Klinik Nörogenetik
Yazanlar: Sibel Aylin Uğur İşeri, Nerses Bebek
Son güncellenme tarihi: 15.07.2020
Genom bilim alanında son on yılda yaşanan teknolojik devrim, nörogenetik hastalıkların tanı ve tedavisine de ciddi katkı sağlamıştır. Yeni nesil dizileme teknolojilerinin ve biyoenformatik analiz stratejilerinin gelişmesi, özellikle tek gen kaynaklı nadir nörolojik hastalıkların genetik tanısını hekim ve hastalar için erişilebilir kılmıştır. Zaten tüm nadir hastalıkların yaklaşık %80’inin genetik kökenli olması ve nadir hastalıkların en az yarısında merkezi veya periferik sinir sistemi tutulumu gözlenmesi, nadir nörogenetik alanına ilgiyi gün geçtikçe arttırmaktadır. Bununla beraber, toplumlarda daha sık rastlanan ve karmaşık genetik altyapıya sahip kompleks nörolojik hastalıklarda dahi gelişen teknolojinin ucuzlaması ile aynı anda yüzbinlerce bireyin genetik analizinin karşılaştırmalı olarak yapılabildiği büyük çıktılı çalışmalar karşımıza çıkmaktadır. Nörogenetiğin bu değişen boyutunda nörologların da söz sahibi olabilmeleri için genetik kavramlara aşina olmaları, yani bir nevi genetik okuryazarlığa sahip olmaları gerekmektedir. Bu bölümde klinik açıdan önem taşıyan nörogenetik kavramlar güncel bir özet halinde ele alınacaktır. Hastalıklara ait klinik ve moleküler genetik bilgiler ilgili bölümlerde ayrıca ele alınmıştır.
İnsan Genom ve Organizasyonu
İnsan genomu
Diğer tüm canlılarda olduğu gibi insanın da genetik bilgisini yeni nesillere aktarabilmek dahil tüm biyolojik süreçlerini gerçekleştirebilmesi için bir genoma ihtiyacı vardır. Genomu kompleks bir ekosistem olarak ele alabiliriz. Bu ekosistemin temelinde genellikle deoksiribonükleik asit (DNA) yapısında olan bir nükleik asit molekülü yatar. Sadece koronavirüsler gibi ribonükleik asit (RNA) virüslerinin genomu RNA molekülünden oluşur. Bu ekosistemde DNA molekülünü paketleyen, eşleşmesini sağlayan, gerektiği yerde aktive eden veya susturan çok sayıda protein ve RNA molekülü de görev alır. Bu ekosistem tüm ökaryotlarda olduğu gibi insanda da büyük oranda hücre çekirdeğinde yerleşik vaziyettedir. Ama genomumuzun bir kısmı da hücrelerimizde değişken sayıda mevcut bulunan mitokondri organellerinde bulunur. Bitkiler için ayrıca genom yerleşkeleri arasında kloroplastlar da sayılmalıdır.
Nükleik asitler
DNA ve RNA molekülleri şeker (DNA’da deoksiriboz ve RNA’da riboz) ve fosfat iskeletine bağlı nitrojenli bazlardan oluşan polimerlerdir. Şeker-fosfat iskeletinde köprü görevi gören fosfat grubu, bir şeker grubunun 3. karbonunu bir sonra gelen şeker grubunun 5. karbonuna fosfodiester bağları ile bağlar. Bu köprü ile nükleik asitlerin polaritesi belirlenir; nükleik asitlerin 5’ uçtan 3’ uca doğru uzadıkları söylenir. Şeker-fosfat iskeletine bağlı nitrojenli bazlar DNA ve RNA’nın kodunu yani anlamını belirler. DNA bazları çift halkalı pürinler (Adenin; A ve Guanin; G), tek halkalı pirimidinler (Timin; T ve Sitozin; C) olmak üzere dört çeşittir. Bunlar İngilizcedeki baş harfleri ile temsil edilir. RNA bazları da 4 çeşittir; A, G ve C bazları ile T yerine urasil (U) bazı bulunur. Birer fosfat, şeker ve nitrojenli baz gruplarından oluşan nükleik asitlerin temel yapıtaşı nükleotid adını alır. DNA molekülü RNA’dan farklı olarak çift zincirli halde bulunur. Birbirine zıt polaritelerde uzanan iki DNA zinciri, nitrojenli bazların arasında oluşan hidrojen bağları ile eşleşir. Kural olarak A ve T iki, C ve G ise birbirine üç hidrojen bağı ile bağlanır. Bu kurala göre DNA’nın iki zinciri birbirini tamamlayıcı yani komplementer niteliktedir. Bu özellik hem DNA dizisinin korunarak aynı şekilde kopyalanmasını (replikasyon) hem de DNA kodunun doğru bir şekilde RNA’ya aktarılmasını (transkripsyon) sağlar (Şekil 1). Genetik araştırmacıları için ise DNA’daki sınırlı bir bölgenin laboratuvar ortamında polimeraz zincir tepkimesi (PZT) ile çoğaltılırken orijinal dizilimdeki gibi olmasını garanti eder.

Şekil 1. DNA’nın yapısı, komplementerliği yanı sıra transkripsiyon ve translasyon aşamaları şematik olarak gösterilmiştir.
Kromozomlar ve taşıdıkları genler
İnsan somatik hücreleri 23 çift olmak üzere toplam 46 kromozom içeren diploid hücrelerdir. Bu çiftlerin 22 tanesi otozomdur. Otozomlar tamamen homolog kromozomlardır. Yani 22 kromozom çiftinin bir seti bireyin annesinden diğer seti de babasından gelir. Son kromozom çifti ise gonozom olarak adlandırılır. Kadınlarda homolog ve XX olan gonozomlar; erkeklerde homolog değildir ve XY olarak adlandırır (Şekil 2). Eşey hücreleri yani gametler ise haploiddir ve tek set halinde 23 kromozomları vardır. Kromozomlar nükleer DNA’nın çeşitli yapısal ve fonksiyonel proteinler ile düzenleyici RNA molekülleri ile kompleks halinde oluşturduğu dinamik ve fonksiyonel birimlerdir. Kromozomların iki uç noktası telomer, orta bölgesi ise sentromer olarak adlandırılır. Tüm kromozomlarımızda yaklaşık 3,6 milyar baz çifti bulunur.


Şekil 2. G bantlama yöntemi
ile elde edilen (a) normal bir kadına ait 46,XX ve (b) normal bir erkeğe ait
46,XY karyotipler.
Kromozomlar üzerinde yer alan ve belki de klinik nörogenetik alanında en çok dikkatimizi çeken bölgeler genlerdir. Gen tanımı yıllar içinde değişim göstererek günümüzde farklı şekillerde ifade edilebilmektedir. Gen klasik tanımı ile RNA üretimi için gerekli bilgiyi içeren DNA bölgesidir. Kromozomların hangi bölgelerinin gen olarak tanındığına dair güncel bilgi GENCODE veritabanından elde edilebilir (https://www.gencodegenes.org/human/stats.html). Bu veritabanına göre 60,000’in üzerinde gen bulunmaktadır (Gencode veritabanı versiyon 34). Bunların ancak 20,000 kadarı protein kodlayan genlerdir; yani mesajcı RNA (mRNA) oluşturarak (transkripsiyon) ribozomlarda protein sentezinde (translasyon) rol alırlar. Kalan genlerin büyük kısmı kodlamayan RNA geni sınıfını temsil eder. Bu genler RNA’ya transkribe edilse dahi proteine dönüşmezler. Yapısal ve fonksiyonel görevleri olan bu RNA’lar önemli biyolojik süreçlerde görev alırlar. Kodlamayan RNA’lar arasında en iyi bilinen gruplar translasyonda rol alan ribozomal ve taşıyıcı RNA’lardır (rRNA ve tRNA). Bunlar dışında mRNA olgunlaşması ve kontrolünü ve hatta DNA molekülünün anlatımını düzenleyen yani epigenetik etkili çok sayıda kodlamayan RNA molekülü bulunur.
İnsan DNA’sında gen ve ilişkili bölgeler genomun yaklaşık %10’unu oluşturur. Kalan %90’lık bölümün çoğu tekrar dizilerinden oluşur.
Protein kodlayan genlere daha detaylı baktığımızda bu gen bölgelerinin DNA üzerinde kodlayan (ekzon) ve kodlamayan (intron) sekansları olduğunu görürüz (Şekil 3). DNA’dan RNA transkripsiyonu gerçekleştiğinde öncül RNA molekülünde ekzonların arası intronlarla doludur. Öncül RNA, mRNA’ya olgunlaşırken hücre çekirdeğinde çeşitli protein ve RNA komplekslerinin yardımı ile kırpılmaya uğrar (RNA splicing). Kısaca intronik sekanslar kırpılıp atılarak ekzonlar birbirine bağlanır. Pek çok gende çok sayıda ekzon bulunur. Tek bir genin alternatif kırpılması yani mRNA oluşurken farklı ekzon kombinasyonları kullanılması ile farklı mRNA’lar dolayısı ile farklı protein ürünleri elde edilebilir. Bu süreç gelişimin farklı evrelerinde ve farklı doku tiplerinde kontrollü olarak gerçekleştirilerek protein çeşitliliğine katkı sağlar.
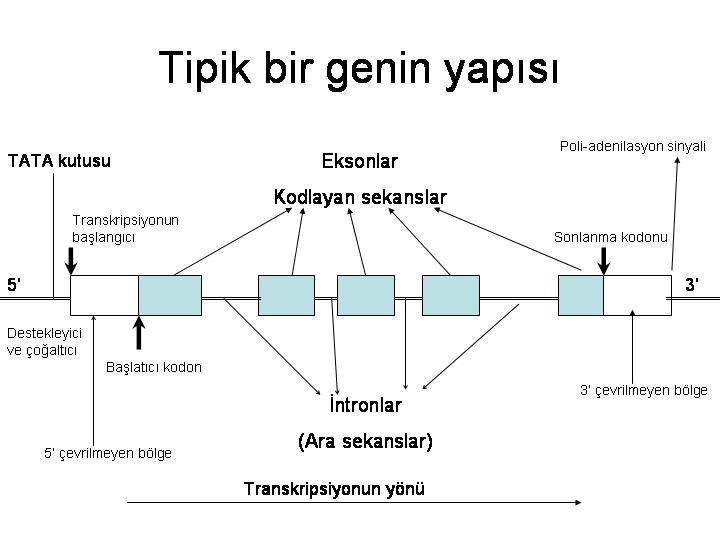
Şekil 3. Bir genin yapısı şematik olarak gösterilmiştir.
Protein kodlayan genlerin anatomisi incelendiğinde baz dizilimlerinin benzer örüntüler halinde olduğunu görürüz. Genlerin temelde ekzon ve intron dizilerinden oluştuğunu zaten belirtilmişti. Ekzonların 5’ kısmında transkripsiyon ile mRNA’ya aktarılan ancak proteine dönüşmeyen bir bölge mevcuttur. Bu bölge 5’ çevrilmeyen bölge (5’ untranslated region; 5'UTR) adını alır. Benzer şekilde ekzonların mRNA’ya geçip protein kodlamayan 3’ kısmına ise 3’UTR denir. Bu alanların içinde veya etrafında genlerin transkripsiyonel aktivitesini düzenleyen özel DNA dizilimleri yer alır. Bu dizilimlerin başında RNA kırpılmasını düzenleyen ekzon ve intronların uçlarında yer alan özel konsensüs diziler sayılabilir. Protein kodlayan genlerin 5’ ucunda RNA polimerazın bağlanması ve transkripsyonun düzenlenmesi için gerekli promoter (başlatıcı) bölge yer alır. 3’ uçta ise mRNA’nın olgunlaşması, kontrolü ve çekirdekten sitoplazmaya geçiş süreçleri için gerekli olan çoklu adenin (poliA) sinyal dizisi bulunur. PoliA sinyal dizisinin transkripsiyona uğramasını takiben mRNA bu bölgeden kesilir ve özel bir polimeraz ile 3’ ucuna pek çok adenin bazı yani bir nevi poliA kuyruğu eklenir. Gen dizisinin içerisinde veya komşuluğunda bulunan bu tarz özel dizilerin yanı sıra gen anlatımlarını çok daha uzak mesafelerden kontrol edebilen “enhancer” (arttırıcı) ve insülatör gibi DNA dizileri de mevcuttur.
Santral Dogma
Moleküler biyolojinin ‘santral dogması’ 1970 yılında Francis Crick tarafından ortaya atılmıştır. Bu hipoteze göre genetik bilginin akışı tek yönlüdür: (i) DNA kendini eşleyebilir yani replike edebilir ve böylece DNA’da saklanan kod korunup sonraki nesillere aktarılabilir (ii) DNA kodu öncelikle mRNA’lara transkripsiyon mekanizması ile aktarılır (iii) Kodlanan mRNA’lar sitoplazmaya geçer ve burada ribozomların yardımıyla kodun proteine translasyonu sağlanır. Ökaryotlarda replikasyon ve transkripsiyon hücre çekirdeğinde gerçekleşirken, translasyon sitoplazmada olur. Prokaryotlarda membran ile sınırlanmış organeller mevcut olmadığı için tüm süreçler sitoplazmada gerçekleşir. Santral dogma hipotezinde bu akış tek yönlüdür. Bu hipotez protein kodlayan genler için büyük ölçüde doğru olsa da bazı güncellemelere gereksinimi vardır. Bu güncellemeler arasında genomu RNA olan virüslerin varlığı, RNA’nın ters transkripsiyon ile DNA’ya dönüşebilmesi, RNA ve proteinlerin hem DNA hem de diğer RNA molekülleri üzerinde epigenetik kontrol yapabilmesi, protein kodlamayan fonksiyonel RNA’ların varlığı ve DNA’dan RNA sentezlendikten sonra çeşitli hücresel mekanizmalarla RNA’nın kodunun yeniden düzenlenmesi sayılabilir.
Biyolojik bir kodlama sistemi: Genetik kod ve protein sentezi
Santral dogmanın klasik akışında yani kodun DNA’dan RNA’ya ardından da proteine aktarılmasında ilk adım RNA polimeraz enziminin genellikle genlerin 5’ ucunda yer alan başlatıcı yani promoter bölgelerini tanımasıdır. Ardından bu enzim DNA’nın 5’ ucundan 3’ ucuna doğru ilerleyerek, DNA’daki kodu RNA’ya kopyalar. Oluşan mRNA ürününün DNA’dan farkları arasında deoksiriboz yerine riboz şekeri içermesi, timin yerine urasil kullanılması ve tek zincir olması sayılabilir. mRNA’ya aktarılan bilgi kodon adı verilen bir nevi üçlü kodlama sistemine sahiptir. Kodlamanın başladığı yerden itibaren her üç baz yani bir kodon bir amino asidi temsil eder. Ribozomlarda özellikle ribozomal RNA’ların (rRNA) enzimatik aktivitesiyle kodonların kodları çözülür, doğru aminoasitler tRNA’lar aracılığı ile taşınır, uç uca birleştirilir ve polipeptidler oluşturulur. mRNA üzerinde bu kodun başlayıp bittiği alana açık okuma çerçevesi (open reading frame; ORF) adı verilir. ORF protein kodlayan genler için neredeyse her zaman metiyonin amino asidini işaret eden ATG (mRNA’da AUG) kodonu ile başlar. ORF’yi amino asit şifrelemeyen 3 çeşit (UAA, UAG, UGA) kodondan biri durdurabilir (stop codon).
DNA'daki dört çeşit nükleotid (A, T, G, C) farklı dizilimler ile üçlü kodonları oluşturabildiğine göre basit bir permütasyon hesabı ile toplam 43 yani 64 farklı kodon bulunabilir. Bunların 3 tanesi amino asit kodlamayan durdurucu kodonlar olduğuna göre toplam 61 adet amino asit kodlayan kodon var demektir. Ancak kodlanan amino asit sayısı sadece 20’dir. Metiyonin ve triptofan amino asitlerinin birer kodonu olmasına rağmen, diğer amino asitler 2 ila 6 farklı kodonca kodlanabilirler (Şekil 4). Bu durum genetik kodun dejenere veya gereğinden fazla olması ile açıklanır, çünkü tek bir amino asit birden fazla kodon tarafından kodlanabilir.
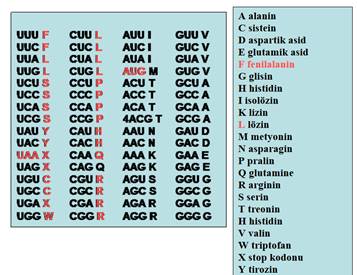
Şekil 4. 64 aminoasidin 3’lü nükleotid kodonları ve 21 aminoasidin kodları görülmektedir.
Bir genden alternatif kırpılmalar ile farklı mRNA ürünleri, dolayısı ile de farklı protein izoformları oluşabileceğini daha önce belirtmiştik. İnsan gibi kompleks genoma sahip organizmalarda protein çeşitliliğine katkı sağlayan diğer bir durum da proteinlerin translasyon sonrası çeşitli mekanizmalar ile kimyasal olarak modifiye edilmeleridir. Seçili amino asitlere küçük kimyasal gruplar eklenmesi yani hidroksilasyon, karboksilasyon, metilasyon, asetilasyon ve fosforilasyon ile yine özel amino asitlere kompleks karbonhidrat veya lipid gruplarının eklenmesi bu kimyasal modifikasyonlar arasında sayılabilir. Ayrıca pepsinojenin aktif olabilmesi için mide asidi varlığında pepsine dönüşmesi örneğinde olduğu gibi bazı proteinlerin de fonksiyonel olabilmesi için engelleyici polipeptit zincirlerinden kurtulmaları gerekmektedir. Proteomik proteinlerin fonksiyon, yapı ve etkileşimlerini farklı doku ve gelişim evrelerinde inceleyen, bu bilgiyi fizyolojik süreçler ve hastalık mekanizmaları ile bağdaştıran bir alandır. Bu alanda yapılan çalışmalar ile ökaryotların evriminde insan gibi memelilere doğru gidildikçe nöral yolaklar dışında immün fonksiyon, sinyal iletimi, homeostaz ve apoptozla ilgili proteinlerin ileri derecede çeşitli olduğu anlaşılmıştır. Yine de pek çok proteinin fonksiyonu henüz tam olarak bilinmemektedir.
Mitokondri genomu
İnsan genomundan bahsederken mitokondriye ait genomdan bahsetmemek büyük bir eksiklik olur. Mitokondri genomu intron içermeyen ve 37 gen kodlayan yaklaşık 16,6 kilobazlık dairesel bir DNA molekülünden oluşur. Bu 37 genin 24 tanesi mitokondriyal tRNA ve rRNA gibi kodlamayan RNA’lar içindir. Kalan 13 tanesi ise mitokondriye ait solunum zinciri ve oksidatif fosforilasyon enzimlerinin alt ünitelerini kodlamaktadır. Ancak mitokondriyal proteinlerin çoğu nükleer genlerce kodlanmakta ve sitoplazmik ribozomlarda sentezlendikten sonra mitokondriye taşınmaktadır. Mitokondriyal genom nükleer genoma kıyasla çok küçüktür ve belki de bu özelliği nedeniyle etkisiz gibi algılansa da tek bir mitokondrinin yaklaşık 2 ile 10 kopya kadar dairesel DNA molekülü içerdiği ve her hücrenin yaklaşık 100 ile 1000 adet mitokondriye sahip olduğu düşünüldüğünde, mitokondrinin tüm genoma katkısının çarpan etkisi daha iyi anlaşılabilir. İnsanlarda mitokondri sayısının en az ve en çok olduğu hücreler sırasıyla yaklaşık 100 mitokondri ile sperm ve 250,000 mitokondri ile de yumurta hücreleridir. Zigot oluşumu sırasında nükleer genomun aksine mitokondri genomu sadece yumurta hücresinden yani anneden kalıtılmaktadır. Sonuç olarak erkekler mitokondriyal DNAlarını sonraki kuşaklara aktaramazlar ve bu nedenle mitokondriye bağlı hastalıklar sadece anneden çocuklara kalıtılabilir.
Genomdaki Varyasyonlar
Toplumlar ve bireyler arasında genetik farklılıklar genom varyasyonları olarak adlandırılır. Yaklaşık 3,2 milyar bazdan oluşan insan genomu bireyler arasında farklılık gösterir. Gelişen dizileme teknolojileri sayesinde farklı popülasyonlardan pek çok kişinin genomu dizilenmiştir. NCBI-dbSNP veritabanının 2017 yılı güncellemesine göre insan genomlarında toplam 324 milyon varyant mevcuttur. Ayrıca 2008-2015 yılları arasında yürütülen ve 26 farklı popülasyondan 2504 kişinin genetik varyasyonlarını listelemiş olan 1000 Genom projesine göre bağımsız 2 kişinin genomu karşılaştırıldığında %0,6’lık bir fark ile yaklaşık 20 milyon baz çiftinde varyasyon görülür (https://www.internationalgenome.org). Bu varyasyonların çoğu nötrdür, bir kısmı ise fiziksel özellikler, metabolik çeşitlilik, çevresel uyaranlara verilen tepkiler gibi bireysel farklılıklar ile ilişkilidir. Ancak pek azı belirli bir hastalıkla ilişkilidir. Bu sebeptendir ki günümüzde hastalıkla ilişkili varyasyonların tespiti bireyin varyasyon havuzundan doğru varyasyonu bir olta ile avlamaya benzer. Doğru oltanın yani varyasyona ait doğru tespit teknolojisin kullanılması son derece önemlidir.
Varyasyonlar ve terminoloji
‘Mutasyon’ ifadesini kullandığımız zaman aslında DNA dizisinde oluşan bir değişimin kalıcı olarak bir sonraki nesle geçtiğini veya hücre bölünmesi yoluyla aktarılabildiğini söylemiş oluruz. Dolayısı ile genetik varyasyonlar çeşitli mutasyon mekanizmaları vasıtası ile ortaya çıkar. Yine de mutasyon terimini hastalıkla ilişkili bir varyasyon tespit ettiğimiz zaman kullanmayı severiz. ‘Hastanın mutasyonu bulundu’ gibi bir ifade yaygın olarak kullanılsa da günümüzde konusunda uzman ekiplerin oluşturduğu konsorsiyumlar mutasyon teriminin hastalık yapıcı varyasyon anlamında kullanılmamasını önermektedirler (Tıbbi Genetik ve Genomik Amerikan Koleji (ACMG) kılavuzu; 2015). Bunun yerine patojenik veya tersi durum için selim varyasyon ifadeleri kullanılmalıdır. Eğer varyasyonun hastalıkla ilişkili olup olmadığına dair yeterince kanıt yok ise o zaman önemi bilinmeyen varyasyon (variant of unknown significance; VUS) sınıfından bahsedilir.
İnsan genomundan, özellikle nükleer genomdan bahsediyorsak insanın diploid bir organizma olduğunu her zaman için aklımızın bir köşesinde tutmalıyız. Diploid olmanın önemi yukarıda da bahsedildiği gibi erkeklerin X ve Y kromozomları hariç tüm nükleer genomumun çift kopya olmasıdır. Dolayısı ile genomdaki bir varyasyonun iki kopyada mı yoksa tek kopyada mı olduğu önem kazanmaktadır. Kromozomlar üzerindeki bölgeleri tanımlarken ‘lokus’ terimi kullanılır. Bireyin farazi bir lokusu annesinden ve babasından gelen 2 ayrı kromozom üzerindedir. Eğer bir lokusta yer alan bir varyasyon hem anneden hem de babadan gelen kromozomlar üzerinde özdeş halde ise birey homozigot, eğer farklı ise heterozigot olarak adlandırılır. Bir erkekte X kromozomu sadece annesinden kalıtılır. Dolayısı ile X kromozomu üzerinde yer alan bir varyant homolog bir eşi olmadığından dolayı erkekler için hemizigottur. Bir lokus üzerinde yer alan bir varyasyonun her bir farklı DNA dizilimi o varyasyonun farklı alelleri olarak bilinir.
Polimorfik ve nadir aleller
Bir varyasyona ait farklı aleller bireyler arasında görülme sıklıkları açısından çeşitlilik gösterebilir. Eğer bir varyasyonunun yaygın aleli hemen hemen tüm bireylerde gözüküyorsa bu lokus monomorfik olarak bilinir. Dolayısı ile bu varyasyona ait diğer aleller toplumda nadir olacaktır. Eğer bir varyasyona ait aleller yaygınsa yani sıklıkları toplumda %1’in üzerinde ise bu aleller polimorfik olarak adlandırılır. Polimorfizim bazen hastalıkla ilişkili olmayan ya da selim olan varyasyon anlamında kullanılır. Çoğu durumda polimorfik alellerin özellikle nadir hastalıkları açıklaması beklenmez ama polimorfik bir varyasyonun kompleks hastalıklar için risk faktörü olabileceği düşünüldüğünde polimorfizim ifadesinin daha dikkatli kullanılması gerekliliği anlaşılabilir. Ayrıca Ailesel Akdeniz Ateşi gibi tek gene bağlı ve kompleks olmayan bir hastalık bazı toplumlar için yaygın hastalık sınıfına girebilir. Bu durumda alel frekansları nispeten yüksek hatta polimorfik varyantların hastalıkla ilişkisi söz konusu olabilir. Günümüzde alel sıklıkları ile ilgili kullanılan yaygın bir değer de minör alel frekansı (MAF) olarak bilinen ve bir varyasyona ait ikinci en sık alelin frekans değeridir.
Varyasyon türleri
Genomumuzdaki varyasyon türlerini üç temel başlık altında inceleyebiliriz: (i) Kısa genetik varyasyonlar, (ii) yapısal varyasyonlar ve (iii) sayısal kromozom anomalileri.
(i) Kısa genetik varyasyonlar (short genetic variation; SGV) tek nükleotid seviyesinde değişimler, küçük çapta insersiyon ve/veya delesyonları ve kısa tandem tekrarları içerir. Kısa tandem tekrarlar yazının ilerleyen kısmında ‘Trinükleotid Tekrar Hastalıkları’ başlığı altında anlatılacaktır. SNP olarak kısaltılan ve snip olarak okunan tek nükleotid seviyesindeki değişimler tüm varyasyon türleri içerisinde belki de en çok çalışılan varyasyon grubu olup, sadece tek bazdaki değişimi işaret eder. Bir bireyde ortalama bin nükleotidde bir sıklıkta yani kaba bir hesapla toplam 4 ila 5 milyon kadar SNP bulunur. İnsanlarda toplam 100 milyonu aşkın farklı SNP olduğu bilinmektedir. SNP’de bir pürin bazı diğer bir pürin bazı ile yer değiştirirse veya aynı durum pirimidin bazları arasında olursa bu durum transizyon adını alır. Pürin ve pirimidin bazları arasında bir değişim söz konusu ise bu bir transversiyondur. İnsersiyon ve/veya delesyonlar baz çiftlerinin bir sekansa eklenmesi veya bu sekanstan eksilmesidir; birkaç baz çiftinden binlerceye kadar değişen boyutlarda olabilir. Bilinen tüm SGV’lere NCBI-dbSNP veritabanı üzerinden erişilebilir (https://www.ncbi.nlm.nih.gov/snp/). SGV’lerin protein üzerine etkileri arasında amino asit değişimine yani yanlış anlama (missense) yol açma veya olması gereken amino asidin yerine erken bir dur kodonu gelmesi (nonsense) sayılabilir. Küçük çapta insersiyon ve delesyonların etkileri ise özellikle okuma çerçevesi açısından önemlidir. Bu insersiyon ve/veya delesyonun etkilediği baz sayısı üç ve üçün katları seviyesinde ise okuma çerçevesi bozulmaz sadece bazı amino asitler eksiktir veya yenileri eklenmiştir (in-frame). Eğer etkilenen baz sayısı 3’ün katlarından değilse bu durumda genetik kodun okuma çerçevesi bozulur (frameshift). Bu tarz çerçeve kaymasına sebep olan varyasyonlar sonucunda genelde erken bir dur kodonuna rastlanır ve gen anlatımı sekteye uğrar. Örneğin distrofin genindeki delesyonlar okuma çerçevesini bozarsa (frameshift) güdük ve sorunlu bir protein üretimine ile ağır Duchenne Musküler Distrofisi’ne neden olur. Okuma çerçevesini koruyan delesyonların varlığı ise genelllikle daha ılımlı Becker Musküler Distrofisi’ne neden olabilir. Distrofin proteinini kodlayan DMD geni X kromozomu üzerinde 2 milyondan fazla baz çiftine yayılan genişliği ve 80'e yakın ekzonu ile bilinen en geniş genlerdendir (Bakınız: Kas ve Nöromusküler Kavşak Hastalıkları). SGV’ler bazen de genlerin kontrol bölgelerine denk gelip gen anlatımını etkileyebilir. Bir SGV kırpılma bölgelerindeki konsensüs dizileri etkiliyorsa bu durumda öncül RNA’nın doğru kırpılmasını, dolayısı ile doğru ORF’nin oluşmasını engelleyebilir.
Yapısal varyasyonlar en az 1 kilobazlık bir DNA bölgesini kapsayan inversiyonlar, dengeli translokasyonlar ve genomik dengesizliklerdir. Genomik dengesizlikler sıkça kopya sayısı değişiklikleri (copy number variations; CNVs) adıyla bilinir ve ilişkili DNA parçasının kaybı veya kazancı sonucunda genomda ölçülebilir bir değişikliğe sebep olurlar (https://www.ncbi.nlm.nih.gov/dbvar/content/overview/).
Sayısal kromozom anomalileri ise anöploidi (ör., monozomi, trizomi), öploidi (ör., tetraploidi) veya monoploidi gibi kromozom sayılarını doğrudan etkileyen durumlardır. Farklı varyasyonları tespit edebilmek için farklı yöntemler uygulamak gerekebilir.
Bir bireye ait varyasyonlar germ hattı veya edinsel yani somatik hücre kaynaklı olabilir. Germ hattı kaynaklı varyasyonların çoğu bireyin anne ve/veya babasından kalıtılır. Bir kısmı ise zigot oluşmadan önce veya oluştuktan sonra kendiliğinden yani de novo olarak oluşur. Ebeveynlerde bulunmayan de novo varyasyonlar eğer zigot oluştuktan sonra ortaya çıktılarsa gelişen birey bu varyasyon açısından mozaik olur. Yani belli dokularında bu varyasyonu taşırken belli dokularında taşımaz. Somatik varyasyon ise germ hücreleri haricindeki vücut hücrelerinde, yani somatik hücrelerde yine de novo olarak ortaya çıkar. Eğer ortaya çıktığı hücre mitoz açısından aktif ise, bu hücreden köken alan hücre hatlarında bu varyasyon gözlenir. Aynı dokuda varyasyona sahip olmayan hücre hatları da bulunacağı için bu doku için sınırlı bir mozaiklik söz konusu olur. Somatik varyasyonlar sonraki kuşağa geçmez, biyolojik sonuçları yalnızca ortaya çıktıkları bireyi etkiler. Kalıtsal olmayan sporadik kanserlerin gelişiminde somatik varyasyonlar rol oynar. Varyasyon germ hücrelerinde mevcut ise bireyin çocuklarına aktarılabilir.
Trinükleotid Tekrar Hastalıkları
Kısa tandem tekrarlar olarak da bilinen mikrosatellitler kısa bir DNA dizisinin art arda yani tandem olarak tekrar etmesini ifade eder. Tekrar eden DNA birimi genellikle 2 ila 5 baz arasındadır. Mikrosatellitler genomumuzun pek çok bölgesinde, özellikle gen dışı alanlarda mevcuttur. Replikasyon ve rekombinasyon sırasında hataya açık olmaları yani mutasyon hızlarının yüksek olmasıyla evrimsel süreçte farklı mikrosatellitlerin pek çok polimorfik aleli ortaya çıkmıştır. Dolayısı ile mikrosatellit profili analizi bireylerin genetik olarak tayininde, özellikle de adlı tıp uygulamalarında DNA parmak izinin ortaya çıkarılmasında sıkça başvurulan bir yöntemdir. Nörogenetik alanında mikrosatellitler için ayrı bir başlık açmamamızın nedeni mikrosatelitllerin bir türü olan trinükleotid tekrar uzamalarının nörolojik hastalıkların bazılarında ortak bir mekanizma olarak karşımıza çıkmasıdır. Bu hastalıklarda, normalde belli bir sayıda tekrar eden CAG, CTG veya GAA gibi üçlü nükleotidler hasta bireylerde çok daha fazla artmış olarak bulunur. Bazen trinükleotid tekrarının ebeveynden çocuğa aktarılırken arttığını görürüz. Bu artış neticesinde çocukta ebeveynine göre hastalık bulgularının daha erken başladığını ve hatta hastalığın seyrinin daha ağır olduğunu görürüz. Bu durum antisipasyon yani genetik beklenti olarak adlandırılır. Antisipasyon nörolojik hastalıklar açısından önem taşımaktadır. Tarihsel olarak bu terim ilk önce otozomal dominant miyotonik distrofide sonraki jenerasyonlarda gözlenen klinik ağırlaşmayı tarif etmek için kullanılmıştır. Tipik olarak bu gene sahip babaların sadece erken yaşta kataraktı vardır. Kız çocuklarında ılımlı kas güçsüzlüğü ve miyotoni, kızların çocuklarında ise zihinsel yetmezlik, miyotoni ve ağır güçsüzlük olabilir. Klinik olarak iyi bilinen, jenerasyonlar boyunca hastalık şiddetinin artması durumunun artık biyolojik temeli bulunmuştur. Kromozom 19 üzerindeki distrofia miyotonika protein kinaz (DMPK) geninin kodlamayan bölgesindeki trinükleotid tekrar genişlemesi sonucu miyotonik distrofi oluşur. Genel popülasyonda normalde 5 ile 34 arası CTG trinükleotid tekrarı vardır. 100 tekrardan fazla genişleme miyotonik distrofi hastalığına neden olur. Tekrar sayısının artması, binlerce tekrarlama daha ağır hastalık formu ile ilişkilidir. Genişleme boyutu babadan oğula değişebilir ve jenerasyonlar arasında artma, antisipasyon denen bu klinik fenomene neden olur (Ayrıca bakınız: Kas ve Nöromusküler Kavşak Hastalıkları).
Kennedy'nin X'e bağlı spinobulber musküler atrofisi, frajil X sendromu, Huntington hastalığı, herediter ataksilerin birçok formu (spinoserebellar ataksi SCA-1, -2, 3,-6, -7, -8, -12 ve -17 ve Friedreich ataksisi) ve dentatorubropallidoluysiyan atrofinin (DRPLA) nadir bir formu gibi birçok nörogenetik hastalığa instabil üçlü tekrarların neden olduğu gösterilmiştir. Şekil 5’de tekrar artışına bağlı ortaya çıkan genetik hastalıklar ve gen üzerindeki yerleşimleri gösterilmiştir.

Şekil 5. Tekrar artışına bağlı nörolojik hastalıklar ve gen üzerindeki yerleşimleri
Huntington hastalığı (HH) gibi tekrar artışına bağlı hastalıkların pek çoğunda, hastalıkla ilişkili genin kodlayan bölgesinde yer alan CAG tekrarının genişlediğini görürüz. CAG kodonu glutamin (Q) amino asidini kodladığı için bu hastalık sınıfı poliQ olarak da bilinir. Tekrar artışı olsa da tekrar eden birim üç bazdır ve okuma çerçevesini bozmaz. Böylece protein ürününde uzun bir poliQ alan üretilir. poliQ bölgesi artışına sahip proteinlerin hücreye zarar veren yeni bir fonksiyonu olur (gain of function).
Bir genişlemenin varlığı hastalık ilişkisini işaret etse de bazen sonuçların yorumu karmaşık olabilir. Genişleme bulunması ile hastalık semptomlarının gelişmesi her zaman aynı anlama gelmeyebilir. Örnek olarak HH’de patojenik alellerde CAG trinüklotid tekrar sayısı 36’dan fazladır. Normal HH alelleri için ise tekrar sayısı 10 ila 26 adet CAG olarak değişir. Eğer bir kişide 27 ila 35 tekrar sayısı tespit edilirse, bu kişi HH açısından gri alandadır ve bu skaladaki tekrar sayısı mutasyon öncesi yani premutasyon olarak bilinir. Bu birey hastalık belirtilerini taşımasa da bireyin premutasyonlu alelleri bir sonraki nesle aktarılırken artış gösterebilir ve hastalığa yol açabilir.
Hastalık fenotipi ile tekrar sayısı arasındaki ilişki her zaman açık olmayabilir. Bu belirsizlik hasta ve hekimi için zorluk yaratabilir. Özet olarak, tekrar boyutunun direkt olarak test edilebilmesi çok önemlidir. Ancak bu kalıtımsal sendromlarda riskli olan kişileri tespit edebilmek, yani bu bilginin prediktif tanı amaçlı kullanılmasında pratik ve etik zorluklar mevcuttur.
Genetik ve fenotipik heterojenite
Genetik heterojenite farklı genlerde yer alan patojenik varyasyonların benzer bir klinik tablo ile ilişkili olması durumunu ifade eder. Örneğin Charcot-Marie-Tooth herediter nöropati tip 1 (CMT1) en az beş farklı gendeki (PMP22, MPZ, LITAF, EGR2, NEFL) farklı patojenik varyasyonların sonucu oluşabilir (Bakınız: Polinöropatiler). Benzer şekilde herediter ataksiler de yüksek genetik heterojenite gösterir (Bakınız: Ataksiler). Dominant, resesif, X’e bağlı ve hatta mitokondriyal olarak kalıtılan 50’nin üzerindeki gen herediter ataksi ile ilişkilendirilmiştir. Klinik muayene genellikle hangi genin etkilendiğini belirleyemez. Dolayısı ile genetik tanı için pek çok genin bir arada taranabileceği yeni nesil dizileme esasına dayalı teknikler gerekir.
Fenotipik heterojenitede ise tek gende yer alan farklı varyasyonlar farklı fenotiplere yol açar. Örneğin periferik miyelin protein 22 (PMP22) 17p12 bölgesinde yer alan ve 22 kilo-daltonluk bir protein kodlayan otozomal bir gendir. PMP22 proteini periferik sinir sistemi miyelininin yaklaşık %2-5’ini oluşturur. PMP22 ile ilişkili farklı nöropatiler tanımlanmıştır: (i) PMP22 gen duplikasyonları bir Charcot-Marie-Tooth herediter nöropati tip 1’e (CMT1A) neden olur. (ii) PMP22 delesyonları basınca duyarlı herediter nöropati (HNPP) ile sonuçlanmaktadır (iii) PMP22’deki nokta mutasyonları etkisine göre CMT1A veya HNPP’ye sebep olabilir (Bakınız: Polinöropatiler).
Varyasyonların tespiti için kullanılan yöntemler
Varyasyonların tespitinde tek gen analizinden tüm genomu hedefleyen tekniklere kadar geniş bir yelpazede farklı yöntemler uygulanabilir. Her bir yöntemin kapasitesini ve kısıtlılıklarını bilmek moleküler tanı hedefine ulaşabilmek için elzemdir. Burada sırasıyla anlatılan yöntemler hedefledikleri alanın hacmine göre küçükten büyüğe yani tek genden tüm genoma doğru sıralanmışlardır.
Tek gen analizi
Tek gen analizi geleneksel moleküler tanı yöntemidir. Klinik olarak en olası genden başlanarak tüm şüpheli genler taranır. Genlerin özellikle ekzonik bölgeleri ile komşuluğundaki kırpılma ile ilişkili intronik bölgelerin polimeraz zincir tepkimesi (PZT) ile çoğaltılması, ardından Sanger yöntemi (Şekil 6) ile dizilenmesi yaygın bir uygulamadır. Aranılan varyasyonun türüne göre Sanger dışı yaklaşımlar gerekebilir. Duchenne tipi kas distrofisinde yaygın olarak gözlenen delesyonların MLPA (Multiplex Ligation-dependent Probe Amplification) yöntemi ile taranması, Unverricht Lundborg hastalığında promoter bölge tekrar sayısı artışının uzun PZT hatta Southern blot yöntemi ile incelenmesi ve amiyotrofik lateral skleroz ile ilişkili C9orf72 gen bozukluğunun tekrara özgü PZT (repeat primed polymerase chain reaction) ile tespit edilmesi sayılabilir.
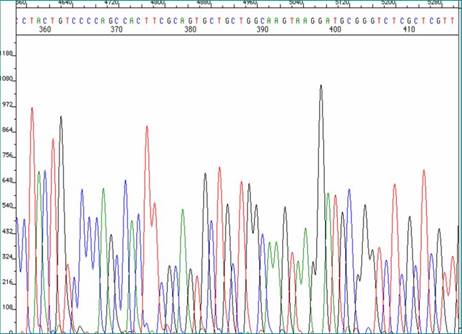
Şekil 6. DNA sekanslama (dizileme) örneği. Her bir farklı renkteki dalga farklı bir baza işaret etmektedir. Normal dizi bilindiğinden farklı olan baz veya bazlar anlaşılabilir.
Yeni nesil dizileme yaklaşımları
Yeni nesil dizileme (YND) yaklaşımları ile farklı hedef bölgeler bir arada dizilenebilir. Yüksek hacimli analizin getirisi olan büyük veri biyoenformatik olarak analiz edilir. YND’nin hedefi genomun farklı bölgeleri olabilir: Panel/hedefli dizilemede ilgilenilen gen grubu, ekzom dizilemede genomdaki tüm ekzonlar, RNA dizilemede çalışılan hücre grubuna ait RNA profili, genom dizilemede ise tüm genom dizilenerek analiz edilir. Bu yöntemler ile elde edilen pek çok varyasyonun arasından hastalıkla ilişkili varyasyonu tespit etmek gerekir. Klinik bilgi, aile hikayesi, hastalığın sıklığı gibi pek çok ipucu doğru varyasyonun bulunmasına yardımcı olabilir. Bulunan varyasyon Sanger dizileme ve çoklu ligasyona bağlı prob amplifikasyonu (MLPA) gibi farklı moleküler yaklaşımlarla konfirme edilir. Ardından mümkün ve gerekli ise aile segregasyonu yapılır ve varyasyonun fonksiyonel etkisi incelenir. Yapılan tüm analizler güncel kılavuzlar doğrultusunda ortak paydada birleştirilir ve varyasyonun hastalıkla ilişkisi analitik olarak belirlenir.
CNV’lerin tespiti
CNV’ler insanlarda genetik çeşitliliği oluşturan ve genomda kantitatif bir dengesizlik oluşturan önemli bir varyasyon grubudur. CNV tespiti “Array” Karşılaştırmalı Genomik Hibridizasyon (Array Comparative Genomic Hybridization; aCGH), SNP “array” ve YND gibi yöntemlerin gelişmesi ile ivme kazanmıştır. CNV’ler çoğunlukla bir fenotiple ilişkili olmasalar da bazı CNV’lerin ciddi klinik etkileri vardır. YND verisinin biyoenformatik araçlar yolu ile CNV’ler açısından değerlendirilmesi epilepsi gibi pek çok nörolojik hastalıkta ilişkili genetik bozukluğu tespit edebilmektedir.
Genetik analize katkı sağlayan klinik ve aile bilgilerinin alınması
Fenotipin tanımlanması
Fenotip bir bireyde gözlenen biyokimyasal, fizyolojik ya da klinik özellikleri işaret eder. Genotip ise belirli bir lokustaki alel içeriğini belirtir. Patojenik bir varyasyonun klinik yansıması bu varyasyon ile ilişkilendirilebilecek herhangi bir fenotipik belirtiyi ifade eder. Bu yansımalar arasında nöbet, zihinsel yetmezlik, deri lezyonları, elektroensefalografi (EEG) bozuklukları, hareket bozukluğu, demans, ya da yavaş sinir ileti hızları sayılabilir. Öncelikle indeks olgunun çok ayrıntılı değerlendirilmesi gerekir. Hastanın özgeçmişinin değerlendirilmesine antenatal dönem ile başlanmalıdır. Hastalık bulgularının başlangıç yaşı, bulguların detaylı tanımlanması, nörolojik ve kognitif bulgular, nörogörüntüleme, elektrofizyoloji (EEG, EMG), nöropsikolojik özellikleri kayıt altına alınmalıdır. Bu bilgilerle sendrom tanımlanmaya çalışılmalıdır. Söz konusu tablonun genetik kökenli mi olduğunun yanıtı her zaman kolay verilemez. Dikkatli aile öyküsü ve aile ağacı çalışması hastalığın kalıtım şeklini ortaya koymaya yardımcı olabilir. Kalıtım şekli özellikle geç başlangıç yaşı olan hastalıklarda üst kuşakların kaybedilmesinden dolayı zor olabilir. Ancak ailede benzer bir öyküsünün olmamasının hastalığın genetik olmadığı anlamına gelmediği akılda tutulmalıdır. Genotip her zaman fenotiple sonuçlanmak zorunda değildir. Yani aynı varyasyonu taşıyan bazı kişiler klinik olarak etkilenmişken diğerleri etkilenmemiş olabilir. Bu durum düşük penetrans terimi ile ifade edilir. Örneğin bilinen patojenik bir varyantı taşıyan 100 kişiden 80’i klinik belirti veriyorsa bu varyasyonun penetransı %80’dir. Penetrans yaşa göre de değişebilir. Huntington hastalığına neden olan gen 20 yaşında %10 penetrans gösterirken, 80 yaşında %90 penetrans gösterebilir. Detaylı klinik inceleme gerektiren diğer bir genetik özellik de varyasyonun değişken ekspresivitesidir. Bu durumda aynı patojenik varyasyona sahip tüm bireyler etkilenmişlerdir ancak klinik bulguları ağırdan hafife kadar uzanan bir skalada değişkenlik gösterir. Hatta subklinik diyebileceğimiz ancak detaylı bir muayene ile anlaşılabilecek vakalar mevcuttur. Örneğin tuberosklerozis’de bazı olgularda klinik tanı ancak dikkatli cilt muayenesi ve beyin manyetik rezonans incelemesi (MRI) ile sağlanabilir. Benzer şekilde asemptomatik ancak belirli bir genetik bozukluğa sahip epilepsili olgularda nöbet olmadan sadece EEG bozuklukları olabilir.
Aile hikayesinin alınması
Aile bilgisi her genetik çalışmada olduğu gibi nadir nörolojik hastalıkların araştırılmasında da son derece önemlidir. Hasta bireylerin ayırıcı tanısının yapılması, aile hikayesinin alınması, özellikle ebeveyn akrabalığı gibi bilgilerin sorgulanması ve aile ağacının doğru çizilmesi son derece önemlidir. Pek çok nörolojik hastalığın geç yaşta başladığı düşünüldüğünde aile ağacı çizilirken bireylerin yaşlarını kaydetmek de son derece önemlidir. İndeks olgunun işbirliği ile tüm aile bireyleri, özellikle aile bilgilerine hakim olmaları nedeniyle yaşlı kadınlar dinlenmelidir. Ulaşılamayanlara telefon veya meslektaş yardımı ile ulaşılmalıdır. Mümkünse o bölgeye gidilmesi birçok bilgiye ulaşılmayı sağlayabilir. Etkilenen ve etkilenmeyen her bireye ulaşılmalıdır. Ailede her birey indeks olgu ile aynı fenotipi göstermeyebilir. Örneğin idyopatik jeneralize epilepsilerde fenotipik değişkenlikler görülebilir, aynı ailede jüvenil miyoklonik epilepsi ve çocukluk çağı absans epilepsili bireylerle karşılaşılabilmektedir. Bu arada beyin sarsıntısı sonrası fokal epilepsi gelişmesi gibi ailesel olgulardan tamamen bağımsız fenotipik özellikler gösteren bireyler de bulunabilir.
Aile ağacının (pedigri) çizilmesi
Hastanın diğer bireyler ile ilişkisini anlaşılır bir şema halinde göstermesi açısından önemlidir. Tüm bu bilgilerle aile ağacı hazırlanmalı, ayrıntılı olmalı, her bireyden alınan bilgi ile hazırlanmalıdır. Tüm bilgiler işaretlenmelidir (İsim, doğum tarihi, ölü doğumlar, düşükler, akraba evlilikleri, evlat edinmeler). Belirli aralıklarla doğumlar, yeni etkilenen bireyler yenilenmelidir. Sonuç olarak hastalığın kalıtım kalıbı belirlenebilir ve moleküler genetik incelemelere daha sağlıklı olarak karar verilebilir. Aile ağacı hazırlanmasında kullanılan semboller Şekil 7’de gösterilmiştir.

Şekil 7. Aile ağacı çiziminde kullanılan bazı semboller örnek bir aile ağacı (pedigri) üzerine yerleştirilmiştir.
Kalıtım kalıbının belirlenmesi: Kalıtım kalıbının doğru bir şekilde belirlenmesi yapılan genetik analize ait verinin değerlendirilmesinde ve tekrarlama riskinin tayininde önem arz eder.

Şekil 8. Farklı kalıtım
kalıpları gösteren aile ağacı örnekleri görülmektedir. (a) Otozomal dominant,
(b) otozomal resesif, (c) X’e bağlı resesif.
1. Otozomal dominant (OD) kalıtım modeli (Şekil 8a): Otozomal varyasyonlar için homolog kromozomlardan herhangi biri üzerinde patojenik bir alelin varlığı görülmesi için yeterlidir. Bu kalıtım şeklinin özellikleri:
- Etkilenen bireyler varyasyon için heterozigottur.
- Etkilenen bireyin her bir çocuğunun %50 hasta olma olasılığı vardır.
- Tüm çocukların, cinsiyete bakılmaksızın etkilenme riski aynıdır. Hastalık sonraki çok sayıda kuşakta ortaya çıkar (vertikal yayılma).
- Azalmış penetrans özelliği gösterebilir, yani bazı bireyler patojenik aleli taşır ama hastalanmaz. Fakat kendi çocuğuna aktarma riski aynıdır (kuşak atlama).
- OD kalıtımda yeni mutasyonlar yaygındır. Yani varyasyon bir çocukta de novo olarak kendi kendine oluşabilir. Özellikle üreme yaşından önce ortaya çıkan ağır veya ölümcül hastalıklar genellikle ebeveynlerde olmayan de novo dominant mutasyonlar ile ortaya çıkar.
- Değişken ekspresivite olabilir.
- Dominant etkili varyasyonlar HH’de örneklendiği gibi anormal bir fonksiyon kazanan (gain-of-function) bir gen ürününe sebep olabilirler. Bazen de dominant varyasyon ile gen ürünü fonksiyon kaybedebilir (loss-of-function). Fonksiyon kaybı olan durumda dominant bir hastalığın ortaya çıkma sebebi tek ve normal kopyadan üretilen gen ürününün yeterli gelmemesidir. Bu durum haployetersizlik (haploinsufficiency) olarak bilinir.
Nörofibromatozis, tuberoskleroz, HH, herediter ataksilerin çeşitli formları ve CMT tip I, miyotonik distrofi, selim neonatal konvulziyonlar, ailesel Alzheimer hastalıklarının bazı formları ve frontotemporal demanslar (FTD) otozomal dominant kalıtımla geçen nörolojik bazı hastalıklara örnektir. OD hastalık için eğer homozigot bireyler varsa bu bireylerin klinik göstergeleri daha ağır olabilir. Herediter hiperkolesterolemide olduğu gibi heterozigotlarda 48 yaşında miyokard infarktüsü, homozigotlarda ise (her iki ebeveynden de patojenik varyasyon kalıtmış olan) 12 yaşında koroner oklüzyon görülebilir. Bu durum dominant ve resesif hastalıkları ayırt etmeyi güçleştirir. HH örneğinde ise homozigot form heterozigot formlardan kötü değildir.
2. Otozomal resesif (OR) kalıtım modeli (Şekil 8b): Bu kalıtım şeklinde patojenik mutasyon bireyde ancak her iki homolog kromozomda varsa birey hasta olmaktadır. Homolog kromozomlardaki her iki varyasyon özdeş olabilir; bu durumda birey homozigottur. Ebeveyn akrabalığı OR kalıtılan hastalıklarda homozigotluk riskini arttırmaktadır (Şekil 8b). Ebeveynlerin akraba değil de aynı yöreden olması da bu riski arttırılabilir. Ülkemizde ortalama %20’yi aşan akraba evliliği oranı unutulmadan mutlaka etkilenmiş bireylerin ebeveyn akrabalığı, aynı köyden olmaları, göçmenlik durumları gibi bilgiler sorgulanmalıdır. OR hastalıkların diğer bir sebebi de birleşik heterozigotluktur (compound heterozygosity). Bu durumda anne ve baba aynı gen için farklı patojenik varyasyonlar için taşıyıcıdırlar. Hasta çocuğa ise bu 2 farklı ancak aynı gene isabet eden patojenik varyasyonlar kalıtılır.
- OR hastalıklarda bir patojenik bir de normal alele sahip bireyler ‘taşıyıcı’ olarak tanımlanır. Heterozigot taşıyıcılar genellikle klinik olarak normaldirler. Nadiren bazı klinik bulgular görülebilir (manifest heterozigot).
- Taşıyıcının çocuğuna mutant geni aktarma olasılığı % 50’dir.
- Hasta bireyin çocuklarının hepsi taşıyıcı olacaktır.
- Kalıtım anne-baba ve cinsiyete göre etkilenmez.
- Her iki ebeveynin de taşıyıcı olması durumunda: (i) Her bir çocuk %25 olasılıkla patojenik varyasyonu homozigot olarak alırlar; (ii) Her bir çocuk %50 olasılıkla taşıyıcı olur; (iii) %25 olasılıkla normal aleller her bir çocuğa homozigot olarak aktarılır ve çocuklar etkilenmez.
- OR hastalıklar genellikle tek jenerasyonda, tipik olarak aynı kuşakta kardeşler arasında görülür (yatay yayılım). Ancak küçük ailelerde özellikle tek çocuklu anne-baba-çocuk üçlülerinde (trio) OR hastalıklar izole ya da sporadik vakalar olarak görülebilir. OR hastalıklar yaygın akraba evliliği olan geniş ailelerde çok sayıda kuşakta da görülebilir. Fenilketonüri, Tay-Sachs hastalığı, Lafora hastalığı, Unverrricht-Lundborg hastalığı, infantil spinal musküler atrofi, Wilson hastalığı, Friedreich ataksisi OR nörolojik hastalıklara örneklerdir.
- Çekinik etkili varyasyonlar genellikle fonksiyon kaybettiren yani ‘loss-of-function’ etkisindedir. Bu durumda ya gen ürününün aktivitesi sekteye uğramış ya da tamamen ortadan kalkmıştır. Taşıyıcı bireylerin genellikle semptom göstermemesinin sebebi tek kopya gen ürününün normal biyolojik süreçlerle uyumlu olmasıdır. Çift kopya varyasyona sahip hasta bireylerde ise ya hiç gen ürünü yoktur ya da tamamen bozulmuş etkidedir.
3. X’e bağlı resesif kalıtım modeli (Şekil 8c): X’e bağlı kalıtılan hastalıklarda genetik bozukluk X kromozomu üzerinde bir gendedir. Heterozigot kadınlar genellikle klinik olarak normaldirler, bazen hafif hastalık bulguları vardır. Erkekler X kromozomu için hemizigotturlar yani sadece anneden gelen tek kopya X kromozomları vardır. Dolayısı ile annede çekinik etkideki patojenik bir varyasyon oğluna aktarılırsa, bu bireyde varyasyonla ilişkili fenotip kendini gösterecektir. X’e bağlı geçişli hastalıklarda hemen hemen normal fenotipli taşıyıcı kadınlardan geçen çok sayıda kuşaklarda etkilenmiş erkekler görülür. Asla erkekten erkeğe geçiş yoktur (Şekil 8c). Menkes’in “kinky” (kıvırcık, dolaşık) saç sendromu, Pelizaeus-Merzbacher hastalığı, frajil X sendromu, Kennedy’nin spinal-bulber musküler atrofisi, Duchenne ve Becker musküler distrofileri ve adrenolökodistrofi X’e bağlı kalıtılan resesif hastalıklara örnektir.
- X’e bağlı resesif varyasyonu taşıyan her kadın taşıyıcıdır ve genellikle klinik bulgu göstermez.
- Varyasyonu taşıyan her erkek hasta olacaktır (Tek bir X kromozomu olduğu için)
- Taşıyıcı annenin tüm çocuklarının varyasyonu kalıtma olasılığı % 50’dir (kızlar bu oranda taşıyıcı, erkekler ise bu oranda hasta olacak şekilde).
- Etkilenmiş babanın kız çocuklarında taşıyıcı (otomatik olarak anormal X kromozomunu kalıtırlar) olma riski %100’dür. Erkek çocukların ise babadan bu varyasyonu kalıtma olasılığı yoktur (çünkü babadan Y kromozomunu alırlar).
X’e bağlı kalıtılan hastalıklarda bazen heterozigot kadın taşıyıcılar, rastlantısal olmayan X inaktivasyonu fenomeni nedeniyle klinik bulgular gösterebilir. Normalde kadınların her bir hücresinde anneden veya babadan gelen X kromozomlarından biri inaktif halde tutulur. Hangi X’in inaktif durumda olacağı rastlantısal olarak embriyonik dönemde belirlenir ve hücreler bölündükçe bir nevi hücre hafızası ile bu bilgi aktarılır. Dolayısı ile kadınlarda tüm hücrelerde ortalama yarı yarıya anne ve babadan gelen X’in aktif olmasını bekleriz. Bu durum kadınların X kromozomu için pratikte mozaik olmasına sebep olur. Rastlantısal olmayan X inaktivasyonu hastalığın etkilediği dokularda olur ve taşıyıcı kadının bu hücrelerinde genetik bozukluğa sahip X kromozomu aktif durumda kalırsa bu kadın hastalık belirtileri gösterebilir.
4. X’e bağlı dominant kalıtım: Bu durumda genellikle heterozigot kadınlarda hastalık gözlenir. Ancak erkekten erkeğe geçiş burada da yoktur. X kromozomu üzerinde yer alan ve dominant GJB1 patojenik varyasyonları ile ilişkili CMT (CMTX1) bu duruma bir örnektir.
- Tek bir mutant kopyayı taşıyan kadın veya erkek etkilenecektir, ancak bu durum genellikle hemizigot erkeklerde daha şiddetlidir
- Etkilenen annenin erkek veya kız çocuklarının % 50’sine mutasyonu kalıtma riski vardır.
- Etkilenen babanın tüm kız çocukları hastadır. Oysa erkek çocukları etkilenmez.
- X dominant hastalıkların çoğu erkeklerde ölümcül olabilir (male lethality). Hatta yaşamla bağdaşmadığı için hiç erkek hasta doğmayabilir. Dolayısı ile ailede sadece kadın hastaların varlığı X-dominant bir hastalığı işaret edebilir.
- X dominant hastalıkların çok özel bir türü patojenik varyasyon sadece kadında ise kendini gösterir. Yani bu varyasyona sahip bir erkek ilginç olarak etkilenmemiştir. Sadece kadınlarda gözüken epilepsi (female restricted epilepsy) bu özel duruma bir örnektir. Bu hastalık X kromozomu üzerinde yer alan PCDH19 geninden kaynaklanır ve Dravet sendromunun bir alt türü olarak bilinir.
5. Y kromozomunun varlığı erkek cinsiyetinin oluşması ile ilgilidir. Bilinen herhangi bir nörolojik hastalık Y kromozom geni ile ilişkili değildir. XYY karyotipli erkeklerde, davranış problemleri görülür ancak zihinsel yetmezlik yoktur.
6. Mitokondriyal kalıtım: Daha önce anlatıldığı gibi mitokondriyal DNA anne yumurtasının sitoplazmasından kalıtılır, ancak spermden kalıtılmaz. Etkilenmiş annenin mitokondri varyasyonunu her bir çocuğuna geçirmesi dolayısı ile de tüm çocuklarının etkilenmesi beklenir. Ancak mitokondri genomunun hücrelerde çoklu olarak bulunduğu unutulmamalıdır. Bir annenin hem varyasyonlu hem de normal mitokondrisi bulunabilir. Mitokondri mozaikliği olarak düşünülebilecek bu durum heteroplazmi terimi ile ifade edilir. Dolayısı ile bu heteroplazmik havuzdan çocuğa ne kadar varyasyon aktarıldığı ve bu varyant mitokondrinin hangi dokularda daha yaygın olduğu değişken olacağı için mitokondriyal hastalıklar için düşük penetrans ve değişken ekspresivite durumu kaçınılmazdır. Mitokondriyal DNA varyasyonları, özellikle mitokondri DNA delesyonları sadece anneden kalıtılmaz; kendiliğinden yani de novo olarak da ortaya çıkabilir. Bu yeni varyasyonun hangi evrede ortaya çıktığı ve takip eden heteroplazmi durumu yine hastalık şiddetini belirleyecektir. Primer mitokondriyal hastalıklar “ragged red’liflerle seyreden miyoklonik epilepsi (MERRF) sendromunu, mitokondriyal ensefalomiyopati, laktik asidoz ve inme (MELAS) sendromunu, Leber’in herediter optik nöropatisini (LHON), Kearns-Sayre sendromunu (miyopati, retinopati, kardiyomiyopati) içerir. Mitokondriyal proteinlerin pek çoğunun nükleer genler tarafından kodlandığını biliyoruz. Bu genlerdeki varyasyonlar tipik Mendelyen kalıtım kalıpları gösterir ve sekonder mitokondriyopatilerden sorumludur. Progresif eksternal oftalmoplejinin bazı formları buna örnektir.
- Kalıtım ve klinik özellikler heteroplazmi oran ve dağılımına göre değişkenlik gösterir.
- Primer mitokondriyal hastalıklar sadece maternal kalıtılır, paternal kalıtılmaz (sitoplazmik kalıtım).
- mtDNA mutasyonu taşıyan annenin tüm çocukları etkilenebilir veya asemptomatik taşıyıcı olabilir.
7. Multifaktöriyel (çok faktörlü) Kalıtım: Bazı klinik tabloların az sayıda genin kümülatif etkisi sonucu ortaya çıktığı tahmin edilmektedir. Çok genli bu kalıtım modeli poligenik olarak bilinir. Poligenik durumlar çevresel faktörlerden de etkilenme eğilimindedir. Bazı tek gen mutasyonlarının latent taşıyıcıları çevresel ajanlara maruz kalınca açığa çıkabilir. Fenobarbital sonrası görülen akut semptomatik intermitan porfirya buna örnektir. Multipl skleroz için de çevresel tetikleyicilerin genetik immünolojik bir yatkınlık ile etkileştiği bir multifaktöriyel kalıtımdan şüphelenilmektedir.
Multifaktöryel hastalıklar genelde nadir değil yaygın hastalıklardır. Nöral tüp defektleri, yarık damak-dudak gibi doğumsal anomaliler ve erişkin yaşta başlayan hastalıkların çoğunluğu bu özelliği gösterirler. Hipertansiyon gibi sistemik hastalıkların yanı sıra pek çok nörolojik hastalık bu gruptadır. Örnek olarak epilepsilerin çoğu, migren, demansiyel sendromların büyük kısmı verilebilir. Aşağıdaki özellikler kalıtım şeklini belirler:
- Genetik yatkınlık ve çevresel faktörlerin etkileşimi söz konusudur.
- Belirgin bir Mendel tipi (tek gen) kalıtım kalıbına uymaz.
- Poligenik (birden fazla genin) etkileşim söz konusu olabilir.
- Aile içinde fenotip için kümelenme olabilir ve akraba evliliği sıklığını arttırabilir.
- Moleküler genetik temeli belirlemek ve tanı testi geliştirmek zordur.
- Yaygın ve çok faktörlü hastalıkların çalışılması pek çok hasta ve sağlıklı örneğin bir arada çalışılabileceği geniş hastalık odaklı konsorsiyumlar ile yapılır. Yaygın hastalık yaygın varyant hipotezi doğrultusunda yaygın SNP’lerin genom çapında yüksek çözünürlüklü olarak incelenmesi ve genom boyu ilişkilendirme analizlerinin yapılması sıklıkla başvurulan bir yöntemdir (Genome wide association studies; GWAS). YND teknolojisinin gelişmesi ve ucuzlaması ile güncel yaklaşım, bu hastalıklara sahip çok sayıda bireyin bir arada dizilenmesi, böylece hem yaygın hem de nadir varyantların hastalık fenotipi ile ilişkisinin bir arada incelenebilmesidir.
İmprinting (Baskılama)
Genetik hastalıklar konusunda bahsedilmesi gereken diğer önemli bir mekanizma da imprinting, yani baskılamadır. Bu durum aslında kadınların iki X kromozomundan birinin epigenetik bir mekanizma ile baskılanmasına benzer. Ancak imprinting mekanizmasında otozomlardaki belirli bölgeler baskılanır ve X inaktivasyonunun aksine rastlantısal değil, hangi ebeveynden gelen bölgenin baskılanacağı her hücre için belli ve aynıdır. Kısaca genomik imprinting ebeveyn kaynağı belli olacak şekilde alelerin susturulması demektir. Böylece tek alele bağlı gen anlatımı sağlar. İnsanlarda otozomların farklı yerlerine yayılmış 82 bölgenin imprinting mekanizmasına maruz kaldığı düşünülmektedir. İmprinting mekanizması aile ağaçlarında bazı hastalıkların kompleks kalıtım kalıpları göstermesine neden olur. Örneğin erkek bir bireyde annesinden gelen homolog kromozom üzerinde patojenik bir varyasyon varsa ve bu bölge anneye göre imprint mekanizması ile baskılanan bir alan ise, patojenik varyasyon bu erkekte sessiz kalacaktır. Yani varyasyonun bu bireyde penetransı olmayacaktır. Ancak bu birey normal bir şekilde yüzde elli ihtimalle çocuklarına bu varyasyonu geçirecektir. Bu varyasyonu babalarından alan çocuklar ise babadan gelen alel imprinting mekanizması dışında kaldığı için hasta olacaklardır. İnsanlarda imprinting mekanizmasının etkili olduğu hastalıklar arasında Prader-Willi, Angelman, Silver-Russell ve Beckwith-Weidemann sendromları sayılabilir.
Genetik Çalışmalara İlişkin Etik Kurallar
Her tür genetik çalışma ve ailenin değerlendirilmesi, genetik incelemelerin planlanması sırasında etik kurallar titizlikle uygulanmalıdır. Başlangıçta indeks olgu ile görüşülmeli, onayı alındıktan sonra aile bireyleri değerlendirmeye alınmalı, özellikle indeks olgunun işbirliği sağlanarak aile bireyleri ile öncelikle onun görüşmesi beklenmelidir. Her birey, hastalık, yapılacak incelemeler, amaç, olası sonuçlar hakkında ayrıntılı olarak bilgilendirilmelidir. Araştırma, hasta ve ailesine kesinlikle amacından büyük gösterilmemeli, umut verilmemeli, olabildiğince açık davranılmalıdır. Hastalığın hangi ebeveynden aktarıldığını söylemek gibi aile içi sorunlara neden olabilecek açıklamalardan kaçınmak ülkemizde daha da önem taşımaktadır.
Her birey sözlü ve yazılı olarak bilgilendirildikten sonra yazılı olur alınmalı, 18 yaşından küçükler için ebeveyn oluru eklenmelidir. Ayrıca bölgesel etik komiteden onay alınması, çalışma hakkında ayrıntılı bilgi verilmesi gereklidir.
Genetik Testler
Genetik test; şüpheli klinik durumlarda genotip, mutasyon, fenotip ve karyotiple ilgili ve insan DNA, RNA, kromozom, protein veya bazı metabolitleriyle ilgili yapılan testlerdir. Bununla birlikte genetik bir hastalığa tanı koymak için her zaman genetik bir test yapılmayabilir. Klinik olarak değerlendirilen ailelerde yapılacak genetik incelemelere klinisyen ve genetikçi birlikte karar vermelidir. Uygun çalışma gruplarının oluşturulması, klinisyenler ve genetikçiler arasında işbirliğinin sağlanması en önemli aşamadır.
DNA izolasyonu tüm genetik incelemeler için gerekli olan DNA materyelinin elde edilmesini sağlar. Her bireyden 10 ml venöz kan EDTA’lı (EtilenDiamin Tetraasetik Asit) tüplere (mor kapaklı) alınmalıdır. Küçük çocuklarda veya kan vermekten çok korkan erişkinlerde tükürüğün toplandığı özel kapların veya ağız içi epitel dokunun kullanımı da mümkündür. DNA, hazır kitler vasıtası ile fazla bekletilmeden kanda ayrıştırılır. Eğer testi yapacak olan genetik laboratuvarının farklı bir önerisi yoksa, EDTA’lı kan +4°C’de, buzdolabının yumurtalık kısmında bekletilebilir, dondurulmaması önerilir. Her aşama, özellikle örneklerin numaralanması veya barkodlanması son derece önem taşır. Bunun dışında tüm süreçler, kanın alınma tarihi, izolasyon öncesi bekleme süresi, taşıma koşulları gibi her adım kayıt altına alınmalıdır. DNA materyali eksi 20-40°C’de saklanabilir.
Günümüzde genetik testler yukarıda da belirtildiği gibi tek gen veya çoklu gen analiz stratejileri ile gerçekleştirilmektedir. Tek gen analizinde sorumlu genin büyük oranda tahmin edilmesi ve beklenen genetik bozukluğa özgü dizileme ve/veya kantitatif ölçümleri içeren bir analiz yöntemi tercih edilmesi gerekir. Çoklu gen analizi tercihi genetik heterojeniteye sahip ve/veya nedeni bilinmeyen hastalıklar için avantaj sağlar. YND teknolojilerinin çoklu gen, ekzom, RNA ve genom ölçekli kullanılması ve verinin klinik genetik işbirliği ile yorumlanması ile genetik tanı testleri gerçekleştirilebilir.
Genomumuzdaki CNV’lerin, yapısal anomalilerin ve kromozomal sayı değişikliklerin tespit edilebilmesi için ise çoğunlukla kromozom analizleri gerçekleştirilir. Klasik bantlama teknikleri ile gerçekleştirilen karyotip analizleri, mikroskop altında görülebilecek kromozom yapısal bozuklukları ile sayısal anomaliler hakkında fikir verir. Kromozomların özel bir alanının işaretlenip mikroskop altında daha net görülmesini sağlayan floresan in situ hibridizasyon (FISH) yöntemi de belirli bir yapısal anomalinin varlığını test etmek için kullanılabilir. Array yöntemleri arasında sayılabilecek karşılaştırmalı genomik hibridizasyon (aCGH) ve genom çapı SNP arrayler ile tüm genom CNV analizi yapılabilir; mikroskop ile tespit edilemeyen yapısal varyasyonlar bulunabilir. YND teknolojilerinden tüm genom dizileme yapısal varyasyonları tespit edebilen alternatif bir yöntem olarak karşımıza çıkar.
Genetik tanı testleri klinik uygulamada şüphelenilen bir hastalığın tanısını doğrulamak, ayırıcı tanıda yer alan hastalıkları dışlamak, sağlıklı bir kişide hastalığın oluşacağını öngörmek (prediktif tanı), taşıyıcıları belirlemek, bir çiftin çocuk kararına destek olmak (prenatal), preimplantasyon ve yenidoğan taramaları için kullanılabilir. Prenatal tanı, genetik hastalığı olduğu bilinen ailenin çocuğuna doğum öncesi tanı verilmesi amacıyla yapılır. Spinal musküler atrofi, Duchenne musküler distrofisi gibi hastalıklarda uygulanabilir. Prediktif tanı ise ailesinde genetik bir hastalık olduğu bilinen ancak belirtilerin henüz ortaya çıkmadığı diğer bireylere önceden bilgi vermek için yapılır. Genetik danışma verilmeyen ve bilgilendirilmiş onay formu olmayan hastaların örnekleri incelemeye alınmamalıdır. Belirtileri ileri yaşlarda ortaya çıkacak hastalıklarda riskli çocuklara korunma önlemleri veya tedavi söz konusu değilse yapılması önerilmez.
Genetik test sonuçları büyük bir özenle değerlendirilmelidir. Pek çok genetik hastalık için henüz etkin tedavi seçeneklerinin geliştirilmemiş olduğu unutulmamalıdır. Test sonucunun negatif olması ilerde hastalığın görülmeyeceği anlamına gelmeyebilir. Tersine, test sonucunun pozitif bulunması da kişinin mutlak hasta olacağını da göstermeyebilir. Ailede genetik hastalık öyküsü olduğunda önce hasta bireyin test edilmesi gerekir. Hangi hastalarda genetik çalışma yapılabileceği, hekimin rolü ve görevleri iyi bilinmelidir. Hastaya sonucu yalnız iken söylemek ve hastalık ile ilgili sorulara hazırlıklı olmak gerekir, ancak test sonuçlarının yalnız kişiyi değil aile bireylerini de etkilediği akılda tutulmalıdır. Testler genetik danışmanlık konusunda deneyimli bir merkezde, psikiyatrın da bulunduğu bir kurul tarafından değerlendirildikten sonra uygulanmalı ve sonuç hastaya sunulurken gerekli bilgiler ve destek verilmelidir. Bu özellikle titizlenilmesi gereken bir konudur. Huntington hastalığında presemptomatik testin pozitif sonucu karşısında intihar girişiminde bulunan hastaların olduğu unutulmamalıdır.
Genetik test sonuçlarının hastaya aktarılmasında dikkat edilmesi gereken diğer bir durum da VUS’lar, yani önemi bilinmeyen varyasyonlardır. Genetik raporlarda VUS olarak gözüken varyasyonların ilerleyen dönemlerde yeni literatür bilgileri ışığında yeniden değerlendirilmesi gerekebilir. YND ile yapılan testlerde çoklu gen incelemesi yapıldığından şans eseri yani ‘incidental’ bulgulara rastlanabilir. Örneğin epilepsisi olan küçük bir kızın analizinde herediter meme kanserine yatkınlık oluşturan patojenik bir BRCA1 varyantına rastlanabilir. Bu bulguların açıklanmasından hasta ve ailesinden alınan rıza formu, hukuki düzenlemeler ve yine ACMG gibi uzman grupların kılavuzları dikkate alınmalıdır.
Genetik Danışmanlık
Genetik danışmada uzmanın tarafsız, yargısız ancak diplomatik bir üslupla karmaşık bir bilgiyi hastaya iletmesi gerekir. Genetik danışmanlıkta ilk basamak kesin tanıyı dikkatlice koymaktır. Doğru tanı konulduktan sonra, genetik danışmanlıktaki ikinci basamak hastayı eğitmektir. Bu, indeks vakanın ve diğer aile üyelerinin ilgili genin kalıtımı için ortalama riskini tahmin etmeyi de içerir. Riskler her zaman bağlam ve bakış açısına göre değerlendirilmelidir. Örneğin her bir gebelikte tüm sağlıklı çiftler %2-4 oranında doğumsal defektli çocuk doğurma riski taşırlar. Belli bir kişinin bir hastalığı kalıtma riskine nasıl tepki vereceği değişebilir, örneğin bir hasta %50 riski kaygı verici bulmazken, diğeri %1 riski bile çok rahatsız edici bulabilir. Risk hesaplamalarına ek olarak danışman beklenen semptom ve bulgular, hastalığın doğal gidişi, ekspresyon değişkenliği ve uzun dönem prognozu hakkında açıklamalar yapmalıdır. Ayrıca mevcut tedavi seçenekleri de tartışılmalıdır. Hekimler semptomların ve birçok hastalığın prognozunun çeşitliliğine alışmış durumdadırlar. Ancak hastalar daha kesin sonuçlar beklemekte ve bu belirsizlikleri anlamakta zorlanabilmektedirler. Hastaya soru sorması için fırsat verilmelidir. Ayrıntılı genetik danışmanlık genellikle aylar ya da yılları kapsayan bir süreçtir. Günümüzde hastalara yaklaşım açısından temel bir konu başlığı haline gelen genetik danışmanlık açısından ülkemizdeki sorun, eğitilmiş kişilerin sınırlı sayıda olması ve genetik danışmaya yönlendirilme konusunda sağlık sistemimizde bir organizasyon eksikliği olmasıdır.
Farmakogenomik
Farmakogenomik; ilaca verilen yanıtta bireyler arasında genetik olarak belirlenen değişkenlik ile ilgili çalışmalar olarak tanımlanabilir. Gerek farmakokinetik (ilacın emilimi, proteine bağlanması, etki yerine taşınması, metabolizması ve atılması, yani vücudun ilaca yaptıkları) gerekse farmakodinamik (belli reseptörlere bağlanarak belli dokularda oluşturulan etkiler yani ilacın vücuda yaptıkları) özelliklerin kişiden kişiye değişiklik göstermesinde genetik varyasyonların da bir rolünün olduğu öngörülmektedir. Genetik altyapı bireylerin hem tedavi cevaplarını etkilemekte hem de ciddi yan etki olasılıklarını gündeme getirmektedir. Farmakogenomik alanında yapılan çalışmalar kompleks hastalıkların analizi gibi çok sayıda bireyin SNP-GWAS veya YND gibi yöntemler ile analizlerini gerektirir. Epilepsi tedavisinde kullanılan ilaçların metabolizmasında görev alan enzimlerin polimorfik “missense” değişimlerinin incelenmesi farmakogenomik yaklaşıma bir örnektir. Bu yaklaşım özellikle ilaç direnç mekanizmalarının aydınlatılmasına katkı sağlayabilir.
KAYNAKLAR:
1. Warman Chardon J, Beaulieu C, Hartley T, Boycott KM, Dyment DA (2015). Axons to Exons: the Molecular Diagnosis of Rare Neurological Diseases by Next-Generation Sequencing, Curr Neurol Neurosci Rep. (9):64. doi: 10.1007/s11910-015-0584-7, PMID: 26289954.
2. Crick F (1970). "Central dogma of molecular biology". Nature. 227 (5258): 561-563. doi: 10.1038/227561a0. PMID 4913914.
3. 1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM ve ark. (2015). "A global reference for human genetic variation". Nature. 526(7571):68-74. doi:10.1038/nature15393. PMID: 26432245.
4. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J ve ark. (2015). "Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology". Genet Med. 17(5):405-24. doi: 10.1038/gim.2015.30, PMID: 25741868.
5. Hunt SE, McLaren W, Gil L, Thormann A, Schuilenburg H, Sheppard D ve ark. (2018). "Ensembl variation resources". Database (Oxford). 2018:bay119. doi: 10.1093/database/bay119, PMID:30576484.
6. Uğur SA (2016). "Kopya sayısı değişikliklerinin SNP array ile tespiti". Türkiye Klinikleri J Pediatr Sci. 12(4):54-59.
7. Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, Huddleston J ve ark. (2015). "An integrated map of structural variation in 2,504 human genomes". Nature. 526(7571):75-81. doi: 10.1038/nature15394, PMID: 26432246.
8. Kuo SH (2019). "Ataxia". Continuum (Minneap Minn). 25(4):1036-1054. doi:10.1212/CON.0000000000000753, PMID: 31356292.
9. Dias R, Torkamani A (2019). "Artificial intelligence in clinical and genomic diagnostics". Genome Med. 11(1):70. doi: 10.1186/s13073-019-0689-8, PMID: 31744524.
10. Epi25 Collaborative (2019). "Ultra-Rare Genetic Variation in the Epilepsies: A Whole-Exome Sequencing Study of 17,606 Individuals". Am J Hum Genet. 105(2):267-282. doi: 10.1016/j.ajhg.2019.05.020, PMID:31327507.
11. Cuellar Partida G, Laurin C, Ring SM, Gaunt TR, McRae AF, Visscher PM ve ark. (2018). "Genome-wide survey of parent-of-origin effects on DNA methylation identifies candidate imprinted loci in humans". Hum Mol Genet. 27(16):2927-2939. doi: 10.1093/hmg/ddy206, PMID: 29860447.