Hareket Bozuklukları
Yazanlar:
Murat Emre, Haşmet A. Hanağası, Hüseyin A.
Şahin, Jale Yazıcı
Son Güncelleme Tarihi : 18.01.2019
Hareket bozuklukları kavramı klinikte
iki ayrı anlamda kullanılmaktadır. Bu kavram bir yandan istemsiz veya anormal
elemanter hareket bozukluklarını ifade ederken bir yandan da bu anormal
hareketlerin altında yatan hastalıkları ifade etmektedir. Örneğin tremor
fenomenolojik olarak iyi tanımlanmış elemanter bir hareket bozukluğudur,
esansiyel tremor ise bu elemanter bozukluğun altında yatan hastalıklardan bir
tanesidir. Bu grup hastalıklarda tutulan fonksiyonel sistem ve anatomik
yapıları göz önüne alarak hareket bozuklukları ile giden hastalıklara ekstrapiramidal sistem hastalıkları veya bazal ganglia hastalıkları
da denilmektedir. Klinikte ortaya çıkan semptomları
normal yapı ve işleyiş çerçevesinde daha iyi anlayabilmek için bu bölümde önce
bazal ganglianın anatomisi ve fizyolojisi kısaca gözden geçirilecektir.
EKSTRAPİRAMİDAL SİSTEM VE BAZAL GANGLİA
Ekstrapiramidal sistem, motor
işlevlerin yürütülmesinde piramidal sistem dışında, ona benzer şekilde anatomik
ve fizyolojik olarak tanımlanabilen ikinci bir sistemin var olduğu
düşüncesiyle, nöroanatominin tarihsel gelişimi esnasında tanımlanmış bir
deyimdir; kelime itibarıyla da "piramidal sistemin dışında kalan
sistem" anlamına gelir. Yani bir anlamda ne olduğuyla değil ne olmadığıyla
tanımlanmış bir sistemdir. Daha sonraki yıllarda anatomik bütünlük anlamında böyle
bir sistemin olmadığı anlaşılmışsa da bu deyim yine de kullanılagelmiştir.
Ekstrapiramidal sistemin ana oluşumu bazal gangliadır.
Motor yolları kabaca
ikiye ayırmak mümkündür: inen yollar ve geri dönen (re-entran yollar). İnen
yollar doğrudan yaptırıcı, uygulatıcı yollar iken (kortikospinal,
kortikopontin, kortikobulber), geri dönen yollar kontrol edici ve modülatör bir
işlev görürler. İki tane ana geri dönen yol ya da devre mevcuttur:
“kortiko-bazal ganglia-talamo-kortikal” yol ve “kortiko-serebellar-talamo-kortikal”
yol. Bu iki ana yol birbirine paralel ve birbirinden bağımsız olarak organize
edilmişlerdir, kendi içlerinde de bağımsız, paralel alt devreler vardır.
Korteksten aşağıya inen yaptırıcı yolların kaynağını primer motor korteks
oluşturur. Korteksten bazal gangliaya ulaşan motor yolların kaynağını ise
lateral premotor alan, suplamanter motor alan ve singulat motor alan oluşturur.
Bu bölgeler yapılacak olan hareketle ilgili motor programların hazırlanması,
sıralanması ve duyusal bilgilerin entegrasyonu (bu
sayede yapılacak olan hareketle ilgili motor emirlerin o ekstremitenin o anki
durumuyla uyumlu hale getirilmesi) görevlerini yürütürler. Bu bölgelerden bilgi
alan bazal ganglia, ekstrapiramidal sistemin ana yapısını oluşturan ve beynin
derinliklerinde simetrik olarak yerleşmiş çekirdek gruplarından oluşur (Şekil 1). Yukarıda da bahsedildiği gibi
bazal ganglia yaptırıcı değil yönlendirici, kontrol edici, modülatör bir işlev
görür. Bazal ganlianın motor, kognitif ve davranışsal işlevleri vardır. Bazal
gangliayı oluşturan ana yapılar Tablo 1'
de Şekil 2 ve Şekil 3’ de
gösterilmiştir.

Şekil 1. Motor sistemin belli
başlı yapıları arasındaki ana ilişkilerin, geri-dönen ve desendan yolların
şematik görünüşü.

Şekil
2. Bazal ganglia ve ilişkili yapıları gösteren
koronal kesit (Washington Üniversitesi Radyoloji Bölümü’nün izniyle alınmıış ve
üzerine eklemeler yapılmıştır)
Tablo 1. Bazal Ganglia
|
·
Striatum
|
|
· Globus pallidus (eksternus ve internus) |
|
· Substantia nigra (pars kompakta ve pars retikulata) |
|
· Subtalamik nukleus (Korpus Luysii) |
Putamen ve kaudat çekirdeğe birlikte neostriatum
adı da verilir. Bazal gangliayı oluşturan bu yapılar içinde birbirine paralel,
ancak birbirinden bağımsız olarak organize olmuş devreler vardır. Bu paralel
devreler içindeki bilgi akışı, bilgi alıp verdikleri bölgeler farklıdır.
Günümüzde, kortiko-subkortikal devreler olarak da adlandırılan bu devrelerden
dört tanesi tanımlanmıştır: iskeleto-motor, okülomotor, asosiyatif ve limbik.
Bu devrelerin her biri serebral korteksin farklı bölgelerinden bilgi alırlar,
bazal ganglia içinde farklı bağlantı yolları izledikten sonra talamus üzerinden
tekrar bilgi aldıkları bölgeye bilgi yollarlar. İskeletomotor
devre, motor korteks, premotor alan, suplamanter motor alan ve paryetal
kortekslerden gelen bilgilerin putamene ulaşmasıyla; okülomotor devre,
prefrontal korteks ve suplamanter göz alanlarından gelen sinyallerin nukleus
kaudatusun gövde kısmına; asosiyatif devre, frontal dorso-lateral ve
frontal-orbital bölgelerden gelen sinyallerin nukleus kaudatusun baş kısmına ve
limbik devre de singulat korteksten gelen sinyallerin ventral striatuma
ulaşmasıyla başlangıçlarını alırlar. Bu sinyaller bazal gangliayı
oluşturan diğer yapılardaki ara istasyonlardan geçip bilgi işlemeye tabi
tutulduktan sonra, işlenmiş bilgi talamus üzerinden aynı bölgelere geri döner.
Her ana devrenin içinde alt devreler de vardır, örneğin iskeleto-motor ana
devrenin alt devreleri farklı motor alanlardan bilgi alıp o bölgeye bilgi
verirler. Bağlantılarına göre hücrelerin ateşleme şekilleri de farklılıklar
gösterir.
Ana görevi kontrol,
ince ayar ve modülasyon olduğu düşünülen bazal ganglia bu işlevleri yerine
getirebilmek için kontrol edip, ince ayarını yapacağı bölgelerden bilgi almak
(aferent uyarılar) ve o bölgelere bilgi vermek (eferent uyarılar) durumundadır.
Bazal ganglianın aferent girdilerinin büyük kısmı frontal korteksten (motor
korteks, premotor alan, suplamanter motor alan, singulat korteks, dorso-lateral
ve orbito-lateral frontal korteks) bir kısmı da paryetal korteksten gelir. Bu
aferent sinyallerin bazal gangliaya giriş yaptığı ana oluşum striatumdur. Bazal
gangliada işlem gören bilgilerin çıkış kapısı da sınırlı olup çıkış sinyalleri
globus pallidus internus (GPi) ve substantia nigra pars reticulata (SNr)
tarafından iletilir. Bazal ganglianın eferent sinyallerinin çok büyük kısmı
talamusa, küçük bir kısmı ise beyinsapındaki pedinkulopontin nukleusa giderler.
Son yıllarda bazal ganglianın iskeleto-motor devresinin iç organizasyonu daha
iyi anlaşılmıştır. Buna göre biribirine zıt ancak birbirini tamamlayan işlevler
gören iki alt devre ya da yol mevcuttur, direkt yol ve indirekt yol. Direkt
yolda korteksten putamene giren sinyaller bazal ganglia içindeki diğer yapılara
uğramadan doğrudan çıkış kapısına yani GPi ve SNr’ya yönelirler ve talamus
üzerinden kortekse geri dönerler. İndirekt yolda ise korteksten putamene giren
sinyaller Globus pallidus eksternus (GPe), subtalamik nukleus (STN) ara
istasyonlarından geçtikten sonra çıkış kapısına, yani GPi /SNr ya yönelirler ve
talamus üzerinden kortekse geri dönerler.
Bazal ganglia hücreleri birbirleriyle
haberleşmek için diğer nöronlar gibi nörotransmiterler ve nöromodülatörler
kullanırlar. Bazal gangliada kullanılan nörotransmiterlerin sayısı oldukça
sınırlıdır. Birçok sinaptik bağlantıda kullanılan, bazal gangliada büyük
miktarlarda bulunan GABA (gama amino bütirik asit) bastırıcı, inhibe edici; bir
aminoasit olan glutamat ise uyarıcı, eksitatör olarak etki ederler. Dopamin D1
tipinde reseptörlerine bağlandığında uyarıcı, D2 tipi reseptörlerine
bağlandığında ise bastırıcı etki yapar. Asetilkolin ise modülatör olarak görev
yapar, yani tek başına uyarıcı ya da baskılayıcı değildir, ancak
uyarılabilirliği değiştirir. Bazal ganglia hücrelerinin birçoğunda bu klasik
nörotransmiterlerin yanında P maddesi, enkefalin gibi nöropeptidler de
bulunurlar.
Bazal ganglianın işlevlerinin tam
olarak ne olduğu ve bu işlevleri ayrıntılarıyla nasıl yerine getirdiği kesin
olarak belli değildir. Ancak yukarıda bahsedilen devrelere uygun olarak ana
stratejik işlevlerinin frontal korteksin büyük kısmının işleyişinin ince ayarı
ve modülasyonu, iskeleto-motor ve okülomotor hareketlerin kontrolu, limbik
sistemin modülasyonu ve asosiyatif işlevlerin modülasyonu olduğu
düşünülmektedir. Hareket bozukluklarına olan ilgisi dolayısıyla bazal
ganglianın motor işlevleri aşağıda biraz daha ayrıntılı olarak ele alınacaktır.
Bazal ganglianın motor işlevleriyle
ilgili bilgiler bir taraftan hayvan deneylerinden, bir yandan da bu yapıları
tutan dejeneratif hastalıklar ya da bu yapılarda oluşan hasarlayıcı lezyonlar
sonucu ortaya çıkan klinik semptomlar göz önüne
alınarak elde edilmişlerdir. Ayrıca fonksiyonel nörogörüntüleme çalışmaları da
bu bilgilerin elde edilmesinde önem taşımaktadır. Dikkat edilmesi gereken bir
konu hayvan deneyleri sonuçlarının insanlara bire bir uygulanamayacağı
gözlemidir. Bu durum büyük olasılıkla kısmen yapısal kısmen de işlevsel
farklılıklardan kaynaklanmaktadır. Bütün bu bilgilerin ışığında bazal
ganglianın motor işlevleri şu şekilde özetlenebilir: Hareketlerin başlatılması
ve planlanması, hareket hız ve büyüklüğünün ayarlanması, öğrenilmiş motor
programların (yürüme, bisiklete binme gibi) otomatik olarak uygulanması,
ardısıra veya eş zamanlı hareketlerin uygulanması, kas tonusunun ayarlanması.
Bazal gangliayı tutan hastalıklarda ortaya çıkan belirtiler bu işlevlerin
değişik ağırlık ve dağılımda bozulması şeklindedir. Bazal ganglianın adı geçen
fonksiyonları bir “ölçekleme” ve “odaklama” işlevi görerek gerçekleştirdiği
düşünülmektedir. Bu düşünceye göre bazal ganglia istirahat halinde talamusa
uyguladıkları sürekli bir inhibisyonla korteksin uyarılabilirliğini kontrol
altında tutar. Bir hareket yapılmak istendiğinde bazal ganglia motor korteksten
medulla spinalise gidecek emirlerin kopyalarını alır, kendisine paryetal
korteksten ulaşan proprioseptif, bilinçsiz derin duyu sinyalleriyle kıyaslar,
gerekli düzeltmeleri talamus üzerinden kortekse geri yollar. Böylelikle
hareketin hız ve büyüklüğünün iskeloto-motor sistemin o anki durumuna ve amaca
uygun olarak istenilen sınırlar içinde kalmasını sağlar, yani “ölçekler”. Diğer
taraftan da kortikal seviyede başlatılan hareket paternini, istenilen
hareketleri, seçilen motor programları direkt yolla kolaylaştırıp
kuvvetlendirirken, ilgili ancak istenmeyen, çelişen motor paternleri indirekt
yolla baskılayarak “odaklar”. Yukarıda bağlantıları anlatılan direkt yol
dopamin D1 reseptörleri tarafından kontrol edilir. Direkt yol aktif hale
geldiğinde oluşan net etki bazal ganglia çıkış sinyallerinin hareketi istenen
kas grubu için azalmasıdır. Bu çıkış sinyalleri nitelik itibarıyla baskılayıcı
oldukları için azalmaları sonucunda talamus daha az inhibe olur ve korteksin
ilgili bölümlerinin talamus tarafından inhibisyonu azalır. İndirekt yol ise
dopamin D2 reseptörleri tarafından kontrol edilir. Bu yol aktif hale geldiğinde
oluşan net etki bazal ganglia çıkış sinyallerinin artması şeklindedir.
Böylelikle talamus daha çok inhibe olur ve talamustan kortekse giden uyarıcı
sinyaller azalır, kortekse bir negatif geribildirim gider. Hareket
bozukluklarından Parkinson hastalığının ve diğer bazı hastalıkların
patogenezinde önemli bir rol oynayan dopaminin bu bağlamdaki rolü ise şöyle
özetlenebilir. Dopamin D1 reseptörlerine bağlandığında direkt yolu uyarır,
böylece bazal ganglia çıkış sinyalleri azalır ve kortekse talamustan giden
pozitif geribildirim artar. Dopamin D2 reseptörlerine bağlandığında ise bu
reseptörleri üzerinde taşıyan hücreler üstünde inhibitör bir etki yapar,
böylece D2 reseptörleri tarafından kontrol edilen indirekt yol inhibe olur.
Uyarıldığında net etki olarak bazal ganglia çıkış sinyallerini arttıran bu
yolun inhibisyonuyla bazal ganglia çıkış sinyalleri azalır, bu yolun sağladığı
talamustan kortekse giden negatif geribildirim baskılanır. Sonuç olarak dopamin
direk yolu uyarıp indirek yolu baskılayarak talamokortikal çıkış sinyallerini
her iki yoldan artırır ve korteks aktive olur.
Direkt ve indirekt yollarla motor
işlevlerin bu şekilde gerçekleştiği varsayımına dayanan model ve bu modeldeki
dopaminin rolü bazal ganglia hastalıklarının bazı semptomlarını
açıklayabilmektedir. Örneğin bu model dopamin eksikliğinde hareketleri
başlatmak ve uygulamakta güçlük olacağını ön görmektedir. Altında dopamin
eksikliği yatan Parkinson hastalığında gerçekten de ana belirtilerden biri
budur. Benzer şekilde bu modele göre STN'un harabiyetinde (normalde STN aktif
hale geldiğinde bazal ganglia çıkış sinyallerini artırır, böylelikle de
talamo-kortikal aktiviteyi baskılar) artmış istemsiz hareketlerin ortaya
çıkması gerekir. Gerçekten de STN'un harabiyetinde klinikte istemsiz
hareketlerle karakterize hemiballismus tablosu ortaya çıkar. Ancak direkt ve
indirekt yollar modeli Parkinson hastalığında görülen rijidite gibi bazı semptomları açıklayamamaktadır.

Şekil 3. Normal insanlarda ve
Parkinson hastalığında bazal ganglia-talamus-korteks bağlantılarının aktivite durumu.
Normalde direkt ve indirekt yollardaki aktivite dengedeyken nigrostriatal
yollardaki (SNc den putamane giden yollar) dejenerasyon
sonucu direkt yoldaki aktivite azalırken indirekt yoldaki artar. Bu
değişikliğin net etkisi bazal gangliadan talamusa ulaşan baskılayıcı
sinyallerin artması şeklindedir.
HAREKET BOZUKLUKLARININ
SINIFLAMASI
Hareket bozukluklarını tabloya hakim olan anormal veya istemsiz hareketin niteliğine göre
hipokinetik (hareketin azaldığı) ve hiperkinetik (hareketin arttığı) sendromlar
olarak iki büyük gruba ayırmak tanıya klinik yaklaşımı kolaylaştırması
açısından yaygın kullanım bulmuştur (Tablo 2). Bu gruplardaki hastalıkları tek
tek incelemeye geçmeden önce hareket bozukluğu olan hastaya fenomenolojik
(belirtiye yönelik) yaklaşım ele alınacaktır.
Tablo 2.
Hiperkinetik ve hipokinetik hareket bozuklukları
|
HİPOKİNETİK HAREKET
BOZUKLUKLARI |
HİPERKİNETİK HAREKET
BOZUKLUKLARI |
|
Akinezi/Bradikinezi Rijidite Donmalar Bloklayıcı tikler Katapleksi ve düşme
atakları Katatoni |
Distoni Tremor Kore Atetoz Ballismus Tikler Miyoklonus Miyoritmi Miyokimi Hemifasiyal spazm Huzursuz bacak sendromu Ağrılı ayak, hareket
eden parmak sendromu Akatizi Paroksismal diskineziler Hiperpleksi Stereotipi |
İstemsiz veya anormal hareket
yakınmasıyla gelen bir hastada ilk yapılacak şey tutulan vücut bölgesi veya
bölgelerinin belirlenmesidir. Hareket bozukluğunun vücudun tek bir bölgesinde,
birbirine komşu birden çok bölgede, birbirinden bağımsız birden çok bölgede ya
da tüm vücutta yaygın olması, distal veya proksimal ağırlıklı olması tutulumun
niteliği, yeri ve altta yatan etyoloji konusunda önemli bilgiler sağlar. İkinci
önemli unsur hareket bozukluğunun istirahat ve harekete bağımlı olup
olmadığıdır. Örneğin esansiyel tremor tipik olarak hareket esnasında ortaya
çıkar, çoğunlukla istirahatte kaybolur, Parkinson tremoru için ise bunun tersi
söz konusudur. Hareketin hızı hareket bozukluğunun fenomenolojik
tanımlanmasında çok önemlidir. Miyoklonus şimşek gibi hızlı, kore hızlı, atetoz
ise yavaş-akıcı hareketlerdir. İstemsiz hareketin ritmik olup olmadığı,
devamlılığı, hangi şartlarda ortadan kaybolduğu diğer önemli unsurlardır.
Örneğin tremor, tanımı itibarıyla ritmik bir harekettir, kore ve genel olarak
miyoklonus ise aritmik hareketlerdir. Bazı hareket bozuklukları sadece belli
postürlerde ortaya çıkarlar veya şiddetlenirler (hasta sadece ayaktayken ortaya
çıkan ortostatik tremor gibi), bu yüzden hareketin postüre bağımlılığı
sorgulanmalıdır. Hareket bozukluklarının çoğu uykuda kaybolurken bazıları
uykuda da devam eder (palatal tremor gibi), bazıları ise sadece uykuya geçerken
ya da uyku esnasında ortaya çıkarlar (huzursuz bacak sendromu,
uykunun periyodik ekstremite hareketleri gibi); dolayısıyla hareketin uykuya
bağımlılığı da gözden geçirilmelidir. Diğer önemli unsurlar istemsiz veya
anormal hareket ortaya çıkmadan önce oluşan duyusal belirtilerin varlığı,
istemsiz hareketin istemli olarak bastırılabilirliği, hareket bozukluğunu
başlatan, kötüleştiren veya düzelten faktörlerin varlığıdır. Örneğin tik
bozukluklarında hastalar tikin oluştuğu bölgede hareketten hemen önce bir
rahatsızlık hissi, bir gerilim hissettiklerini, hareketi bu hissi rahatlatmak
için istemli olarak kendilerinin yaptıklarını söyler ve bu hareketi belli bir
müddet istemli olarak baskılayabilir, ancak bu baskılama kalktığında tik
hareketini kısa bir süre daha sık yaparlar. Distonilerde distonik hareketin
olduğu bölgeye uygulanan duyusal bir uyarı (örneğin spazmodik tortikolliste
çeneye hafifçe dokunma gibi) istemsiz hareketin kısmen de olsa baskılanmasını
sağlar. Hareket bozukluklarının teşhisinde ilk basamak yukarıda sözü edilen
bütün bu unsurları göz önüne alarak temel hareket bozukluğunu fenomenolojik olarak
tanımlamak ve adlandırmaktır (tremor, distoni, bradikinezi vb).
HİPOKİNETİK
HAREKET BOZUKLUKLARI
Bu gruptaki hastalıklara akinetik-rijid sendromlar da denir.
Bu hastalıklarda hakim tablo hareketlerin azalması,
güçleşmesi, yavaşlaması ve eşlik eden kas tonusundaki artıştır. Akinetik-rijid sendromların prototipi Parkinson sendromudur.
Parkinson sendromunu
oluşturan semptomlar bradikinezi-akinezi, rijidite, bazen eşlik eden postüral
instabilitedir. Bu belirtilere istirahat tremoru eşlik edebilir, ancak bu şart
değildir. Bradikinezi hareketleri başlatmakta
güçlük, uygulamada yavaşlık ve zorluk, genel olarak hareketlerin fakirleşmesi
olarak tanımlanır. Kelime itibariyle akinezi
hareketleri hiç yapamama olarak tanımlansa da pratikte sıklıkla bradikinezi ile
eş anlamlı olarak kullanılır. Rijidite
agonist ve antagonist kasların eşzamanlı olarak
kasılmalarına bağlı olarak ortaya çıkan, o bölgenin pasif hareketi esnasında
bir kurşun boruyu bükercesine devamlı bir direnme şeklinde kendini gösteren kas
tonusundaki artışı ifade eder. Postüral instabilite
ayakta veya otururken normalde otomatik olarak devreye giren, alınan vücut
pozisyonun devamını sağlayan postüral reflekslerin bozulması veya kaybıdır.
Postüral instabilite sonucu hastalar oturdukları yerden desteksiz kalkmada
zorluk çekerler, otururken veya ayakta spontan veya hafif itmeler, tökezlemeler
sonucu öne, arkaya veya yana düşme eğilimi gösterirler. Düşmenin yönüne göre
buna antero-, retro- veya lateropulsiyon denir. Parkinson sendromunda
görülen tremor tipik olarak istirahat halinde
ortaya çıkan, tutulan ekstremitenin kullanılmasıyla geçici de olsa kaybolan,
belli bir postürün korunması esnasında tekrar ortaya çıkan, yavaş frekanslı
(4-6 Hz), kaba bir tremordur. Sıklıkla başparmak ve işaret parmağının birbirlerine
ritmik sürtmesi olarak ortaya çıkar ve bu niteliğiyle hap yapma veya para sayma
tremoru olarak da adlandırılır. Parkinson sendromu
tanısı koymak için tüm bu belirtilerin bir arada olması gerekmez. Bazı
hastalarda sadece bradikinezi ve rijidite saptanıp akinetik-rijid sendrom tanısı konurken diğer hastalarda buna tremor veya
postüral instabilite ya da her ikisi eklenebilir, bazı hastalarda ise tabloya
tremor hakim olabilir. Donmalar akinezinin bir formudur. Tipik olarak yürümenin
durmasına yol açacak şekilde alt ekstremitelerde görülse de üst ekstremitelerde
veya konuşmada da olabilir. Parkinson hastalığında ileri dönemde görülürken, Parkinson
artı sendromlarda daha erken evrede görülür.
Parkinson sendromunu etyolojiye yönelik olarak dejeneratif ve semptomatik
olarak iki ana gruba ayırmak mümkündür (Tablo 3). Dejeneratif kökenli
Parkinsonizm tablosu da üçe ayrılabilir:
İdyopatik Parkinson hastalığı (İPH), Parkinson artı (Parkinson plus) sendromları ve Parkinsonizmin eşlik edebildiği diğer
dejeneratif hastalıklar. Bu hastalıklarda altta yatan patoloji dejeneratif
hücre kaybıdır ve etyolojik bir başka sebep saptanmaz. Parkinson artı sendromlarının bu isimle anılmasının sebebi bu gruptaki
hastalıklarda yukarıda belirtilen klasik dört bulguya ilave bulgu ve
belirtilerin saptanmasıdır. Göz hareket bozukluklarından otonomik bulgulara,
ataksiden demansa kadar değişen ve geniş bir spektrum oluşturan bu ilave
bulgular tek tek hastalıklar anlatılırken irdelenecektir. Ayrıca Tablo 3’de
görüldüğü gibi pek çok dejeneratif kökenli hastalığa Parkinsonizm bulguları
eşlik edebilir. Semptomatik Parkinson sendromunda ise
altta yatan etyopatogenez bazal ganglianın doğrudan beyin hasarı yaratan ya da
ikincil olarak beyni etkileyen hastalıklar sonucu tutulmasıdır. Başka bir deyişle
bu durumda Parkinsonizm bulguları altta yatan bir başka patolojinin semptomlarını oluştururlar ve çok çeşitli sebeplere bağlı
olarak ortaya çıkabilirler.
Tablo 3. Parkinson sendromunun
sınıflaması
|
Nörodejeneratif 1- İdyopatik Parkinson hastalığı 2-
Parkinson artı sendromları ·
Multisistem atrofi ·
Progresif supranükleer palsi ·
Kortikobazal dejenerasyon
·
Lewy cisimcikli demans 3- Parkinsonizmin eşlik edebildiği diğer
dejeneratif hastalıklar · Spinoserebellar
ataksi tip 2,3,17 · Huntington
hastalığı · Striopallidodentat
kalsinozis (Fahr Hastalığı) · Hemiparkinson-hemiatrofi
sendromu · Frontotemporal
demans-parkinsonizm kompleksi · Dentatorubropallidolusiyan
atrofi · Nöronal
intranükleer inklüzyon hastalığı · Pallidal dejenerasyonlar · Striatal nekrozla
giden mitokondriyal hastalıklar · Beyinde demir
birikimi ile giden nörodejeneratif hastalıklar (Pantotenatkinaza eşlik eden
nörodejenerasyon ve diğerleri) · Beyinde manganez
birikimi ile giden nörodejeneratif hastalık · Nöroakantositoz |
Semptomatik 1- Vasküler (küçük damar hastalığına bağlı
subkortikal ensefalopati, multi-lakünler, bazal ganglia ve beyinsapının
hemorajileri ve infarktları, kavernomlar, arteriovenöz malformasyonlar ) 2- Normal basınçlı hidrosefali 3- Yer kaplayıcı lezyonlar 4- İlaca bağlı (dopamin reseptör blokerleri [antiemetikler, nöroleptikler], dopamin boşaltıcı ilaçlar, kalsiyum kanal blokerleri,
valproik asit, lityum, flunarizin) 5- İntoksikasyonlar
(karbon monoksit, manganez, potasyum permanganat/efedrin kötü kullanımı, cıva
ve diğer ağır metaller, organik çözücüler,
karbon disulfid, 1-metil-4-fenil-1,2,3,6-tetrahidrobiyopteridin [MPTP], siyanid, metanol) 6- İnfeksiyonlar
(ensefalitler, prion hastalıkları, nörosifiliz, toksoplazmoz AIDS) 7- Metabolik nedenler (hipoksi,
hipoparatiroidizm, ekstrapontin miyelinolizis, kronik karaciğer hastalığı,
Wilson hastalığı) 8- Kafa travması,
dementia pujilistika (boksörlerde) 9- Demiyelinizan hastalıklar 10- Fonksiyonel [psikojenik] parkinsonizm |
“Bloklayıcı
tikler”, tik bozukluğunun bir bileşeni olabilir ve sürdürülmekte
olan hareketin kısa sürelerle durmasıdır. Düşme atakları
postüral kas tonusunda çözülme veya bacakta anormal kas kontraksiyonuna bağlı
oluşan, bilinç kaybının olduğu ya da olmadığı ani düşmelerdir. Düşme
ataklarının semptomatik nedenlerinden biri olan “katapleksi”
ise hemen sadece narkolepsi sendromunun bileşeni
olarak gülme ya da emosyonel tonusun arttığı durumlarda veya spontan olarak da
ortaya çıkabilen ani düşme ataklarıdır. Kataplekside bilinç kaybı olmaz ancak
hastalar düşme esnasında konuşamazlar. “Katatoni” bir
postür veya pozisyonun uzun sürelerle sabit olarak sürdürülmesidir. Klasik
olarak psikotik hastalıkların bir özelliğidir ancak major depresyonda da
görülebilir. Hasta istenen hareketi çok yavaş yaparken spontan hareketleri
normal hızında yapar.
İDYOPATİK
PARKİNSON HASTALIĞI
Parkinson hastalığı nörodejeneratif hastalıklar arasında
Alzheimer hastalığından sonra en sık rastlanılan ikinci hastalıktır. Değişik
ülke ve ırklarda yapılmış epidemiyolojik çalışmalar bu hastalığın prevalansının
genel popülasyonda binde 2-3, 55 yaş üzerinde ise %1
civarında olduğunu göstermiştir. Seksenli yaşlarda sıklığı %3-4’e kadar
çıkabilir. Hastalığın başlangıç yaşı ortalama 50-60 yaş aralığında olup prevalansı
ilerleyen yaşla artmakla birlikte genç yaşlarda da başlayabilir; tüm hastaların
%5'inde hastalık 40 yaşından önce başlar. Bu durumda genç başlangıçlı, 20 yaşın
altında başlayan hastalarda ise jüvenil Parkinson hastalığından söz edilir.
Parkinson hastalığındaki patolojik
değişiklikler substantia nigra pars compacta'daki melanin içeren dopaminerjik
hücrelerin kaybı, kalan hücrelerin içinde de Lewy cisimciği olarak
adlandırılan, ağırlıklı olarak alfa-sinüklein ve ubikuitin adı verilen
proteinler içeren küresel inklüzyon cisimciklerinin saptanması şeklindedir.
Klinik belirtilerin ortaya çıkması için bu bölgedeki dopaminerjik hücre
kaybının %60-70 seviyelerinde gerçekleşmesi gerekmektedir. Parkinson
hastalığının patolojik olarak kesin teşhisinin konulması için mutlaka bu
değişikliklerin saptanması gereklidir. Elimizdeki bilgi birikimi hastalığa ait
ilk patolojik bulguların olfaktor bulbus ve beyinsapı yapılarından başladığını,
daha sonra substantia nigra pars compacta’ya yayıldığını ve hastalığın ileri evrelerinde
ise kortikal yapılara ulaştığını göstermektedir.
Hastalığın etyolojisi ve hücre kaybına
yol açan patofizyolojik olayların niteliği henüz tam olarak anlaşılamamıştır.
Sadece çevresel faktörlerin ya da sadece genetik faktörlerin hastalığa sebep olduğu
yönündeki görüşler hastalığın tarihsel gelişimi içinde zaman zaman ağırlık
kazanmış olsa da şu an hakim olan görüş her iki
faktörün de rol oynadığı yönündedir. Bu görüşe göre sporadik Parkinson
hastalığı genetik yatkınlık taşıyan insanlarda çeşitli çevresel faktörlerin
etkisi sonucu ortaya çıkmaktadır. Son zamanlarda bulunan genetik ve
biyokimyasal veriler ışığında hastalığın patogenezinde, hücre içinde yıpranmış
proteinlerin yıkım sürecinde oluşan aksamaların rol oynadığı görüşü ağırlık
kazanmıştır. Bu görüşün ana kaynağı bugüne kadar saptanan, monojenik kalıtımsal
Parkinson hastalığının çoğu tipinde sorumlu genlerin yıpranmış proteinlerin
yıkım sürecinde rol alan molekülleri kodluyor olmasıdır. Bu görüşle uyumlu olan
bir başka bulgu da hastalığın patognomonik bulgusu olan Lewy cisimciklerinin
içinde çeşitli protein birikintilerinin saptanmasıdır. Bu süreçteki ana
mekanizmanın ise, yıpranmış proteinlerin yıkımından sorumlu olan
ubikuitin-proteozom yolunun genetik ve/veya çevresel nedenlerle hasara uğraması
olduğu düşünülmektedir. Parkinson hastalığında bugüne kadar gösterilebilmiş,
üzerinde az çok fikir birliğine varılmış biyokimyasal anormalliklerden biri
mitokondriyal işlev bozukluğudur; bu da hücrenin enerji metabolizmasını
aksatarak enerjiye bağlı tüm süreçleri (örneğin hücre içi protein trafiği ve
yıkımı) etkileyebilir. Ayrıca lizozomal disfonksiyon (ki bu da protein
yıkımında rol oynar) ve inflamasyonun da Parkinson hastalığının
etyopatogenezinde rolü olduğu düşünülmektedir. İleri yaş, birinci derecede
akrabalarda Parkinson hastalığı olması, ciddi kafa travması hikayesi
olması ve kırsal alanda yaşamak, epidemiyolojik çalışmalarda tutarlı olarak
ortaya çıkan risk faktörleridir. Kırsal yaşamın tarımsal ilaçlara maruziyeti
arttırdığı, bu ilaçların ise dopaminerjik hücrelerde ölüme yol açtığı
düşünülmektedir. Çalışmalarda saptanan koruyucu faktörler ise sigara kullanımı
ve belli bir miktarın üzerinde kahve içimidir. Bu faktörlerin hangi
mekanizmalarla hastalık riskini arttırdığı ya da azalttığı henüz kesin olarak
bilinmemektedir.
Parkinson hastalığının genetik
özellikleri son yirmi yıl içinde çok daha iyi anlaşılmaya başlamıştır. Genç
başlangıçlı Parkinson hastalarında yapılan, tek yumurta ikizleriyle çift
yumurta ikizlerinin karşılaştırıldığı bir çalışmada, tek yumurta ikizlerinden
birinde hastalık saptandığında diğerinde de hastalığın ortaya çıkma riskinin
çift yumurta ikizlerine kıyasla anlamlı olarak arttığı saptanmıştır. Bu bulgu
hastalıkta genetik faktörlerin etkili olduğu yönünde kuvvetli bir kanıt sağlamıştır.
Bu tarihe kadar parkinsonizm ile ilgili en az 23 tane lokus ve bunlardan
19’unda hastalık ile ilişkili gen saptanmıştır. Mutasyonu halinde parkinsonizme
yol açabilen genler ve lokusları Tablo 4’de
görülmektedir. Bu lokuslardan bir kısmı tartışmalıdır. Ailevi Parkinson
hastalığı tüm Parkinson hasta popülasyonunun
%10-15'inden azını oluşturmaktadır. Ayrıca bugüne kadar saptanan genetik
bozukluklar ailevi Parkinson hastalarının ancak bir kısmını
açıklayabilmektedir. Bu bulgu Parkinson hastalığına sebep olan başka genetik
mutasyonların varlığını düşündürmektedir. Bunun yanında sporadik Parkinson
hastalığı için pek çok genetik risk lokusu çeşitli ilişkilendirme
çalışmalarında bulunmuştur. Bunlardan en önemlisi Glukoserebrosidaz (GBA) gen mutasyonlarıdır. Gaucher hastalığı, GBA geninin otozomal dominant mutasyonları sonucu ortaya
çıkar. Bu gendeki heterozigot mutasyonlar ise Parkinson hastalığı için önemli
bir risk faktörüdür. Bu mutasyonu taşıyan Parkinson hastalarında süreç daha
hızlı bir motor kötüleşme gösterir ve kognitif bozukluk sıktır.
Tablo 4. Ailevi
Parkinson hastalığı ve ilişkili genler
|
Lokus |
Lokalizasyon |
Gen |
Kalıtım |
|
PARK1 |
4q22.1 |
α-sinüklein |
OD |
|
PARK2 |
6q26 |
Parkin |
OR |
|
PARK3 |
2p13 |
Kesin
değil |
OD |
|
PARK4 |
4q22.1 |
α-sinüklein |
OD |
|
PARK5 |
4p13 |
UCHL1 |
OD |
|
PARK6 |
1p36 |
PINK-1 |
OR |
|
PARK7 |
1p36.23 |
DJ-1 |
OR |
|
PARK8 |
12q12 |
LRRK2 |
OD |
|
PARK9 |
1p36.13 |
ATP13A2 |
OR |
|
PARK10 |
1p32 |
Kesin
değil |
? |
|
PARK11 |
2q37.1 |
GIGYF2 |
OD |
|
PARK12 |
Xq21-25 |
Kesin
değil |
X’e bağlı |
|
PARK13 |
2p13.1 |
HTRA2 |
OD |
|
PARK14 |
22q13.1 |
PLA2G6 |
OR |
|
PARK15 |
22q12.3 |
FBOX7 |
OR |
|
PARK16 |
1p32 |
Kesin
değil |
? |
|
PARK17 |
16q11.2 |
VPS35 |
OD |
|
PARK18 |
3q27.1 |
EIF4G1 |
OD |
|
PARK19 |
1p31.3 |
DNAJC6 |
OR |
|
PARK20 |
21q22.1 |
SYNJ1 |
OR |
|
PARK21 |
2p13
|
DNAJC13 |
OD |
|
PARK22 |
7p11.2 |
CHCHD2 |
OD |
|
PARK23 |
15q22.2 |
VPS13C |
OR |
(OD: Otozomal dominant, OR: Otozomal
resesif)
Parkinson hastalığının ana klinik
belirtileri yukarda da belirtildiği gibi bradikinezi, rijidite, tremor ve
postüral instabilitedir. Parkinson hastalığı teşhisi koyabilmek için bu
bulgulardan mutlaka hareketlerde yavaşlamanın yani bradikinezinin olması
gerekir. Bradikineziye ek olarak istirahat tremoru veya rijiditenin bulunması
Parkinson hastalığının tanısını koydurur. Parkinson hastalığı teşhisi koymak
için bu dört bulgunun da bir arada olması gerekmez. Postüral instabilitenin
hastalığın erken basamaklarında görülmesi beklenmez, bulunduğu zaman
parkinsonizm yapan başka hastalıkları düşünmek gerekir. Hastalık hemen her
zaman asimetrik olarak, vücudun bir yarısında başlar. İlk belirtiler sıklıkla
bir ekstremitede istirahat tremoru, bir elin özellikle ince hareketlerde
beceriksizleşmesi, tutuklaşması, yavaşlaması ya da tüm hareketlerin, özellikle
yürümenin yavaşlaması, vücudun öne doğru eğilmesi şeklindedir. Bu belirtiler
kural olarak sinsi başlayıp yavaş ilerlerler, zamanla hastalık vücudun diğer
yarısına da geçer. Hastalar spontan olarak ya da sorulduğunda yazılarının
değişip küçüldüğünü, düğme iliklemek ya da açmak gibi ince işlerde zorluk
çektiklerini, gece yatakta dönmenin, yerlerinden kalkmanın güçleştiğini, hareketlerinin,
yürümelerinin yavaşladığını, ayaklarını sürterek yürüdüklerini, harekete
başlamada güçlük çektiklerini buna karşın kaba kas kuvveti gerektiren işlerde
güçsüzlük farketmediklerini söylerler. Terleme, salivasyon ve derinin
yağlanması artabilir, hastalığın başlangıcında omuz, sırt, ekstremite ağrıları
olabilir. Omuz ağrısı başlangıç bulgusu olarak oldukça sıktır, bu tip
hastaların teşhisi ağrıya odaklanıldığı için gecikebilir. Hasta yakınları yüzün
donuklaştığını, sesin hafif çıktığını ve konuşmanın anlaşılmasının güçleştiğini
bildirebilirler. Parkinson hastalığının premotor bulguları olarak da kabul
edilen REM uykusu davranış bozukluğu (uykunun rüya safhası olan REM uykusu
sırasında canlı rüyalara bağlı olarak konuşma, bağırma, atma, sıçrama, çırpınma
şeklinde hareketler), koku duyusunun kaybı (anosmi) ve konstipasyon hastaların
çoğunda görülür. Bu bulgular motor bulguların başlamasından yıllarca önce
ortaya çıkabilirler. Uygun zihinsel testlerle bakıldığında hastaların
birçoğunda hafiften ağıra kadar uzanan zihinsel işlev bozuklukları saptanır.
Özellikle yaşlı hastalarda ve hastalığın ilerleyen yıllarında hastaların bir
kısmında Parkinson hastalığı demansı gelişebilir, bu oran hastalığın 15.-20.
yıllarında %50-80 gibi çok yüksek rakamlara ulaşabilir.
Muayenede
bradikinezinin ifadesi olarak maske yüz, hipofonik, zor anlaşılan bir konuşma,
ardısıra veya tekrarlayıcı el, ayak hareketlerini başlatmada güçlük, yavaşlama,
beceriksizlik, hareketin amplitüdünde azalma ve değişkenlik saptanır;
ekstremite bulguları sıklıkla asimetriktir. Hasta özellikle alçak
sandalyelerden güç kalkar, yürümeyi başlatmada güçlük çekebilir, yürümesi küçük
adımlarla, yavaş, gövde öne doğru eğiktir, ayaklarını sürüyebilir ve bir ya da
her iki kolunu sallamaz. Kas gücü normal sınırlardadır ve piramidal bulgulara
rastlanmaz. Hastada düşük frekanslı, kaba bir istirahat tremoru görülebilir, bu
tremor tipik olarak istemli hareketler esnasında kaybolur, belli bir postürün
korunması esnasında tekrar ortaya çıkabilir, genelde hastanın işlevselliğini
bozmaz. Tremor tek taraflı ya da bir tarafta baskın olabileceği gibi bacak ve
çeneyi tutmak üzere yaygın da olabilir. Tutulan ekstremitelerin pasif hareketi
esnasında rijidite tarzında, yani bir kurşun boruyu bükercesine sürekli bir
dirençle karşılaşılır, rijidite el bileğini hareket ettiren kaslarda olduğunda
dişli çark belirtisi alınır. Bununla kastedilen el bilekten itibaren yukarı
aşağı hareket ettirildiğinde birbirini döndüren iki dişli çarkın her dişlisinin
atlayışında görülen kesik kesik bir direnç hissinin alınmasıdır. Benzer bir
duyguyu rijidite olmadan, muayene edilen elde mevcut olan bir istirahat tremoru
da verebilir. Rijidite karşı ekstremitenin kuvvetle innerve edilmesi (örneğin
karşı eli yumruk yapma) esnasında artar. Postüral instabilite mevcutsa hasta
spontan olarak, özellikle yokuş veya merdiven çıkarken ya da inerken düşebilir.
Muayene esnasında ayaklar hafif ayrık şekilde dururken hafiçe öne, arkaya veya
yana doğru itelendiğinde de kendisini yakalamakta güçlük çeker, tutulmazsa blok
halinde itilen yöne doğru yıkılabilir. Diğer nörolojik muayene bulguları
arasında baskılanamayan nazopalpebral refleks (burun köküne vurulduğunda
refleks olarak oluşan göz kırpmanın istemli olarak baskılanamaması), artmış
“snout” refleksi (dudakların üzerine vurulduğunda dudakların öne doğru
büzülmesi), pozitif palmomental refleks (avuç içi kuvvetle çizildiğinde o taraf
submental bölgenin kasılması) gibi canlı yüz refleksleri sayılabilir.
Parkinson hastalığının semptomatik
tedavisindeki ana yaklaşım azalmış dopaminerjik geçişi arttırmaya yönelik bir
yerine koyma tedavisidir. Bu amaçla en sık kullanılan ilaçlar levodopa içeren
preparatlardır. Levodopa dopaminin ön maddesidir, dopamin kan beyin bariyerini
geçmediğinden onun yerine levodopa verilir. Levodopanın çevre dokularda
dopamine çevrilerek periferik yan etkilerin ortaya çıkmasını önlemek ve beyine
geçen levodopa miktarını arttırmak amacıyla levodopa bu maddenin dopamine
çevrilmesini sağlayan enzim olan dopa dekarboksilazın periferik tip inhibitörleri
(benserazid veya karbidopa) ile birlikte kullanılır. Bu maddeler kan beyin
bariyerini geçmedikleri için levodopanın beyinde dopamine çevrilmesini
engellemezler. Levodopa preparatları Parkinson hastalığında en etkili
ilaçlardır. Ancak yüksek dozlarda ve uzun süre kullanıldıklarında, zamanla
alınan motor cevapta dalgalanmalar, hastanın doza bağlı veya ondan bağımsız
olarak açılıp kapanması, istemsiz hareketler (diskineziler) gibi yan etkiler
ortaya çıkarlar. Bu yüzden Parkinson hastalığının modern tedavisinde özellikle
de hasta erken yaşta ise bu preparatlarla tedaviye başlamayı geciktirme eğilimi
vardır. İleri evredeki hastalarda “açık-kapalı” dalgalanmalar ve diskinezilerle
oral tedaviler ile başa çıkılamıyorsa invazif tedavilerden intraintestinal levodopa
karbidopa jel tedavisi uygulanabilir. Bu tedavideki amaç incebağırsak yoluyla
devamlı dopaminerjik stimülasyon sağlanması ve böylelikle “açık-kapalı”
dalgalanmaların en aza indirilmesidir. Dopaminerjik geçişi artırmaya yönelik
olarak kullanılan ikinci grup ilaçlar dopamin agonistleridir. Bu ilaçlar
dopamin reseptörlerine bağlanarak dopaminin etkisini taklit ederler.
Bromokriptin, kabergolin, piribedil, ropinirol, pramipeksol gibi ilaçlar bu
gruptandır. Parkinson hastalığı tedavisinde bu ilaçlardan yan etki profili göz önüne alınarak non-ergot türevi olan piribedil,
ropinirol ve pramipeksol kullanılmaktadır. Bir başka dopamin agonisti olan
apomorfin etki süresinin kısalığı ve yanlızca subkutan uygulanabilmesi
nedeniyle “açık-kapalı” dalgalanmaları olan ileri evre Parkinson hastalarında
kullanılmaktadır. Ancak agonistlerin etkisi genelde levodopa kadar güçlü
olmadığından ve bu ilaçlar genelde yavaş titrasyona ihtiyaç gösterdiklerinden
bazen, özellikle hızla etki alınmak istenen ağır hastalarda tedaviye doğrudan
levodopa preparatlarıyla başlamak gerekebilir. Dopamin agonistlerinin yan
etkileri genelde levodopaya göre daha fazladır. Tedavide
kullanılan diğer ilaçlar bazal gangliada dopamin ile asetilkolin arasında
normalde varolan ve asetilkolin lehine bozulan dengeyi tekrar sağlamak için
verilen antikolinerjik ajanlar (biperiden, bornaprin, triheksifenidil) ve
dopaminin sinaptik aralıkta daha uzun süre kalmasını sağlamaya yönelik olarak
verilen, dopamini yıkan ana enzim olan monoaminooksidaz-B'yi (MAO-B) inhibe
eden ajanlardır (selejilin ve rasajilin). Antikolinerjikler yaşlı
hastalarda yan etkilerinden dolayı çok istisnai durumlar haricinde
kullanılmamalıdır. Rasajilin ve selejilin hastalığın başlangıcında, az ve hafif
bulguları olan hastalarda başlangıç tedavisi olarak bir süre tek başlarına
kullanılabilirler. Bir başka ilaç grubu ise çevre dokularda ve beyinde
levodopayı metabolize eden bir diğer enzim olan katekol-O-metil transferazı
(COMT) inhibe eden ve böylelikle beyine geçen levodopa miktarını arttıran ve
levodapanın yarı ömrünü uzatan ajanlardır (entakapon, tolkapon). Entakaponun
levodopa ve karbidopa ile kombine edildiği bir üçlü kombinasyon
da mevcuttur. Tedavide bugünkü yaklaşım, eğer hastanın yaşı 65-70 yaşından az
ve eşlik eden ciddi bir sistemik hastalığı veya demansı yoksa tedaviye bir
MAO-B inhibitörü veya dopamin agonistiyle başlayıp ihtiyaç halinde bir levodopa
preparatı eklemek yönündedir. Son yıllarda Parkinson hastalığının cerrahi
tedavisi de tekrar güncellik kazanmıştır. Seçilen cerrahi hedefler ön plandaki semptomlara göre değişir ve talamus, globus pallidus
internus ve subtalamik nükleusu içerir. Uygulanan teknik ise ya bu bölgelerin
yüksek frekanslı ses dalgalarıyla yakılması ya da hedef bölgelere yerleştirilen
derin elektrodlar vasıtasıyla uygulanan yüksek frekanslı elektrik uyarılarla
(derin beyin stimülasyonu) bu bölgedeki anormal aktivitenin değiştirilmesidir.
Derin beyin stimülasyonu yöntemi için günümüzde en çok tercih edilen hedef
subtalamik nukleustur.
İdyopatik Parkinson hastalığı kural
olarak yavaş ilerleyen bir hastalıktır. Dopaminerjik tedavinin kullanılmaya
başlamasıyla hastaların prognozu anlamlı olarak düzelmiş ve ortalama yaşam
beklentileri uzamıştır. Bununla birlikte prognoz hastadan hastaya ciddi
farklılıklar gösterir. Hastaların yaklaşık üçte biri tüm hastalık boyunca
tedaviye cevap verip önemli bir kısıtlama olmadan yaşamlarını sürdürürken bir
kısmında tedavi cevabı zamanla bozulur, bir kısmı ise başlangıçtan itibaren
tedaviye kısıtlı cevap verirler. Bu son gruptaki hastaların büyük bir kısmının,
başlangıçta klinik olarak idyopatik Parkinson hastalığından ayırt edilemese
dahi aşağıda bahsedilecek Parkinson artı sendromlarına dahil
olduğu düşünülmektedir.
PARKİNSON
ARTI SENDROMLARI
Bu gruptaki hastalıkların ortak özellikleri
Parkinson sendromunun klasik bulgularının
(bradikinezi, rijidite, tremor, postüral refleks bozuklukları) yanına başka
bulgu ve belirtilerin eklenmesidir, Parkinson artı ismi de bu ilave bulgulara
atfen konulmuştur ve hastada klasik bulgulara ilave bulgular olduğunu ifade
eder. Bu grupta kimileri oldukça nadir görülen birçok hastalık vardır,
deskriptif bir deyim olduğu için de grubun sınırlarını net olarak çizmek mümkün
değildir. Bu hastalıklardan aşağıda kısaca bahsedilecektir.
Multisistem
Atrofi
Bu hastalık Parkinson artı sendromları arasında en sık görülenlerden biridir.
Multisistem atrofi (MSA) deyimi patolojik anatomik bulguları birbirine
benzeyen, klinik bulguları kısmi örtüşme gösteren birkaç klinik tabloyu
tanımlamak için kullanılır. Geçmişte striatonigral dejenerasyon,
sporadik olivopontoserebeller atrofi ve Shy-Drager sendromu olarak adlandırılan
tablolar bugün tek bir isim altında toplanıp multisistem atrofi olarak
anılmaktadırlar. Buna sebep, yukarıda da bahsedildiği gibi bu tabloların patolojik
özelliklerinin benzerliğidir. Bu hastalarda substantia nigra, striatum,
inferior oliva, pons, medulla spinalisin otonom sinir sistemine ait olan
intermedyolateral kolonu ve Onuf nukleusunda hastadan hastaya değişen
miktarlarda nöron kaybı ve gliozis gözlenir. Oligodendrosit hücrelerinin
sitoplazmasında biriken anormal sinüklein proteini içeren inklüzyon
cisimcikleri bu hastalığın hücresel düzeydeki patognomik özelliğidir. Kesin
patolojik teşhis için bu cisimciklerin görülmesi şarttır, ancak bunlar nadiren
başka nörodejeneratif hastalıklarda da görülebilir ve MSA'ya özgün değildir.
Hangi dokunun ne ölçüde tutulduğuna göre de klinik tablo, ekstrapiramidal
(parkinsonizm), serebellar (ataksi, dismetri), pontin (piramidal, okülomotor)
ve otonomik bulguların (inkontinans, ortostatik hipotansiyon) değişen ölçüde
birlikteliği olarak ortaya çıkar.
Başlangıçta birbirinden farklı
antiteler olarak tarif edilen üç değişik klinik tablo MSA adı altında
toplandıktan sonra da bu tablolara MSA'nın üç alt-tipi gözüyle bakılıyordu. Son
olarak gelinen nokta MSA'yı MSA-P (MSA-Parkinsonizm) ve MSA-C
(MSA-Serebellar) olarak ikiye ayırıp
orijinal ismi Shy-Drager olan tabloyu ayrı bir antite olarak görmeme
yönündedir. Bunun sebebi otonomik bulguların az ya da çok tüm MSA hastalarında
bulunması ve böylece ayırt edici bir özellik taşımamasıdır. Ancak sadece
belirgin otonomik disfonksiyonun olduğu “saf otonomik yetmezlik” olarak
adlandırılan ve esas olarak otonomik yapılarda yoğun sinüklein patolojisinin
bulunduğu bir hastalık bulunmaktadır.
MSA-Parkinsonizm olarak tanımlanan tablo ön planda parkinsonizmin
bulunduğu, otonomik bulguların az ya da çok eşlik etiği, serebellar bulguların
ise ya hiç bulunmadığı ya da geri planda olduğu tablolardır. MSA-Serebellarda
ise ön planda serebellar ve otonomik bulgular olup parkinsonizm bulguları geri
plandadır.
MSA-Parkinsonizm (eski deyimle
striatonigral dejenerasyon) orta-ileri yaşlarda bir
parkinsonizm tablosuyla başlar ve başlangıçta idyopatik Parkinson hastalığından
ayırmak çok güç olabilir. Tipik olarak bu hastalar dopaminerjik tedaviye ya hiç
cevap vermezler, ya kısmi cevap verirler ya da başlangıçta iyi olan tedavi
cevabı kısa süre içinde kaybolur. Parkinsonizm tablosunda sıklıkla rastlanan
özellikler aksiyal rijidite, postüral bozukluklar ve antekollis olarak
adlandırılan başın öne eğilmesidir. Başlangıçtan itibaren ya da zamanla
otonomik bulgular tabloya eklenir. Bu bağlamda idrar inkontinansı, erektil
impotans, ortostatik hipotansiyon, terleme bozuklukları görülebilir. Ortostatik
hipotansiyon hastanın ayakta durmasını olanaksız kılacak kadar ağır olabilir,
hasta ayağa kalktığında baş dönmesi ve fenalaşma hissi tarif eder. Bazı
hastalarda ataksi gibi serebellar bulgular da saptanabilir. MSA-Serebellar
tipte (eski deyimle sporadik olivopontoserebeller atrofi) hakim
olan bulgular sinsi başlayıp yavaş ilerleyen gövde ve yürüme ataksisi,
dismetri, göz hareketleri bozuklukları gibi serebellar ve pontin bulgulardır.
Otonomik şikayetler, piramidal bulgular ve rijidite,
bradikinezi gibi parkinsonizm bulguları tabloya eşlik edebilir. MSA’nın her iki
klinik tablosunda da hastaların çoğunda REM uykusu davranış bozukluğu görülür.
Kranyal MR bulguları
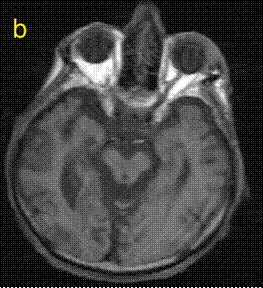
hastalığın tanısı için yardımcı olabilir. Orta serebellar pedunkül ve pons dejenerasyonu sonucunda ponsta “haç şeklinde çörek-hot cross
bun belirtisi” olarak adlandırılan haç benzeri görüntü oluşur: Bu bulgu en çok
MSA’da görülse de bu hastalık için patognomonik değildir (Şekil 4a).
Putamen atrofisinde, putamen posterolateralinde hiperintens yarık benzeri
çerçeve görülebilir (Şekil 4 b,c).
Özellikle MSA-C’de serebellar ve beyinsapı atrofisi de görülebilmektedir Şekil 4a).
MSA’da görülen parkinsonizmde levodopa
preparatları ve dopamin agonistleri kullanılabilir, ancak yukarıda bahsedildiği
gibi cevap sıklıkla kısıtlıdır. Bazen amantadin diğer dopaminerjik ilaçlardan
daha etkili olabilir. Ağır ortostatik hipotansiyon olan hastalarda sıkı varis
çorapları, ya da vücudda su ve tuz tutulmasını sağlayan fluorokortizon veya
midodrin kullanılabilir. MSA’nın prognozu Parkinson hastalığından daha kötüdür,
teşhisten sonra ortalama yaşam süresi 5 ile 10 yıl arasındadır.



Şekil 4. a-
Multisistem atrofili bir hastada “haç şeklinde çörek” belirtisi (ok). Beyin
sapı ve serebellumdaki atrofiye dikkat ediniz. b ve c- putaminal
yarık belirtisi oklarla gösterilmiştir.
Progresif
Supranükleer Palsi
Prevalansı yüksek olmamakla birlikte
Progresif supranükleer palsi (PSP), Parkinson artı sendromları
arasında göreceli olarak sık rastlanılanlardan biridir. İlk tanımlayan
araştırıcıların adına atfen bu hastalık eskiden Steele-Richardson-Olszewski
hastalığı olarak isimlendirilmiştir.
Patolojisindeki belirleyici özellik beyinsapının merkezi gri
maddesindeki değişik yapılarda, bazal gangliada özellikle pallidum, subtalamik
nukleus, substantia nigra gibi oluşumlarda nöron kaybı ve gliozis, bu
bölgelerdeki nöronların içinde de tau proteini içeren nörofibriler yumakların
görülmesidir; bazen patoloji diğer bazal ganglia yapılarını ve hatta bazı
kortikal bölgeleri tutacak şekilde yaygınlaşabilir.
PSP’nin klinik tablosu akinetik-rijid
bir parkinsonizmle birlikte supranükleer tipte bakış felci ve subkortikal tipte
bir demans olarak özetlenebilir. Hastalığın başlangıcında dengesizlik ve sık,
sebepsiz düşmeler oldukça karakteristiktir. Hastalığın başlangıcından itibaren
görünürde bir sebep olmadan ortaya çıkan düşmeler PSP’yi düşündürmelidir.
Konuşma erken dönemde etkilenebilir, ses alçak tonda, konuşma dizartrik ve zor
anlaşılır hale gelir. Hastalar gözlerini oynatamamaktan, görme güçlüğü veya
çift görmeden yakınabilir. Kişilik değişiklikleri, mental yavaşlama, dikkat ve
buna bağlı bellek bozuklukları erken dönemde ortaya çıkabilir. Muayenede
özellikle aksiyal tipte, yani boyun ve gövde kaslarını tutan bir rijidite ya da
distoni ve postüral refleks bozuklukları, ekstremitelerde bradikinezi saptanır.
Bazı hastaların klinik tabloları hastalığın ilk yıllarında Parkinson
hastalığına benzeyebilir (asimetrik başlangıç, istirahat tremorunun varlığı,
iyi L-dopa cevabı, yavaş progresyon). Bu
hastalarda zamanla klasik PSP bulguları ortaya çıkar. Bazı hastalarda
yavaşlamış dil hareketleri, canlı yüz refleksleri, kontrolsuz, duygusal
komponenti olmayan ağlama ve gülme ile karakterize psödobulber parezi
görülebilir. Bu hastalık için tipik olan göz hareket bozukluklarıdır.
Hastalarda özellikle vertikal bakış kısıtlanır, hasta sıklıkla aşağıya, bazen
de yukarıya ve hastalık ilerledikçe yanlara istemli olarak bakamaz, bakmak
istediğinde gözler yerine başını tüm olarak o yöne çevirir. Hastanın başı pasif
olarak aşağı ve yukarı oynatıldığında gözlerin serbestçe aşağı-yukarı hareket
ettiği gözlenir (taş bebek fenomeni), bu da göz
hareketlerinin nükleer seviyede sağlam olduğunu ancak supranükleer kontrolün
bozulduğunu gösterir. Nöropsikolojik incelemede hastaların özellikle dikkat,
mental hız ve yürütücü işlevlerde bozulma gösterdiği gözlenir.
Kranyal MRG’de mezensefalon atrofisi,
mezensefalon/pons oranında azalma, üst beyin sapındaki atrofi nedeniyle orta
sagittal kesitte karakteristik olarak görülen “hummingbird” (sinek kuşu)
bulgusu ayırıcı tanıda yardımcı olur (Şekil 5 a ve b).

Şekil 5. a-
PSP’li bir hastada “hummingbird” bulgusu ok ile gösterilmiştir, b- Aynı hastadaki mezensefalon atrofisi görülmektedir
Klasik PSP tablosundaki hastalar
dopaminerjik tedaviye ya hiç yanıt vermezler ya da az oranda fayda görürler.
Bazı hastalar amantadin veya antikolinerjiklerden kısmen faydalanabilirler.
Parkinson hastalığına benzer klinik fenotipte olan hastalar ilk yıllarda
dopaminerjik tedaviye cevap verebilir. Sürekli ve yavaş bir ilerleme gösteren
hastalığın prognozu kötüdür, başlangıçtan itibaren ortalama yaşam süresi 5-10
yıl arasındadır.
Kortikobazal
Dejenerasyon
Adından da anlaşılacağı gibi bu
hastalık serebral korteks ve bazal ganglianın birlikte tutulumu ile
karakterizedir. Nöropatolojik özelliği beynin özellikle frontal ve paryetal bölgelerindeki
kortikal piramidal nöronların şişmesi, balonlaşması ve akromazisi
(soluklaşması) ile karakterize asimetrik kortikal atrofi, substantia nigra pars
compactadaki melanin içeren hücrelerin kaybı ve ilave olarak daha birçok
subkortikal, beyinsapı ve bazen serebellar yapılarda hücre kaybı ve gliozis
olarak özetlenebilir. Etkilenen dokularda hiperfosforile tau birikimi ve
astrositik plaklar görülür.
Kortikal bölgelerin ve bazal ganglia
yapılarının birlikte tutulmasına bağlı olarak bu hastalıkta çeşitli kortikal ve
ekstrapiramidal bulgular bir arada görülürler. Bunun yanısıra en önemli özellik
çoğu vakada bulguların belirgin ölçüde asimetrik olmasıdır. Hastalık orta-ileri
yaşlarda kortikal veya ekstrapiramidal bulgularla başlayabilir. Ekstrapiramidal
bulgular genellikle asimetrik rijidite, bradikinezi, distoni olarak ortaya
çıkan akinetik-rijid bir sendromdur. Rijidite ve
distoni hastanın elini kullanmasını çok güçleştirecek ağırlıkta olabilir. Buna
sıklıkla postüral instabilite ve dizartri eklenir, bulgular zamanla diğer
tarafa da yayılırlar. Kortikal bulguların en sık ve en belirgin olanı
apraksidir. Apraksi bazen ağır tutulum gösteren tarafta rijidite ve distoniden
dolayı muayene edilemez, daha az tutulan tarafta daha kolay gösterilebilir.
Bunun yanında “yabancı el” (alien limb) olarak adlandırılan, bir ekstremitenin
vücuda ait olmadığı hissi ve bu ekstremitenin istemdışı, iradeden bağımsız,
bazen amaçlı gibi görünen hareketleri ortaya çıkabilir. Diğer kortikal bulgular
miyoklonus ve kortikal tipte duyu kusurunu (elin üzerine yazılan harfleri,
içine konulan eşyaları tanıyamama) içerir. Hastaların birçoğunda diseksekütif sendrom olarak da adlandırılan frontal tipte bir demans
oluşur. Bazen supranükleer tipte bir bakış felci gelişebilir ve PSP ile karışabilir.
Görülebilecek diğer bulgular canlı yüz refleksleri, yakalama refleksi gibi
ilkel refleksler, disfaji bazen de serebellar belirtilerdir.
Kranyal MR’de
asimetrik frontoparyetal atrofi görülür (Şekil 6).
Beynin flurodeoksiglukoz-PET ve SPECT incelemelerinde frontoparyetal bölgede
asimetrik hipometabolizma görülür.
Kortikobazal dejenerasyon
hastaları dopaminerjik tedaviye iyi cevap vermezler. Miyoklonus klonazepamdan,
distoni botulinum toksini enjeksiyonları ve
baklofenden yararlanabilir. Kontraktür gelişmesini önlemek için fizyoterapi ve ergoterapi önemlidir. Hastalık başladıktan
sonra sürekli ve yavaş bir şekilde ilerler, prognozu kötüdür, başlangıçtan
itibaren yaşam beklentisi 6-8 yıldır.


Şekil 6. Kortikobazal
dejenerasyonlu bir hastada sağda daha belirgin olan
asimetrik frontoparyetal atrofi ve bununla uyumlu olarak sağ lateral
ventrikülün daha geniş olduğu görülmektedir.
Lewy
Cisimcikli Demans
Lewy cisimcikli demans ekstrapiramidal
belirtilerle demansın bir arada olduğu bir başka hastalıktır. Patolojik
özelliği Parkinson hastalığında tarif edilen hücre kaybının, gliozis ve Lewy
cisimciklerinin bazen serebral kortekse, bazen diğer beyinsapı ve limbik
bölgelere bazen de bu yapıların tümüne yayılmasıdır.
Bu hastalık önce parkinsonizm ile ya da
önce demansiyel bulgularla başlayabilir, bu iki grup bulgudan biri bir süre
tabloya hakim olabilir, ancak teşhis için hastada her
iki sendromun bir arada bulunması gerekir. Parkinsonizm tablosu Parkinson
hastalığındakinden farklı değildir, bradikinezi, rijidite, tremor, postüral
instabilite görülebilir, düşmeler göreceli olarak daha sıktır. Demans
diseksekütif tipte, yani frontal loba atfedilen işlevlerin ön planda tutulması
şeklindedir; dikkat bozukluğuna ikincil olarak bellek bozukluğu ve sıklıkla
görsel-mekansal işlevlerin bozukluğu da görülür.
Hastalığın en önemli özelliklerinden birisi klinik tablonun dalgalanmalar
göstermesidir, hastalar bazı günlerde, bazen de aynı gün içinde belirgin olarak
daha iyi veya daha kötü olurlar. Diğer belirtiler arasında dopaminerjik
tedaviden bağımsız olarak ortaya çıkan görsel halüsinasyonlar,
klasik nöroleptiklere aşırı duyarlılık, senkoplar, artmış uyuklamalar
sayılabilir.
Lewy cisimcikli demans hastalarının
parkinsonizm belirtileri en azından başlangıçta dopaminerjik tedaviden
yararlanır, zamanla bu cevap azalıp kaybolabilir. Demans belirtilerinin
Alzheimer hastalığında kullanılan asetilkolinesteraz inhibitörlerinden
yararlanabileceği bildirilmiştir. Halüsinasyonların ve diğer davranışsal semptomların tedavisinde klasik nöroleptiklerden
(haloperidol gibi) kaçınılmalı, gerekirse klozapin veya ketiapin gibi atipik
nöroleptikler kullanılmalıdır.
Parkinsonizm tablosunun eşlik ettiği
diğer nörodejeneratif hastalıklar Tablo 3’de
verilmiştir. Bu hastalıkların bazılarından bu bölümün ilerisinde ve kitabın
diğer bölümlerinde bahsedilmiştir.
Beynin
hücresel yapısını doğrudan etkileyen dejeneratif hastalıkların dışında beynin
işleyişini, anatomisini, nöronal bağlantılarını ikincil olarak etkileyen birçok
durumda parkinsonizm görülebilir. Bunlar arasında ilaçlar, zehirlenmeler,
vasküler nedenler, hipoksi, travma, infeksiyonlar,
normal basınçlı hidrosefali ve beyin tümörleri gibi yer kaplayıcı lezyonlar
sayılabilir. Diğer bir semptomatik parkinsonizm nedeni psikojenik
parkinsonizmdir. Semptomatik
parkinsonizm nedenlerinden göreceli olarak sık rastlananları ayrıntılara
girmeden kısaca ele alınacaktır.
İlaçlara bağlı parkinsonizm en sık nöroleptik
ilaçların (örn., haloperidol) kullanımı sonucu görülür
ve bu ilaçların dopaminerjik reseptörleri bloke etmesi sonucu ortaya çıkar.
Benzer özellik taşıyan dopamini boşaltıcı ilaçlar, antiemetikler, kalsiyum
kanal blokerleri ve valproik asit de parkinsonizme yol açabilir. Belirtiler
genelde subakut yerleşir, tabloya genelde akinetik-rijid bir sendrom
hakimdir ve belirtiler kural olarak ilacın kesilmesiyle birkaç ay içinde kaybolurlar.
Özellikle
küçük damar hastalığı sonucu bazal gangliayı besleyen perforan arterlerin
tıkanmasıyla vasküler tipte bir parkinsonizm
ortaya çıkabilir, tipik olarak bulgular alt ekstremitelerde, yürüme ve dengede
daha belirgindir ve dopaminerjik tedaviye iyi cevap vermezler. Karbon monoksit
zehirlenmesi veya yaygın hipoksi (ağır sistemik hipotansiyon gibi) sonucu
akinetik-rijid bulguların ön planda olduğu bir parkinsonizm tablosu ortaya
çıkabilir, benzer tablolar maden işçilerinde manganez zehirlenmesi sonucu
tanımlanmıştır.
Ağır
kafa travmaları
sonrasında özellikle akinezinin ön planda olduğu, bazen rijiditenin de
katıldığı tablolar görülebilir; bunun bir alt-tipi tekrarlayan kafa
travmalarının sonucu olarak boksörlerde gelişen, tabloya demansın da katıldığı
dementia pugilistica (punch-drunk) sendromudur. İnfeksiyonlara bağlı olarak
gelişen parkinsonizmin prototipi geçen yüzyılın
başında salgın olarak ortaya çıkan von Economo ensefaliti sonrası görülen
parkinsonizmdir. Bunun dışında viral, bakteriyel, fungal infeksiyonlar sonucu
nadiren parkinsonizmin geliştiği de bildirilmiştir. Normal
basınçlı hidrosefali beyin-omurilik sıvısının (BOS) geri-emiliminde
oluşan bozukluklara bağlı olarak BOS retansiyonu, ventriküllerin genişlemesi ve
yakınından geçen liflere baskı yapması sonucunda gelişir, klinik tablosu
özellikle vücudun alt yarısını, postürü ve yürümeyi etkileyen parkinsonizm,
diseksekütif tipte demans ve idrar inkontinansı olarak ortaya çıkar. Bazal
ganglia motor bölgelerinde oluşan tümörler ve diğer yer kaplayıcı lezyonlar
parkinsonizm tablosuna yol açabilirler. Kronik subdural hematomlarda bazen
yürüme ve denge bozuklukları, bradikinezi gelişip parkinsonizmi andırabilir.
Yukarıda
sözü edilen bütün bu sebepler parkinsonizmin ayırıcı tanısında göz önünde
bulundurulmalı ve klinik şüphe varsa uygun inceleme metodları kullanılarak
araştırılmalıdır.
HİPERKİNETİK
HAREKET BOZUKLUKLARI
Bu
gruptaki hareket bozukluklarının ortak özelliği tabloya istemdışı,
kendiliğinden oluşan hareketlerin hakim olması, istemli
hareketlerin yürütülmesi sırasında kontrolsüz kasılmaların, amaç dışı
hareketlerin ortaya çıkması ya da her iki tip bozukluğun bir arada
bulunmasıdır.
DİSTONİ
Distoni
istemsiz, süreğen yahut tekrarlayıcı, bükücü, döndürücü nitelikte kas
kasılmaları nedeniyle geçici ya da kalıcı anormal postür ve hareketlere yol
açan bir hareket bozukluğudur. Amaca uygun motor davranışların yapılabilmesi ya
da belli bir postürün korunabilmesi için birbirlerine zıt çalışan kasların
(agonist ve antagonist kaslar) eşzamanlı kasılmaları
sonucu gelişir. Kasılma genellikle yavaş, döndürücü niteliktedir. Fakat koreik
veya miyoklonik distoni olarak adlandırılan ani ve hızlı kasılmalar da
görülebilir. Distoni özellikle hastalığın başlangıcında, tutulan vücut bölgesi
istemli olarak kullanıldığında ortaya çıkar veya fazlalaşır. Diğer bir vücut
bölgesinin istemli hareketleri esnasında da distonik bölge daha çok kasılır
("overflow" ya da "taşma" fenomeni).
Uykuda ise, hareket bozukluklarının çoğunda olduğu gibi, kaybolur. Distonik bölgeye
uygulanan hafif bir duyusal uyarı esnasında (örneğin spazmodik tortikolliste
çeneye uygulanan hafif bir bası esnasında) distonik kasılmalar geçici olarak
kaybolur; buna "duyusal hile" (sensory trick) adı verilir.
Klinik
semptomatolojinin nispeten heterojen oluşu – hastalık şiddetinin hafif olması,
günlük yaşamı çok etkilememesi, fokal yahut jeneralize tutulum, başlangıç
yaşındaki değişkenlik vs. – distoni ile ilgili epidemiyolojik verilerin de
değişken olmasına yol açsa da sıklığı tüm popülasyonda
100.000’de 15-30, 50 yaş üstü popülasyonda ise 100.000’de 732 olarak
bildirilmektedir. Kadınlar erkeklere göre iki kat daha fazla etkilenmektedir.
Distoniler,
geçmiş yıllarda, başlangıç yaşına, vücuttaki tutulum yerine, edinsel ya da
kalıtsal oluşuna göre çeşitli kriterler temel alınarak
sınıflandırılmıştır. Tablo 5’de
sunulan son sınıflandırma 2013 yılında Hareket Bozuklukları Cemiyeti tarafından
oluşturulmuştur. Bu sınıflamada distoniler iki eksen halinde ele alınmaktadır.
Eksen I başlangıç yaşı, tutulum yeri ve yayılım tarzı gibi klinik özellikleri,
eksen II sinir sistemi patolojisi, kalıtımsal yönü gibi etyolojik özellikleri
temel almaktadır. Distonik sendromla karşılaşan
hekimin bu sınıflandırmayı gözönünde bulundurması, tercih edilecek tanısal
girişim ve tedavi seçenekleri bakımından önemli ipuçları sağlar.
Tablo 5. Hareket Bozuklukları Cemiyeti distoni sınıflaması
|
Eksen I |
Eksen II |
|
Yaş İnfantil: 0-2 yaş Çocukluk çağı: 3-12 yaş Ergenlik: 13-20 yaş Erken erişkin: 21-40 yaş İleri erişkin: 40 yaş ve üstü Tutulum yeri Fokal Segmental Multifokal Jeneralize Hemidistoni Zamansal patern Statik İlerleyici Değişken (paroksismal, diurnal
…) Eşlik eden özellikler İzole Diğer hareket bozuklukları ile
birlikte Diğer nörolojik/sistemik
belirtilerle birlikte |
Sinir sistemin patolojisi Dejeneratif durumla birlikte Dejeneratif durum yok Yapısal lezyon var Kalıtımsal geçiş Otozomal
dominant Otozomal
resesif X’e bağlı Mitokondriyal Edinsel Kafa
travması İlaçlar/toksinler İnfeksiyon Psikojen İdyopatik Sporadik Ailesel |
Burada
sınıflamada gösterilen tüm distoni türlerinden söz edilmeyecek, klinik pratikte
sık rastlanılan distoni türleri aşağıda vurgulanacaktır.
İdyopatik Distoniler: İdyopatik distoni, her yaş grubunda görülebilen, altta yatan başka
bir hastalık bulunmayan distonilerin genel ismidir. Başlangıç yaşı 1. ve 4.
onyılda tepe noktasına ulaşır. Birinci onyılda başlayan ve çocukluk çağı
distonileri başlığı altında toplanan distoniler, genellikle bir ekstremiteden,
sıklıkla da bacaklardan başlayıp aylar ya da yıllar içinde diğer
ekstremitelere, bazen kranyal kaslara yayılır. Ancak üst ekstremiteler veya
kranyal alandan başlayan erken yaş başlangıçlı distoniler de olabilir.
Kırk yaşın
üzerinde başlayan distoniler sıklıkla fokal veya segmental özelliktedir,
öncelikli olarak da kranyoservikal kasları tutar. Nadiren, çocukluk çağında
fokal/segmental, erişkinde ise ekstremitelerde başlayan distoniler görülebilir.
Nöroloji pratiğinde, erişkin yaş distonilerini klinik görünüm ve tutulan kas
gruplarına göre adlandırma, idyopatik torsiyon distonisi (İTD) terimini ise
çocukluk çağında başlayan distoniler için kullanma eğilimi vardır.
Çocukluk
çağında görülen İTD’lerin çoğu otozomal dominant kalıtımla geçmektedir. Bu tip
distonilerin, özellikle Musevi kökenli ailelerde daha sık görülen bir tipinde
sorumlu gen 9. kromozomun uzun koluna lokalize edilmiş
ve mutasyonu sonucu distoniye sebep olan bu gene DYT1
geni adı verilmiştir. DYT1 geni bir
ısı şoku ATP bağlayıcı protein olan torsin A’yı kodlar. DYT1
mutant genini taşıyan ailenin her bireyinde hastalık ortaya çıkmamaktadır. Bu
durum genin penetransının düşük olmasından (%30-40) ileri gelmektedir. Bugüne
kadar genetik bir bozukluğun saptanamadığı çoğu İTD’lerinin klinik görünümleri
az çok birbirine benzer. Hastalık genellikle alt ekstremitelerden, yavaşça
başlar, özellikle yürüme esnasında bacakların distonik kasılmaları göze çarpar,
yürüme güçleşir ve düşmeler görülebilir. Zamanla kasılmalar aksiyal, kranyal
kaslara da yayılır ve jeneralize olabilir. Jeneralize ve şiddetli olgularda
yürüyüş, konuşma ve kalıcı postür bozuklukları gelişir. Distonik kasılmalar
istemli hareket, stres ve yorgunluk ile artar, ağrı ise nadirdir.
Dopa Yanıtlı Distoni (DYD, Segawa Hastalığı): Otozomal dominant geçişli olan bu nadir hastalıkta 14. kromozomun
uzun kolunda bulunan GTP siklohidrolaz
geninde çeşitli mutasyonlar saptanmıştır. Genin penetransı %30-40 civarındadır.
Ayrıca 11. kromozomda bulunan tirozin hidroksilaz
genindeki mutasyonlar da benzer ama daha hafif bir klinik tabloya sebebiyet
verebilmektedir. Hastalık çocukluk çağında ve genellikle alt ekstremitelerde
distoni ile başlar. Parkinsonizm ve piramidal bulgular da eşlik edebilir.
Kranyal alan tutulumu şimdiye kadar bildirilmemiştir. En önemli özelliği
yakınmaların diurnal dalgalanma göstermesi (sabah yakınmalar belirgin olarak azken
gün içinde belirgin kötüleşme görülmesi) ve düşük doz L-dopa (50-100 mg/gün)
tedavisine çok iyi yanıt vermesidir. Nadiren L-dopa dozunu yükseltmek gerekir.
Tanısal açıdan da önemli olan bu dramatik özellik, mutant genin, dopamin
sentezi için gerekli olan enzimlerden birini (eritro-dihidrobiopterin
triptofaz) kodlamasından kaynaklanır; buna bağlı olarak sentez edilen dopamin
miktarında azalma vardır. Gün boyu dopamin kullanıldıkça zaten normalden az
olan dopamin depoları çabuk tükenir ve yakınmalar akşama doğru artar. Çocukluk
çağında karşılaşılan her türlü primer distonide, DYD olasılığına karşı mutlaka
L-dopa tedavisi denenmelidir. Ayrıca yine dopa yanıtlı distoni spektrumunda yer
alan hastalıklardan tirozin hidroksilaz
genindeki mutasyonlar daha hafif formda bir hastalık yapar. Hastalık infantil
ve erken yaş başlangıçlıdır.
Primer
distoniler arasında farklı genetik ve klinik özelliklere sahip hızlı
başlangıçlı distoni-parkinsonizm (ATP1A3 geni),
X'e bağlı distoni-parkinsonizm (TAF1 geni) ve
miyoklonik distoni (SGCE geni)
hastalıkları da bulunmaktadır. Ayrıca yeni saptanan idyopatik distoni genleri
de vardır. Bunlar arasında THAP1, ANO3, GNAL, CIZ1 ve TUBB4
genleri sayılabilir. Bu yeni mutasyonlara sahip hastaların klinik fenotipleri
farklılıklar göstermektedir.
Fokal Distoniler
Bu tip
distonilerde vücudun sadece bir bölgesi tutulur ve distoni tutulan vücuT
bölgesine göre adlandırılır. Daha çok erişkinlerde görülen bu tip distonilerin
sık karşılaşılanlarından aşağıda bahsedilecektir.
Blefarospazm: Palpebral ve periorbital kasların, tekrarlayıcı ve istemsiz
kasılmaları sonucu oluşan göz kapanması şeklinde ortaya çıkan bir fokal
distonidir. Çoğunlukla 50-60 yaş arasında başlar ve kadınlarda erkeklere oranla
3 kez fazla görülür. Kasılmalar daima bilateraldir. Yalnızca göz kırpma
frekansının artması şeklinde hafif olabileceği gibi, işlevsel körlüğe yol
açacak kadar sık ve şiddetli göz kapanmaları da olabilir. Uykuda kaybolan
kasılmalar, emosyonel stres, okuma, yukarı veya aşağı bakış sırasında ve parlak
ışıkta artar. Bazı manevralarla kasılmanın şiddeti azaltılabilir ya da
durdurulabilir. Esneme, alna bastırma, çiğneme, şarkı söyleme gibi hastadan
hastaya değişen bu manevralara "duyusal hile" (sensory trick) adı
verilir. Blefarospazm çoğu hastada idyopatiktir. Ancak beyinsapı, diensefalon
ve striatum lezyonlarına bağlı semptomatik blefarospazm olarak veya PSP gibi
nörodejeneratif hastalıklarla birlikte görülebilir.
Oromandibulolingual Distoni: Çiğneme ve dil kaslarının istemsiz, distonik kasılmalarıdır.
Bu kaslarda distoni tek tek olabileceği gibi bahsedilen kasların tümünde de
olabilir. Temporal, masseter ve medyal pterigoid kaslar alt çeneyi kapatan,
lateral pterigoid kas ise çeneyi açan kaslardır. Ağırlıklı olarak tutulan kasa
göre alt yüz bölgesinde şekil ve işlev bozukluğu ortaya çıkar; çene konuşma
veya yeme esnasında kilitlenip açılamaz ya da ağız istemsiz olarak açılır,
hasta ağzını kapatamaz, çene öne veya yana kayabilir. Benzer şekilde dilde
yanlara, yukarı ve aşağı doğru kasılma olur, bazen dil ağız içinden dışarı
çıkabilir. Platisma kasının tutulumu sonucu boyunda kasılmalar görülebilir.
Masseter kaslarının tutulumu trismus ve bruksizme (diş gıcırdatma) yol açar.
Dudaklarda büzülme, bazen ağız komissürlerinde yanlara doğru çekilme
görülebilir. Kasılmalar bazen ağrılıdır. Çiğneme ve yutma zorluğuna bağlı
olarak beslenme bozukluğu gelişebilir. Özellikle dil kaslarının tutulumu, dilin
katkısı ile çıkan l, k, r gibi harflerin, perioral kasların tutulumu dudakların
katkısı ile çıkan b, f, m, p gibi harflerin telaffuzunu zorlaştırır,
dolayısıyla konuşma bozulur. Bu hastalar ciklet çiğnemek, dilaltına leblebi
koymak, çeneye dokunmak gibi manevralarla kasılmaların şiddetini
azaltabilirler.
Oromandibulolingual
distoni blefarospazm ile birlikte görüldüğünde, kranyal sinirlerce innerve
edilen kasların distonisi anlamında kranyal distoni (Meige sendromu)
olarak adlandırılır.
Larengeal Distoni (Spazmodik Distoni): Larenks kaslarının distonisidir. Sıklıkla 30-50 yaşları
arasında ortaya çıkar, kadınlarda erkeklerden fazla görülür. Tuttuğu kas
grubuna göre iki tipi vardır ve klinik olarak bunları birbirlerinden ayırmak
çoğu kez mümkündür. Adduktor tip larengeal distoni vokal kordların aşırı
adduksiyonuna bağlıdır. Hasta konuşurken zorlanır, ses boğuk, kısık çıkar, hasta
normal başladığı bir cümlenin sonuna doğru boğulur gibi olur, sık sık nefes
almaya gereksinim duyar. Larengeal distonilerin çoğu bu tiptedir. Abduktor
larengeal distoni ise posterior krikoaritenoid kasların konuşma sırasında
anormal kasılmaları sonucu vokal kordların uzun süre açık kalması nedeniyle
gelişir. Hasta ses çıkarmakta zorlanır, adeta fısıldar tarzda konuşur. Buna
karşın gülme, esneme gibi aynı kas gruplarının aktivasyonunu gerektiren
eylemlerde sorun yaşanmaz. Bu nedenle larengeal distoniyi "eyleme
özgü" (task-specific) distoniler sınıfına koyanlar da vardır. Nadiren her
iki kasın birlikte kasıldığı da görülür. Yakınmalar emosyonel stres ile artar.
Diğer çoğu distonik sendromun tersine yakınmaları
hafifleten etkili duyusal hile yoktur.
Servikal Distoni (Spazmodik Tortikollis): Boynun ve başın normal postürünü koruyan ve hareketini sağlayan
kasların distonisidir. En sık görülen idyopatik distoni tiplerinden birisidir.
Daha çok 40 yaşın üzerinde görülür ancak gençlerde de olabilir. Kadınlarda erkeklerden
2 kat fazladır. Yakınmalar hasta olan tarafta hafif bir gerginlik ve çekilme
hissi ile başlayıp aylar bazen yıllar içinde yerleşebilir. Ağırlıklı olarak
tutulan kasın işlevine göre baş karşı tarafa dönebilir (tortikollis); yana
(laterokollis), öne (anterokollis) veya arkaya (retrokollis) doğru eğilebilir.
Bazen bu distonik kasılmaların kombinasyonları
görülebilir. Tutulan taraftaki omuz hafif eleve olur. Kasılmalar sıklıkla
ağrılıdır. Baş sağlam tarafa doğru çevrildiğinde distonik olan tarafın aktivitesinin
aşırı olması nedeniyle postürü korumak mümkün olmaz ve baş distonik tarafa
doğru yavaşca döner. Bu esnada tremor da görülebilir (distonik tremor). Hasta
alna, çeneye, enseye elini koyarak (duyusal manevra) yakınmalarını azaltabilir.
Kasılmalar herhangi bir motor aktivite esnasında artabilir, uykuda ise
kaybolur.
Eyleme Özgü Distoniler (“Task”- Spesifik
Distoniler): Yalnızca
belli bir motor işlev esnasında ortaya çıkan distonilerdir. Aynı kasların
kullanımını gerektiren başka bir eylemde görülmezler, bu nedenle de
"eyleme özgü" (task-specific) olarak adlandırılırlar. Genellikle üst
ekstremiteyi tutan bu ilginç sendromun en sık görülen
formu yazıcı krampıdır. El ve önkol kaslarını
tutan kasılmalar yalnızca yazı yazarken ortaya çıkar. Başlangıçta kalem
parmaklar arasında istemsiz olarak kuvvetlice sıkılır, bilek ve dirsek arasında
gerginlik ve kasılma hissedilir. Zamanla parmaklarda hiperekstansiyon veya
hiperfleksiyon; bilekte pronasyon, supinasyon, ulnar ya da radyal deviyasyon
gelişir. Buna bağlı olarak yazı şekli iyice bozulur, okunamaz hale gelir.
Hastalar bazen diğer elleri ile yazmaya alışırlar. Fakat %20 oranında zamanla o
tarafta da benzer semptomlar gelişebilir. İstirahatte
hiçbir yakınma yoktur, makas kullanmak, örgü örmek, kağıt
para saymak gibi aktiviteler rahatlıkla yerine getirilebilir. Bu hastaların bir
kısmı kalın kalemle veya kara tahtaya yazı yazarken ciddi sorun yaşamazlar.
Eyleme
özgü distoniler, müzisyen, terzi gibi günlük pratiği sınırlı bir kas grubunun
uzun süreli ve tekrarlayıcı aktivasyonuna dayanan meslek sahiplerinde de
görülebilir. Yakınmaların şiddeti nedeniyle meslek değiştirenler dahi vardır.
Paroksismal Diskineziler: Distonik kasılmaların eşlik edebildiği paroksismal
diskineziler zaman zaman, ataklar halinde ortaya çıkar. Paroksismal
diskineziler son yıllardaki veriler ışığında üç grup halinde
sınıflandırılmıştır. Her grup kendi içinde idyopatik ve semptomatik olarak
ikiye ayrılır. Semptomatik paroksismal diskinezi nedenleri arasında multipl
skleroz, kafa travması, hipoparatiroidizm, perinatal
hipoksik ensefalopati, serebral infarkt, hipoglisemi, ensefalitler, diyabet,
hipertiroidizm ve anoksi gibi nedenler sayılabilir.
Paroksismal Kineziyojenik Diskinezi (PKD, Paroksismal
Kineziyojenik Koreoatetoz, Paroksismal Kineziyojenik Distoni): En sık rastlanan paroksismal hareket bozukluğudur.
Genellikle 5-15 yaş arasında başlar. İstemsiz hareketler daima belli istemli
bir hareket girişimini izleyerek ortaya çıkar. Hiperventilasyon ve stres
atakları kolaylaştırabilir. Bir ya da birden çok ekstremiteyi tutan istemsiz
hareketler ekstremitelerin distonik kasılmaları şeklinde olabileceği gibi
koreoatetoid veya ballistik de olabilir. Ataklar genelde beş dakikadan kısa
sürerler, ancak gün içinde onlarca kez tekrarlayabilirler. Epileptik nöbetlerle
karıştırılabilen PKD atakları sırasında bilinç kaybı olmaz ve atak aralarında
nörolojik muayene normaldir. Erkeklerde daha fazla görülen primer (idyopatik)
PKD vakalarının çoğu otozomal dominant kalıtımla geçer, ancak sporadik olanları
da vardır. Aile öyküsü bulunan vakalarda PKD, PRRT2
mutasyonları (Proline-rich transmembrane protein 2) sonucu ortaya çıkar. PRRT2
mutasyonları benign (nonfebril) familyal infantil konvülziyonlar, infantil
konvülziyon koreoatetoz sendromu, hemiplejik migren,
paroksismal tortikoliz, migren, epizodik ataksi ve homozigot mutasyonlarda
kognitif yıkım kliniği ile de ortaya çıkabilir. Antikonvülzif ilaçlara iyi
yanıt vermesi PKD'nin önemli bir özelliğidir. PKD ve benign familyal infantil
konvülziyonlar aynı hastada veya aynı ailelerdeki farklı bireylerde birlikte
görülebilir.
Paroksismal Non-Kineziyojenik Diskinezi (PNKD,
Paroksismal Non-kineziyojenik Distoni, Paroksismal Non-Kineziyojenik
Koreoatetoz): PKD'de
olduğu gibi genellikle ekstremitelerde ve bazende yüzde distonik, koreoatetoid
veya ballistik kasılmalar vardır. PKD'den asıl önemli farkı atakların
kendiliğinden başlaması, istemli bir hareket girişimini takip etmemeleridir.
Ataklar daha seyrek ancak daha uzundur; günde 3-4 defa gelen ve 5 dakikadan
uzun süren atakların sona ermesi bazen 4-5 saati alır. Yorgunluk, stres, açlık
ve heyecan atakları kolaylaştırabilir. Çocukluk çağında biraz daha sık olmakla
beraber her yaşta görülebilir. Erkeklerde kadınlara oranla daha sıktır.
İdyopatik olan PNKD'lerin çoğu otozomal dominant kalıtımla geçer. Miyofibrilogenez regülatör gen–1
mutasyonlarının PNKD ile ilişkili olduğu bulunmuştur. PNKD sporadik de
olabilir, ancak bu olguların psikojenik distoniden ayrımı son derece zordur.
Birkaç ailede PNKD lokusunun 2. kromozom üzerinde olduğu bildirilmiştir.
Egzersizin Neden Olduğu Paroksismal Diskinezi
(EPD): EPD uzamış
egzersiz sonrası ortaya çıkan paroksismal hareket bozukluğudur. 5-15 dakika
koşma veya yürüme, ekstremitelerin uzun süre pasif hareketi gibi egzersizler
istemsiz hareketleri ortaya çıkartabilir. Genellikle çocukluk çağında başlayan
hastalığın atak sıklığı ayda 1-5'den, günde 1-2'ye kadar değişebilir. Soğuk,
stres veya menstruasyon ile ataklar artabilir. İstemsiz hareketler esas olarak
bacağı veya ayağı tutar; ancak kollarda, yüz, boyun ve/veya gövdede olabilir.
Glukoz taşıyıcı tip 1 eksikliği sendromuna yol SLC2A1 mutasyonları mental retardasyon, epilepsi gibi pek
çok farklı klinik tablonun yanında EPD’ye neden olabilir.
Hemifasyal spazm: Gerçek anlamda bir distoni olmamakla birlikte fenomenolojik
benzerliğinden dolayı bu bölümde ele alınan hemifasyal spazm yüzün bir
yarısında görülen, sürekli ya da ara ara ortaya çıkan, semi-ritmik
kasılmalardır. Esas olarak fasyal sinirin çeşitli nedenlerle aşırı
uyarılmasından kaynaklanır. En fazla göz etrafında görülen kasılmalar sıklıkla
aynı taraf yanağa, üst-alt dudağa ve platismaya yayılır. Tek taraflı oluşu ve
yayılım özelliği nedeniyle blefarospazmdan ayrılır. Kasılmalar mimik
hareketleri sırasında artar, uykuda devam edebilir. Zamanla tutulan tarafta kas
zaafı ve kontraktür ortaya çıkabilir.
Semptomatik Distoniler
Serebrovasküler
hastalıklar, infeksiyonlar, demiyelinizan, metabolik ve doğumsal hastalıklar
ile travma, intoksikasyon gibi, bazal ganglia ve
bağlantılarını tutan her türlü merkezi sinir sistemi patolojisi semptomatik
distoniye yol açabilir. En sık rastlanılan semptomatik distoni nedenlerinden
biri olması ve tedavi edilebilir olması açısından Wilson hastalığından aşağıda
ayrıntılı olarak söz edilecek diğer semptomatik distoniler ise ilgili
bölümlerde ele alınacaktır.
Wilson Hastalığı (Hepatolentiküler Dejenerasyon)
Merkezi
sinir sistemini tutan kalıtımsal metabolik hastalıklar arasında en iyi
bilinenidir. 1912'de Kinnier Wilson tarafından tanımlanmıştır. Bu hastalık
bakır metabolizması bozukluğuna bağlıdır. Besinle alınan bakır birçok enzimatik
süreçte kullanılmakta ve gereksinimden fazlası, esas olarak karaciğerde
seruloplazmine bağlanarak safra yolları ile atılmaktadır. Wilson hastalığında
(WH) ihtiyaç fazlası bakırın atılımı bozulmuştur. Kesin olmamakla birlikte,
patofizyolojik mekanizmanın karaciğerde bakırın seruloplazmine bağlanmasını
sağlayan enzimin bozukluğuna dayandığı düşünülmektedir. Otozomal resesif geçen
hastalığın geni 13. kromozomun uzun kolundadır. Bu gen bakır taşıyan ATPaz
enzimini kodlamaktadır. Enzim eksikliğinde bakır seruloplazmine bağlanamamakta
ve karaciğerden atılamamaktadır. Dolayısıyla serumda düzeyi yükselen bakır
karaciğere, beyne, böbreğe çökmekte ve klinik belirti ve bulgulara yol açmaktadır.
Hastalık
genellikle 10-20 yaş arasında başlar, fakat daha geç yaşta başlayan olgular da
vardır. WH'de belirti ve bulgular serumda serbest dolaşan bakırın çöktüğü doku
ve organların işlevlerini bozması ile ortaya çıkar. En sık tutulan organ karaciğerdir.
Bu tutulum serum transaminazlarında hafif artıştan akut veya kronik hepatite
kadar değişik tablolara yol açabilir. Hastalarda bulantı, kusma, kilo kaybı,
çeşitli kanamalar görülebilir.
Nörolojik
tutulum hastaların %50'sinde ilk belirtidir. Bazal gangliaya çöken bakırın
yarattığı işlev bozukluğu sonucunda tremor, distoni, rijidite, akinezi gibi
değişik ekstrapiramidal bulgular ortaya çıkabilir. Kore, tik ve miyoklonus
nadir görülürken tremor en sık bulgudur. İstirahat, aksiyon veya postüral tremordan
her biri tek başına ya da bir arada bulunabilirler. Tremor ekstremitelerin hem
distalini hem de proksimalini tutabilir, amplitüdü değişkendir. Kolların
proksimalinde, yüksek amplitüdlü tremor olduğunda ya da kollar öne doğru
uzatılıp eller gerildiğinde ellerin bilekten ani olarak aşağıya düşmesi ve
tekrar aynı pozisyona dönmesinden dolayı hasta sanki kanat çırpıyormuş gibi
görünebilir (“wing-beating” veya “flapping” tremor). Yüz kaslarının distonik
tutulumuna bağlı olarak hastanın yüzünde sırıtır gibi bir görünüm vardır. Dil
ve farenks kaslarının tutulumu konuşma ve yutma bozukluklarına yol açar. Hasta
bazen tükrüğünü yutamaz, salyası ağız kenarından akar. Larenks kaslarının
tutulumu nedeniyle hasta fısıldar tarzda konuşur, anlaşılması çok zor olabilir.
Ekstremite ve aksiyal kaslardaki rijidite ve distoni, yürüyüşü olumsuz etkiler
ve postürü bozar. Bazen tipik parkinsonizm tablosu gelişebilir. Serebellar
sistem tutulumu hastaların %25'inde görülür, yürüme ve konuşma bozukluğuna
sebep olabilir veya katkıda bulunabilir. Nadiren epileptik nöbetler de tabloya
katılabilir. Somatik nörolojik belirtilerle hemen hemen eş zamanlı olarak
davranışsal sorunlar ve mental durum değişiklikleri de ortaya çıkar. Bu
değişiklikler çok hafif belirtilerle seyredebileceği gibi ciddi sorunlara da
yol açabilirler. Okul başarısının azalması çoğu hasta yakınının dikkatini çeken
ilk belirti olabilir. Davranış ve kişilik değişiklikleri, duygudurum
bozuklukları, psikotik belirtiler, dikkat, konsantrasyon
gibi zihinsel işlevlerde bozulma çeşitli derecelerde görülebilirler.
Serumda
dolaşan bakırın korneanın desement tabakasına çökmesi sonucu, korneada
kahverengi-yeşil bir halka görülür. Hastalık için patognomonik olan bu
bulguya Kayser-Fleischer halkası (KFH)
denir (Şekil 7a ve 4b). Bazen çıplak
gözle dahi farkedilebilecek kadar belirgin olabilirse de klasik bir göz
muayenesi ile çoğu kez saptanamaz ve gözün yarık-lamba (slit-lamp) ile
muayenesi gerekir. Klasik bilgi olarak nörolojik belirtiler ortaya çıktığında
KFH'nin mutlaka mevcut olduğu söylense de, bazen nörolojik tutulumlu bir Wilson
hastasında bu bulgu saptanmayabilir. Üriner, endokrin, kas-iskelet sistemleri
de tutulabilir ve bu sistemlere ait laboratuvar ve klinik bulgular ortaya
çıkabilir.
Şekil 7a ve b. Kayser-Fleischer Halkası (KFH). Wilson hastalığı tanısı konan
hastada çıplak gözle görülebilen KFH Şekil 4b’de KFH ok uçları ile işaretlenmiştir.
Çocukluk ve adölesan dönemde ekstrapiramidal sistem tutulumu ile
seyreden her türlü klinik nörolojik tabloda WH mutlaka araştırılmalıdır. Klinik
olarak WH şüphesi belirdiğinde, kesin tanı ve ayırıcı tanı için laboratuvar
incelemelerine başvurulur. Tipik bir tabloda serum seruloplazmin düzeyi düşük,
serumdaki serbest bakır düzeyi ise yüksek olarak bulunur. Serum total bakır
miktarı azalmış seruloplazmin miktarına bağlı olarak sıklıkla düşük bulunur.
Ancak her iki parametrenin de normal olduğu olgular vardır. Hastaların hemen
tümünde 24 saatlik idrarda bakır atılımı artmıştır; bu bulgu diğer ikisine göre
daha duyarlı bir ölçümdür. Korneada KFH'nin saptanması, WH de dahil olmak üzere bakır metabolizma bozukluğunu gösteren
önemli bir bulgudur. Karaciğer fonksiyon testleri bozuk bulunabilir. Ancak
kesin tanı karaciğer biyopsisi ile karaciğer kuru dokusunda bakır miktarının
artmış bulunması ile konulur. Klinik olarak WH düşünüldüğünde, diğer olası
tanılar ekarte edilmişse, kan-idrar bulguları normal olsa bile karaciğer
biyopsisi incelemesinde ısrarlı davranılmalıdır. Yardımcı inceleme
yöntemlerinden bilgisayarlı tomografi ve manyetik
rezonans görüntülemesinde bazal ganglia lokalizasyonuna uyan bölgelerde sinyal
değişiklikleri, nekroz saptanabilir. WH tanısı konan hastaların aile
bireylerinin de hastalık açısından taranması gerekir.
WH’de
tedavinin ana hedefi vücuttaa fazla miktarda bulunan bakırın
uzaklaştırılması/atılması ilkesine dayanır. Bunun için bakır içeren fındık,
ceviz, mantar, kabuklu deniz ürünleri gibi besinler diyetten çıkarılmalıdır.
Çinko, bağırsaklardan bakır emilimini azaltmak amacıyla kullanılır, direkt
etkisi olmayıp, bakırın bağırsak epitel hücrelerinde bağlanıp daha sonra gaita
ile atılımını sağlayan metallothion'in sentezini artırarak etki göstermektedir.
WH'de en sık kullanılan şelatör ilaçlar D-penisilamin ve trientin'dir. Bu
maddeler serumda dolaşan ve dokulara çökmüş halde bulunan bakıra bağlanır,
oluşan penisilamin-bakır kompleksi idrarla atılır.
Tedaviye başladıktan sonra hastaların bir kısmında nörolojik bulgularda bozulma
görülebilir. Kesin nedeni bilinmemekle birlikte bu bozulmanın dokulardan
çözülen bakırın kana geçmesi, oradan da merkezi sinir sistemine geçerek yeniden
çökmesine bağlı olduğu düşünülmektedir. Bu hastaların yarısı eski haline
dönmekte, yarısında ise düzelme olmamaktadır. Bu yüzden hasta ilaç başlanmadan
önce olası bir kötüleşme hakkında mutlaka bilgilendirilmeli ve tercihen
hastanın ilk birkaç haftayı hastanede geçirmesi sağlanmalıdır. Wilson hastalığı
fulminan hepatite yol açmışsa karaciğer transplantasyonu
denenebilecek bir tedavi seçeneğidir; ancak bu evredeki bir hastada mortalite
oranı çok yüksektir.
Semptomatik
distonilerde tedavi nedene yöneliktir. İdyopatik distonilerde ise distoninin
tipine göre antikolinerjik, antidopaminerjik, baklofen, benzodiazepin grubu
ilaçlar ve botulinum toksini kullanılmaktadır. Genelde jeneralize distonilerde
ilaç, fokal distonilerde ise botulinum toksini ilk tercih olarak seçilmektedir.
İdyopatik jeneralize distonilerde derin beyin stimülasyonu ameliyatları da
önemli bir seçenektir.
Son
yıllarda rutin kullanımı yerleşen ve özellikle fokal distoni tedavisinde bir
çığır açan botulinum toksini etkisini nöromüsküler ileti bloğu yaratarak
gösterir. Toksin distonik kasılmadan sorumlu olan kas içine enjekte edilmekte,
etkisi 1-2 haftada başlayıp 3-6 ay sürmekte ve daha sonra uygulama
tekrarlanmaktadır. Diğer ilaçlardan yarar görmeyen jeneralize distonilerde ya
da ağız içi/larinks gibi kritik bölgelere yapılacak uygulamalarda en aktif kas
EMG ile saptanmalı ve o kasa yahut kaslara botulinum toksini yapılmalıdır. En
önemli yan etkisi toksinin komşu kaslara yayılarak geçici zaaf oluşturmasıdır.
Tekrarlanan uygulamalarda, nadir de olsa toksine karşı oluşan antikorlar
nedeniyle etkinlik azalabilir.
Özellikle
çocukluk çağında ve alt ekstremitelerde başlayan distonilerde ilk düşünülmesi
gereken ilaç L-dopa preparatlarıdır. Bu tip hastalarda dopaya yanıtlı distoniyi
dışlamak için mutlaka L-dopa ile bir tedavi denemesi yapılmalıdır. Bunun
dışında antikolinerjik ilaçlar jeneralize distoni tedavisinde ilk tercihi
oluştururlar. En sık kullanılan antikolinerjikler; triheksifenidil,
bornaprin ve biperidendir. Düşük dozda başlayıp, alınan yanıta göre
doz artırılabilir. Bu grup ilaçların distonideki etki mekanizmaları tam olarak
bilinmemektedir. Tek başına ya da diğer ilaçlarla birlikte kullanılabilirler.
Uykuya eğilim, ağız kuruluğu, görme bulanıklığı ve idrar yapmada zorluk en sık
yan etkileridir. Özellikle yaşlı hastalarda konfüzyonel duruma da yol
açabilirler. Benzodiazepinler merkezi sinir sisteminde inhibitör bir
nörotransmiter olan GABA'nın etkisini artırırlar. En sık kullanılan
benzodiazepinler klonazepam ve diazepamdır. Sedasyon en önemli yan etkileridir.
GABA sistemi üzerinden etkili bir başka ilaç olan baklofen benzer şekilde
nöronal eksitabiliteyi azaltır. Baklofen genellikle ekstremite distonilerinde
tercih edilir. Yorgunluk, kas zayıflığı ve sedasyon en sık rastlanan yan
etkileridir. Klinik etkilerini antidopaminerjik bir mekanizmayla gösteren
klasik nöroleptiklerin hemen hepsinin distoniye neden olma olasılığı
bulunmasına rağmen, paradoksal olarak bazı distonilerde bu grup ilaçlar yararlı
olmaktadır. Son yıllarda geliştirilen ve düşük ekstrapiramidal yan etki profilleri nedeniyle atipik nöroleptik diye adlandırılan
klozapin, olanzapin gibi ilaçlar diğer ilaç tedavilerine dirençli fokal ya da
jeneralize distonilerde kullanılmakta ve bazen başarılı sonuçlar alınmaktadır.
Tetrabenazin de gerek fokal gerekse jeneralize distonilerde, dopamin antagonisti olarak kullanılan bir diğer ilaçtır.
Derin
beyin stimülasyon (DBS) ameliyatları jeneralize distonilerde mutlaka akla
gelmelidir. Özellikle DYT1 mutasyonuna sahip olan hastalarda dramatik iyileşme
olan vakalar bildirilmiştir. Distoni için DBS’de hedef Globus pallidus
interna’dır.
Paroksismal
diskinezilerin tedavisinde genellikle ilk seçenek antiepileptiklerdir
(karbamazepin, valproat, fenitoin, benzodiazepinler, vb.). Antiepileptiklere
cevapsız vakalarda karbonik anhidraz inhibitörleri (asetazolamid), L-dopa
ve nöroleptikler denenebilir.
Kore
aritmik, hızlı, sıçrayıcı veya akıcı, basit veya kompleks
özellikte genellikle ekstremitelerin distalini tutan küçük amplitüdlü istemsiz
hareketler olarak tanımlanır. Eğer bu istemsiz hareketin amplitüdü büyükse ve
ekstremitenin proksimalini tutuyorsa buna ballizm adı verilir. Koreik ve
ballistik hareketler bazen bir arada bulunurlar veya bir hastalığın seyri
esnasında birbirlerini izleyebilirler.
Koreik
hareketler bazen çok belirsiz olabilir ve özellikle çocuklarda normal
hareketlerle içiçe geçebilirler. Anormal solunum sesleri, sırıtma ve
ekstremitelerde hipotoni koreye eşlik edebilir. Ballizm çoğunlukla hemoraji
veya infarkt gibi serebrovasküler hastalıklar sonucu ortaya çıkar. Ballizm
vücudun bir yarısına sınırlıysa hemiballizm, her iki vücut yarısında
görülüyorsa biballizm, bir ekstremiteye sınırlıysa monoballizm adını alır.
Koreik ve
ballistik hareketler için putamen, globus pallidus ve subtalamik nukleus kritik
yapılardır; ancak ballistik hareketler daha çok subtalamik nukleustaki patoloji
sonucu ortaya çıkarlar.
Kore-ballizmi
altta yatan sebebin niteliğine göre primer ve sekonder olmak üzere iki ana
gruba ayırmak mümkündür. Primer kore-ballizm doğrudan beyni tutan
nörodejeneratif ya da herediter hastalıklar sonucu ortaya çıkar. Buna karşın
sekonder kore-ballizm beyni ikincil olarak etkileyen bozukluklar ya da
hastalıklar sonucu ortaya çıkar ve altında pek çok neden yatabilir. Aşağıda
kore-ballizme sebep olabilecek hastalıklar liste halinde verilmiştir (Tablo 6).
Tablo 6. Kore-ballizm yapabilen hastalıklar
|
Primer Kore-Ballizm Huntington benzeri hastalık
1,2,3,4 Striatodentat kalsinozis Mc Leod sendromu Ataksi telanjiektazi Wilson hastalığı Nöroferritinopati Selim kalıtsal kore Beyin demir birikimi Tip I Nöronal lipofüksinozis Pantotenat kinazla ilişkili kore |
Sekonder Kore-Ballizm Henoch-Schönlein purpurası Behçet hastalığı -Yapısal nedenler Arteriyovenöz malformasyon Hepatik ensefalopati, hepatoserebral dejenerasyon Porfiri Kernikterus -Toksinler ve İlaçlar Talyum Toluen Antikonvülzanlar Steroidler Digoksin -İnfeksiyonlar
ve diğer nedenler Difteri Lyme hastalığı Creuzfeldt-Jacob hastalığı Sarkoidoz Serebral palsi |
Aşağıda
primer ve sekonder koreye neden olan hastalıklardan göreceli olarak sık
görülenler ele alınacaktır.
Huntington Hastalığı
Huntington
hastalığı (HH) veya Huntington koresi genelde korenin baskın olduğu hareket
bozukluğu (kore, distoni, parkinsonizm), psikiyatrik bulgular ve demansın eşlik
ettiği herediter nörodejeneratif bir hastalıktır. Hastalık 4. kromozomun kısa
kolundaki huntingtin proteinini kodlayan IT-15 veya huntingtin geninin mutasyonu sonucu olur. HH otozomal
dominant geçişlidir ve çok büyük oranda ailevidir; yeni mutasyonlar sonucu
sporadik vakaların ortaya çıkması çok nadirdir. HH çoğunlukla 4. veya 5.
onyılda başlar ve kadın-erkek eşit olarak etkilenir. Hastalık yaklaşık %10
oranında 20 yaşından önce (jüvenil HH) ve %10 oranında da 60 yaşından sonra
ortaya çıkar.
HH motor,
psikiyatrik ve kognitif tutulum bulguları ile seyreder. Erişkin başlangıçlı HH
genellikle hafif motor koordinasyon güçlüğü, beceriksizlik veya yerinde
duramama ile başlar. Daha sonra belirgin koreik hareketler klinik tabloya hakim olur. Kore hastalığın en belirgin bulgusu olmasına
rağmen, parkinsonizm bulguları da hastaların hemen hemen hepsinde görülür.
Zamanla hastalar akinetik-rijid hale gelirler ve distonik postür ortaya
çıkabilir. Yürümede güçlük ve düşmeler sıktır. Okülomotor bulgular HH'nın erken
belirtisi olabilir ve hareket bozukluğu olmadan nörolojik muayenede
bulunabilir. Sakkadik göz hareketlerinde yavaşlama, göz hareketine başlamada
güçlük ve yavaş izleme hareketinde duraksamalar başlıca okülomotor bulgulardır.
Dizartri ve disfaji hastalığın ileri basamaklarında görülür, anartriye kadar
ilerleyebilir ve aspirasyona sebep olabilir. Terminal dönemde hasta yatağa
bağımlı hale gelir. Ölüm aspirasyon pnömonisinden veya diğer tıbbi komplikasyonlardan olur. Bulguların ortaya çıkmasından sonra
ölüme kadar geçen ortalama yaşam süresi genellikle 15-20 yıl arasındadır.
Kognitif
ve psikiyatrik bulgular hareket bozukluğundan önce görülebilir. Kognitif
bozukluk ilk olarak sıklıkla hastanın mental esnekliğinde, reaksiyon hızında
azalma ile ortaya çıkar ve ön planda frontal işlev bozukluğu ile
karakterizedir. Hastalık ilerledikçe global bir mental
yıkım, dolayısıyla demans tablosu gelişir. HH'nda en sık görülen psikiyatrik
bozukluk depresyondur. Bundan başka mani, obsesif-kompülsif
bozukluk, iritabilite, anksiyete, ajitasyon, impulsivite ve apati olabilir.
Hastalarda intihara da sık rastlanır.
Jüvenil
başlangıçlı HH (Westphal varyantı)
erişkin başlangıçlı hastalıktan birkaç özelliği ile ayrılır. Jüvenil
başlangıçlı hastalarda görülen hareket bozukluğu genellikle koreden çok
parkinsonizmdir. Kore hastalığın seyrinde hiç görülmeyebilir. Hastalığın
başlangıç bulguları sıklıkla dikkat güçlüğü, davranış ve kişilik değişikliği,
okul performansında düşme, distoni, bradikinezi ve bazen de tremordur.
Epileptik ataklar ve hızlı seyirli demans da görülebilir. Bu varyantın ölüme
kadar geçen hastalık süresi erişkin vakalardan daha kısadır.
HH’de en
belirgin patoloji kaudat nukleus ve putamende olan atrofidir. Daha az oranda
globus pallidus, korteks, talamus ve subtalamusta da atrofi görülür.
Mikroskopik olarak korteks, kaudat nukleus ve putamende nöron kaybı vardır ve
buna gliozis eşlik eder. Striatumdaki orta büyüklükteki dikensi nöronlarda
büyük oranda kayıp gözlenir. Nörogörüntülemede ilerleyici korteks ve kaudat
nukleus atrofisi göze çarpar.
HH’ye
neden olan 4. kromozomun kısa kolundaki mutasyon, huntingtin
geninde normalden fazla CAG trinükleotid tekrarına ve bunun sonucunda da
huntingtin proteinindeki glutamin miktarında artışa neden olur. CAG tekrarı
40'ın üzerinde olursa HH ortaya çıkar. CAG tekrar sayısı 30-35 arasında ise
premutasyon, 36-39 arasında ise düşük penetranslı hastalık söz konusu olur. Patolojik
CAG tekrar sayısı normal sınıra yaklaştıkça, hastalığın ortaya çıkış süresi de
genellikle uzar. Huntingtin geninin ürünü olan huntingtin proteininin görevi
henüz bilinmemektedir. HH’nin kesin tanısı huntingtin
genindeki CAG tekrar sayısındaki artışın gösterilmesi ile konur. Asemptomatik
bireyler için prediktif ve prenatal tanı da mümkündür.
HH'nin
tedavisi günümüzde sadece semptomatiktir. Korenin tedavisi için dopamin
reseptör blokerleri (haloperidol, pimozid, flufenazin, klozapin, olanzapin,
ketiapin, risperidon vb.), benzodiazepinler, dopamin depolarını boşaltan
ilaçlar (rezerpin, tetrabenazin), valproat kullanılabilir.
Dopaminerjik nörotransmisyonu azaltan ilaçlar hastalığın ileri evrelerinde ve
jüvenil başlangıçlı hastalarda parkinsonizmi arttırabileceğinden çok dikkatli
kullanılmalıdır. Hastada bradikinezi ve rijidite ağırlıklı ise dopamin yararlı
olabilir ancak dopaminin koreyi arttırabileceği de unutulmamalıdır. Son
yıllarda diğer ilaçların etkisiz kaldığı koreik-diskinetik hastalarda derin beyin
stimülasyonunun yararlı olduğuna ilişkin yayınlar bulunmaktadır. Depresif semptomlar için antidepresanlar kullanılabilir.
Sydenham Koresi (Romatik kore, St. Vitus Dansı, Kore
Minör)
Sydenham
koresi (SK) akut romatizmal ateşin majör kriterlerinden
biridir. Hastalık A grubu streptokokların neden olduğu farenjit epizodundan sonra haftalar içinde gelişir. Bu süre
infeksiyonun başlangıcından 6 ay sonraya kadar uzayabilir ve bazı hastalarda
infeksiyon öyküsü alınmayabilir. SK genellikle 5-15 yaş arasında başlar, ancak
erişkin yaşlarda da görülebilir. Kızlarda erkeklere göre iki kat daha sıktır.
Obsesif-kompülsif bozukluk, iritabilite, öğrenme güçlüğü ve hiperaktivite sık
başlangıç bulgularıdır. Bu bulgular koreden önce ortaya çıkabilir, nadiren
ensefalopati ve konfüzyon da görülebilir, dizartri, basit vokal tikler tabloya
eklenebilir. Kore ani veya basamaksal olarak başlar ve hızlıca jeneralize olma
eğilimindedir; ancak hastaların %20'sinde hemikore olarak kalabilir. Kas tonusu
genellikle azalmıştır ve bu bazen ciddi olarak hastayı yatağa bağlayabilir.
Korenin en şiddetli olduğu dönem olan 3-6 ay geçtikten sonra hastalık kendi
kendini sınırlar ve 6-9 ay içinde düzelme eğilimi gösterirse de nadir olarak
kore daha uzun sürebilir. Bazı çocuklarda hatta erişkin bireylerde tekrarlayan
kore atakları olabilir. Gebelik, oral kontraseptif kullanımı ve yeni
streptokokal infeksiyonlar atakları tetikleyebilir.
SK’nın
patofizyolojisinde otoimmün mekanizmaların rol oynadığı düşünülmektedir.
Otoimmün hipoteze göre A grubu streptokoklara karşı yapılan antikorlar,
muhtemelen genetik olarak duyarlı kişilerde subtalamus ve kaudat nukleustaki
nöronlarla çapraz antikor reaksiyonu oluştururlar. Bazı hastaların
kortikosteroidler, plazmaferez ve immünglobulinlerden yararlanması bu hipotezi
desteklemektedir.
Streptokok
infeksiyonunu araştırmaya yönelik tetkikler (antistreptolizin-O, diğer
antistreptokokal antikorlar, A grubu streptokoklar için pozitif boğaz kültürü,
lökositoz) infeksiyonu göstermek için yararlıdır. Ancak kore genellikle
infeksiyonu takiben, bazen de uzun süre sonra ortaya çıktığı için bu tetkikler
normal bulunabilir. BOS analizi nadiren görülen hafif lenfositoz dışında
normaldir.
Streptokok
infeksiyonlarının çocuklarda kore olmadan da tikler, obsesif-kompülsif
bozukluk ve diğer psikiyatrik bozukluklara yol açabildiği düşünülmektedir.
Syndenham koresinin tedavisinde valproik asit ve nöroleptikler kullanılabilir.
Kore hafif ise tedavi gerekmez. Ağır ve ilaçlara dirençli vakalarda
kortikosteroidler, intravenöz immünglobulin ve plazmaferez gibi immünsupresif
tedavilerin etkili olduğu bildirilmiştir. Aktif streptokokal infeksiyon
yirmibir yaşına kadar penisilinle tedavi edilmelidir.
Nöroakantositozis
Nöroakantositozis,
kore, distoni, parkinsonizm, oromandibuler diskineziler, motor ve vokal tikler
gibi zengin nörolojik bulguların eşlik ettiği nadir nörodejeneratif bir
hastalıktır. Çoğunlukla otozomal resesif geçişle uyumlu aile öyküsü vardır.
Hastalık genellikle 3. veya 4. onyılda başlar ancak her yaşta görülebilir.
Ekstremitelerdeki kore ve orofasyal diskineziler en göze çarpan klinik
özelliklerdir. Vakaların üçte birinde nöbetler ve yarısında demans vardır.
Periferik aksonal nöropati ve amiyotrofi olabilir. Hastalarda depresyon, anksiyete,
paranoid delüzyonlar, obsesif kişilik, kişilik
değişiklikleri, apati ve impulsivite gibi psikiyatrik bulgular görülebilir.
Yukarıda
bahsedilen klinik bulgularla beraber periferik yaymadaki akantosit sayısının
artışına bağlı olarak tanı koyulur. Hastaların büyük çoğunluğunda kas enzimleri
artmıştır. Postmortem incelemelerde kaudat nukleus ve putamende belirgin
atrofi, gliozis ve nöron kaybı görülür. Ayrıca periferik sinirlerde aksonal dejenerasyon vardır. Hastalığa VPS13A genindeki
mutasyon neden olur. Klinik özellikler, uygun aile öyküsü ve periferik yaymada
artmış akantositlerin görülmesi nöroakantositozu düşündürür. Western blot ile
eritrosit membranında korein proteinin olmadığının gösterilmesi kesin tanıyı
koydurmaktadır. Nöroakantositozisin kesin bir tedavisi yoktur. Hareket
bozukluklarına ve psikiyatrik bulgulara semptomatik olarak yaklaşılabilir.
İyi Huylu Herediter Kore
İyi huylu
herediter kore otozomal dominant geçişli nadir bir hastalıktır. Genellikle
erken çocukluk çağında başlar. Çoğunlukla koreik hareketlerden başka nörolojik
bulgu yoktur, bazen distoni eklenebilir ve tanımı itibarıyla hastalık
ilerleyici değildir. Bazen kore erişkin hayatta tamamıyla düzelir ve çoğunlukla
tedaviye ihtiyaç göstermez.
Gebelik Koresi (Korea Gravidarum)
Gebelik
esnasında ortaya çıkan koredir. Hastaların üçte birinden fazlasında geçirilmiş
Sydenham koresi öyküsü bulunur. Koreye, gebelikteki progesteron ve östrojen
düzeylerindeki yükselmeler sonucu bazal ganglia metabolizmasında olan
değişikliklerin rol açtığı düşünülmektedir. Benzer şekilde kore, östrojen veya
progesteron alan kadınlarda da ortaya çıkabilir. Gebelik koresi nadiren fetusu
da etkiler ve genellikle doğumdan sonra antikoreik ilaç kullanmaya gerek
olmadan düzelir.
Sistemik Lupus Eritematozus ve Antifosfolipid Sendromu
Sistemik
lupus eritematozuslu (SLE) hastaların % 5'inden azında kore görülür. Kore,
SLE'nin tek ve ilk bulgusu olabilir. SLE tanısı korenin başlangıcından yıllar
sonra konabilir. Kore genellikle hastalığın erken evrelerinde ortaya çıkar ve
hemikore daha sık görülür. Korenin ortaya çıkış nedeni bilinmemektedir. SLE’ye
eşlik eden kore steroidlere cevap verebilir. Benzer şekilde antifosfolipid
antikor sendromunda da kore görülebilir.
Kore-Ballizm Tedavisi
Kore-ballizmin
tedavisi esas nedene yönelik olarak yapılmalıdır. Eğer hareket bozukluğu günlük
yaşam aktivitelerinde kısıtlanmaya neden oluyor ya da yaşam kalitesini
etkiliyorsa ilaç tedavisi ile kore-ballizm baskılanabilir. Yan etkiler göz
önünde tutularak ilaç tedavisine uzun süre devam etmekten kaçınılmalıdır.
Koreik ve
ballistik hareketlerin semptomatik tedavisi temelde dopaminerjik
nörotransmisyonun azaltılmasına dayanır. Dopamin reseptör blokerleri
(haloperidol, pimozid, flufenazin, olanzapin, risperidon, ketiapin, klozapin)
ve presinaptik dopamin depolarını boşaltan ilaçlar (tetrabenazin, rezerpin) bu
grupta yer alırlar. Bunlardan başka benzodiazepinler (klonazepam, diazepam,
oksazepam) ve antikonvülzanlar (fenitoin, karbamazepin, valproik asit) tedavide
kullanılabilir. Ballizm ilaç tedavisine oldukça dirençli olabilir. Dirençli
vakalarda talamotomi yahut derin beyin stimülasyonu gibi cerrahi girişimler
düşünülebilir.
TREMOR
Tremor bir
vücut parçasını hareket ettiren resiprokal kasların alternan veya senkron kasılması sonucu oluşan istemsiz, ritmik
osilasyonlardır. Tremor, etkilenen alana (baş, çene, dirsek, vokal kordlar, alt
veya üst ekstremite, vücut), hareketle ilişkisine (istirahatte, postüral,
aksiyonel, intansiyonel), frekansına (düşük: 4 Hz'den az; orta: 4-7 Hz; yüksek
7 Hz'den fazla) ve genliğine (ince, kaba) göre tanımlanır. Bu özelliklerin
saptanması, özellikle de tremorun hareket ve postürle bağlantısı etyolojik
tanıya yönelmek açısından önemlidir.
Aşağıda
hareket ve postüre bağımlılığı yönünden tanımlanan tremor çeşitleri, bunların
ortaya çıktığı durumlar tarif edilecek ve nispeten sık görülen tremor nedenleri
kısaca özetlenecektir.
Hareket ve Postüre Bağımlılığı Açısından Tremor Çeşitleri
İstirahat Tremoru
Etkilenen
bölgede herhangi bir istemli kas kasılması, istemli hareket girişimi ya da
yerçekiminin etkisi olmadan vücudun bir parçasında görülen tremordur. Tremor
hareket esnasında kaybolur, ekstremiteyi yeni bir pozisyonda sabit tutunca
yeniden başlar. En sık ellerde ve ekstremitelerin distal kısımlarında görülür.
Hareketin niteliğinden dolayı bazen para sayma ya da hap yapma tremoru olarak
da adlandırılır. İstirahat tremoru kural olarak Parkinson hastalığı ve
parkinsonizme neden olan diğer durumlarda ortaya çıkar. Tedavide en yararlı
ilaçlar antikolinerjikler, L-dopa preparatları ve dopamin agonistleridir.
Cerrahi girişimlerden talamus veya subtalamusa yönelik derin beyin stimülasyonu
veya lezyon uygulanması tremora karşı oldukça etkilidir.
Postüral Tremor
Kişi
istemli olarak yerçekimine karşı koyduğunda, örneğin kollarını öne doğru uzatıp
ellerini gerdiğinde ortaya çıkar, o postür korunduğu sürece de devam eder.
Frekansı ve genliği altta yatan nedene göre değişir. Aşağıda detaylı olarak ele
alınacak olan esansiyel tremor hastalığı için tipiktir.
Aksiyon Tremoru (Kinetik Tremor)
Bir
ekstremiteyi aktif olarak kullanırken, hareket esnasında ortaya çıkar. Genelde
yüksek frekanslıdır ancak genliği az ya da çok olabilir, genlik hareket
esnasında hep aynı kalır. Postüral tremor gibi aksiyon tremoru da en sık
esansiyel tremor hastalığında görülür.
İntansiyon Tremoru
Bu tip
tremorun en büyük özelliği istemli hareket esnasında ortaya çıkması ve hedefe
yaklaşırken tremorun genliğinin çok artmasıdır. Örneğin parmak-burun testinde
hasta parmağını burnuna yaklaştırdıkça tremorun genliği artar ve hasta hedefi
bulamaz. İntansiyon tremoru düşük frekanslı, kaba bir tremordur ve kural olarak
serebellum ve bağlantılarının tutulması sonucu ortaya çıkar. Dizartri ve
nistagmus tremora eşlik edebilir. Tedavisi oldukça güçtür. İzoniazid veya
talamotomi yararlı olabilir.
Rubral Tremor (Ortabeyin ya da Holmes tremoru)
Rubral
tremorun özelliği istirahatte görülmesi ve hareketle belirgin olarak
artmasıdır. Bu özelliğiyle rubral tremor istirahat, postüral ve aksiyon tremorunun
bir kombinasyonudur. Frekansı düşük, kaba, düzensiz
bir tremordur ve proksimal kaslar distal kaslardan daha fazla etkilenir. Rubral
tremor orta beynin tegmentum bölgesindeki nukleus ruber veya bağlantılarının
hasarı sonucu ortaya çıkar, en sık multipl skleroz ve serebrovasküler
hastalıklarda görülür. Tremora ek olarak hemen daima hemiparezi, kranyal sinir
felci gibi diğer orta beyin hasarı bulguları vardır. Rubral tremorun tedavisi
oldukça güçtür. L-dopa, izoniazid, klonazepam, klozapin ve talamusa yönelik DBS
veya lezyon ameliyatları tedavi seçenekleri arasındadır.
Etyoloji ve Klinik Belirtilere Göre Tremor
Fizyolojik Tremor
Fizyolojik
tremor parmakların veya kolların aktif innervasyonu sonucu, normal kas
kasılmasına bağlı olarak tüm insanlarda ortaya çıkan, hızlı (10-12 Hz), düşük
genlikli postüral bir tremordur, aksiyonda ve istirahatte görülmez ve patolojik
bir anlam taşımaz. Mikrocerrahi, saat tamirciliği gibi ince işlerle
uğraşanlarda daha çok göze çarpar. Artmış fizyolojik tremor fizyolojik tremorla
aynı frekanstadır ancak genliği daha büyüktür. Mental nedenler (anksiyete,
stres, yorgunluk), metabolik nedenler (ateş, tirotoksikoz, hipoglisemi),
ilaçlar (nöroleptikler, beta adrenerjik agonistler, valproik asit, lityum,
steroidler, dopamin agonistleri, antidepresanlar), alkol yoksunluğu, toksinler
(cıva, kurşun, arsenik) ve bazı gıda maddeleri (kafein) artmış fizyolojik
tremora yol açabilir.
Esansiyel Tremor
En sık
rastlanılan hareket bozukluğudur. Esansiyel tremor (ET) genellikle asimetrik ve
postüral bir tremor olarak başlar, zamanla karşı tarafa yayılır. Tremor sadece
özel bir pozisyonda ortaya çıkabilir. Pozisyonun açısı değişince tremorun
şiddeti de farklılaşabilir. Kural olarak aksiyonla artıp istirahatte geçer,
ancak nadiren istirahatte de görülebilir. ET frekansı genelde hızlıdır, ancak
4-12 Hz aralığında farklı frekanslar görülebilir. ET herhangi bir vücut
parçasında olabilir (eller, baş, bacaklar, ses telleri). Elde baskın olunca
yeme, içme ve yazı yazma bozulabilir.
Hastalığın
tanısı bazen tesadüfen konulur veya hasta tremorun yarattığı mekanik ve sosyal
kısıtılık artınca doktora başvurur. Tremor dışındaki nörolojik muayene sıklıkla
normaldir. Bazı olgularda hafif ekstrapiramidal sistem bulguları ile yürütücü
işlev, dikkat, görsel mekansal işlev bozuklukları
saptanabilir. Hastaların yarısından fazlasında penetransı değişken aile öyküsü
vardır. Hastalık her yaşta olabilir fakat sıklığı yaşla beraber artar. Ailevi
vakalar genellikle erken yaşta başlar. Çoğu zaman tremor iyi huyludur, yavaş
ilerler, dalgalanabilir ve yaşam süresini kısaltmaz. Hastalar baskın ellerini
değiştirmek zorunda kalabilirler. Hastalığın ileri evrelerinde istemli
hareketler aşırı titremeden dolayı oldukça kısıtlanabilir. ET’nin önemli
ayırıcı özelliklerinden biri vakaların çoğunda alkolün kısa süreli olumlu
etkisidir. Yorgunluk, kafein, sinir sistemi stimülanları, artmış katekolamin
düzeyleri, ısı ve emosyon değişiklikleri, heyecanlanma, sinirlenme, stres,
topluluk içinde olma tremoru fazlalaştırabilir. Uykuda tremor kesilir. ET bazen
bradikinezi, rijidite ve istirahat tremoru gibi Parkinson hastalığı
bulgularıyla birlikte görülür. Bu durumda her iki hastalığın birlikte
bulunduğunu düşünmek gerekir. ET’nin patofizyolojisi bilinmemektedir. Şimdiye
kadar santral ve periferik mekanizmalar suçlanmıştır.
ET’nin
kesin tedavisi yoktur. Ağır bileklik ve su içerken ağır bardak kullanma gibi
yöntemler tremoru azaltarak günlük yaşam aktivitelerinde iyilik sağlayabilir.
Tedavide ilk tercih edilen ilaçlar beta-adrenerjik blokerlerdir. Bu grupta en
sık propranolol kullanılır ve hastaların yarısından fazlasında semptomatik
yarar sağlar. Dramatik düzelme az oranda görülür. Hastaların bir kısmı bu ilaca
cevapsızdır. Propranolol etkisine karşı zamanla direnç gelişebilir. Kalp
yetmezliği, A-V blok, astım ve göreceli olarak diyabette kontrendikedir. Yan
etkileri başağrısı, sedasyon, yorgunluk, depresyon ve baş dönmesidir. ET
tedavisinde ikinci seçilecek ilaç primidondur. Primidon, propranololdan daha
etkili bir ilaç olmasına rağmen başlangıçtaki akut yan etkileri ve yüksek
dozlarda gelişen ilaç intoleransı nedeniyle kullanımı sınırlıdır. Yan etkileri
bulantı, kusma, ataksi ve sedasyondur. İlaca kural olarak çok düşük dozlarda
başlanmalıdır. Primidonun etkisi zaman içinde kaybolabilir. Propranolol ve
primodondan başka pek çok ilaç ET tedavisinde denenmiştir. Benzodiazepinler
(klonazepam, diazepam), karbonik anhidraz inhibitörleri (asetozolamid),
antikonvülzanlar (gabapentin, fenobarbital, topiramat, zonisamid), trazodon,
flunarizin, amantadin ve atipik nöroleptikler (klozapin, olanzapin) bunlar
arasında sayılabilir. Ancak yukarıda sayılan ilaçlarla kontrollü çalışma sayısı
çok azdır ve etkileri hakkında sonuçlar değişkendir. Botulinum toksini bazı el,
ses ve baş tremorlarında etkilidir.
İlaç
tedavisine cevapsız vakalarda cerrahi girişim uygulanabilir. ET’nin cerrahi
tedavisinde hedef olarak talamusun ventral intermedyal nukleusu veya ventral
anterior nukleusu seçilir. Son yıllarda DBS ameliyatları komplikasyon
oranlarının daha az olması nedeniyle tercih edilmektedir. Bu yöntem talamotomi
ile karşılaştırıldığında kalıcı nöronal hasar yapmaması ve az komplikasyon oranıyla daha avantajlıdır.
Ortostatik Tremor
Ortostatik
tremor hasta ayağa kalktıktan birkaç saniye sonra bacaklarda görülen, vücudun
sallanmasına yol açan bir tremor çeşididir. Hasta ayakta durduğu müddetçe devam
eder, hastanın düşmesine yol açabilir. Otururken veya destek alındığında tremor
ortaya çıkmaz, yürürken azalır. Ortostatik tremor genellikle 6.-7. onyıllarda
görülür ve çok hızlı frekanstadır (15-16 Hz). Klonazepam bazen de propranolol
ortostatik tremorda etkilidir. Bazı vakalarda gabapentinin etkinliği
bildirilmiştir.
Primer Yazıcı Tremoru
Sadece
yazı yazarken ellerde ortaya çıkan pronasyon-supinasyon şeklinde 5-6 Hz.
frekanslı bir tremordur. Esansiyel veya distonik tremorun bir alt tipi olduğu
düşünülmektedir. Tremora bazen distoni de eşlik edebilir. Sadece belirli bir
eylem esnasında ortaya çıkan başka tremorlar da olabilir (piyano çalarken,
raket tutarken vb.). Primer yazma tremorunda propranolol gibi klasik medikal
tedavilerden başka botulinum toksini de faydalı olabilir.
İzole Ses Tremoru
Tremor
sadece sese sınırlıdır, konuşurken ses titremesi şeklinde ortaya çıkar. Bazen larengeal
spazmodik distoni veya esansiyel tremor bağlamında görülebilir.
İzole Çene Tremoru (Geniospazm)
Otozomal
dominant geçişli sadece çenede tremor ile seyreden bir hastalıktır. İzole çene
tremorunun prognozu iyidir ve ileri yaşlarda spontan düzelebilir.
Distonik Tremor
Hızlı
distonik kasılmalar sonucu ortaya çıkan, sıklıkla distoniye ait
bükücü-döndürücü postür gibi diğer bulguların da eşlik ettiği bir tremor
çeşididir. Tutulan vücut parçası istemli olarak distoninin zorla çevirdiği
tarafa doğru döndürülürse tremor azalır ya da kaybolabilir. İstirahatte
görülebilir, hareketle artma olabilir. Bu tür tremorda esas olarak distoni
tedavisine yönelik tedaviler kullanılmaktadır.
Periferik Nöropati ve Tremor
Periferik
nöropatilere tremor eşlik edebilir. Tremor herediter nöropatilerde nispeten sık
görülmesine rağmen her çeşit nöropatide ortaya çıkabilir. Periferik
nöropatilerde görülen tremor düzensiz veya ritmik, proksimal veya distal
yerleşimli olabilir. Mekanizmasının ekstremitedeki güçsüzlük sonucu artmış
fizyolojik tremora ikincil veya bozulmuş gerilme refleksi ile ilişkili olduğu
düşünülmektedir.
Palatal Tremor (Palatal Miyoklonus): Yumuşak damağın istemsiz hareketidir. Tek ya da çift taraflı
olabilir. Serebellumun dentat çekirdeği, mezensefalondaki rubral çekirdek ve
bulbusdaki inferior oliva arasında bulunan ve Guillain-Mollaret üçgeni olarak
adlandırılan bölgenin etkilenmesine bağlıdır. Klinik olarak yumuşak damağın,
ritmik veya semi-ritmik, hızlı kasılmalara bağlı olarak tremoru görülür. Bazen
yumuşak damağın yanı sıra dil, farenks ve yüz kasları da kasılmalara
katılabilir. Palatal tremor istemli olarak durdurulamaz, kural olarak
başladıktan sonra bir daha kaybolmaz. Esansiyel ve semptomatik
olmak üzere iki tipi vardır. Esansiyel tipinde her kontraksiyonda östaki
borusunun kapanmasına bağlı olarak kulakta bir klik sesi işitilir, semptomatik
de ise bu ses ortaya çıkmaz. Semptomatik tipinde genellikle tremor dışında
serebellar ve beyinsapı bulguları da saptanır, kasılmalar esansiyel olanın
tersine uykuda kaybolur. Semptomatik olanda tedavi nedene yöneliktir. Esansiyel
olanda ise antikolinerjikler, benzodiazepinler, antikonvülzanlar ve botulinum
toksini denenebilir, ancak tedaviye genellikle dirençlidir.
Psikojenik Tremor
Ani
başlangıçlıdır, bir psikolojik travmayı izleyerek
başlayabilir, değişken karakterdedir ve sıklıkla bilinen organik tremor
tablolarından hiçbirine uymaz. Kinetik, postüral, istirahat veya her üçünün kombinasyonu olabilir. Kadınlarda daha sık görülür.
Psikojenik tremoru psişik stres altında artan fizyolojik veya esansiyel tremor
ile karıştırmamak gerekir.
TİK BOZUKLUKLARI
Tikler
genelde kısa, ritmik olmayan, stereotipik ve amaçsız görünen, normal hareket
zemininde bir veya birkaç kas grubunu tutan, ani, istemsiz hareketler (motor
tik) veya sesler (vokal tik) olarak tanımlanır. Tikler, koreik ve distonik
hareketleri taklit edebilir. Oldukça stereotipik olmaları, istemli olarak kısa
bir süre de olsa bastırılabilmeleriyle diğer hareket bozukluklarından
ayrılırlar. Hasta istemli olarak kısa bir süre tiklerini baskılayıp sonra
kendisini rahat bıraktığında tiklerde artma görülür. Bir başka önemli ve
ayırıcı özellik hastanın aslında bu hareketleri istemli olarak, içten gelen
dayanılmaz bir hareket etme isteğini ya da o bölgede hissettiği ve iyi
adlandıramadığı bir rahatsızlık hissini gidermek için yaptığını söylemesidir.
Tikler çocukluk ve adölesan dönemde sıktır. Yaş ilerledikçe tikler bir plato
çizme ve kaybolma eğilimindedir.
Motor ve
vokal tiklerin çeşitli formları vardır. Kabaca basit ve kompleks
tikler olarak ikiye ayrılırlar. Basit motor tikler göz kırpma, göz kayması,
sırıtma, ağız açma, omuz silkme gibi izole hareketlerdir. Kompleks motor tikler
dokunma, hafifçe vurma, koklama, el çırpma, ekopraksi (aynı hareketi
tekrarlama), kopropraksi (küfür şeklinde hareketler) gibi daha koordine ve
karışık, bazen amaçlı gibi görünen hareketlerdir. Basit vokal tiklere örnek
olarak artiküle olmayan basit, ilkel sesler, boğaz temizleme veya burun çekme gösterilebilir.
Kompleks vokal tikler anlamlı ve uzun kelimeler veya seslerdir. Buna örnek
olarak şarkı söyleme, ıslık çalma, ekolali (başkasının söylediği kelimeleri
tekrar etme), palilali (kendi söylediklerini tekrar etme) ve koprolali (küfür
etme) sayılabilir. Bazı tikler distoniyi taklit ederler ve distonik tik olarak
adlandırılırlar. Distonik tikler blefarospazm, distonik boyun dönmesi ve karın
kaslarında gerilme şeklinde olabilir. Motor ve vokal tiklerden başka nadir
görülen duyusal tikler de vardır. Bunlar genellikle bir vücut bölgesine sınırlı
basınç, gıdıklanma veya sıcaklık gibi rahatsız edici duyulardır.
Tiklerin
nörobiyolojik mekanizması tam olarak bilinmemektedir. Primer tik bozukluğu olan
bireylerin beyinlerinin postmortem incelemesinde yapısal ve histolojik olarak
özellik yoktur. Tiklerin dopaminerjik ilaçlarla artması ve dopamin blokerleri
ile yapılan tedavi ile azalması patofizyolojide santral dopamin
reseptörlerindeki aşırı duyarlılığın rol oynadığını düşündürmektedir. Bu
düşünce ile uyumlu olarak pozitron emisyon tomografisi
gibi fonksiyonel görüntüleme çalışmalarında presinaptik dopamin sinir
terminallerinde artmış yoğunluk gösterilmiştir.
Tik
bozuklarına yol açan nedenler primer ve sekonder olarak ikiye ayrılabilir (Tablo 6).
Tablo 7. Tik bozuklukları
|
Primer
tik bozuklukları Gilles
de la Tourette sendromu Kronik
motor-vokal tikler (bir yıldan uzun süren tikler) Geçici
tik bozuklukları (bir aydan fazla, ancak bir yıldan az süren tikler) |
|
Sekonder
tik bozuklukları Doğumsal o Huntington
hastalığı o Nöroakantositozis o İdyopatik
torsiyon distonisi o Kromozom
anormallikleri Edinsel o İlaçlar
(stimülanlar, nöroleptikler, antikonvülzanlar, L-dopa, doğum kontrol hapları,
trisiklik antidepresanlar) o Travma o İnfeksiyöz
hastalıklar (Creutzfeldt-Jacob hastalığı, Sydenham koresi, viral
ensefalitler) o Gelişimsel
(statik ensefalopati, mental retardasyon, otizm) o İnme o Dejeneratif
hastalıklar (İdyopatik Parkinson hastalığı, progresif supranükleer felç) o Toksik
(karbonmonoksit zehirlenmesi) o Hiperekpleksia |
Aşağıda en
sık görülen primer tik bozukluklarından olan Gilles de la Tourette sendromundan (GTS) kısaca bahsedilecektir.
Gilles de la Tourette Sendromu
GTS
sıklıkla davranışsal ve psikiyatrik bozuklukların eşlik ettiği motor veya vokal
tikler ile karakterize bir hastalıktır. GTS’nin görülme sıklığı ve ağırlığı
adölesan dönemde tepe noktasına ulaşır. Tikler genellikle erişkin yaşa gelince
düzelir veya oldukça hafifler. GTS erkeklerde yaklaşık 3 kat daha fazla
görülür. Hastaların pek çoğunda otozomal dominant geçişle uyumlu aile öyküsü
vardır. Son zamanlarda yapılan genetik çalışmalarda bazı lokuslar
gösterilmiştir. Çoğu hasta özellikle sosyal ortamlarda tiklerini birkaç
dakikadan yarım saate kadar baskılayabilir. Baskılanan sürenin sonunda tikler
daha şiddetli olarak geri döner. Tüm semptomlar
fiziksel ve psikolojik stresle artar. Çoğu zaman GTS hafiftir ve fazla
fonksiyonel bozukluğa yol açmaz.
GTS’li
hastaların yarısı obsesif-kompülsif davranış bozukluğu
gösterirler. Obsesif-kompülsif bozukluk tiklerden daha fazla kısıtlılığa neden
olabilir. Obsesyonlar tekrar eden kalıcı fikirler, düşünceler, impulslar veya
hayallerdir. Kompülsiyonlar obsesyonlar sonucunda
gelişen tekrarlayıcı ve kasıtlı davranışlardır. Kompülsiyonların kesin
kuralları vardır. Sık görülen kompülsiyonlar tekrar tekrar kontrol etme, yatma ritüelleri, sayma ve aşırı temizlik (tekrar tekrar el
yıkama) gibi örnekleri içerir.
GTS’li
hastaların yarıya yakınında görülen diğer bir bulgu hiperaktif davranışın eşlik
ettiği dikkat bozukluğudur. Ayrıca GTS’ye disleksi, antisosyal davranış,
depresyon, uyku bozuklukları ve kişilik değişikleri eşlik edebilir.
Aşağıda
GTS'nin DSM IV tanı kriterleri görülmektedir.
Birden
fazla motor ve vokal tikler.
Tiklerin
hemen hemen her gün olup, 1 yıldan uzun sürmesi. Tiksiz zamanların 3 aydan
fazla sürmemesi.
Tiklere
bağlı olarak sosyal, mesleki ve diğer önemli işlevsel alanlarda bozulma.
21
yaşından önce başlangıç.
Tiklerin
ilaçlara veya başka bir tıbbi hastalığa bağlı olmaması.
GTS’de tiklerin ve davranışsal semptomların
ağırlığı hastadan hastaya değişir. Tedavi hedef semptoma
göre seçilmelidir. İlk olarak hasta, aile bireyleri, eş ve okul personeli
hastalık hakkında bilgilendirilmelidir. Tikler ve davranışsal problemler
hastanın yaşam kalitesini etkilemiyor, sosyal ortama uyumda problem
yaratmıyorsa ilaç tedavisi gerekmeyebilir. İlaç dozları yavaş titre edilmeli ve
doz semptomların düzeldiği en düşük seviyede
tutulmalıdır. Tikler, obsesif-kompülsif bozukluk ve
hiperaktivite-dikkat bozukluğunun tedavisi için seçilen ilaçlar farklıdır.
Tikler zaman zaman ortaya çıkıp, kaybolabileceğinden uzun süre ilaç tedavisine
devam etmek gerekmeyebilir. Tikler için dopaminerjik tonusu azaltan ilaçlar
(haloperidol, pimozid, sulpirid gibi dopamin reseptör blokerleri veya
tetrabenazin gibi dopamin deposunu boşaltan ilaçlar), atipik nöroleptikler
(klozapin, olanzapin, risperidon, ketiapin, ziprasidon, aripiprazol),
noradrenerjik tonusu azaltan ilaçlar (klonidin) en etkili seçeneklerdir. Ancak
haloperidol, sulpirid gibi nöroleptikler tardif diskineziye yol
açabileceklerinden dikkatli kullanılmalıdırlar. Klonazepam gibi
benzodiazepinler, kalsiyum kanal antagonistleri,
trisiklik antidepresanlar ve lityum diğer tedavi seçeneklerini oluştururlar.
Çoğu hastada tiklere eşlik eden obsesif-kompülsif
bozukluk serotonin geri alınım inhibitörlerine (fluoksetin,
sitalopram, sertralin, paroksetin v.b) iyi cevap verir. Son yıllarda
DBS de ilaca dirençli olgularda başvurulan bir tedavi seçeneğidir.
İrkilme (Hiperekpleksi) Sendromları
Bir tik
bozukluğu olmamakla birlikte bazı benzerliklerinden dolayı bu bölümde ele
alınan irkilme sendromları (hiperekpleksi) ani,
beklenmeyen bir duyusal stimulusa (ses, dokunma) karşı artmış irkilme refleksi
ile karakterize bir grup bozukluktur. Beyinsapı lezyonları, perinatal hipoksi,
talamik lezyonlar, ilaçlar ve Tourette sendromu
semptomatik hiperekpleksi yapabilir. İrkilme sendromlarından
en sık görüleni ailesel irkilme hastalığı veya familyal hiperekpleksi,
beklenmeyen taktil veya akustik stimulusa karşı artmış irkilme reaksiyonu ve
neonatal dönemde kas rijiditesinin görüldüğü otozomal dominant bir hastalıktır.
Hastalık 5. kromozomun uzun kolunda lokalize olan,
inhibitör glisin reseptörlerinin alfa-1 subünitesini kodlayan gendeki nokta
mutasyonu sonucu oluşur. Klonazepam hiperekpleksi tedavisinde etkilidir. Eğer
hasta bu ilacı tolere edemiyorsa valproik asit önerilebilir.
MİYOKLONUS
Miyoklonus
ani, kısa süreli (<100ms), şimşek çakar gibi hızlı, atma, sıçrama şeklindeki
bir istemsiz harekettir. Genelde düzensiz atmalar şeklinde ortaya çıkmakla
birlikte ritmik de olabilir. Miyoklonus ekstremiteleri, yüzü ve gövdeyi
tutabilir ve istemli olarak engellenemez. Belli bir kas grubunun aniden
kasılması pozitif miyoklonus, bir postürü sürdüren
kasların aniden gevşemesi ise negatif miyoklonus
olarak adlandırılır. Tonus çözülmesi olarak da tarif edilen negatif miyoklonusa
asteriksis de denir.
Miyoklonik
kasılmalar istirahat zemininde spontan oluşabileceği gibi, sadece belirli bir
duyusal uyarı ile (refleks miyoklonus) ya da istemli hareketler esnasında
ortaya çıkabilirler (aksiyon miyoklonusu). Miyoklonik hareketler,
kaynaklandıkları anatomik oluşumlara, etkiledikleri vücut bölgelerine ve
etyolojilerine göre sınıflandırılmaktadırlar (Tablo
8). Buna göre miyoklonik hareketler tek bir bölgeyi tuttuğunda
fokal, bütün vücuda yayıldığında jeneralize, birbirine komşu birkaç spinal kord
segmentinden innerve olan kas gruplarını tuttuğunda segmental, birbirinden uzak
vücut bölgelerini etkilediklerinde ise multifokal miyokloniden sözedilir.
Miyoklonik kasılmalara yol açan elektriksel boşalımların kaynaklandığı yapıya
göre kortikal, subkortikal, beyinsapı ve spinal miyoklonusdan söz edilir.
Kortikal miyoklonus klinik olarak distal yerleşimli, 10-50 ms süreli, fokal
atmalar şeklinde görülür. Spinal miyoklonus daha çok gövde ve karın kaslarını
tutar. Subkortikal miyoklonus ise korteks ve spinal kord arası bölgelerden
kaynaklanır, jeneralize olma ve ektsremitelerin proksimalini tutma
eğilimindedir. Bu tip miyoklonik atmaların kaynağını epileptik deşarjlar oluşturur.
Progresif miyoklonik epilepsi diye adlandırılan, altında değişik etyolojilerin
yattığı bir grup hastalıkta miyoklonik kasılmalar ile birlikte epileptik
nöbetler, demans ve serebellar sendrom bulguları
görülür. Miyokloni genç yaşta, tek başına epileptik bir fenomen
olarak (jüvenil miyoklonik epilepsi) veya idyopatik jeneralize epilepsinin
diğer nöbet formlarıyla bir arada görülebilir. Epileptik miyoklonustan kitabın
epilepsi bölümünde daha ayrıntılı bahsedilecektir (Bakınız: Epilepsi bölümü).
Tablo 8. Miyoklonusun sınıflandırılması
|
I- Tutulan bölgeye göre |
II- Kaynağına göre |
III- Etyolojiye göre |
Non-epileptik Miyoklonus
Bu tipte
miyoklonik atmaları olan hastalarda eşlik eden epileptik deşarjlar saptanmaz.
Fizyolojik, esansiyel ve semptomatik tipleri vardır.
Fizyolojik miyoklonus: Normal insanlarda görülen, herhangi bir patolojik duruma
atfedilemeyen miyoklonik sıçramalardır. Uykuya dalış sırasında, anksiyetede ve
aşırı egzersiz sonrasında görülen miyokloniler bu grubun klasik örnekleridir.
Esansiyel miyoklonus: Zaman zaman miyoklonik kasılmaların ortaya çıktığı bu
tabloda genelde miyoklonus dışında patolojik bulgu ve belirti yoktur. Hastalık
genellikle yaşamın ilk 20 yılı içinde başlar, statik gidişli veya çok yavaş
ilerleyicidir. Miyoklonik sıçramalar üst ekstremitelerde daha belirgin olmak
üzere tüm vücutta görülebilir ve hareketle artar. Miyoklonik atmalara bazen tremor
(miyoklonik tremor) veya distoni (miyoklonik distoni) de eşlik edebilir. Bu
atmalar yaygın ve şiddetli olduğunda günlük yaşam aktivitelerini ciddi derecede
bozabilir. Atmaların alkole olumlu yanıt vermesi tipiktir ve bazen tanı
konulmasına da yardımcı olur. Kalıtımsal formu otozomal dominant geçer, ancak
gen penetransı değişkendir. Tedavide klonazepam, sodyum valproat, pirimidon
gibi ilaçlar kullanılır.
Semptomatik miyoklonus: Karaciğer yetmezliği, üremi, elektrolit dengesizliği,
intoksikasyon, kafa travması, iskemik ensafalopati,
nörodejeneratif hastalıklar, merkezi sinir sistemi infeksiyonları, prion
hastalıkları ve statik ensefalopati gibi hastalıkların seyrinde, asıl
hastalığın bulgularına ilave olarak miyoklonus görülebilir. Bu tip miyoklonik
atmalar fokal, jeneralize veya segmental olabilir ve genelde hareketle
artarlar. Post-hipoksik ensefalopatiye bağlı olarak gelişen, genelde
jeneralize, refleks ve aksiyon miyoklonilerinin ön planda olduğu tabloya
Lance-Adams sendromu adı verilir. Dış uyaranlarla belirgin
bir biçimde artan miyoklonik sıçramalar ekstremitelerde, yüzde ve gövdede
görülebilir. Tedavide sodyum valproat, klonazepam ve pirasetam kullanılır.
ATAKSİLER
Ataksi primer motor işlevlerdeki bir
kayba (felç gibi) bağlı olmaksızın ortaya çıkan hareket koordinasyon bozukluğu
olarak tanımlanır ve sıklıkla geniş tabanlı, dengesiz yürüyüşü tarif etmekte
kullanılır. Ataksi serebellum ve bağlantılarındaki tutulumlardan, spinal kord
lezyonlarından, periferik duyu kayıplarından veya bu üç durumun kombinasyonundan kaynaklanabilir. Ataksiler basitçe edinsel
ve herediter ataksiler olarak ikiye ayrılabilir. Edinsel ataksilere alkolizm,
serebrovasküler hastalıklar, vitamin eksiklikleri, multipl skleroz, otoimmün
hastalıklar, primer veya metastatik tümörler, paraneoplastik sendromlar
gibi yukarıda sözü edilen yapıları tutan birçok etyoloji neden olabilir.
Herediter ataksiler sıklıkla yavaş ilerleyici, spinal ve/veya serebellar
atrofinin eşlik ettiği tablolardır. Bu hastalıkların hepsinde serebeller ataksi
değişmez bulgudur ve nesilden nesile geçiş özelliklerine göre
sınıflanabilirler. Bazen genetik geçiş niteliğinin saptanamadığı, yeni
mutasyonların yol açtığı sporadik herediter ataksiler de görülebilir.
Ataksilerin herediter ve edinsel nedenleri kitabın ilgili bölümünde
anlatılmıştır (Bakınız: Ataksiler).
UYKU
İLE İLİŞKİLİ HAREKET BOZUKLUKLARI
Uyku, fizyolojik ve geçici bir
bilinçsizlik durumudur. Uyku esnasında gözlerin hareketli olduğu ve EEG’de
uyanıklık durumundaki bulgulara benzer deşarjların bulunduğu evreye REM uykusu,
EEG deşarjlarının yavaşladığı, göz hareketlerinin kaybolduğu evreye ise non-REM
uykusu denir. Uyku, non-REM evresi ile başlar ve 60-90 dk. sonra
REM evresine girilir. REM evresi yaklaşık 10-20 dk. sürer.
Normal bir gece uykusu boyunca bu iki evre periyodik olarak birbirini izler.
Dinlendirici uyku, derin uyku veya yavaş dalga uykusu da denilen non-REM
uykusudur. Yaşla birlikte non-REM süresi de kısalır.
Hareket bozukluklarının büyük çoğunluğu
uykuda kaybolurken bazı hareket bozuklukları sadece uykuya geçer veya uyku
esnasında ortaya çıkarlar. Bu bölümde bu tip hareket bozukluklarından kısaca
bahsedilecektir.
Paroksismal
Nokturnal Distoni
Non-REM uykusunda ortaya çıkan ve
genellikle alt ekstremitelerde görülen distonik, bazen koreoatetoid nitelikte
istemsiz hareketlerdir. Herhangi bir yaşta başlayabilir. Kasılmalar 2-60 dk
sürer ve bir gecede 3-4 kez tekrarlayabilirler. Atak sırasında hasta
uyanabilir, atak sonrasında tekrar uykuya dalar. Distonik kasılmalar esnasında
hasta bazen garip sesler de çıkarabilir. Rutin EEG’de epileptik deşarjlar
görülmemekle birlikte bu sendromun epileptik kökenli
olduğu düşüncesi de vardır. Tedavide karbamazepin veya benzodiazepinler
kullanılabilir.
Huzursuz
Bacak (Restless Legs) Sendromu
Huzursuz Bacak Sendromu (HBS) yattıktan kısa bir süre sonra alt
ekstremitelerde ortaya çıkan ve genellikle gerginlik, uyuşma, batma, yanma,
iğnelenme veya tam tarif edilemeyen garip bir rahatsızlık hissi biçiminde ifade
edilen bir yakınmadır. Hasta bu yakınmalarını gidermek için bacaklarını hareket
ettirir veya kalkıp yürür, ancak bu kez bacakların hareket ettirilmesi uykuya
dalmayı geciktirir. Çoğunlukla ileri yaşlarda görülür ve %50 oranında otozomal
dominant kalıtımlıdır. Hastalığın penetransı değişkendir. Esansiyel HBS'de
nörolojik muayenede patolojik bulgu saptanmaz. Gebelik, diyabet, koksartroz,
malabsorbsiyon sendromu, demir eksikliği anemisi,
karsinoma ve periferik nöropati seyrinde görülen ve semptomatik HBS olarak
adlandırılan tipinde ise belirtilen durumlara özgü bulgular saptanır. Tedavi,
semptomatik olanda nedene yöneliktir. Esansiyel HBS’nin santral-spinal
dopaminerjik geçişte meydana gelen bir yetersizliğe bağlı olduğu
düşünülmektedir. Bu görüşe uygun olarak tedavide en etkili ilaçlar yatmadan
önce verilen L-dopa preparatları ve dopamin agonistleridir. Ancak dopaminerjik
preperatlara zamanla direnç gelişmesi ve bulguların artarak gündüz saatlerine
de kayabilmesi nedeniyle dikkatli olarak kullanılmalıdır. Bazı vakalarda
pregabalin veya gabapentin gibi antiepileptikler ilk tedavi seçeneği olarak
düşünülebilir. Yeterli cevap vemeyen hastalarda klonazepam gibi
benzodiazepinler ve kodein gibi opiat türevleri kullanılabilir.
Uykunun
Periyodik Ekstremite Hareketleri (Periodic Limb Movements of Sleep)
Non-REM uykusu sırasında özellikle diz,
kalça ve ayak bileğinde ortaya çıkan, periyodik, istemsiz fleksiyon
hareketleridir. Bu hareketler her iki bacakta, bazen de kollarda
görülebilirler. Miyoklonik atmalara oranla daha uzun süreli ve amplitüdü daha
düşük olan hareketler birkaç dakikada birden saatte bire kadar değişen
aralıklarla tekrarlanırlar ve sıklıkla uyku kalitesini bozarlar. Yaşlılarda
daha sık rastlanan bu sendrom kalıtımsal olabilir. HBS
olan hastaların %50'sinde uykunun periyodik ekstremite hareketleri de vardır.
Yalnızca bacaklarda ağrı ve ayak parmaklarında istemsiz hareketler ile giden
sınırlı bir formu da görülebilir. Bu hastalarda nörolojik muayene normaldir.
Semptomatik olduğunda, etyolojik faktörler HBS'dekilerle aynıdır. Bu sendromun da santral dopaminerjik geçişteki bir yetersizlik
sonucu olduğu düşünülmektedir ve tedavi yaklaşımı da HBS'nda olduğu gibidir.
(Bakınız: Uyku Hastalıkları
Bölümü)
İLAÇ
KULLANIMINA BAĞLI OLARAK ORTAYA ÇIKAN HAREKET BOZUKLUKLARI
Başta nöroleptikler
olmak üzere, çeşitli ilaçların kullanımı hipokinetik veya hiperkinetik hareket
bozukluklarının ortaya çıkmasına neden olabilir. Farklı mekanizmalar söz konusu
olmakla birlikte bu tip hareket bozukluklarından genelde dopamin
reseptörlerinin aşırı derecede blokajı ya da
stimülasyonunun sorumlu olduğu düşünülmektedir. Bu bozukluklar ortaya
çıktıkları zamansal süreç açısından üç ayrı grupta incelenebilirler.
Akut distonik reaksiyonlar:
Daha çok genç ve duyarlı kişilerde, ilaç alınımını izleyen saatler içinde
ortaya çıkar. Hasta ve çevresini korkutucu bir biçimde gelişir. Özellikle baş,
boyun, yüz, dil, ağız çevresinde distonik ve diskinetik nitelikte istemsiz
hareketler oluşur. Bunlara sıklıkla okülojirik krizler eşlik edebilir. Panik
içinde hastayı acil polikliniğe getiren bu klinik tabloda yapılacak ilk şey
sebep olan ilacı kesmek ve hastaya bu tablonun herhangi bir şey yapılmasa da
kısa süre sonra kendiliğinden geçeceğini anlatmaktır. Parenteral antikolinerjik
ilaç (örneğin biperiden ampul uygulanması düzelmeyi hızlandırır. Nöroleptikler,
nöroleptik özelliği taşıyan antiemetikler ve kalsiyum kanal blokerleri bu tip
tablolara yol açabilirler. Fizyopatolojik mekanizması tam açıklanmamış olmakla
birlikte bu tablodan ani ve aşırı dopamin reseptörü blokajı
sonucu striatal dopaminerjik ve kolinerjik sistem arasında oluşan dengesizlik
sorumlu tutulmaktadır.
Subakut gelişen parkinsonizm veya akatizi: Bu
tablolar çoğunlukla nöroleptik kullanımından sonraki birinci ile üçüncü ay
içinde gelişir. Dopamin reseptör blokajı yapan
antiemetikler ve kalsiyum kanal blokerleri de bu tabloya yol açabilir.
İatrojenik parkinsonizm adı da verilen tabloda nöroleptik veya yukarıda
bahsedilen ilaç kullanımını izleyen haftalar içinde, maske yüz, bradikinezi,
küçük adımlarla yürüme gibi parkinsonizmin daha çok akinetik-rijid belirtileri
görülür, nadiren buna tremor da eklenebilir. Mekanizma dopamin reseptörlerinin blokajı sonucu dopaminerjik geçişin azalmasıdır. Sorumlu
ilacın kesilmesi ve antikolinerjik ilaçların uygulanmasıyla bulgular yavaş
yavaş geriler. Nöroleptik kullanımı sonucu, erken dönemde ve dozla ilintili
olarak ortaya çıkabilen bir diğer tablo akatizidir. Klinik olarak subjektif
huzursuzlukla birlikte, sürekli hareket etme isteği, yerinde duramama vardır.
Bunlara sıklıkla stereotipik hareketler eşlik eder. Fizyopatolojisi
bilinmemektedir. Tedavide sorumlu ilacın kesilmesi gerekir, ilaveten antikolinerjikler
veya benzodiazepinler de uygulanabilir.
Tardiv sendromlar:
Uzun süreli nöroleptik veya dopamin reseptör blokajı yapan ilaç kullanımı
sonucu ortaya çıkan, bazen geri dönüşümsüz olan istemsiz hareketlerdir. Kronik
ilaç kullanımını gerektiren psikozlarda, tedavi başlangıcından aylar, yıllar
sonra ortaya çıkarlar. Genellikle yüz, ağız çevresi, ekstremiteler ve gövdede,
sıklıkla stereotipik nitelikte istemsiz hareketler vardır. Mekanizmadan dopamin
reseptörlerinde kronik blokaj sonucu gelişen aşırı
duyarlılık sorumlu tutulmaktadır. Tardiv diskinezi çiğneme, yutma gibi çeşitli
orofasyal koreik, stereotipik hareketlerin ön planda bulunduğu tablolardır.
İstemsiz hareketlerin niteliği başlıca distonik olduğunda tabloya tardiv
distoni adı verilir. İdyopatik olanlara kıyasla retrokollis ve geriye doğru
olan gövde distonisi daha sık görülür. Benzer şekilde tardiv akatizi, tardiv
tremor, tardiv miyoklonus tabloları tanımlanmıştır. Tedavide sorumlu ilacın
kesilmesi dışında tetrabenazin gibi dopamin depolarını boşaltıcı ilaçlar,
selektif dopamin reseptör blokerleri ve klozapin gibi atipik nöreleptikler
kullanılabilir.
FONKSİYONEL
HAREKET BOZUKLUKLARI
Fonksiyonel kökenli hareket
bozuklukları parkinsonizm, distoni, kore. miyoklonus, tremor, tik gibi her türlü
hareket bozukluğunu taklit edebilirler. Bazı ayrıntıların dikkatli bir biçimde
irdelenmesi tanıda yanılgı payını en aza indirir. Yakınmaların başlangıcının
ani olması, psişik bir travmayı takiben ortaya
çıkması, ciddi handikaplar yaratması, alışılmış biçimin dışına taşan bir
görünüm sergilemesi, anatomik ve patofizyolojik kalıpların dışına çıkması,
hastanın dikkati dağıtılınca yakınmaların düzelmesi, kendiliğinden iyileşme
dönemlerinin bulunması, plasebo tedavisine olumlu yanıt alınması, ikincil
kazançların varlığı ve hastada psikopatoloji lehine belirtilerin sezilmesi
hareket bozukluğunun fonksiyonel olduğunu düşündürür. Bunun yanı sıra nörolojik
muayene tamamen normaldir.
Kaynaklar:
Fahn S, Jankovic J. Principles and
Practice of Movement Disorders. Philadelphia. Churchill
Livingstone/Elsevier, 2011.
Jankovic J, Tolosa E. Parkinson's
Disease and Movement Disorders. Philadelphia. Lippincott Williams&Wilkins,
2015.
Koller WC, Melamed M. Parkinson's
Disease and Related Disorders Volume I-II: Handbook of Clinical Neurology Series.
Amsterdam. Elsevier Health Sciences, 2007.
Sawle G. Movement Disorders in Clinical
Practice. Oxford. Isis Medical Media Ltd, 1999.
Watts Rl, Koller WC. Movement
Disorders. Neurologic Principles and Practice. New York. McGraw-Hill, 2006.