Dejeneratif Ataksiler
Yazan: Başar Bilgiç
Son Güncelleştirme Tarihi: 08.06.2019
Dejeneratif ataksiler, isminin de ifade ettiği şekilde, “ataksi”nin ana ve hakim bulgu olduğu “dejeneratif” doğalı bir hastalık grubudur. Prevalansı hakkında yapılan az sayıdaki çalışma nadir bir hastalık olduğunu ortaya koymaktadır. Yaklaşık yüze yakın neden dejeneratif ataksiye yol açmaktadır. Özellikle moleküler biyoloji ve genetik alanındaki gelişmeler ile son dönemde bu sendroma yol açan hastalıkların anlaşılması, tanısı ve sınıflanmasında kayda değer gelişmeler kaydedilmiştir.
Dejeneratif ataksiler, sınıflamada genel bir uzlaşı olmasa da, “kazanılmış dejeneratif ataksiler” ve “herediter dejeneratif ataksiler” olarak iki ana başlık altında değerlendirilebilir.
KAZANILMIŞ DEJENERATİF ATAKSİLER
İnme, tümörler, demiyelinizasyon, infeksiyonlar, travma, hipoksi, toksikasyon, vitamin eksiklikleri ve immün aracılıklı tutulum sonucu gelişen ataksilere “kazanılmış ataksiler” denir. Bunlar içinde toksik ve immün aracılıklı ataksiler dejeneratif ataksi tablosu şeklinde kendini gösterir.
“Alkolik serebellar dejenerasyon” toksik nedenli bir dejeneratif ataksi alt tipidir. Daha çok erkeklerde görülen bu hastalıkta kronik alkol maruziyeti sonucu serebellumda vermisin anterior kısmı ve komşuluğundaki serebellar hemisferlerde dejenerasyon izlenir ve klinik olarak hastalarda ön planda gövde ataksisi gelişir. Patogenezde alkolün doğrudan toksik etkisi ile birlikte B1 vitamininin eksikliği de rol almaktadır. Toksik nedenli ataksiler iyatrojenik nedenli de olabilir. Epilepsi tedavisinde kullanılan “fenitoin”’in kronik kullanımı serebellar dejenerasyona yol açıp epilepsi hastalarında ataksi gelişmesine neden olabilir.
İmmün aracılıklı dejeneratif ataksilerin en bilineni subakut seyirli “paraneoplastik serebellar dejenerasyon (PSD)” dur. Tüm paraneoplastik sendromlar içinde polinöropati ile birlikte en sık görülen form PSD’dir. En sık ilişkili olduğu kanser tipi küçük hücreli akciğer kanseri olsa da Hodgkin lenfoması, meme, over ve çok nadiren de prostat kanseri ve kondrosarkom ile birlikte görülebilir. PSD ile birlikte en sık saptanan oto antikor anti-Yo’dur ama anti-Hu, anti-CV2, anti-Tr ve anti-Ma gibi diğer antikorlar ile de birlikte olabilir. Herhangi bir maligniteye eşlik etmeden de immün-aracılıklı ataksiler izlenebilir. Bunlara örnek olarak glutamik asit dekarboksilaza (GAD) yönelik anti-GAD antikorları ve glutamat reseptörlerine yönelik mGluR1 antikorları aracılı ataksiler sayılabilir.
Çölyak hastalığı seyrinde barsak tutulumu bulguları dışında hastalarda ataksi gelişebilir (gluten ataksisi). Bazı hastalarda ise Çölyak hastalığının barsak tutulumu olmaksızın izole ataksi olabilmektedir. Hastalarda ataksi genellikle sinsi bir şekilde başlasa da PSD’yi telkin edecek şekilde subakut başlangıçlı da olabilir. Nörolojik tutulum bulgusu olarak ataksi tek başına olabileceği gibi, miyoklonus, palatal tremor, kore, miyelopati ve epileptik nöbetler ile birlikte de görülebilir. Nörogörüntülemede serebellum atrofik olarak izlenmektedir. Bazı hastalarda bilgisayarlı tomografide oksipital alanda kortiko-subkortikal yerleşimli kalsifikasyonlar görülebilir. Gluten içermeyen diyet ile belirgin düzelme ve hatta kür sağlanan bu hastalığın ayırıcı tanısı önemlidir. Bu nedenle sporadik ataksi hastalarında gluten duyarlılığı sorgulanmalı, diğer etyolojilere ait bir ipucu olmadığı durumlarda tanısal amaçlı gliadin, endomisyum ve transglutaminaz antikorlarının varlığı araştırılmalıdır. Şüphe halinde ise barsak biyopsisi yapılmalıdır.
Özellikle kırk yaşından sonra başlayan kronik seyirli progresif ataksisi olan hastalarda multi sistem atrofi (MSA) hastalığı da sık rastlanan bir dejeneratif ataksi nedenidir. Eşlik eden parkinsonizm, otonom bulgular ve REM-uyku davranış bozukluğunun olması bir sinükleinopati olan bu hastalığı akla getirmelidir. (Bkz. Hareket bozuklukları bölümü)
HEREDİTER DEJENERATİF ATAKSİLER
Herediter dejeneratif ataksiler kalıtım tipine göre sınıflanırlar: otozomal resesif kalıtımlı, otozomal dominant kalıtımlı, X’e bağlı kalıtımlı ve mitokondriyal kalıtımlı. Otozomal dominant kalıtımlı ataksilerin görülme sıklığı yüz binde 1 ila 5 arasında iken otozomal resesif kalıtımlı ataksiler yüz binde 3 oranında izlenirler. En sık izlenen otozomal dominant kalıtımlı ataksiler spinoserebellar ataksi (SCA) 1,2 ve 3 iken, otozomal resesif kalıtımlı ataksiler içinde ise Friedreich ataksisi, ataksi okülomotor apraksi ve ataksi-telanjiektazi ön plana çıkmaktadır. Mitokondriyal ataksilere POLG1 mutasyonu ile giden ataksi ve nöropati-ataksi-retinitis pigmentoza (NARP) sendromu örnek verilebilirken, X’e bağlı kalıtılan ataksilere adrenolökodistrofi, frajil-x ataksi sendromu örnek gösterilebilir. Otozomal resesif kalıtımlı ataksi ile seyreden hastalıkların ana klinik özellikleri, başlangıç yaşları ve ilişkili mutasyonlar Tablo 1’de verilmiştir.
Otozomal Resesif Kalıtımlı Dejeneratif Ataksiler
Friedreich Ataksisi
Nikolaus Friedreich’ın* tanımladığı otozomal resesif kalıtımlı bu hastalık en sık görülen resesif geçişli ataksidir ve başlangıç genellikle çocukluk veya yetişkinliğin erken döneminde olur. Ataksi ile beraber arka kordon ve arka kök ganglionunun dejenerasyonuna bağlı duysal bulgular ve arefleksi de izlenir. Seyirde disfaji, alt ekstremite kaslarında güçsüzlük ve atrofi gelişir. Nörolojik bulgular dışında skolyoz, pes cavus gibi ayak deformiteleri, kardiyomiyopati, diabetes mellitus ve sağırlık izlenebilir. Hastalığın başlangıcından ortalama 15 yıl sonra yürüme yetisi ortadan kalkar. Kognitif işlevler genellikle korunur. Son yıllarda daha geç başlayan ve daha selim seyreden, sıklıkla reflekslerin korunmuş olduğu yetişkin bir formun olduğu da anlaşılmıştır.
Kranyal MR’de serebellumda belirgin atrofi izlenmezken atrofi kendini medulla spinaliste gösterir. Transkraniyal ultrasonografi ile serebellumun dentat çekirdeğinde hiperekojenite gösterilebilir. Elektrofizyolojik incelemelerde hastaların büyük bir çoğunluğunda duysal aksiyon potansiyelleri alınamaz, buna karşın sinir ileti hızları normaldir. Duysal ve görsel uyandırılmış potansiyellerde de latans uzaması ve amplitüd azalması saptanabilir.
Dokuzuncu kromozomda yerleşik frataksin geninde GAA trinükleotid tekrarı bu hastalıkta patolojik olarak artmıştır. Bu artışa bağlı oksidatif fosforilasyon ve demir metabolizmasında rol alan frataksin proteininin sentezi bozulur. Günümüzde kesin tanı genetik inceleme ile bahsi geçen trinükleotid artışının gösterilmesi ile konulmaktadır.
Friedreich ataksisi ortalama 15-20 yıl süren ilerleyici bir hastalıktır. Kardiyak tutulum en önemli mortalite nedenidir.
Kesin
tedavisi olmayan bu hastalıkta küçük çaplı çalışmalar ile idebenon, deferipron,
koenzim Q10, 5-hidroksitriptofan ve interferon-gama moleküllerinin kısmi etkileri
olduğu gösterilmiş olsa da birinci sınıf kanıt düzeyine sahip bir tedavi
henüz mevcut değildir.

*Nikolaus
Friedreich
(1825-1882): Alman patolog ve nörolog. Würzburg ve Heidelberg Üniversiteleri’nde
kas hastalıkları, beyin tümörleri ve ataksiler konusunda çalışmıştır. Kendi
adını taşıyan ataksi
hastalığını 1863 yılında tanımlamıştır. Öğrencileri Kussmaul, Schultze ve
Erb’in yetişmesinde önemli katkıları olmuştur. (Kaynak: Heidelberg Üniversitesi
Resim Kolleksiyonu https://heidicon.ub.uni-heidelberg.de)
Ataksi Okülomotor Apraksi
Bu hastalık sıklıkla çocukluk çağında ortaya çıkmaktadır ve hastalarda ataksi ile birlikte sakkadları başlatamama ve istemli göz hareketlerini kontrol etmede zorluk şeklinde tanımlanabilecek okülomotor apraksi olmaktadır. Hastalar gözleri ile hedefe yönelemediklerinden çoğu kez hedefi izlemek için fazladan baş ve boyun hareketleri yaparlar. Uzakdoğu gibi bazı coğrafyalarda en sık ataksi nedeni olan bu hastalığın dört tipi tanımlanmıştır. Tüm tipler fenotipik benzerlikler gösterse de sorumlu mutasyon ve etkilenen proteinler her tipte farklıdır.
Ataksi Okülomotor Apraksi Tip 1 (AOA1): Çocukluk çağında ataksi ve onun hemen ardından okülomotor apraksi bulguları ile kendini belli eden, görece yavaş ilerleyici bu hastalıkta refleksler alınamaz ve aksonal motor polinöropati ataksiye eşlik eder. Seyirde distoni, miyoklonus ve kore gibi hareket bozuklukları ve kognitif tutulum izlenebilir. Kranial MR’de serebellar atrofi görülmektedir. Hastalarda hipoalbuminemi ve hiperkolesterolemi, kreatinin kinaz düzeylerinde artış izlenebilmektedir. Alfa-fetoprotein düzeylerinde artış olmaz. AOA1, 9. kromozomda yer alan ve aprataksin proteinini kodlayan APTX genindeki mutasyonlar sonucu ortaya çıkar. Bazı mutasyon tipleri ile klinik fenotip arasında ilişki saptanmıştır. Kore ile giden hastalarda A198V mutasyonu daha sık izlenirken, delesyonlar daha kötü bir klinik seyir ve kognitif yıkımla ilişkili bulunmuştur.
Ataksi Okülomotor Apraksi Tip 2 (AOA2): Başlangıç yaşı çocukluk çağı olsa da yetişkinlikte de ortaya çıkabilen bu tipte hastaların sadece yarısında okülomotor apraksi olmaktadır. Duysal-motor tipte bir polinöropati ise tüm hastalarda izlenmektedir, buna karşın nöropati AOA1’dekine göre daha geç ortaya çıkmakta ve daha hafif bulgular ile seyretmektedir. Piramidal bulgular, distoni, kore ve tremor, amenore, hipogonadizm izlenebilir. Kranial MR’de serebellum atrofik olarak izlenir ve alfa-fetoprotein düzeyleri hastalarda tipik olarak artmıştır. Bazı hastalarda kolesterol, immunglobulin G ve immunglobulin A ve kreatininkinaz düzeyleri artmış olarak bulunmaktadır. Dokuzuncu kromozomda yer alan senataksin proteinin kodlayan SETX genindeki mutasyonlar bu hastalıktan sorumludur. Senataksin, DNA replikasyonu, transkripsiyonu ve tamirinde rol alan bir DNA/RNA helikaz proteinidir. Bu gendeki bazı mutasyonlar juvenil başlangıçlı motor nöron hastalığına da yol açabilmektedir.
Ataksi Okülomotor Apraksi Tip 3 (AOA3): Suudi Arabistan’da bir ailede PIK3R5 genindeki mutasyona bağlı olarak ortaya çıktığı bildirilen bu tipte, ataksi ve daha geç ortaya çıkan okülomotor apraksiye erken yaşta başlayan ciddi bir polinöropati eşlik etmektedir. Alfa-fetoprotein düzeyleri artmıştır. Diğer hareket bozuklukları ve kognitif tutulum bildirilmemiştir.
Ataksi Okülomotor Apraksi Tip 4 (AOA4): Bu form Portekizli ailelerde PNKP genindeki mutasyonlara bağlı olarak bildirilmiştir. Fenotip olarak AOA1’e oldukça benzemektedir.
Ataksi - Telanjiektazi
Beş yaş öncesi en sık görülen dejeneratif ataksi tipi olan bu hastalık genellikle ilk on yıl içinde ilerleyici ataksi ve okülokütanöz telanjiektaziler ile kendini gösterir. Bu bulgulara polinöropati, arefleksi, okülomotor apraksi, kore, distoni ve miyokloni eşlik edebilir. Derin tendon refleksleri zamanla azalır ve alınamaz, piramidal bulgular ise çoğunlukla izlenmez. Hastalarda zamanla belirginleşen bir intansiyonel tremor izlenir ve bu bulgu ayırıcı tanı açısından oldukça yardımcıdır. İlerleyici olan bu hastalıkta, hastalar ergenlik döneminde yürüme yetilerini kaybederler. Hastaların yaklaşık %30’unda hafif düzeyde mental retardasyon vardır. Kranial MR’de serebellum atrofiktir ve ak madde içinde milimetrik parlamalar izlenebilir. Ataksi-telanjiektazi hastalarında nörolojik bulgular dışında immün sistem zayıflamıştır ve hastalarda sinopulmoner infeksiyonlar sıktır. Ayrıca bu hastalarda lösemi ve lenfoma gibi kanserlerin görülme sıklığı kayda değer şekilde yüksektir. Hastalarda timüs bezi gelişmemiş bulunabilir. Terapötik ve diagnostik dozlardaki X-ışınları bu hastalarda radyasyon yan etkileri ortaya çıkmasına yol açabilir ve bu durumdan dolayı tetkik istenirken dikkatli olunmalıdır. Hastaların alfa-fetoprotein düzeyi artmıştır ve immunglobulin M düzeyleri artıp immunglobulin A düzeyleri düşmüş olarak bulunabilir. On birinci kromozomda yerleşmiş ATM genindeki mutasyonlar hücre bölünmesi ve DNA tamiri ile ilişkili ATM proteinin işlevini bozarak hastalık semptomlarına yol açar. Bu protein ayrıca TP53 ve BRCA1 gibi tümör baskılayıcı genler ile de ilişkilidir.
E Vitamini Eksikliği İle Birlikte İzlenen Ataksi
Antioksidan özelliği olan ve yağda eriyen bir vitamin olan E vitamini eksikliğinde nörodejenerasyon süreçleri tetiklenmekte ve hastalarda diğer bulgular ile birlikte nörolojik bulgular ortaya çıkmaktadır. Hastalardaki ana bulgu ataksidir. Friedreich ataksisi hastalarındaki gibi ataksi dışında arefleksi ve polinöropati de olmaktadır. Ayrıca retinitis pigmentoza, skolyoz ve kardiyomiyopati de görülebilmektedir.
Sekizinci kromozomda yer alan alfa-tokoferol transfer proteinini kodlayan gendeki mutasyonlar, E vitamininin emilim sorunundan ziyade karaciğer işlevlerinde bozukluğa yol açmaktadır. Tedavide yüksek doz E vitamini ile stabilizasyon ve bazen de semptomlarda gerileme izlenmektedir.
Abetalipoproteinemi (Bassen-Kornzweig hastalığı)
Yağ emilim bozukluğu olan bu hastalık, dördüncü kromozomda yer alan mikrozomal trigliserid transfer protein (MTTP) genindeki mutasyonlar sonucu ortaya çıkmaktadır. Bebeklik çağında ishal, steatore ve kilo kaybı ile yağda eriyen vitaminler olan A, D, E ve K vitaminlerinin eksikliği gelişir. Çocukluk veya ergenlik çağında ataksi gelişen bu hastalıkta tremor, tikler ve motor zaaf gibi başka nörolojik bulgular da izlenebilir. Derin tendon refleksleri azalmıştır ve bazı hastalarda polinöropati gelişebilir. Kranial MR incelemesinde ise serebellar atrofi genellikle izlenmez. Nörolojik bulgular dışında kifoskolyoz, pes cavus gibi iskelet deformiteleri ile göz muayenesinde retinitis pigmentoza ve periferik yaymada akantositler saptanır. Kan biyokimyasında LDL ve VLDL düzeyleri düşüktür. Erken tanı ve vitamin desteği ile bulgular stasyoner bir seyir gösterebilir. Tedavi edilmeyen hastalar 40 yaş civarında tekerlekli sandalyeye bağımlı hale gelirler.
Koenzim Q10 Eksikliği İle Birlikte İzlenen Ataksi
Mitokondriyal solunum zincirinde elektron taşıyıcı görev yapan Koenzim Q10’un eksikliği heterojen bir klinik başlangıç ve seyir gösterir. Kas tutulumu, Leigh sendromu, nefritik sendromu ve ataksi şeklinde fenotipler ile kendini gösterebilir. En sık görülen ataksik fenotipte çocukluk çağında başlayan ataksiye, piramidal bulgular, nistagmus, derin tendon reflekslerinde azalma ve epileptik nöbetler eşlik eder. Bazı hastalarda polinöropati, mental retardasyon, hipogonadizm ve kas zaafı da izlenebilir. Kranial MR’da serebellum atrofiktir. COQ2, COQ4, COQ6, COQ8A ve COQ8B genlerindeki mutasyonlar Koenzim Q10 eksikliğine yol açmaktadır. Koenzim Q10 tedavisi ile bazı hastaların semptomlarında dramatik iyileşme izlenirken, bazı hastalar daha kısmi bir cevap vermektedir. Koenzim Q10 tedavi dozu hakkında uzlaşı yoktur. 100mg/gün ile 3000mg/gün arasında Koenzim Q10 dozu ile cevap alınmış vakalar literatürde mevcuttur.
Charlevoix-Saguenay Ataksisi
Kanada’nın Quebec bölgesinde ilk kez tanımlanan bu hastalık ülkemizde de görülebilmektedir. Hastalarda ilk on yıl içinde spastisite ile birlikte ataksi ortaya çıkmaktadır (spastik ataksi). Derin tendon refleksleri, eğer tabloya polinöropati eklenmez ise, canlıdır ve taban derisi refleksi ekstansör cevaplıdır. Distal kaslarda amiyotrofi, nistagmus ve dizartri sıklıkla mevcuttur. Retinal hipermiyelinizasyon ve mitral kapak prolapsusu eşlik edebilir. Kranial MR’de vermis atrofisi ile birlikte ponsta sinyal değişiklikleri ve servikal spinal MR’de ise medulla spinaliste atrofi izlenir. On üçüncü kromozomda bulunan SACS genindeki mutasyonlar bir şaperon protein olan sacsin proteininin işlevini bozmakta ve hastalığa yol açmaktadır.
Serebrotendinoz Ksantomatozis
Günümüzde nedene yönelik tedavi olanağının olduğu bu hastalığın erken tanısı önem kazanmıştır. Bu nadir lipid depo hastalığında CYP27A1 gen mutasyonlarına bağlı olarak bir mitokondriyal enzim olan stero 27-hidroksilazın eksikliği olur ve kolesterolün ekstraksiyonu için gerekli safra asidlerinin sentezinde bozulma olur. Buna bağlı olarak bağ dokusu ve sinir sisteminde kolesterol ve kolestanol birikir. Hastalarda ise bebeklik döneminde tekrar eden ishaller ve yenidoğan döneminde görülen uzamış sarılık hastalığın ilk bulguları olabilir. İlk onyıl içinde katarakt gelişmesi sıktır. Hastalığa adını veren ksantomlar genellikle 20 yaştan sonra belirir ve en çok Aşil tendonunda izlenir (Şekil 1). Patella, dirsek, el ve boyunda da ksantomlar görülebilir. Nörolojik bulgular ise genellikle otuzlu yaşlarda ortaya çıkar. En sık görülen nörolojik semptomlar ekstrapiramidal bulgular (distoni, parkinsonizm), ataksi ve kognitif tutulum bulgularıdır. Nöbetler, polinöropati ve spastisite de görülebilir. Kranial MR’de diffüz atrofi ile birlikte hiperintens lezyonlar görülür. Özellikle serebellumun dentat çekirdeğinde izlenen simetrik parlamalar bu hastalığın patognomonik bulgusudur (Şekil 2). Laboratuvar incelemelerinde serum kolestanol düzeyi yükselmiştir. Kenodeoksikolik asid tedavisinin başlanması ile semptomlarda stabilizasyon ve regresyon olmaktadır. Özellikle erken dönemde tanının konmasının ardından tedavi ile ileri yaşlarda ana engellilik nedeni olan semptomlar önlenebilir.

Şekil 1. Serebrotendinoz ksantomatoziste görülen Aşil tendonu ksantomları
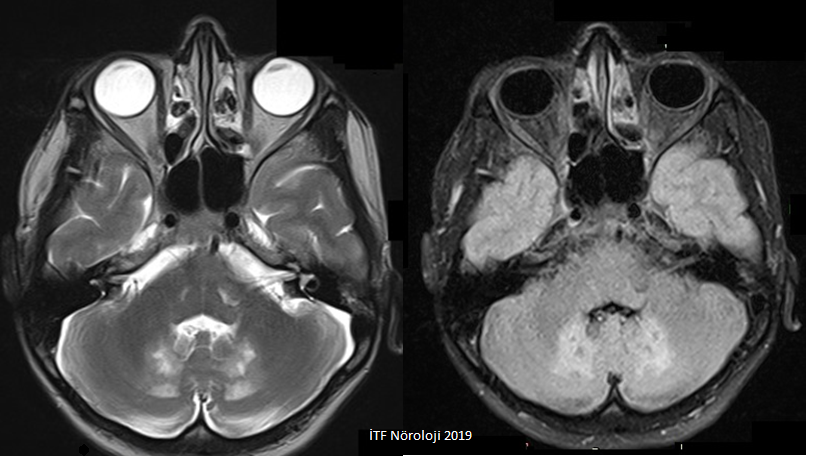
Şekil 2. Nörolojik tutulumu olan bir serebrotendinoz ksantomatozis hastasının kranial manyetik rezonans incelemesinde serebellumun dentat çekirdeklerinde simetrik parlama izlenmektedir.
Otozomal Resesif Serebellar Ataksi Tip 1
Bu formda ilk vakalar Charlevoix-Saguenay hastalığı gibi Kanada’nın Quebec eyaletinde tanımlanmıştır. İlerleme hızının yavaş olduğu ve çocukluk çağından sonra başlayan bu hastalıkta genellikle saf serebellar bulgular izlenir. Bazı hastalarda başlangıç otuzlu, kırklı yaşlarda olabilir. Kanada dışında ülkemizde ve başka ülkelerde de görülebilen bu hastalıkta, Kanada’lı hastaların aksine üst motor nöron tutulum bulguları ve pes cavus izlenebilir. Beyin görüntülemede serebellum ve beyinsapında atrofi belirgindir. Hastalık, 6. kromozomda bulunan SYNE1 genindeki mutasyonlara bağlı olarak ortaya çıkmaktadır.
Tablo 1. Başlıca resesif kalıtımlı ataksiler
|
Hastalık |
Gen |
Eşlik eden bulgular |
Sıklıkla izlenen başlangıç yaşı |
|
Friedreich ataksisi |
Frataksin |
Arefleksi, derin duyu bozukluğu, skolyoz, pes kavus, diabetes mellitus, kardiyak tutulum |
Geç çocukluk, yetişkinlik |
|
Vitamin E eksikliği ile birlikte görülen ataksi |
a-TTP |
Titubasyon, görme bozukluğu |
Çocukluk |
|
Ataksi okülomotor apraksi, tip 1 |
Aprataksin |
Okülomotor apraksi, ılımlı kognitif yıkım |
Çocukluk |
|
Ataksi okülomotor apraksi, tip 2 |
SETX |
Okülomotor apraksi, kognitif yıkım, alfa-fetoprotein düzeyinde artış |
Geç çocukluk, erken yetişkinlik |
|
Ataksi-telanjiektazi |
ATM |
Okülokütanöz telanjiektaziler, kanser riskinde artış |
Çocukluk |
|
Charlevoix-Saguenay |
SACS |
Spastisite, polinöropati |
Çocukluk |
|
Abetalipoproteinemi |
MPT |
Yağlı dışkı, akantositoz, retinopati |
Geç çocukluk, erken yetişkinlik |
|
KoenzimQ10 eksikliği ile birlikte görülen ataksi |
COQ |
Piramidal bulgular, epileptik nöbetler |
Çocukluk |
|
Otozomal resesif serebellar ataksi Tip 1 |
SYNE1 |
Saf ataksi, ılımlı seyir |
Çocukluk ve yetişkinlik |
|
Serebrotendinoz ksantomatozis |
CYP27 |
Ksantomlar, kognitif yıkım, periferik nöropati, katarakt, MR’de serebellar hiperintensiteler |
Genellikle yetişkinlik |
Otozomal resesif kalıtımlı ataksilerin heterojen bir hastalık grubu olması nedeni ile tanısı zor olabilir. Günümüzde genetik tanı için tüm genom - ekzom dizileme dışında özgün ataksi panelleri geliştirilmiştir ve ataksi ile giden resesif kalıtımlı ataksiler bu paneller ile taranabilmektedir. Fakat birçok genin taranması bir ekonomik yük yarattığından bu yöntem kolay ulaşılabilir değildir. Çeşitli klinik ve laboratuvar bulguları üzerinden çalışan algoritmalar kullanılarak etyoloji için bir filtreleme yapılabilir. Bunlara bir örnek web üzerinden erişilebilen Radial isimli algoritmadır (http://www.radial-ataxia-algorithm.com).
Otozomal Dominant Kalıtımlı Dejeneratif Ataksiler
Otozomal dominant kalıtım tarzı ile kendini gösteren bu hastalıkların çoğunu SCA tipleri oluşturmaktadır. Bununla birlikte ataksinin belirgin bulgu olduğu dentatorubropallidoluysian atrofi (DRPLA), epizodik ataksiler ve bir prionopati olan Gerstmann–Sträussler–Scheinker hastalığı da aynı kalıtım tarzını gösterir.
SCA gerek klinik fenotip gerekse de altta yatan genetik bozukluklar açısından oldukça heterojendir. SCA tipleri, ayırt edici klinik özellikleri ve sorumlu mutasyonlar Tablo 2’de verilmiştir. Dünyada en sık izlenen SCA alt tipi SCA3’dür ama sıklık konusunda coğrafi farklıklar da bildirilmiştir. Ülkemizde yapılmış ve henüz yayınlanmamış bir çalışmanın verileri SCA2’nin diğer tiplere göre daha sık olduğunu düşündürtmektedir. SCA’larda çekirdek bulgular ataksi, nistagmus ve dizartridir. Hastalarda SCA tiplerine göre ekstrapiramidal bulgular, oftalmoparezi, kognitif bozukluklar, periferik sinir tutulumu gibi ilave bulgular da olabilir.
Günümüzde 50’ye yakın SCA alt tipi genetik olarak tanımlanmıştır. Bu alt tiplere yönelik olarak 1982 yılında Harding tarafından önerilen klinik temelli sınıflama halen geçerliliğini korumaktadır. Bu sınıflamada, saf serebellar bulgular ile giden bir grup (SCA 5,6,11,23, 26,30,37, 41,45), retinal dejenerasyon ile giden bir grup (SCA7) ve piramidal, ekstrapiramidal ve amiyotrofi bulguları ile giden bir grup (SCA 1-4,8,12-15,17-22,25,27,28,31,32,34-38,42-44, 46,47) bulunmaktadır. SCA alt tiplerinde ataksi ile beraber izlenen ön plandaki nörolojik bulgular Tablo 3’de gösterilmiştir.
Tanımlanan mutasyonlar toksik işlev kazanma, mitokondriyal işlev bozukluğu, kanalopati, otofaji bozuklukları ve transkripsiyon bozuklukları gibi çeşitli sorunlara yol açmaktadır. Heterojen bir hastalık grubu olan SCA’da izlenen moleküler işlev bozuklukları üzerinden moleküler temelli sınıflamalar da önerilmiştir ama henüz yaygın şekilde kullanılmamaktadır.
Tablo 2. SCA tipleri, ayırt edici klinik özellikleri ve sorumlu mutasyonlar
|
Hastalık adı |
Klinik özellik |
Gen |
Protein/Mutasyon |
|
SCA 1 |
Piramidal ve ekstrapiramidal bulgular, periferik nöropati, oftalmoparezi |
ATXN1 |
Ataksin – 1 CAG tekrarı |
|
SCA 2 |
Piramidal ve ekstrapiramidal bulgular, periferik nöropati, sakkadlarda yavaşlama, oftalmoparezi, demans (nadir) |
ATXN2 |
Ataksin – 2 CAG tekrarı |
|
SCA 3 Machado-Joseph) |
Piramidal bulgular, ekzoftalmus, oftalmoparezi, ekstrapiramidal bulgular, amiyotrofi |
ATXN3 |
Ataksin – 3 CAG tekrarı |
|
SCA 4 |
Duysal aksonal nöropati, piramidal bulgular |
? |
? |
|
SCA 5 |
Geç başlangıçlılarda saf serebellar bulgular, erken başlangıçlılarda piramidal bulgular ile birlikte |
SPTBN2 |
β III Spectrin Nokta mutasyonu |
|
SCA 6 |
Saf serebellar bulgular, genellikle 50 yaş ve sonrası başlangıç |
CACNA1A |
Cav 2.1 CAG tekrarı |
|
SCA 7 |
Retinopati, oftalmoparezi, piramidal bulgular |
ATXN7 |
Ataksin – 7 CAG tekrarı |
|
SCA 8 |
Spastisite, piramidal bulgular, spastik ve ataksik dizartri, vibrasyon hissinde azalma |
ATXN8OS/ |
Saf poliglutamin proteini CAG/CTG tekrarı |
|
SCA 9 |
Ekstapiramidal bulgular, oftalmoparezi, piramidal bulgular, arka kolon tutulum bulguları, santral demiyelinizasyon |
? |
? |
|
SCA 10 |
Nöbetler, kognitif bulgular, piramidal bulgular, polinöropati |
ATXN10 |
Ataksin-10 |
|
SCA 11 |
Saf serebellar bulgular |
TTBK2 |
Tau tubulin kinaz-2 Nokta mutasyonu |
|
SCA 12 |
Üst ekstremitede tremor, refleks canlılığı |
PPP2R2B |
Protein fosfataz PP2A CAG tekrarı |
|
SCA 13 |
Saf serebellar ya da mental retardasyonla beraber |
KCNC3 |
Kv3.3 Nokta mutasyonu |
|
SCA 14 |
Nadiren kore ve kognitif bulgular |
PRKCG |
Protein kinaz C gama Nokta mutasyonu |
|
SCA 15/16 |
Nadiren baş tremoru ve kognitif bulgular |
ITPR1 |
Inosital 1,4,5-trifosfat Reseptör Nokta mutasyonu |
|
SCA 17 |
Kore, distoni, demans, psikiyatrik bulgular, refleks canlılığı |
TBP |
TATA box-binding protein CAG tekrarı |
|
SCA 18 |
Genellikle erken başlangıçlı, amiyotrofi, arka kolon tutulum bulguları |
? |
? |
|
SCA19/22 |
Hafif ataksi, postüral tremor, kognitif bozukluk |
KCND3 |
Kv4.3 Nokta mutasyonu |
|
SCA 20 |
Spazmodik disfoni, palatal tremor |
? |
? |
|
SCA 21 |
Erken başlangıçlı, yavaş seyir, parkinsonizm, kognitif bulgular, reflekslerde azalma |
? |
? |
|
SCA 23 |
Geç başlangıçlı, saf serebellar bulgular |
PDYN |
Prodinorfin Nokta mutasyonu |
|
SCA 25 |
Duyusal nöropati, arefleksi |
? |
? |
|
SCA 26 |
Saf serebellar |
? |
? |
|
SCA 27 |
Baş tremoru, orofasial diskineziler, kognitif bulgular, psikiyatrik bulgular |
FGF14 |
Fibroblast büyüme faktörü 14 Nokta mutasyonu |
|
SCA 28 |
Erken başlangıçlı, yavaş seyir, oftalmoparezi, ptoz, piramidal bulgular |
AFG3L2 |
ATPaz ailesi gen 3-benzeri 2 (ATPase family gene 3-like 2) Nokta mutasyonu |
|
SCA 29 |
Konjenital ve ilerleyici değil, vermis atrofisi |
? |
? |
|
SCA 30 |
Geç başlangıçlı, saf serebellar |
? |
? |
|
SCA 31 |
Geç başlangıçlı, saf serebellar
|
BEAN1 ve TK2 |
NEDD4 ile ilişkili beyin ekspresyonu (Brain expressed, associated with NEDD4) Nokta mutasyonu |
|
SCA 32 |
Azospermi, erken başlar ise kognitif bulgular |
? |
? |
|
SCA 34 |
Deride eritem, keratoz, nistagmus, reflekslerde azalma |
ELOVL4 |
Çok uzun zincirli yağ asitleri uzaması benzeri -4 (Elongation of very long chain fatty acids-like 4) Nokta mutasyonu |
|
SCA 35 |
Psödobulber felç, piramidal bulgular |
TGM6 |
Transglutaminaz 6 Nokta mutasyonu |
|
SCA 36 |
Alt motor nöron tutulumu, dilde atrofi |
NOP56 |
Nükleolar protein 56 İntronik GGCCTG tekrarı |
|
SCA 37 |
Vertikal göz hareketlerinde bozukluk |
? |
? |
|
SCA 38 |
Genellikle saf serebellar, aksonal nöropati eşlik edebilir |
ELOVL5 |
Çok uzun zincirli yağ asitleri uzaması benzeri -5 Nokta mutasyonu |
|
SCA 39 |
Spastisite, horizontal bakış kusuru |
? |
? |
|
SCA 40 |
Spastisite |
? |
? |
|
DRPLA |
Demans, kore, miyoklonik epilepsi |
ATN1 |
Atrofin I CAG tekrarı |
Tablo 3. Ataksi ile birlikte seyreden nörolojik bulgular ve izlendiği SCA alt tipleri (Kaynak 3’ten değiştirilerek)
|
Polinöropati Oftalmoparezi Görme kaybı İşitme kaybı Piramidal bulgular Kognitif bulgular Psikiyatrik sorunlar Parkinsonizm Kore Distoni Miyoklonus Tremor Nöbetler Yüz ve dilde fasikÜlasyon
|
SCA 1,2,3,4,18,25,38,43,46 SCA 2,3,28,40 SCA 7 SCA 31,36 SCA 1,3,7,8,10, 14,15,17,35,40,43 SCA 2,8,13,17, 19,21,36,44, DRPLA SCA 2,17 SCA 2,3,10,14,17,19,21 SCA 17,27,DRPLA SCA 3,14,17,20,35 SCA 14 SCA 12,15,27 SCA 10,19 SCA 36 |
Dentatorubropallidoluysian atrofi (DRPLA) ataksi dışında demans, nöbetler ve kore ile giden ve otozomal dominant kalıtımlı bir hastalıktır. Erken ya da geç yaşta başlayabilir. Japonya’da daha sık görülen bu hastalığın ülkemizde çok nadir olduğu düşünülmektedir. Erken başlangıçlı olgular progresif miyoklonik epilepsiye benzeyebilir. Geç başlangıçlı olgular ise daha çok Huntington hastalığı benzeri bir klinikle seyreder. On ikinci kromozomdaki ATN1 genindeki artmış CAG tekrarı sonucunda poliglutaminlenmiş atrofin I proteini nukleusta birikir ve hastalığa yol açar.
Epizodik ataksiler, iyon kanallarındaki sorunlara bağlı olarak epizodik şekilde dengesizlikle seyreden otozomal dominant kalıtımlı hastalıklardır. Yedi tipi olan bu hastalıkta en sık epizodik ataksi tip 1 ve epizodik ataksi tip 2’ye rastlanır. Epizodik ataksi tip 1, bir potasyum kanalı genindeki (KCNA) mutasyona bağlı olarak gelişir ve hastalıkta dakikalar veya saniyeler süren gün içinde defalarca tekrarlayan ataksi, dizartri, tremor ve kramplar izlenir. Ataklar dışında hasta normaldir. Emosyonel stres, kafein, alkol, egzersiz ve diğer hastalıklar atakları tetikleyebilir. Epizodik ataksi tip 2, voltaja bağımlı kalsiyum kanal genindeki (CACNA1A) mutasyonlar sonucu meydana gelir. Ataklar tipik olarak saatler-günler sürer. Asetazolamid ile ataklar önlenebilir. Hastalarda ataklardan bağımsız olarak kalıcı nistagmus ve ılımlı serebellar bulgular ile görüntülemede ilerleyici serebellum atrofisi saptanabilir. Epizodik ataksi tip 3, tip 1’e oldukça benzer. Ataklar günde 1-2 kez genelde yarım saatten kısa sürer ve atak sırasında baş dönmesi ve tinnitus olabilir. Asetazolamid bazı hastalarda atakları önleyebilmektedir. Bu tipte sorumlu gen henüz saptanamamıştır. Diğer tiplere oldukça nadir rastlanılır.
Otozomal dominant kalıtımlı ataksilerde kesin tanı genetik analiz yapılarak ilgili mutasyonun gösterilmesi ile konulmaktadır. Çok özel bir klinik bulgu (örneğin, retinopati, azoospermi, konjenital başlangıç gibi) eşlik etmediği sürece dominant kalıtım hikayesi olan bir hastada en sık rastlanan ve değişken klinik bulgular ile seyreden SCA 1,2,3 taraması yapılmalıdır. Bu hastalıklar ekarte edildiği durumlarda klinik bulgulara göre diğer SCA tiplerine hedeflenmeli ya da geliştirilmiş ataksi panelleri ile ataksiye yol açabilecek hastalıklar araştırılmalıdır. Eşlik eden nöropatiyi göstermek için elektrofizyolojik incelemeler, olası bir retinopatiyi gösterme amacı ile göz muayenesi yapılması hem tanısal hem de prognoz açısından yararlıdır.
Semptomatik tedavi için çeşitli tedaviler denenebilir. Çeşitli dejeneratif ataksi tiplerinde 2,4 aminopridin, riluzol, valproat, tirotropin salgılatıcı hormon, amantadin ve pirasetam ile ilgili kısmi olumlu sonuçlar gösteren küçük çaplı çalışmalar mevcuttur. Resesif kalıtımlı birçok atakside olduğu gibi etkinliği birinci sınıf kanıt düzeyinde gösterilen bir tedavi yöntemi henüz mevcut değildir. Fizyoterapi her hastada ve hastalığın her evresinde asla ihmal edilmemesi gereken bir tedavi yöntemidir.
Günümüzde yeni geliştirilen genetik temelli tedavi yöntemlerinden olan “antisense” oligonükleotidler ile proteinlerin toksik etkisi azaltılabilmekte veya modifiye edilebilmektedir. Laboratuvar ortamında “antisense” oligonükleotidler ile hedef proteinlerin ekspresyonu azaltılabilmektedir. Hayvan çalışmalarında SCA2 ve SCA3’deki patolojik ataksin 2 ve ataksin-3 proteininin toksik parçaları bu yöntemle temizlenebilmiştir. Bu yöntemler insanlar üzerinde de denenmeye başlanmıştır. SCA 3 ve SCA7’de RNA temelli tedavi yöntemleri de umut vaat etmektedir.
Sonuç olarak nadir bir hastalık olan herediter dejeneratif ataksilerde detaylı bir fenotipleme yapıldıktan sonra tanısal amaçlı genetik analiz aşamasına geçilmelidir. Mutasyon saptanması durumunda saptanan mutasyonun kalıtım tarzına göre ailelere genetik danışmanlık verilmelidir. Progresyonun takibi, eşlik edecek diğer bulguların saptanması ve yönetilmesi ve gelecekte kullanılması muhtemel tedavi yöntemlerinin önerilebilmesi amacı ile hasta ve mutasyon taşıyıcılarının düzenli takibi önem taşımaktadır.
Kaynaklar
1. Akbar, Ashizawa. Ataxia. Neurol Clin 2015 ;33:225-48.
2. Sullivan R, Yau WY, O'Connor E, Houlden H. Spinocerebellar ataxia: an update. J Neurol 2019;266:533-44.
4. Soong BW, Morrison PJ. Spinocerebellar ataxias. Handb Clin Neurol 2018;155:143-74.
5. Sun YM, Lu C, Wu ZY. Spinocerebellar ataxia: relationship between phenotype and genotype - a review. Clin Genet 2016 ;90:305-14.