan ayak sendromu) ile seyreden uzunluğa bağımlı aksonal bir polinöropati görülebilir. Hastalarda genelde birden çok mikronutrisyonda yetersizlik bulunur ve yerine koyma ile ancak kısmi iyileşme görülür. Ayrıca başta karpal tünel sendromu ve fibuler sinirin fibula başı düzeyinde kompresyon nöropatisine bağlı düşük ayak tablosu olmak üzere mononöropatilerin de bariatrik cerrahi sonrası sık olarak görülebileceği akılda tutulmalıdır.
Alkolik Polinöropati
Yetersiz diyet, bozulmuş absorbsiyon ya da alkolün metabolizasyonu için artmış gereksinim nedeniyle gelişen tiamin ve diğer B grubu vitaminlerin yetersizliği sonucunda ortaya çıktığı düşünülmektedir. Gelişmiş batı ülkelerinde beslenme yetersizliğine bağlı başlıca polinöropatiler alkolizm nedeniyle ortaya çıkanlardır. Ön planda yaygın distal aksonal hasarla seyreden bu nöropatinin başlıca semptomları, alt ekstremite distallerinde belirgin yanıcı, batıcı ağrı ve paresteziler ile buna eklenen distal kas zaafı ve kramplardır. Muayenede ekstremite uçlarında duyu azalması ve ağrılı aşırı duyarlık, tendon reflekslerinin kaybı ve hafif distal kas zaafı bulunur. İlerlemiş olgularda pozisyon duyusu kaybına bağlı ataksi ile alkolik serebellar ataksi birlikte görülebilir. Alkolik nöropatiye neden olacak alkol alım miktarını belirlemek güçtür. Bununla birlikte, hafif-orta miktarda alkol alan, iyi beslenen ve kronik karaciğer hastalığı belirtileri taşımayan bir hastada alkolik polinöropati bulunma olasılığı düşüktür. Bu durumdaki bir hastada diğer polinöropati nedenlerinin de araştırılması gerekir. Alkolik nöropati tedavisinin başlıca bileşenleri, alkol alımının kesilmesi, dengeli beslenmenin sağlanması ve bunun tiamin uygulaması ile desteklenmesidir. Şiddeti gastrointestinal semptomları olan hastalara tiamin parenteral olarak verilmelidir.
Kritik Hastalık Polinöropatisi
Yoğun bakım ünitelerinde oldukça sık karşılaşılan bir sorundur. Genellikle nörolojik olmayan bir hastalık nedeniyle yoğun bakım ünitesinde yatmakta olan, sepsis ve multipl organ yetersizliği bulunan hastalarda ortaya çıkar. Sıklıkla hastanın solunum cihazından ayrılmasındaki güçlük nedeniyle fark edilir. Birçok kez septik ensefalopati bulguları olan, sedasyon uygulanmış ve entübe edilmiş durumda bulunan bu hastaların nörolojik değerlendirilmesinde güçlükle karşılaşılır. Genel bir gevşek felç vardır, tendon refleksleri yaygın olarak kayıptır. İyi muayene edilebilen hastalarda ekstremite distallerinde eldiven-çorap şeklinde duyu kusuru saptanabilir. Kranial sinirler genellikle tutulmaz, nadiren iki yanlı fasiyal zaaf görülebilir. Otonom sinir sistemi tutulması nadirdir.
Yaygın distal akson hasarıyla seyreden bu polinöropatiden sistemik inflamatuvar yanıt sendromunun sorumlu olduğu düşünülmektedir. Bu sendromda infeksiyon, travma vb. gibi nedenlerle faaliyete geçen hümoral ve sellüler mekanizmaların, çeşitli organların mikrosirkülasyonunu etkileyerek bu organlarda yetersiz perfüzyona ve multipl organ yetersizliğine neden olduğu düşünülmektedir. Elektromiyografi ve sinir iletim incelemelerinde aksonal polinöropati ile uyumlu bulgular saptanır. BOS bulguları normaldir. Genel durumu düzelip yoğun bakımdan çıkarılabilen hastalarda polinöropati aylar içinde yavaş bir düzelme gösterir.
Kronik hastalık polinöropatisinin ayırıcı tanısına giren hastalıklardan birisi GBS, diğeri ise kritik hastalık miyopatisidir. Kritik hastalık miyopatisi, yoğun bakım ünitesinde yüksek doz kortikosteroid ve nöromüsküler bloker ilaç uygulanan hastalarda akut olarak ortaya çıkar. Kasın histopatolojik incelemesinde kalın filamentlerin kaybı izlenir. Serum CPK düzeyi artmış olabilir. Elektrofizyolojik incelemelerde duyusal sinir iletimleri normaldir. İğne elektromiyografisinde fibrilasyon ve pozitif diken potansiyelleri şeklinde patolojik spontan aktivite görülebilir (bu açıdan kritik hastalık nöropatisinden farklılık göstermez) ancak hastada bir derece istemli ya da refleks kas kasılması sağlanabilirse ‘miyopatik’ motor ünite potansiyelleri tanı konmasını sağlar. Elektrofizyoloji laboratuvarlarında yaygın olarak kullanıma girmemiş bir yöntemle, kas liflerinin doğrudan elektriksel uyarımı ve ilgili sinirin uyarımı ile elde edilen motor yanıtların büyüklükleri arasındaki orana dayanarak yoğun bakım nöropatisi ve miyopatisi oldukça başarılı şekilde ayırt edilebilmektedir. Benzer klinik tablolar nöromüsküler bloker ilaçların atılma bozukluğuna bağlı olarak da ortaya çıkabileceğinden elektrofizyolojik incelemelere nöromüsküler iletim bozukluğunu araştıran yöntemler (ardısıra uyarım testleri ve tek lif elektromiyografisi) eklenmelidir.
Görüldüğü gibi, kritik hastalık nöropatisi ve miyopatisini birbirlerinden ayırmak birçok hastada son derece zordur. Ayrıca kritik hastalık nöropatisi ve miyopatisine ait özellikleri birlikte taşıyan çok sayıda hasta olduğu bilinmekte, sistemin inflamatuvar yanıt sendromunun yoğun bakım miyopatisine de yol açabildiği düşünülmektedir. Bu nedenle her iki tabloyu kritik hastalık nöromiyopatisi olarak isimlendirme yönünde bir eğilim vardır.
Yoğun bakımda ortaya çıkan yaygın kas zaafı tablolarının ayırıcı tanısına giren tüm hastalık durumlarının oldukça tipik elektrofizyolojik bulguları vardır. Bu nedenle elektrofizyolojik yöntemler bu hastalıkların tanınmasında temel bir rol oynar. Ancak, yoğun bakım ünitelerinde bu incelemelerden yararlı sonuçlar almakta güçlüklerle karşılaşılabilir. Bir yandan yoğun bakım ünitelerinin elektrikle çalışan çok sayıda aletle donatılmış olması elektrofizyolojik test cihazlarında artefaktlara yol açarken, diğer yandan hastaların incelemeye işbirliği yapamaması ya da birçok kez ödemli olan ekstremiteleri güvenilir kayıtlamalar yapmayı zorlaştırabilir. Tanımlanmış metodlara uygun yapılan frenik sinir motor iletim çalışmaları ve diyafram kasının iğne elektromiyografisi, hastanın ventilatörden ayrılamama sebebinin kritik hastalık nöropati/miyopatisine bağlı olduğunu ortaya koymada yardımcı olabilir. Kas biyopsisi, endikasyonunun titizlikle değerlendirilmesi ve kas patolojisi konusunda tecrübeli bir merkeze gönderilmesi halinde, yoğun bakım miyopatisinin tanınmasında yararlı olabilir.
Kronik Kriptojenik (İdyopatik) Duyusal Polinöropati
Edinsel duyusal ya da duyusal-motor polinöropatilerin büyük kısmında iyi bir araştırma ile polinöropati nedeni ortaya konulabilir. Ancak bir grup hastada bütün araştırmalara rağmen böyle bir neden belirlenemez. Polinöropatiler içinde bu kriptojenik grubun oranı eski serilerde %50-70 civarındayken, yakın zamanlardaki çalışmalarda -yeni polinöropati nedenlerinin tanımlanması ve gelişen diyagnostik yöntemlerin katkısıyla- %11-31 arasında verilmektedir. Kriptojenik nöropatisi olan hastalardan bir bölümünün belirti, bulgu ve seyirleri açısından oldukça homojen özellikler gösterdiği dikkati çekmiş, bu olgular “kronik kriptojenik (idyopatik) duyusal polinöropati” ismi altına toplanmıştır. Bu tabloya neden olabilecek mekanizmalar halen bilinmemekle birlikte bazı otoimmün süreçlerin rol oynayabileceği üzerinde durulmaktadır.
Genellikle 50 yaşın üzerinde olan (ortalama başlangıç yaşı 60,4, erkek/kadın oranı: 3:2) bu olgularda ön planda duyusal aksonal bir polinöropati söz konusudur. Alt ekstremite distallerinden başlayıp çok yavaş şekilde yükselen yanıcı, geceleyin artan ağrı ve uyuşmalar başlıca yakınmayı oluşturur. Tendon refleksleri korunmuş ya da ekstremite distallerinde azalmıştır. Kas kuvvetsizliği nadirdir. Elektrofizyolojik incelemelerde sinir iletimleri normal ya da duyusal aksiyon potansiyelleri düşük amplitüdlü bulunur. Genellikle ince sinir liflerinin tutulduğu bu polinöropatilerde kantitatif duyusal testler ve deri biyopsileri tanı için yol gösterici olabilir. Bu grubun en önemli özelliği prognozunun oldukça iyi olması, hastalığın çok yavaş progresyon göstermesi, hatta bazı hastalarda fazla kısıtlılığa yol açmayan bir düzeyde platoya ulaşabilmesidir. Ancak bazı hastalarda aylar-yıllar içerisinde fark edilir bir progresyon olabilir, başlangıçta ince lif nöropatilerinde görülen tipte duyusal yakınma ve bulgular varken giderek propriyoseptif duyu bozukluğu ve ılımlı distal motor güçsüzlük katılabilir.
Yukarıdaki tanımlamadan görülebileceği üzere, kronik idyopatik duyusal nöropati tanısı bir dışlama tanısıdır. Hastalara bu tanıyı koyarken duyusal ağırlıklı aksonal nöropatiye neden olan tüm nedenler ayrıntılı şekilde araştırılmalı, bu araştırmaların negatif kaldığı olgular hem klinik seyirleri açısından gözlem altında tutulmalı hem de gereğinde uygun aralıklarla tanı testleri tekrarlanmalıdır (tanı testinin başlangıçta negatif kalıp sonraki takipte pozitifleşmesi olasılığı nedeniyle).
Kronik kriptojenik
(idyopatik) duyusal polinöropatinin tedavisinde nöropatik ağrı yakınmalarının
ilaçla tedavisi ön sırada yer alır. Hastanın ağrıları, yaşam aktivitelerindeki
kısıtlılıklar ve geleceğe ilişkin korkularının yarattığı depresyonla hem
ilaçlar yardımıyla hem de hastalığın genellikle iyi olan seyri hakkında aydınlatıcı
bilgiler vererek mücadele etmek gerekir.
Periferik Sinir
Hipereksitabilite Sendromları
Periferik sinir
hipereksitabilite (PSH) sendromları motor sinir
liflerinden doğan spontan deşarjlar ile oluşur ve bu durum kas aktivitesinde
artışa neden olur. Artmış kas aktivitesine bağlı biyoelektriksel faaliyet
genellikle elektrofizyolojik olarak kayıt edilebilir. PSH sendromlarını primer ve sekonder sendromlar olarak
alt sınıflara ayrılmak mümkündür. Nöromiyotoni (Isaacs sendromu), kramp-fasikülasyon sendromu ve Morvan sendromu
gibi primer PSH sendromları genellikle altta yatan başka bir periferik sinir
hastalığı olmaksızın görülür. Primer PSH sendromu
olan bazı hastalarda periferik sinirlerin özellikle terminal segmentlerinde
yerleşen voltaj kapılı potasyum kanal kompleksine karşı bir otoimmün yanıtın
geliştiği gösterilmiştir. Bu yanıtın hastalığın altında yatan patolojik mekanizmayı
başlattığı düşünülmektedir. Hastalarda kas seyirmesi ve katılığı gibi belirti
ve bulguların yanısıra kaslarda kramp, gevşeme güçlüğü (psödomiyotoni), terleme
artışı gibi bulgular ve daha nadiren hafif pozitif duyusal semptomlar görülür. Morvan sendromunda
merkez sinir sisteminin etkilenmesine bağlı belirtiler de ortaya çıkar. Nöromiyotonide
ve Morvan sendromu hastalarının büyük bir bölümünde
terleme artışı, kardiyovasküler (taşikardi ve hipertansiyon gibi) ve gastrointestinal
(konstipasyon ve diyare gibi) otonom semptomlar bulunur. Sekonder PSH sendromları ise genellikle periferik sinir sisteminin
fokal ya da yaygın olarak etkilendiği inflamatuvar demiyelinizan polinöropatiler
veya tuzaklanmaya ya da radyasyona bağlı fokal nöropatilerde görülebilmektedir.
Bu hastalarda PSH ile ilişkili klinik ve elektrofizyolojik bulgular sıklıkla
muayene sırasında rastlantısal olarak saptanır. PSH sendromları
klinikopatolojik ve etyolojik açıdan ise immün aracılı, genetik (esas olarak
potasyum kanal alt birimlerinde görülen mutasyonların neden olduğu) ve diğer
çeşitli faktörlere bağlı olanlar şeklinde ayrılabilir.
Nöromiyotoninin
başlıca semptomları aylar içerisinde sinsi bir
şekilde ilerleyen kaslarda seyirme ve sertliktir. Kas seyirmeleri genellikle
kas gruplarının düzensiz ve senkron olmayan, deri
altındaki solucansı hareketleri şeklinde gözlenen miyokimilerden ibarettir.
Bazı hastalarda ise sık fasikülasyonların olduğu dikkati çeker. Seyirmeler
genellikle kol ve bacaklarda görülür, fakat yüzde veya gövdede de olabilir.
Kaslardaki sertlik ve kramplar da daha çok ekstremitelerde görülmekle birlikte
zaman zaman kavşak ya da gövde kaslarına yayılır. Kas seyirme ve kasılmaları
uykuda devam eder, hedef kasa kürar veya botulinum toksini enjeksiyonu ile azalır veya kaybolur. Bu özellikler
patolojik elektriksel aktivitenin periferik sinir liflerinden kaynaklandığını
göstermiştir. Hastalarda sık olarak görülen terleme artışının artmış kas
lifi aktivitesine bağlı olarak geliştiği düşünülmüştür. Fakat özellikle Morvan
sendromunda başka otonom semptomların da görülmesi
nedeniyle hastalığın otonom lifleri de etkilediği söylenebilir. Yine kas
lifi aktivitesindeki artışa bağlı olarak baldır ya da önkol kaslarında hipertrofi
gelişebildiği bilinmektedir. Hastaların 1/3’ünde, parestezi veya elektriklenme
şeklinde pozitif duyusal semptomlar görülmektedir.
Bir diğer primer PSH sendromu olan kramp fasikülasyon
sendromunda kramp ve seyirme semptomları bulunur fakat nöromiyotonide olduğu
kadar ağır derecede kas sertliği görülmez. Yani Isaacs sendromu ile kramp fasikülasyon sendromu arasındaki
başlıca farklılık niteliksel değil niceliksel gibi görünmektedir.
Morvan sendromunda
ise nöromiyotoni ve otonom semptomlara eşlik eden merkez sinir sistemi tutulmasına
işaret eden semptomların (halüsinasyon, konfüzyon,
nöbet ve insomni gibi) olduğu görülür. Bu özellikleriyle Morvan sendromu klinik planda otoimmün limbik ensefalit ile
nöromiyotoni arasında bir yere yerleşmektedir. Bu hastalıkta fatal familyal
insomnide görülen ve “agrypnia excitata” adı verilen, uyanıklığın azaldığı,
mental konfüzyon, halüsinasyon, motor ajitasyon,
rüyaları taklit eden kompleks motor hareketler ve otonomik aktivasyonun olduğu
uzun süreli ağır bir total insomni tablosu da görülebilir. Merkez sinir sistemini
işaret eden belirtilerin ve otonom semptomların
uyku-uyanıklık ritmi ve otonomik fonksiyonları düzenleyen subkortikal yapıların
kortikolimbik kontrolünde azalmaya bağlı olarak geliştiği düşünülmektedir.
Nöromiyotoni
hastalarının yaklaşık olarak %30-40’ında memeli beyin dokusundan elde edilen
ve α-dendrotoksin
ile işaretli voltaj kapılı potasyum kanal kompleksine
karşı immün yanıt oluşturan antikorların bulunduğu gösterilmiştir. Son dönemde
yapılan immünopresipitasyon ve kitle spektrometrisi analizleri, bu antikorların
özellikle voltaj kapılı potasyum kanallarıyla sinir hücre duvarında kompleks oluşturan proteinler olan kontaktin ilişkili
protein-2 (CASPR2) ve lösin-zengin, glioma inaktive 1 (LGI1) ile reaksiyona
girdiğini göstermiştir. Morvan sendromunda voltaj
kapılı potasyum kanal antikorları, özellikle de CASPR2, %90’ın üzerinde oranda
pozitif bulunmakta ve hastaların yaklaşık olarak yarısında tabloya timoma
eşlik etmektedir. Kramp fasikülasyon sendromunda
daha seyrek olarak antikor pozitifliği ve malignite varlığı görülmektedir.
LGI1 antikorları ise genellikle limbik ensefalit hastalarında pozitif bulunmaktadır.
Primer PSH sendromlarının tanısında yararlanılan elektrofizyolojik
incelemelerde en sık karşılaşılan bulgular iğne elektromiyografisinde saptanan
sık fasikülasyonlar, miyokimik ve nöromiyotonik deşarjlardır. Ayrıca motor
ileti ya da F dalga incelemeleri esnasında “supramaksimal” uyarı ile elde
edilen bileşik kas aksiyon potansiyelini izleyen ardışık boşalımlar olduğu
da görülebilmektedir (Şekil 16). Motor ve duyusal iletim incelemeleri
genellikle normal olmakla birlikte, bazı hastalarda hafif aksonal nöropatiyle
uyumlu bulgular saptanmaktadır.
Primer PSH sendromlarının semptomatik tedavisinde antikonvülzan
(karbamazepin, fenitoin, valproat ve lamotrijin gibi) ilaçlar ve bazen asetazolamid
yararlı olabilir. Hastalığın hızlı ve ağır seyrettiği olgularda ise plazmaferez,
IVIg ya da yüksek doz IV prednizolon ile immün modülatör tedavi gerekmektedir.
Ek olarak PSH sendromlarının habis tümörler (başta
timoma olmak üzere, akciğer karsinomları ve daha az sıklıkta lenfomalar)
ve diğer otoimmün hastalıklar (myastenia gravis ve diğerleri) ile ilişkili
olabileceği bilindiğinden altta yatan olası malign ve/veya disimmün hastalıkların
da araştırılması ve tedavisi önem taşımaktadır.
PSH sendromları ile ayırıcı tanıya giren Schwartz-Jampel
sendromu, ya da diğer adıyla “kondrodistrofik miyotoni”; kas sertliği, fasial
dismorfizm ve iskelet deformiteleri (boy kısalığı, kifoz, kalça displazisi
ve yalancı kırıklar) ile karakterize nadir bir otozomal resesif hastalıktır.
Hastalık bazal membranın ana bileşenlerinden biri olan “perlecan”ı kodlayan
HSPG2 gen mutasyonlarına bağlı olarak gelişir. Sık görülen
bulguları kaslarda sertlik, gevşeme güçlüğü, hipertrofi ve kontraktürlerdir.
İletim incelemeleri normaldir. İğne elektromiyografisinde istirahatte yüksek
frekanslı tekrarlayıcı deşarjlar görülür. Schwartz-Jampel sendromunda görülen deşarjların sinir liflerinden,
preterminal sinir lifleri ve nöromüsküler kavşağın “remodelling”ine bağlı
olarak ortaya çıkabileceği ileri sürülmüş olmakla birlikte, anormal deşarjların
karakteristik özellikleri presinaptik kökenli oldukları konusunda şüpheye
yol açmaktadır.
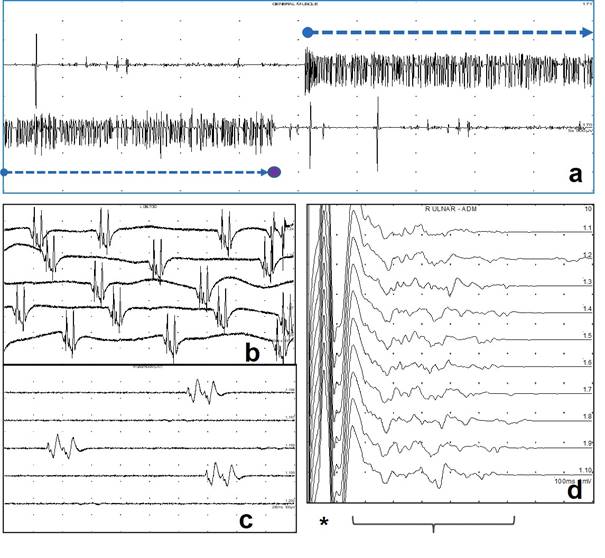
Şekil 16. Periferik sinir
hipereksitabilite sendromlarında elektrofizyolojik
bulgular. a)Ani başlangıç ve bitişi olan uzun süreli, yüksek frekanslı nöromiyotonik
boşalım. Alt alta kaydedilmiş iki trase üzerinde kesikli okla işaretlenmiştir; b ve c) Ritmik/semiritmik olarak tekrarlayan ikili, üçlü
potansiyellerden oluşan miyokimik boşalımlar; d) F yanıtı incelemesinde ekran
duyarlılığının yüksek olması nedeniyle kayıt trasesine sığmayan bileşik kas
aksiyon potansiyelini (*) izleyen tekrarlayıcı boşalımlar (yatay parantezin
içerisinde).
Bu bölümdeki
histopatolojik resimler ‘İstanbul Tıp Fakültesi Nöroloji Anabilim Dalı Nöromüsküler
Hastalıklar İnceleme Laboratuvarı’ arşivinden alınmıştır (Prof. Dr. Yeşim
Gülşen Parman)
KAYNAKLAR
1.
Amato AA, Russell
JA (Ed). Neuromuscular Disorders, 2. Baskı (Kindle Edition). New York: McGraw-Hill
Education, 2016
2.
Aminoff MJ, Josephson
AS (Ed). Neurology and General Medicine. 5. Baskı. Philadelphia: Churchill
Livingstone-Elsevier, 2014
3.
Barohn RJ, Amato
AA. Pattern-recognition approach to neuropathy and neuronopathy. Neurol Clin. 2013;31:343-61
4.
Becker DA, Balcer
LJ, Galetta SL. The Neurological complications of nutritional deficiency
following bariatric surgery. J Obes. 2012;2012:608534
5.
Bella IR. Acute
inflammatory demyelinating polyradiculoneuropathy. İçinde: Deymeer F (ed).
Neuromuscular Diseases: From Basic Mechanisms to Clinical Management. Monogr
Clin Neurosci. Basel: Karger, 2000: 147-162
6.
Bosch EP, Smith
BE. Disorders of peripheral nerves. İçinde: Bradley WG, Daroff RB, Fenichel
GM, Jankovic J (Eds). Neurology in Clinical Practice. 4. Baskı. Philadephia:
Butterworth Heinemann, 2004: 2299-2402
7.
Cakar A, Durmus-Tekce
H, Parman Y. Familial amyloid polyneuropathy. Noro Psikiyatr Ars. 2019;56:150-156
8.
Callaghan BC, Price RS, Feldman EL. Distal symmetric polyneuropathy:
A review JAMA. 2015 24;314:2172-81
9.
Donofrio PD.
Guillain-Barré Syndrome. AAN Continuum (Peripheral Nerve and Motor Neuron
Disorders No 5). 2017; 23: 1295-1309
10.
Doets AY, Jacobs
BC, van Doorn PA. Advances in management of Guillain-Barré syndrome. Curr
Opin Neurol. 2018;31:541-550
11.
Feldman EL, Callaghan
BC, Pop-Busui R, ve ark. Diabetic neuropathy.
Nat Rev Dis Primers 2019;5:41.
12.
Gwathmey KG,
Jovanovich N. Sensory loss and neuropathic pain. İçinde: Silvestri NJ (Ed)
Neuromuscular Disorders. A Symptoms and Signs Approach to Differential Diagnosis
and Treatment. New York, Demos Medical, 2018 : 163-199
13.
Gwathmey KG,
Tracy JA, Dyck PJB. Peripheral nerve vasculitis: Classification and disease
associations. Neurol Clin 2019;37:303-333
14.
Hughes RAC. Peripheral
neuropathy. Brit Med J. 2002;324:466-469
15.
Iqbal Z, Azmi
S, Yadav R, ve ark. Diabetic peripheral neuropathy:
Epidemiology, diagnosis, and pharmacotherapy. Clin Ther 2018;40:828-849
16.
Küçükali CI,
Kürtüncü M, Akçay Hİ, Tüzün E, Öge AE. Peripheral nerve hyperexcitability
syndromes. Rev Neurosci. 2015;26(2):239-51.
17.
Levine TD, Saperstein
DS. Laboratory evaluation of peripheral neuropathy. Neurol Clin 2013:31:363-376
18.
Mauermann ML.
Paraproteinemic Neuropathies. AAN Continuum (Peripheral Nervous System Disorders
No 5) 2014;20: 1307-1322
19.
Oge AE, Yazici
J, Boyaciyan A, ve ark. Peripheral and central
conduction in n-hexane polyneuropathy. Muscle Nerve 1994;17:1416-1430.
20.
Parman Y. Hereditary
neuropathies. Current opinion in neurology 2007;20:542-547.
21.
Pestronk A (2016).
Neuropathy. Differential diagnosis. http:// neuromuscular.wustl.edu/naltbrain.htm
22.
Pop-Busui R,
Boulton AJ, Feldman EL, ve ark. Diabetic Neuropathy:
A Position Statement by the American Diabetes Association. Diabetes Care
2017;40:136-154.
23.
Ramchandren S.
Charcot-Marie-Tooth Disease and Other Genetic Polyneuropathies. Continuum
(Minneap Minn). 2017;23(5, Peripheral Nerve and Motor Neuron Disorders):1360-1377.
24.
Vriesendorp FJ.
(2018). Guillain-Barré Syndrome in Adults: Clinical Features and Diagnosis.
In J. A. Melin (Ed.), UpToDate. Retrieved May 31, 2020, from https://www.uptodate.com/contents/guillain-barre-syndrome-in-adults-clinical-features-and-diagnosis
25.
Watson JC, Dyck
PJB. Peripheral Neuropathy: A Practical Approach to Diagnosis and Symptom
Management. Mayo Clin Proc. 2015;90:940-951.
26.
Willison HJ,
Jacobs BV, van Doorn PA. Guillain-Barré Syndrome. Lancet 2016; 388: 717-27
27.
Yeh WZ, Dyck
PJ, van den Berg LH, et al. Multifocal Motor Neuropathy: Controversies and
Priorities. J. Neurol Neurosurg Psychiatry 2020; 91: 140-148. doi:10.1136/jnnp-2019-321532