KAS VE NÖROMÜSKÜLER KAVŞAK HASTALIKLARI
Yazanlar: Piraye Oflazer, Hacer Durmuş-Tekçe, Feza Deymeer
Güncelleme tarihi: 23.07.2020
İSKELET KASI
Hareketin son ortak yapısı olan iskelet kasları (çizgili kaslar), kas lifi adını alan hücrelerden oluşurlar (Şekil 1). Olgunlaşmış kas hücresi olan kas lifi, boyu birkaç santimetreye kadar ulaşabilen, iğ görünümünde ve çok çekirdekli bir yapıdadır. Çok sayıda ve çevrelerindeki bazal ve retiküler laminalar (endomizyum) ile birbirinden ayrılmış olan kas lifi toplulukları kas fasikülünü oluşturur. Endomizyumda kapillerler ve intramüsküler sinir lifleri bulunur. Fasiküller ise birbirinden perimizyum adı verilen bağ dokusu ile ayrılır. Perimizyal alanda, arteryol ve venüllerle birlikte kas liflerine yönelmiş motor sinir demetleri bulunur. Birkaç fasikülü, epimizyal bağ dokusu çevreler. Epimizyumda ise daha büyük çaplı damarlar vardır.

Şekil 1. Çizgili kasın şematize edilmiş yapısı.
Diğer mezodermal kökenli hücrelerden farklı olarak kas lifi kendisine gelen uyarıyı alan, uyarlayabilen, yayan ve buna kasılma ile yanıt verebilen bir hücredir. Bu nedenle uygun özellikte ve dayanıklılıktadır, yoğun enerji kullanır. Kendisinden beklenen fonksiyonlar kas lifi yapısının özgün olmasını gerektirir.
Kas lifi membranı sarkolemma adını alır. Sarkolemma, iki katmanlı lipid yapısındadır. Bu iki katmanın içine gömülü olarak proteinler yerleşmiştir (integral membran proteinleri). Lipid katmanların dışta olanı bazal lamina ile içte olanı ise hücre plazması ile komşuluktadır. Sarkolemma üzerindeki en önemli yapılardan birisi, motor sinir akson terminalinin sinaps yaptığı, nöromüsküler kavşağın postsinaptik bölgesidir.
Nöromüsküler kavşak yapısına bir bütün olarak bakalım (Şekil 2): Tek bir kas lifini bir motor sinir terminali innerve eder. Genellikle her kas lifinde bir adet olan nöromüsküler kavşak, motor sinir terminali (presinaptik bölge), kas lifi membranı (postsinaptik bölge, motor son plak) ve aralarında bulunan bazal laminadan (sinaptik aralık) oluşur. Sinir terminalinde çok sayıda, asetilkolin (ACh) molekülü içeren sinaptik veziküller, membranın ‘serbestleşme’ bölgeleri, yani ‘aktif’ bölgeler bulunur. Kas membranı ise sinir terminali ile bağlantı kurduğu postsinaptik bölgede, kıvrımlar yaparak çok özelleşmiş bir yapı halini alır. Kıvrımların nedeni membranın, bu bölgede alanını çok genişletmesi ve sinir-kas iletisinden, olabilecek en fazla şekilde yararlanmasıdır. Kıvrımların tepe kısımlarında nikotinik asetilkolin reseptörleri (n-AChR) bulunur. Bu reseptörler, sinir terminalinden salınan ACh moleküllerini yakalamak, kullanmak ve kendisine gelen bu kimyasal uyarıyı alarak membranda depolarizasyon dalgasının oluşmasını başlatmakla Bu son plak bölgesinin hemen altındaki subsarkolemmal alanda, hücre veya yalnızca nöromüsküler kavşakta ifade edilen birçok protein, ayrıca mitokondriler yoğunlaşmış olarak bulunurlar.
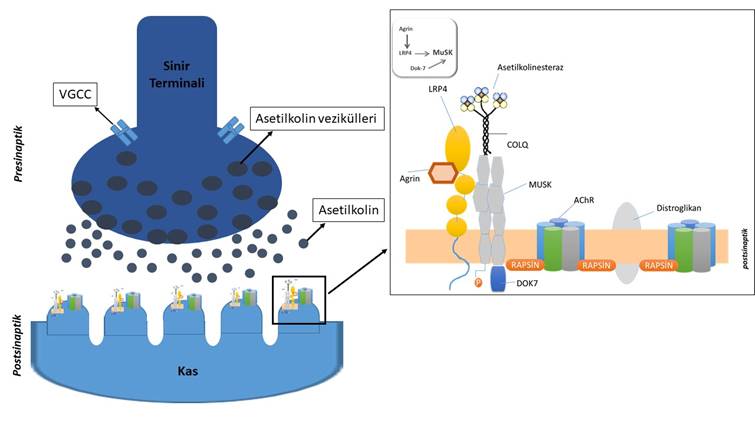
Şekil 2. Nöromüsküler kavşak. Aksiyon potansiyeli sinir terminaline ulaştığında voltaj kapılı Ca kanalları açılır ve Ca sinir terminaline girer. Bunun etkisi ile asetilkolin (ACh) ihtiva eden veziküller presinaptik hücre membranı ile birleşir ve ACh sinaptik aralığa salınır. ACh’in ACh reseptörlerine (AChR) bağlanması sonucu kanal açılır ve Na hücre içine girerek kas membranı boyunca yayılacak olan aksiyon potansiyelini başlatır. Nöromüsküler kavşakta rolü olan diğer proteinler: Agrin, presinaptik sinir ucundan salınır ve postsinaptik LRP4’e bağlanır, LRP4 de MuSK’a bağlanıp onu fosforilize ederek aktifleştirir. DOK7 de MuSK’u aktive eden bir proteindir. Fosforile MuSK, rapsin aracılığıyla AChR’yi etkileyerek AChR’nin NMK’ta postsinaptik yarıklarda kümelenmesini sağlar. Bu kümelenmenin nöromüsküler bileşkedeki iletinin sağlıklı şekilde sürdürülmesi için gerekli olduğu bilinmektedir. ColQ proteininin de MuSK’u sinapsa bağlı tutarak MuSK’un fonksiyonu ile ilişkili olduğu düşünülmektedir.
VGCC: “voltage gated calcium channel”, MuSK: “muscle specific kinase”, LRP4:“ low density lipoprotein 4”, COLQ:”collajen Q”, DOK7:” Docking protein 7”
Sarkolemma üzerinde ayrıca “caveola” isimli invaginasyonlar bulunur. Bu invaginasyonlar, membranın devamı niteliğinde olan transvers tübüler (T-tübül sistemi) sistemle hücre içine devam eder ve sarkoplazmik retikuluma bağlanır. Sarkoplazmik retikulum, kas hücresinin kalsiyum deposudur, kasılma için gerekli kalsiyum buradan sitoplazmaya sağlanır.
Kas membranının yapısal bütünlüğünü ve sağlamlığını sağlayan proteinlerin başında distrofin gelmektedir (Şekil 3). İkili ters sarmal yapısında olan her bir distrofin molekülü, N- ucu ile subsarkolemmal yerleşimli a -aktinin aracılığı ile hücre içine, C- ucu ile ise, ektraselüler matriksteki laminin aracılığı ile hücre dışına bağlanır. Ekstraselüler matriks ile bağlantı, yine membranda bulunan distrofinle ilişkili proteinler (sarkoglikanlar ve distroglikanlar) aracılığı ile olur. Böylece kas membranı iki yandan sağlam olarak hücre içine ve hücre dışına bağlanmış olur. Bu bağlanma çok gereklidir, çünkü kas hücresi, diğer hücrelerden farklı olarak, kasılabilen bir hücredir ve bu kasılma sırasında membran çok fazla yük altında kalarak gerilir. Membranın bu bütünlüğü ve bağlanması bozulduğunda dayanıklılığı ortadan kalkar ve yırtılması kolaylaşır.

Şekil 3. Çizgili kas membran proteinleri.
{kind=link}
{kind=link}
Kas lifi sitoplazması sarkoplazma adını alır. Sarkoplazma içinde mitokondriler, lizozomlar, ribozomlar, endoplazmik retikulum, Golgi organı, sarkoplazmik retikulum, T-tübülleri, lipid damlacıkları, glikojen partikülleri, bazı proteinler ve tümüyle kas hücresine özgü olan miyofilamentler bulunur. Miyofilamentler, kas lifinin kasılmasını sağlayan fibriler proteinlerdir, kalın ve ince filamentlerden oluşurlar. Kalın filamentlerin ana proteini miyozin, ince filamentlerinki ise aktin proteinidir (Şekil 1). Yapı ve işlevleri başka proteinler tarafından da belirlenen bu miyofilamentler, kas lifinin uzun eksenine paralel yerleşmişlerdir ve lif boyunca tekrarlar oluştururlar. Bu tekrar birimlerinin her biri bir kasılma ünitesidir ve sarkomer adını alır (Şekil 4). Her sarkomer kas lifinin uzun eksenine dik, titin ve nebulin proteinlerini de barındıran Z-bantlarından başlar. Kontraktil miyofilamentlerin bu diziliş biçiminin ışık geçirgenliği iskelet kasına çizgili bir görünüm verir.

Şekil 4. A) Bir Z bandından diğerine uzanan sarkomer kasın kasılma ünitesidir.
B) Sarkomerlerden oluşan iskelet kasının çizgili görünümü.
Kas lifleri, birbirinden farklı biyokimyasal ve fizyolojik özelliklere sahip iki ana gruptan oluşur. Tip-1 kas lifleri kan akımı ve oksijenlenmesi çok iyi, mitokondri ve lipidden zengin, dolayısı ile aerobik koşullarda çalışan, uzun süreli ve/veya yavaş ama fazla güç gerektiren hareketleri yaparken (maraton koşusu, vs.) kullandığımız, geç yorulan kas lifleridir. Buna karşılık Tip-2 kas lifleri, kan akımı ve oksijenlenmesi daha az olan, genel olarak anaerobik koşullarda da kasılabilen, glikojenden zengin, daha çok hızlı ve ani hareketleri yapmamızı sağlayan (hızlı koşmak, vs.) ve çabuk yorulan kas lifleridir. Tip-2 lifleri, kendi içinde Tip-2A, 2B ve 2C olarak ayrılırlar. Kas liflerinin bu özellikleri birkaç faktör tarafından belirlenir. Bu faktörlerin başında, her bir kas lifini innerve eden motor nöronun tipi gelmektedir. Normal bir kasta kas liflerinden birbirine komşu olanlar, farklı tipteki motor nöronlar tarafından innerve edilir ve böylece uzaysal dağılımda Tip-1 ve Tip-2 lifleri mozaik oluşturur. Böylece aynı kas içinde hem çabuk ve kısa süreli kasılabilecek, hem de yavaş kasılıp hareketi sürdürebilecek kas lifleri birlikte bulunur. Bu birliktelik, bir hareketin başlatılabilmesi ve sürdürülebilmesi açısından önemlidir.
Kas lifleri, asıl fonksiyonları olan kasılma sırasında, enerji ihtiyacı içindedir. Bu enerjiyi üç ana yakıtı, yani glikojeni, lipidleri ve gerekirse proteinleri kullanarak, bu sırada da ATP üreterek elde ederler. Her üç yakıttan elde edilen enerjinin ana kısmı mitokondri içindeki sistemler aracılığı ile elde edilir (sitrik asit döngüsü, beta-oksidasyon vs.). Son ortak enerji üretim yolu ise yine mitokondridedir, burada yer alan oksidatif fosforilasyon en yüksek miktardaki enerjinin üretildiği süreçtir.
Kas lifinin kasılmasının başlaması motor sinir terminalindeki elektriksel uyarının biyokimyasal uyarıya dönüşmesiyle sinaptik yoldan ve ACh aracılığı ile olur. Son plak bölgesindeki sinapsa verilen ACh molekülleri, reseptör ile birleşir ve buradaki katyon kanalları açılarak sodyum kas hücresi içine girer (iyon kanallarının özellikleri ile ilgili bilgi daha sonra verilecektir). Bu katyon değişimi 0mV düzeyinde bir dengeye ulaştığında postsinaptik membranda depolarizasyon başlar (son plak potansiyeli, endplate potential, EPP). EPP amplitüdü aksiyon potansiyeli (AP) için gereken eşiğe ulaşırsa kas lifi boyunca yayılan AP oluşur yani depolarizasyon sarkolemma boyunca yayılır. Miyofibril eksitasyonu ve kas kontraksiyonu için bu depolarizasyonun sarkolemma dışında, hücre içine doğru transvers tübül sistemlerinden geçerek sarkoplazmik retikuluma ulaşması gerekmektedir. Bu bölgelerin depolarizasyonundan sonra tübüllerin sisterna bölgelerindeki dihidropiridin reseptörleri tübüllerdeki uyarıyı, protein eşleşmesi yoluyla, intraselüler komşuluklarındaki sarkoplazmik retikulum ryanodin reseptörlerine ulaştırırlar. Sarkoplazmik retikulumdaki ryanodin reseptörleri aracılığı ile ise sarkoplazmaya, miyofibrillerin çevresine Ca++ salınır. Ca++ salınmadan önce, yani istirahat halinde iken aktin üzerindeki miyozin bağlantı noktaları troponin tarafından kapatılmış haldedir. Ortama Ca++ salındığında, miyofilament düzenleyici proteinlerden tropomiyozin, aktinin duruşunda bir uzaysal değişim yapar ve tropomiyozin bağlanma noktaları değişir. Böylece, miyozin bağlanma noktaları açılmış olan aktin, miyozin üzerinde, ATP’nin hidrolizasyonu ile oluşan ADP ve Pi bağlantısı aracılığı ile kayarak kontraksiyonu gerçekleştirir (Şekil 1). Bu sırada kasın boyu kısalır ama hacmini korumak için eni genişler. Eğer uygulanan kasılma uyarısı çok fazla ise kas kısalmadan kontraksiyon gerçekleşir. Gevşemenin oluşabilmesi için Ca++ ‘un yeniden ortamdan sarkoplazmik retikulum içine çekilmesi ve aktin-troponin bağlantısının, istirahat haline dönmesi, yani aktinin miyozini tanıyan noktalarının kapatılması gerekir.
İYON KANALLARI
Kas lifi membranındaki lipidler büyük ölçüde hidrofobiktir ve hidrofilik iyonların geçişine elverişli değildir. İyonlar hücre içine ancak membrandaki özel gözeneklerden (kanallardan) geçerek girebilirler. İyon kanalları, iyonların hücre membranından geçmesini sağlamak üzere bir gözenek (‘pore’) oluşturan ve membranın tüm kalınlığı boyunca yerleşmiş olan proteinlerdir. İyon kanallarının en önemli özelliği spesifik bir iyonu tanıyıp seçmeleridir. İyon kanallarından geçiş pasiftir: hücre membranının iki tarafı arasındaki elektriksel potansiyel ve konsantrasyon farkı, söz konusu olan iyonun belli bir yönde geçişini sağlar.
İyon kanallarının genellikle iki veya daha fazla farklı durumu vardır. Örneğin, sodyum kanalları açık ve kapalı halde bulunabilir. Kapalı oldukları zamanlarda da iki farklı durumda olabilirler: istirahat hali (potansiyel olarak aktive olabilirler) ve inaktivasyon hali (refrakter, aktive edilemezler). Bir durumdan bir diğerine geçiş kanalın ‘kapı’ (gate) görevi yapan kısımlarının açılıp kapanmasıyla olur. Kanalların birçoğunun açılıp kapanması bazı dış faktörlerin etkisiyle olur (kapılı kanallar). Bu faktörler elektriksel (voltaj-kapılı), kimyasal (ligand-kapılı) veya mekanik (mekanik-kapılı) olabilir. Bazı kanallar ise herhangi bir dış faktörün etkisi olmaksızın açık halde bulunurlar (kapısız kanallar).
Kapılı kanalların en önemlileri voltaj kapılı kanallardır. Bunlarda, hücre membranında spesifik elektriksel potansiyel farkları sonucu kanallar açılır. Voltaja bağımlı kanallardan potasyum kanalı (VGKC), sodyum kanalı ve kalsiyum kanalının (VGCC) aynı kökten geldiği düşünülür ve temel yapıları birbirine benzer. İyon seçiciliği ve voltaj fark edicilik fonksiyonlarını başlıca α subünitesi üstlenmiştir. Bir α subünitesinin temel motifi 6 transmembran segmentten (S1-S6) oluşan bir yapıdır. Membrandaki voltaj değişimini fark edenin bu transmembran segmentlerden dördüncüsü (S4) olduğu düşünülmektedir. Potasyum kanalının α subünitesi tek bir temel motiften ibarettir. Sodyum ve kalsiyum kanallarının bir α subünitesi ise temel motifin 4 kez tekrarından oluşur. Sodyum ve kalsiyum kanallarında iyonların içinden geçtiği kanalın gözeneğini oluşturan tek bir α subünitesi iken potasyum kanalında 4 subünite gözeneği oluşturur. Her üç kanalda da α subünitesi dışında başka subüniteler de bulunur. Klor kanalının yapısı bu üç kanalınkinden farklıdır.
Kas iyon kanalı hastalıkları adı altında toplanan bir grup herediter hastalıkta bozukluk iskelet kasının klor, sodyum, kalsiyum ve potasyum kanallarındadır. Birçoğu 19. yüzyıldan beri bilinen bu hastalıkların iskelet kasındaki voltaj kapılı iyon kanallarının bozukluğundan kaynaklandığının anlaşılması iki önemli alandaki gelişme sonucu olmuştur: Modern elektrofizyoloji ve moleküler biyoloji. Bin dokuz yüz ellili yıllarda keşfedilen özel elektrofizyolojik incelemeleri kullanarak daha sonraki yıllarda bu hastalıklardan bazılarının klor, bazılarının ise sodyum iyon kanalı bozukluğu sonucu oluştuğuna dair, gen araştırmalarına da ışık tutan, değerli bilgiler elde edilmişti. Bin dokuz yüz doksanlı yıllarda bu hastalıkların gerçekten de tahmin edilen iyon kanalını kodlayan genlerdeki mutasyonlara bağlı olarak ortaya çıktığı anlaşıldı ve önceden tahmin edilemeyen yeni genetik bilgiler eklendi. Voltaj kapılı iyon kanallarının, çok nadir de olsa, otoimmün hastalıkları da bulunmaktadır.
Ligand kapılı iyon kanallarında spesifik bir ligandın proteine bağlanması gerekir. Kas hastalıkları açısından en önemli reseptör iyon kanalı nikotinik asetilkolin reseptörüdür (AChR). Bu kanal erişkinde iki α, bir β, bir δ, bir ε olmak üzere beş subüniteden oluşur, fetusta ε yerine γ subünitesi bulunur. Asetilkolin yapışma yeri α subünitelerindedir. Asetilkolin ile birleşme sonucu açılan kanal fazla selektif olmaksızın sodyum ve potasyum iyonlarını geçirir. Bu kanalın genetik defektleri konjenital miyastenik sendromlarının bazı tiplerine yol açar, otoimmün hastalığı ise miyastenia gravis’tir.
Genetik bozukluklar ve otoimmün saldırıların yanı sıra bazı doğal toksinlerin de iyon kanallarını hedef aldığı bilinir: Tetrodotoksin voltaj kapılı sodyum kanallarını, alfa bungarotoksin nikotinik asetilkolin reseptörünü bloke eder. Birçok ilaç kanallar üzerinden etkisini gösterir.
KAS HASTALIKLARINDA BELİRTİ VE BULGULAR
Güçsüzlük
Kas hastalıklarının birincil belirtisi kas güçsüzlüğüdür. Kas hastaları genellikle yokuş ve merdiven çıkma, oturduğu yerden kalkma, yürüme, kollarını kaldırıp yükseğe uzanma, başını yıkamada güçlük gibi, kök kaslarında güçsüzlük nedeni ile ortaya çıkan yakınmalarla doktora başvururlar. Bazen bunlara gözkapaklarını açma veya kapama, değişik yönlere bakma, yüzün mimik hareketlerini yapma, çiğneme, yutma, başını yastıktan kaldırma gibi hareketleri sağlayan kasların güçsüzlüğü de eşlik eder. Birçok kas hastalığının ilerlemiş dönemlerinde tabloya ayak ve el kasları gibi distal kasların güçsüzlüğü de eklenir. Ancak bazı kas hastalıklarında bu distal kasların tutulumu hastalığın en erken ve en belirgin bulgusu olabilir (distal miyopatiler). Bazı kas hastalıklarının ise kendine özgü güçsüzlük dağılımı vardır; bu dağılımlara rastlandığında öncelikle o hastalıkların varlığı düşünülmelidir. Örneğin yüzün mimik kasları, skapulayı yerinde tutan kaslar ve humerus çevresi kaslar, özellikle de asimetrik biçimde tutulmuşsa fasyoskapulohumeral distrofiyi, frontal, yüz kasları ile birlikte sternokleidomastoid kas ve distal (özellikle tibialis anterior) kasların tutulması öncelikle miyotonik distrofiyi düşündürür.
Birçoğu kalıtımsal olan kas hastalıklarında güçsüzlük yıllar, hatta on-yıllar içinde gelişir, hastalık kronik gidişlidir. Edinsel kas hastalıklarında, örneğin inflamatuvar miyopatilerde ise zaafın gelişimi subakuttur; hasta birkaç hafta veya ay içinde ciddi fonksiyon kaybına ve özürlülük durumuna ulaşır. Rabdomiyoliz, malign hipertermi gibi durumlarda ise güçsüzlük saatler içinde ilerler ve en çok birkaç gün içinde en üst düzeyine ulaşır. Bazı durumlarda güçsüzlük hastada akut ve epizodik olarak yerleşir ve tekrarlar. Periyodik paraliziler bu son duruma örnektir. Miyastenia gravis’te olduğu gibi bazen güçsüzlük gün içinde değişkenlik gösterir (fluktuasyon). Birçok miyopatide kalp kası ve solunum kaslarının, değişik derecelerde tutulması söz konusudur.
Egzersiz İntoleransı
Belirli bir hareketi yapmakla, o hareketi yapan kasta gelişen ve normalde olmaması gerektiği ölçüde olan yorgunluğa egzersiz intoleransı denir. Bazen bu yorgunluğa ağrı da eşlik edebilir. Genellikle metabolik hastalıkların seyri sırasında görülür. Yorgunluğu doğuran hareket, hareket sırasında enerji elde etmek için kullanılan metabolik yollardan hangisinin tutulduğuna bağlıdır. Örneğin bir hastada yürürken egzersiz intoleransı gelişiyor ancak bu hasta hızlı koşarken rahatsız olmuyorsa bu durum öncelikle, Tip-1 kas lifleri ile yavaş hareketleri yapmamızı sağlayan lipid metabolizması yolunun bozukluğunu düşündürmelidir. Buna karşılık hastada aynı yakınma örneğin seri ve hızlı yapılan yer silme gibi bir kol hareketinde, kol kaslarında ortaya çıkıyorsa bu kez başlıca Tip-2 kas liflerinin kullandığı glikojen metabolizmasının bozuk olduğu düşünülmelidir. Bu yolların veya son ortak enerji yolu olan oksidasyon-fosforilasyon sisteminin bozukluğunda, egzersiz intoleransına serum laktat, piruvat, amonyak, miyoglobin düzeylerinde, her bir metabolik bozukluğa özgü olan değişiklikler de eşlik edebilir.
Bitkinlik
Miyastenia gravis ve kas-sinir kavşağının diğer hastalıklarında bir hareketi yapmakla o kasta, bazen başka kaslarda da, çok ileri derecede yorgunluk (tükenme) ortaya çıkar. Bu yorgunluk, başlangıçta normal veya daha iyi güç oluşturan kasın hareketinde devamlılığın sağlanmasındaki güçlük nedeni ile ortaya çıkar; yani hasta, hareketi devam ettiremez. Bu durum, çok hareket edip yorgunluk hissetmemizden farklı bir motor tükenme halidir. Hasta muayene edildiğinde fonksiyon azalması veya kaybı, çok belirgin olarak tespit edilir. Burada bozukluk sinir-kas kavşağındadır ve sorun, ACh ile n-AChR arasındaki iletişimin, biri veya diğerinin defektine bağlı olarak yeterli düzeyde olmamasıdır.
Atrofi
Kas hastalıklarında atrofi, periferik sinir hastalıklarındakine oranla çok daha geç gelişir. Bunun nedeni kas liflerinin tek tek hastalanması ve ancak zaman içinde, yeterince kas lifi tutulduktan sonra, kasın tüm kitlesini etkileyecek ölçüde küçülmesine, yani atrofiye neden olmasıdır.
Hipertrofi
Gerçek hipertrofi, miyotoni gibi kasılmanın belirgin, gevşemenin ise zor olduğu durumlarda görülür. Miyotoniye kas zaafının eşlik etmediği durumda (myotonia congenita), hipertrofi nedeni ile hasta “Herkül” görünümündedir.
Psödohipertrofi
Adından anlaşılacağı gibi, yalancı hipertrofidir (Şekil 5). Hastalık nedeni ile henüz kaybedilmemiş kas liflerindeki hipertrofiye bağ ve yağ dokusundaki artış da eşlik eder ve kas kitlesi bu nedenle artar. En sık baldırlarda gastroknemius-soleus kas grubunda, bazen kuadriseps kasında, bazen de başka kaslarda görülür. Aynı miyopati nedeni ile çevredeki kasların atrofik durumda olması bu genişlemiş görüntüyü abartılı hale getirir.

Şekil 5. Duchenne müsküler distrofili bir hastada baldır psödohipertrofisi.
{kind=link}
Kemik Veter Refleksleri (KVR)
Kas hastalıklarında KVR de, o kastaki zaaf ve atrofi ile orantılı olarak ve zaman içinde azalır veya kaybolur. Buna karşılık polinöropatilerde KVR birden veya çok kısa sürede kaybolur.
Miyotoni
Klinik olarak miyotoni güçsüzlüğün tersine, kasılmanın normal veya oldukça iyi, kasıldıktan sonra gevşemenin güç olduğu bir durumdur. Bu gevşeme güçlüğü o kasın, istirahatten sonraki ilk hareketlerinde çok belirgindir, aynı hareket tekrarladıkça harekette rahatlama görülür, çünkü gevşeme güçlüğü azalır. Örneğin hasta sabah yataktan ilk kalktığında, durakta otobüs beklerken otobüsün geldiğini görünce aniden koşup yakalamaya çalıştığında, başını aniden bir yöne çevirdiğinde, suskunken birden konuşmaya başladığında, eli ile bir şey tuttuğunda veya sıktığında, yemek yemeye başladığında ilgili harekette tutukluk hisseder, tutukluk bir süre sonra düzelir. Bu tutukluk bazen romatizmal hastalıklar, özellikle sabah tutukluğu ile karıştırılabilir. Miyotoni, myotonia congenita’da olduğu gibi, bazı hastalıklarda tek belirtidir; bazen de kalıcı kas güçsüzlüğü ile beraberdir; miyotonik distrofi buna örnektir.
Kontraktür
Aynı eklemin etrafındaki kasların kuvveti birbirinden farklı olduğunda o eklem, belli bir pozisyonda durma eğilimi gösterir, ilgili kasta kısalma olur ve zaman içinde eklemin bu pozisyonu sabitleşir ve kontraktür gelişir (Şekil 6). Kas hastalıklarında bu kontraktürün gelişmesi genellikle yıllar alır. Kontraktür oluşuncaya kadar tendonda gelişen ve giderek artan sertlik, egzersizle veya eklemi ters pozisyonda tutan atellerle önlenebilir veya azaltılabilir. Ancak sabit kontraktür geliştikten sonra o eklem, ancak cerrahi girişimle düzeltilebilir. Bazı miyopatiler kontraktür oluşturmaya özellikle eğilimlidir. Bu durumlarda kontraktür, kasların tutulumundan bağımsız olarak, oldukça erken gelişir. Bu hastalıklarda kontraktürlerin dağılımı da özellik gösterir. Bu hastalıklara örnek olarak Emery-Dreifuss kas distrofisi, “rigid spine” hastalığı, Bethlem miyopatisi gösterilebilir.

Şekil 6. Dirsekte kontraktür. Kontraktür nedeni ile hasta dirseğini açamamaktadır.
Kas Ağrısı (Miyalji)
Jeneralize kas ağrıları başlıca kaslardaki inflamatuvar, nekrotizan, toksik veya metabolik süreçleri akla getirir. Metabolik hastalıklardaki ağrı genellikle veya yalnızca egzersizle ilişkilidir. Seyrek olarak distrofinopati, kaveolinopati gibi kas distrofilerinde de egzersizle kas ağrıları, özellikle baldır ağrıları görülebilir. Buna karşılık polimiyozit ve dermatomiyozite bazen, rabdomiyolize ise hemen her zaman eşlik eden miyaljiler sürekli ağrılardır. Bu ağrılar, ancak patolojik sürecin gerilemesi ile ortadan kalkarlar. Lokal kas ağrıları ise infeksiyon, lokal inflamasyon, infarkt, travma, hematom veya tümörlerde görülür. Kas ağrılarını eklem ve kemik ağrılarından ayırmak önemlidir. Eklem ağrıları lokal ve ekleme sınırlıdır, o eklemin hareketi ile artar. Kemik ağrıları ise daha derin, şiddetli, şiddeti sürekli aynı kalan, birçok zaman geceleri artan ağrılardır.
Kramp
Kramp kas hastalığından çok bir periferik sinir hastalığını düşündürür. Buna rağmen, özellikle egzersizle ortaya çıkan kramplarda ayırıcı tanıda metabolik miyopatileri, distrofinopati, kaveolinopati ve disferlinopati gibi kas distrofilerini de düşünmek gerekir. Kas miyofosforilaz eksikliğinde (McArdle hastalığı) ortaya çıkan kramp, egzersizi yapan kastadır ve elektrofizyolojik olarak sessizdir.
Diğer Kasların Zaafı
Kas hastalığı olduğu düşünülen bir hasta, özellikle geceleri nefes darlığı veya nefesinin yetmediğinden söz ediyorsa miyastenia gravis, asit maltaz eksikliği, polimiyozit mutlaka tanı olasılıkları arasında olmalıdır. Yutma güçlüğü ve ses kısıklığı da benzer durumları akla getirmelidir. Belirgin çift görme, oftalmoparezi ise, yine bu belirtileri gösteren mitokondriyal miyopatiler veya okülofaringeal distrofiden önce miyastenia gravis’i düşündürmelidir. Boyun ekstansör kaslarının zaafına bağlı gelişebilecek başın öne düşmesi miyastenia gravis, polimiyozit gibi hastalıkları akla getirmelidir. Birçok miyopatide ise boyun fleksiyon zaafı görülür.
Sistemik Bulgular
Anamnez alınırken bazı soruların hastaya özellikle sorulması çok yardımcı olabilir. Örneğin rabdomiyoliz olduğu düşünülen bir hastada o sırada veya daha önceleri idrar renginde koyulaşma olduğunun anlaşılması tanıya çok yardımcı olur. Bazı ilaçların (anti-hiperlipidemik veya anti-hiperkolesterolemik ilaçlar, siklosporin-A, ipeka, amiodaron, kolşisin, klorokin, d-penisilamin vs.) kullanımı ve bazı toksinlere maruz kalmanın da miyopati yaratacağı bilinmeli ve hasta bu açıdan da sorgulanmalıdır. Bazı sistemik hastalıkların da miyopati yapacağı bilinmeli ve hasta bu sistemik hastalıklar açısından da incelenmelidir. Bu hastalıklar arasında en sık düşünülmesi gereken hipotiroididir. Sinir sisteminin başka bölümlerinin tutulması da tanı olasılıkları yelpazesini daraltır. Örneğin, miyopatisi olan bir hasta epileptik nöbetler geçirdiğinden söz ediyor veya mental retardasyon gösteriyorsa mitokondriyal hastalıklar akla gelebilir.
Başlangıç Yaşı
Hastalığın başlangıç yaşının öğrenilmesi hastalığın gidiş hızını ve doğasını anlamak için gereklidir. Çok yavaş ve sinsi ilerleyen hastalıklarda hastanın ve ailesinin hastalığın başlangıç yaşını hatırlaması sıklıkla zor olur. Bu nedenle çoğu zaman hastaya ve ailesine geçmiş yıllara ait hatırlatmalar yapmak gerekebilir. Hastalık belirtilerinin başlangıcı yenidoğan dönemi, çocukluk veya ergenlik döneminde ise öncelikle herediter miyopatiler, genç ve orta erişkinlik döneminde ise öncelikle edinsel miyopatiler, geç erişkinlik ve yaşlılık döneminde ise öncelikle toksik-iyatrojenik miyopatiler akla gelmelidir.
Aile Öyküsü (Soygeçmiş)
Birçoğu genetik olarak kalıtılan kas hastalıklarında aile öyküsü, anamnezin vazgeçilmez bir parçasıdır. Soyağacı, anne-baba akrabalığı (akraba değillerse ailelerin aynı köy, kasaba veya şehirden olup olmadıkları), ailedeki ölüm nedenleri, ailede aynı hastalığı taşıyan bireyler ve yaşları/cinsiyetleri, ailedeki hasta olmayan bireyler ve yaşları/cinsiyetleri mutlaka kaydedilmelidir. Otozomal dominant geçiş belli hastalıkları, otozomal resesif geçiş başkalarını, X’e bağlı resesif geçiş ise diğer bazı kas hastalıklarını düşündürür. Ayrıca hastalığın başlangıç yaşı, ailede hasta olan bireylerde klinik tablonun homojen olup olmamasına ait bilgiler de tanıya yaklaşım açısından yararlıdır.
KAS HASTALIKLARINDA MUAYENE
Kas hastalığına eşlik edebilecek bulgulardan yararlanmak için nörolojik muayenenin tam olarak yapılması gerekir. Örneğin, miyopatisi olan bir hastada merkezi sinir sisteminin tutulduğunu gösteren bulguların varlığı bir mitokondriyal hastalığı akla getirebilir. Ancak kası ilgilendiren özelliklerin muayenesi ayrı bir dikkat gerektirir. Bir başka dikkat edilmesi gereken nokta, atrofi, hipertrofi, skolyoz, vs. gibi, tespiti görsel olarak yapılabilecek değişiklikler ve bunların dağılımını görebilmek amacı ile hastanın, mümkünse, iç çamaşırları ile kalacak şekilde soyunması ve muayenenin böyle yapılmasıdır. Aşağıda yalnızca bu muayene tekniklerine değinilecektir.
Kas Gücü Muayenesi
Herhangi bir cihaz kullanmadan kas gücünün test edilmesi sırasında (manual muscle testing) tüm dünyada yaygın olarak kullanılan değerlendirme sistemi, [Medical Research Council (MRC)] skalasıdır. Bu sistemde, hareketin yerçekimi bağlamında değerlendirilmesi önem taşır. Her bir kas bu prensip dikkate alınarak test edilmelidir. Doğru yapılmış kas gücü test sonuçları, zaafın dağılımını doğru verecek ve tanıya yardımcı olacaktır. Dikkat edilmesi gereken bir nokta, test edilmesi birçok zaman unutulan gluteus maksimus kasının, kas hastalığı olduğu düşünülen bir bireyde, diğer kasların gücü tam olsa bile, muayene edilmesinin unutulmaması ve boyun fleksiyon zaafının mutlaka hasta sırtüstü yatırılarak test edilmesidir.
Yukarda anılan MRC sistemine göre kas gücü aşağıdaki gibi değerlendirilir:
5/5 (Tam) kas gücü: Test edilen kas, yerçekimine karşı hareketini tamamladıktan sonra, kendisine uygulanan tam karşı kuvvete, tam bir dirençle karşılık veriyor.
4/5 kas gücü: Test edilen kas, yerçekimine karşı hareketini tamamladıktan sonra, kendisine uygulanan tam karşı kuvvete, direnç gösterebildiği halde, yeniliyor.
3/5 kas gücü: Test edilen kas, yerçekimine karşı hareketini tamamladıktan sonra, kendisine uygulanan karşı kuvvete, hiçbir direnç gösteremeden yeniliyor.
2/5 kas gücü: Test edilen kas, ancak yerçekimi etkisi kaldırıldığında hareketini tamamlıyor.
1/5 kas gücü: Test edilen kas, yerçekimi etkisi kaldırıldığında bile hareketini tamamlayamıyor, yalnızca kasılma gösteriyor.
=0= kas gücü: Test edilen kas hiçbir şekilde hareket veya kasılma gösteremiyor.
Ayrıca kalça kasları ve aksiyal kasların zaafını göstermek için hastaya yere çömelmesi, ardından da ayağa kalkması söylenir. Hastanın, hiçbir yere tutunmadan kalkabilmesi gerekir. Bunun dışındaki her durum, değişik derecelerde Gowers belirtisinin varlığını gösterir (Şekil 7). Örneğin Duchenne tipi kas distrofisi olan bir çocuk, ayağa kalkabilmek için önce vücudunu öne alır, başını dikleştirir ve ardından önce bacaklarına, sonra uyluk proksimaline, sonra da karnına elleri ile dayanarak, yani adeta kendi vücuduna tırmanarak dik duruma gelebilir. Yattığı yerden kalkmak isteyen bir hasta, abdominal kasları zayıf ise önce yana döner, ardından dirseğine dayanarak oturur duruma gelebilir. Ayak distal dorsifleksör kaslarında, muayene ile saptanamayacak kadar hafif zaaf olsa da hasta, topukları üzerinde yeterince kalkamaz.

Şekil 7. Proksimal zaafı olan bir hastada vücuduna dayanarak ayağa kalkma sekansları (Gowers belirtisi).
Hareketli Görüntü: Gowers belirtisi
Bitkinlik Ortaya Çıkaran Manevralar
Bitkinlik, bir kasta tekrarlanan hareket sonucu ortaya çıkan ve muayene ile tespit edilebilen aşırı yorgunluk hali olduğuna göre, hastada kas gücü tam olsa bile aşağıdaki manevralarla bitkinlik ortaya çıkarılabilir. Bu manevralar daha çok miyastenia gravis’te kullanılır:
Gözkapaklarını yormak için hasta, başı doktorun eli ile sabitleştirilerek zorlu olarak yukarı, hatta yukarı-dışa baktırılır. Bitkinlik söz konusu ise, çoğu zaman 1 dakikaya kalmadan göz kapaklarından biri veya her ikisinin düşmeye başladığı gözlenir. Bazen buna hastada çift görme de eklenebilir. Ayrıca bir nefeste hastanın kaça kadar saydığı değerlendirilebilir. Kollardaki bitkinlik kolları öne uzatıp böylece bekleterek, bacaklardaki bitkinlik ise hastaya çömelip kalkma hareketinin tekrarlatılması ile ortaya çıkarılabilir. Miyastenia gravis’te göz kapağı üzerine, dayanılabilecek sıcaklıkta bir su tüpünün bir süre tutulması ptoz yaratabilir (tersine ptozu olan bir miyastenik hastada da içinde buz olan bir havlu ile birkaç saniye dokunulması ptozu düzeltebilir).
Miyotoni Aranması
Gevşeme güçlüğünü gösterebilmek için hastanın o sırada kullanmadığı, istirahatte olan bir kası kastırılır ve sonra gevşetmesi istenir; bu sırada gevşemenin ne kadar güç olduğu gözlenir (aksiyon miyotonisi). Ardından aynı hareket birkaç kez tekrarlatılır ve giderek hareketin rahatladığı görülür. En çok kullanılan yöntem hastanın bir elini veya gözkapaklarını sıktırmak ve bir süre böyle sıkılı tuttuktan sonra aniden açtırmaktır. Bu sırada açılma hareketinin güçlüğü ve hareketin tekrarlanması ile hareketin daha rahat yapılabildiğinin gözlenmesi gerekir (Şekil 8). Bir başka yöntem de, tenar kaslar, önkol kasları en sık olmak üzere herhangi bir kasın üzerine refleks çekici ile bir kez vurup beklemektir. Miyotoni var ise kasılmış olan kas bir süre bu kasılı pozisyonda, hatta bazen çökük olarak (kayık belirtisi) kalır (perküsyon miyotonisi).

Şekil 8. Aksiyon miyotonisi olan hastanın elini açma hareketinin sekansları.
İskemik Önkol Egzersiz Testi
Metabolik hastalıkların tanınmasına bazen yardımcı olabilen bu testte hastanın koluna tansiyon aletinin manşonu geçirilir ve sistolik kan basıncı ölçülür. Manşon bu basınçta 1 dakika süreyle sıkılı tutulur ve bu süre içinde hastanın, bir lastik balonu veya kendi elini sürekli olarak sıkıp bırakması sağlanır. Testin sonunda egzersiz durdurulur ve manşon açılır, hemen ardından el veya kolda güçsüzlük, önkolda sertlik (kontraktür) gelişip gelişmediğine bakılır. Bu bulguların gelişmesi bir metabolik bozukluğu gösterir. Ancak çok hassas olmadığından, testin negatif olması metabolik hastalık olmadığı anlamına gelmez. Günümüzde test, rabdomiyoliz gelişme olasılığına karşı, non-iskemik olarak da yapılmaktadır. Elbette teste başlamadan önce hastaya ne yapılacağının anlatılması, testin başarısı açısından önemlidir.
Duruş ve Yürüyüş Muayenesi
Hasta ayakta dururken dikkati çeken bir özellik (hiperlordoz, skolyoz vs.), başını veya vücudunu öne eğebilme kapasitesi (rigid spine’da eğilemez) mutlaka kaydedilmelidir. Kas hastalığı olan bireylerin birçoğu yürürken kalçalarını iki yana sallarlar; bu arada vücudun ağırlık merkezini uygun duruma getirmek için lordozlarını artırırlar. Bu yürüyüş biçimi, “ördekvari” olarak nitelenir. Zaafın özelliğine göre başka yürüme bozuklukları da gözlenebilir. Örneğin distal miyopatilerde hasta, aynen bir polinöropatide olduğu gibi, stepaj yaparak yürür.
Hareketli görüntü: “Ördekvari” yürüyüş.
Kas Kitlesindeki Değişiklikler
Soyunarak kol ve bacakları açığa çıkmış olan hastada atrofinin dağılımı, psödohipertrofinin bulunup bulunmaması tanı olasılıklarını azaltabilir, tanıya daha çok yaklaştırabilir. Bu nedenle nöromüsküler muayene mutlaka hasta soyunduktan sonra yapılmalıdır. Örneğin, biseps, triseps gibi humerus çevresi kaslarla birlikte skapula çevresi kasların ve yüz kaslarının atrofisi, hasta kollarını öne uzattığında veya yana açtığında skapulanın omuzdan yukarı çıkması (fikse olamaması, “humping”) fasyoskapulohumeral distrofiyi, temporal kaslarla sternokleidomastoid kasların atrofisi miyotonik distrofiyi, proksimal kas kitleleri ve ayak kasları oldukça iyi iken alt bacaktaki atrofi ise distal miyopatileri akla getirir. Alt bacak kaslarının atrofisi arka grupta ise Miyoshi isimli kas distrofisi öncelikle düşünülmelidir.
Baldır kaslarının psödohipertrofisi genellikle dikkati çeker; hasta parmak ucuna kaldırıldığında iyice belirginleşir (Şekil 9). Psödohipertrofinin görüldüğü durumlarda, örneğin metabolik bir miyopati (Pompe hastalığı dışında) düşünülmemelidir; bu bulgu herhangi bir kas distrofisi için tipiktir.

Şekil 9. Baldır kaslarında hafif ve asimetrik psödohipertrofi, (parmak ucunda yükselmekle belirginleşiyor).
Diğer Bulgular
Yukarda yazılanlara eşlik eden başka bulgular da değişik kas hastalıklarının öncelikli olarak düşünülmesini sağlayabilir. Örneğin, yüzde veya vücuttaki dismorfik bir özellik, çene yapısındaki bozukluk, yüksek damak gibi göstergeler daha çok konjenital miyopatileri düşündürür. Gözkapaklarındaki ödem ve hafif eritem (“heliotropi”), yüzdeki kırmızı döküntü (Şekil 10), boyunda “V” şeklinde eritem (“V” belirtisi), el-dirsek-diz eklem ekstansör yüzlerindeki eritem, ellerde kabalaşma ve tırnak yatağı değişiklikleri dermatomiyozit düşündürmelidir. Kaslar içinde sert kalsifiye nodüller ise daha çok çocukluk çağı dermatomiyozitinde görülür.

Şekil 10. Dermatomiyozitli bir hastada göz çevresinde heliotrop ödem ve yüzde “rash”.
KAS HASTALIKLARINDA LABORATUVAR İNCELEMELERİ
Kas Enzimleri
Kas hastalıklarının tanısında kas enzimlerinden kreatin fosfokinaz (CPK veya CK), aldolaz ve laktik dehidrogenazın (LDH) serum düzeyleri kullanılmakla birlikte, pratikte en sık kullanılan CK düzeyidir. Normal şartlar altında egzersiz, kasa uygulanan bir travma veya enjeksiyon CK değerini bir miktar yükseltebileceğinden, serum CK değerine hastanın dinlendiği koşullarda ve EMG veya kas biyopsisi yapılmadan önce bakılmalıdır. Serum CK düzeyinin laboratuvar normalleri değişebildiğinden, elde edilen değerin normalin üst sınırının kaç katı olduğunun veya sayısal değer yazılıyorsa yanına mutlaka laboratuvar normalinin kaydedilmesi yararlıdır.
Kas hastalıklarında serum CK düzeyi genellikle yükselir. Bu artış özellikle kas membranı bozuklukları ve kas nekrozu ile giden miyopatilerde çok yüksek değerlere ulaşabilir. Örneğin distrofiler, miyozitler, asit maltaz eksikliği, Miyoshi miyopatisi, rabdomiyoliz, CK düzeyini en çok yükselten durumlardır. Bu durumlarda CK normalin 5-10 katından 50-100 katına (5-10X, 50-100X) kadar çıkabilir. Diğer miyopatilerde CK hafif-orta derecede yüksek veya tümüyle normaldir. Kas membranının bozulmadığı, değişikliklerin hücre içine sınırlı kaldığı, örneğin konjenital miyopatiler gibi, durumlarda CK değerinin, bir miyopati olmasına rağmen normal olabileceği unutulmamalıdır. Buna karşılık bazı nöropatik/nöronopatik süreçlerde de hafif CK yükselmesi olduğundan, hafif ölçülerdeki yükselmelerde dikkat etmek gerekir. Ayrıca, kas hastalığı olmadığı halde serum CK düzeyini yükselten bir durum hipotiroididir. Hipertiroidi ise CK değerini düşürebilir; böylece bir miyopatide CK değeri daha yüksek olabilecek iken o değerin altında bulunabilir. Metabolik miyopatilerde ise aynı kişide ve değişik zamanlarda CK değeri bazen normal, bazen az yüksek, bazen ise çok yüksek (dalgalı) olabilir ve bu durum hastalığın miyozit ile karıştırılmasına yol açar.
Elektromiyografi (EMG)
Bir hastada kas hastalığı olup olmadığını gösteren en önemli incelemelerden biri EMG’dir ve miyopatiyi motor nöron ve periferik sinir hastalıklarından ayırmaya yarar. Bir hastada CK değeri çok yüksek ise o hastada mutlaka bir miyopati olacağından, bu durumda EMG yapılması gerekmez (ancak, eşlik eden bir polinöropati olup olmadığını anlamak istiyorsak gerekebilir). Buna karşılık, bir hastada hafif yüksek veya normal CK değeri varsa, bu durumun diğer nörojenik hastalıklarda da, miyopatilerde de olabilmesi nedeni ile ayırıcı tanı amacıyla mutlaka EMG yapmak gerekir. Motor ünite potansiyellerinin kısa süreli ve düşük amplitüdlü olması ve erken katılım göstermesi miyopati bulgularıdır. Nekrozla giden bazı miyopatilerde fibrilasyon, pozitif diken, psödomiyotonik boşalım gibi iritasyon bulguları da gözlenir. Bazı kas hastalıkları dışında EMG genellikle miyopatinin cinsini ayırt edemez. Ancak EMG’de miyotonik boşalımların bulunması, miyotoni ile giden hastalıklar için tanı koydurucudur. Belli frekanslarda ardı sıra uyaran verilerek alınan bileşik kas aksiyon potansiyellerindeki amplitüd değişiklikleri ise, bulundukları zaman miyastenia gravis ve Lambert-Eaton miyastenik sendromu için tanı koydurur. Tüm bunlar normal bulunursa veya çok hafif klinik bulgular varsa, yapılacak tek lif EMG incelemesi miyastenia gravis tanısı koydurabilir (Ayrıca bakınız: Nörolojide Laboratuvar İncelemeleri/Sinir İleti İncelemeleri ve Elektromiyografi)
Genetik İnceleme
Birçoğu genetik geçişli olan kas hastalıklarında, hastalık genlerinin birçoğunun bulunmuş olması nedeni ile defekt gösterildiği zaman tanıyı %100 kesinleştiren DNA incelemeleri önem kazanmıştır. Yeni nesil dizileme teknikleri ile aynı anda tüm hastalık genlerine bakma olanağı (klinik ekzom analizi), tüm ekzom dizileme ve tüm genom dizileme mümkün hale gelmiş ve çok daha fazla kalıtımsal miyopati kesin teşhis edilmeye başlanmıştır. Bazı durumlarda, miyopatinin cinsini ayırt etmek için kas biyopsisi gibi invazif bir yöntemin kullanılması gerekliliği de azalmıştır.
İncelemeler EDTAlı (mor kapaklı) tüplere, 10cc miktarında alınan venöz kanda yapılmaktadır. Bu amaçla hastadan alınan kanın oda ısısında 72 saat içinde, incelemeyi yapabilen bir laboratuvara ulaştırılması yeterlidir. Her inceleme gibi DNA incelemesinde de kan gönderirken hastalık öyküsünün özetinin ve düşünülen tanı olasılığının yazılı olarak eklenmesi, tüp üzerine hastanın isminin doğru yazılması önemlidir. Genetik tanının konması tanının kesinleştirilmesinden daha geniş boyutta önem taşımaktadır. Genin bilindiği durumlarda aile içindeki taşıyıcı bireylerin saptanması, fetusta 16. haftada hastalık olup olmadığının belirlenmesi (prenatal tanı) mümkün olabilmektedir. Özellikle yaşam süresini çok kısaltan hastalıklarda, hasta birey kaybedilmeden önce genetik materyalin elde edilmesi bu açıdan önemlidir (Ayrıca bakınız: Nörolojide Laboratuvar İncelemeleri/Klinik Nörogenetik).
Kas Biyopsisi
Birçok kas hastalığının, bazen genetik defekti gösterilmiş bile olsa, cinsinin ayırt edilmesi için kas biyopsisi yapılması gerekebilir. Kas biyopsisi, lokal anestezi ile insizyonel olarak veya iğne biyopsisi şeklinde yapılır. Diğer patolojik incelemelerden farklı olarak alınan parçayı parafine gömmek yerine sıvı azotta soğutulmuş izopentan içinde dondurmak ve kası mümkün olduğu kadar aslına uygun saklamak gerektiğinden parça alındıktan sonra laboratuvara ulaştırılma koşulları, geleneksel biyopsilerden farklıdır. Bu teknikler birçok standart patoloji laboratuvarında uygulanamamaktadır.
Her şeyden önce bilinmesi gereken kural, kas biyopsisinin, inflamatuvar hastalıklar gibi immünsupresif tedavi başlanması zorunlu durumlarda, tedaviye başlamadan önce yapılması gerekliliğidir. Her durumda biyopsi yapılacak kasın seçimi büyük önem taşır. Genellikle orta dereceli (4/5 - 3/5 kas gücü gösteren) zaafı olan proksimal bir kas (biseps, deltoid, triseps, kuadriseps gibi) oldukça bilgi verici olur. Ancak bazı durumlarda bu kuralın dışına çıkılır. Örneğin distal miyopatilerde distal bir kas (tibialis anterior, önkol ekstansör veya fleksör grup), metabolik miyopatilerde ise en çok tutulmuş ve/veya en az tutulmuş kas daha aydınlatıcı olabilir. İnsizyonel biyopsi genellikle daha çok tercih edilen bir yöntemdir. İnsizyonla istenen kasa ulaşıldıktan sonra kasın hiç örselenmemesi çok önemlidir. Bir klamp ile iki yandan kıstırılan kas, klampın dışından kesilerek çıkarılır. Alınan parçanın en az 0,5X0,5X1,00cm olmasına ve uzun eksenin, kasın uzun eksenine paralel olmasına dikkat edilmelidir. Daha da önemlisi, alınan parçanın, hiçbir sıvı içine konmadan gazlı bez içine konmasıdır. Laboratuvara ulaşıncaya kadar materyalin kurumaması için gazlı bez serum fizyolojik ile nemlendirilir. Alınan parçanın tercihan 30 dakika, en çok 4-5 saat içinde laboratuvara ulaştırılması gerekir. Ulaşım 3-4 saati bulacak ise parçanın, gazlı bez içinde, termosa konarak gönderilmesi uygundur. Materyali laboratuvara gönderirken mutlaka hasta hakkında etraflıca bilgiyi de beraberinde göndermek, değerlendirme açısından önem taşır. Materyal laboratuvara ulaştıktan sonra -160-180 0Cde dondurularak -80 0Cde saklanır ve sonra alınan kesitlere standart, enzim histokimyasal boyama ve gerektiğinde immünhistokimyasal boyama teknikleri uygulanarak mikroskop altında değerlendirilir. Bu yöntemlerle birçok kas hastalığının tanısı konabilir. Gerektiğinde ayrı işlem gören kas parçası elektron mikroskobunda incelenir. Bazen dondurulmuş parçanın bir bölümü biyokimyasal incelemeler veya protein kalitesini araştıran incelemelere (Western blotting), bazen de genetik incelemeye (özellikle mitokondriyal hastalıklarda) tabi tutulur.

Şekil 11.Kas biyopsisinde dikkat edilmesi gerekenler (“YAP”lar ve “YAPMA”lar).
Diğer Laboratuvar İncelemeleri
Bazı edinsel veya kalıtımsal kas hastalıklarının sistemik hastalıklar olduğunu unutmamak gerekir. Örneğin polimiyozit, dermatomiyozit gibi hastalıklar tek başlarına olduklarında bile akciğer veya kalbi tutabilirler, ayrıca sistemik inflamasyon bulguları verebilirler (eritrosit sedimantasyon hızı yüksekliği gibi). Bazen başka bağ dokusu hastalıkları ile birlikte olabilirler. Bu nedenle bu hastalıkların araştırılması gerekir. Kalıtımsal miyopatilerden mitokondriyal hastalıklar multisistemik olabilir. Glikojen veya lipid depo hastalıkları başlıca karaciğer, kalp, bazen merkezi sinir sistemini tutabilir. Bu hastalıklarla karşılaşıldığında, tutulması muhtemel sistemler incelenmelidir. En önemlisi, birçok miyopatinin kalp kasını da etkileyebileceği bilinmeli ve miyopatili hastalarda kardiyolojik inceleme mutlaka EKG, ekokardiyografi ve ritim Holter incelemelerini kapsamalı, hastalar yıllık izleme alınmalıdır. Solunum kaslarının tutulma olasılığına karşı bu hastalar yıllık solunum fonksiyon testleri, hastalığın ilerlemiş dönemlerinde ise polisomnografi ve saturasyon testleri ile izlenmelidir. Bazı metabolik miyopatilerde iskemik laktat testi, kan, idrar ve beyin omurilik sıvısında organik ve aminoasit (örneğin laktat) düzeyi işe yarayabilir. Gerektiğinde doku ve/veya kanda enzim defektleri biyokimyasal olarak saptanabilir.
KAS HASTALIKLARI
KAS DİSTROFİLERİ
Kas distrofileri kas liflerinin histopatolojik olarak tekrarlayıcı yıkım (nekroz), tamir (rejenerasyon) süreçlerini birlikte gösteren, sonuçta nekrozun galip gelmesi ile kas lifi kaybı, bu kaybın yerini alan endomizyal ve perimizyal yağ ve bağ dokusu artışı ile karakterize genetik hastalıklarıdır. Değişik ağırlıkta, ancak ilerleyici seyrederler.
Kas distrofilerini, klinik özelliklerine göre başlıca iki ana grupta ele almak mümkündür (Tablo 1).
{kind=link}
Tablo 1. Kas Distrofileri Sınıflaması.
{kind=link}
|
KAVŞAK TİPİ KAS DİSTROFİLERİ “LIMB GIRDLE MUSCULAR DYSTROPHIES” (LGMD)
|
|
|
X’e BAĞLI RESESİF GEÇİŞLİ (Xp21) (Distrofinopatiler) |
a) Duchenne tipi kas distrofisi (DMD) b) Becker tipi kas distrofisi (BMD) c) Açık distrofinopati taşıyıcıları d) Diğer (X’e bağlı dilate kardiyomiyopati, mental retardasyon, translokasyonlar, Turner sendromu, Mc Leod sendromu) |
|
OTOZOMAL DOMİNANT GEÇİŞLİ
|
LGMD 1A’dan LGMD 1N’ye kadar |
|
OTOZOMAL RESESİF GEÇİŞLİ
|
LGMD 2A’dan LGMD 2Z’ye kadar |
|
ÖZEL KAS ZAAFI DAĞILIMI VEYA KLİNİK TABLO GÖSTEREN KAS DİSTROFİLERİ
|
|
|
X’e BAĞLI RESESİF GEÇİŞLİ
|
a) Emery-Dreifuss kas distrofisi (Otozomal dominant ve resesif formları da var) |
|
OTOZOMAL DOMİNANT GEÇİŞLİ |
a) Miyotonik distrofi (DM) b) Fasyoskapulohumeral distrofi (FSHD) c) Okülofaringeal kas distrofisi (OFMD) |
|
OTOZOMAL RESESİF GEÇİŞLİ
|
a) Konjenital kas distrofileri b) Diğer |
KAVŞAK TİPİ KAS DİSTROFİLERİ
Başlıca ve öncelikle kalça ve omuz kavşağı kaslarının tutulumu ile seyreden kas distrofileridir. Sürecin çok ilerlemesi ile diğer iskelet kaslarına da yayılırlar. Bu bölümde kavşak tipi kas distrofilerinin ağır ve hafif formlarına klasik örnekler olmak üzere Duchenne tipi kas distrofisi (DMD) ve Becker tipi kas distrofisi (BMD) ayrıntılı olarak anlatılacak, otozomal geçişli kavşak distrofilerinde bu bulgulardan farkların belirtilmesi ile yetinilecektir.
X’e BAĞLI RESESİF GEÇEN KAVŞAK TİPİ KAS DİSTROFİLERİ (DİSTROFİNOPATİLER)
Xp21 geni tarafından kodlanan ve kas hücrelerinde sarkolemma altında yerleşmiş olan distrofin proteininin kalitatif veya kantitatif bozuklukları sonucu ortaya çıkan bir grup kas distrofisidir.
Xp21 distrofin geninde hastalığa neden olan en sık mutasyon tipi, genin değişik bölgelerindeki delesyonlar (olguların %50-60’ında), daha az oranda duplikasyonlar, daha da seyrek olarak nokta mutasyonlardır. Mutasyon sonucu protein hiç üretilemez ise DMD ortaya çıkar, klinik tablo oldukça ağırdır. Eğer var olan mutasyon bir miktar distrofin üretebiliyorsa bu durumda BMD ortaya çıkar, klinik tablonun ağırlığı genellikle yapılabilen distrofinin miktarı veya büyüklüğü ile doğru orantılıdır.
Duchenne tipi kas distrofisi 1/3500 erkek doğumda bir görülür. Görülme sıklığı toplumlara göre değişkenlik göstermemekle birlikte, prenatal tanının yapılabildiği toplumlarda ailevi olguların sayısı giderek azalmış, olguların büyük çoğunluğu yeni mutasyonlarla oluşmaya başlamıştır.
Hastalık erkek çocuklarda görülür, ailenin kadınları taşıyıcı olabilirler. Eğer rastlantısal X-inaktivasyonu sırasında, içinde hasta allelin aktif olduğu kas hücresi sayısı yeterli olursa, kadın taşıyıcılarda bu taşıyıcılık açık klinik tablo haline dönüşebilir.
DMD, sarkolemmada distrofinin hiç bulunmadığı durumda ortaya çıkar. Çocuğun doğumu ve ilk yıl içindeki gelişme aşamaları tümüyle normaldir. Genellikle çocuklar normal yaşta yürümeye başlarlar. Yürümeye başladıktan sonra çocuğun yavaş yürümesi “ağırkanlılığa”, sık sık kucak istemesi “şımarıklığa”, sık düşmesi ise “beceriksizliğe” atfedilir. Baldırlar, çocukluktan itibaren genellikle şiş ve serttir (psödohipertrofi) (Şekil 9). Genellikle yürüme ve merdiven çıkma zorluğu fark edildiğinde hekime başvurulur. Sıklıkla çocuğun aldığı tanı “pes planus” veya “konjenital kalça çıkığı” olur. Ailelere sorulduğunda çocuğun hiçbir zaman koşamadığı, yaşıtlarına göre yavaş olduğu öğrenilir. Bazen baldır hipertrofisi erken yaşta dikkati çeker. Nedenli veya nedensiz düşmeler oldukça sıktır. Hastanın herhangi bir nedenle (herhangi bir infeksiyon, tonsillektomi, sünnet, fraktür) birkaç gün istirahati yürüme yetisini geriletir, çok erkenden yürümenin durmasına bile yol açabilir. Bu nedenle bu çocukları uzun süreli dinlenmeden mümkün olduğu kadar uzak tutmak, sürekli ayakta durmaya ve yürümeye teşvik etmek gerekmektedir. Yaklaşık 4 yaş civarına kadar bu merdiven çıkma, yürüme bozukluğu dikkati çekse de, 4-8 yaş arası çocuğun vücudunun büyüme hızı hastalığın ilerleme hızını geçer ve aile göreceli bir iyileşmeden söz edebilir. Bu durum tümüyle yanıltıcıdır. Yaklaşık 8 yaştan sonra hastalığın gerçek ilerleme hızı kendini gösterir ve omuz kavşağının da tutulduğu fark edilmeye başlar. Erken veya geç olarak hasta parmak ucunda yürümeye eğilimli olur ve giderek bu durum Aşil tendonunun kontraktürüne yol açar. Yürüyüş hiperlordotik ve paytaktır. Merdiven-yokuş çıkma, oturduğu yerden kalkma, yürüme, giderek ayakta durma, önce dayanma yoluyla, giderek bir başkasının desteği ile sürdürülmeye başlanır. Bu destek de yetmediğinde hastalar tekerlekli iskemleye bağlanırlar. Bu genellikle 9-11 yaş arasında olmakla birlikte hastalar en geç 13 yaşlarına kadar bu bağımlılığa ulaşırlar (ENMC tanı kriterleri). Kollar da güçsüzleşmeye başlamıştır ve bu da ilerler. Hastalar kol güçsüzlüğünden fazla yakınmazlar, ancak bir bardak suyu eline alıp içmek, yemeğini kendisi yemek gibi işlevler giderek etkilenir ve yardım gerektirir. Baştan beri zayıf olan boyun fleksör kasları ve karın kasları hastanın yattığı yerden kalkmasında, giderek artan güçlük yaratır. Bu nedenle hastalığın erken dönemlerinde yan dönerek ve uzun sürede oturur duruma gelen hasta, sonraki yıllarda yataktan yardımsız kalkamaz olur. Tekerlekli iskemleye bağımlı olduktan sonra hastada genellikle hızla skolyoz gelişir. Bu skolyoz da ilerleyicidir ve solunum işlevini, solunum kaslarının tutulmasından beklenenin ötesinde, bozar. Çocukluk döneminde dikkate alınmayabilecek kardiyolojik bulgular giderek ilerler ve 16 yaştan sonraki hemen bütün hastalarda kardiyomiyopati bulguları saptanır. Hasta 18-19 yaşına geldiğinde bir yandan yatağa bağımlılığın getirdiği sorunlar, diğer yandan azalmış solunum kapasitesi nedeni ile, genellikle araya giren bir akciğer infeksiyonu ile solunum sıkıntısı yaşar. Taşınabilir solunum cihazı kullanmadıkça hastalar genellikle 20-25 yaş arasında kaybedilirler.
CK, hiç klinik bulgunun olmadığı neonatal dönemde bile çok yüksektir (20-100 kat). Bebeklikte, herhangi bir sağlık sorunu sırasında yapılan tetkiklerde karaciğer enzimlerinin yüksek bulunması durumunda, bir karaciğer hastalığı düşünmeden önce hastada CK düzeyi bakılması, çocuğu karaciğer biyopsisi gibi invazif bir incelemeden kurtarır. Zira CK yüksekliğine genellikle karaciğer enzim düzeylerinde yükseklik de eşlik eder. Hastalığın ileri dönemlerinde, kas lifi dışına CK çıkaracak kas miktarı çok azaldığından, CK daha düşük düzeylerde bulunur, bazen normale yaklaşır. Hastaların yaklaşık %10’unda orta derecede zeka geriliği, birçoğunda disleksi mevcuttur.
Becker tipi kas distrofisi, sarkolemmada distrofinin bulunduğu, ancak az miktarda veya normalden kısa bir protein yapısında olduğu durumda ortaya çıkar. Belirtiler 5-55 yaş arasında başlar. Distrofinin çok kısa olduğu bazı olgularda başlangıç yaşı DMD’dekine yakın olabilir. Hastalığın seyri de DMD’dekine oranla daha yavaş veya selimdir. CK her dönemde çok yüksektir. Hastaların birçoğu başlangıçta baldırlarda şişlik (psödohipertrofi) ve egzersizle baldırlarda ağrı ve kramplardan yakınırlar. Bu dönemde hiç güçsüzlük olmayabilir veya yalnızca gluteus maksimus kasında zaaf bulunabilir. Bu nedenle tüm kasların test edilmesi, gluteus maksimus kasının ise hasta yüzükoyun yatarken test edilmesi önemlidir. Becker tipi kas distrofisinde gluteus maksimus kasının tek başına veya iliopsoas kasından daha belirgin olarak tutulması bu hastalığı, klinik olarak kendisine çok benzeyen spinal müsküler atrofiden klinik planda ayırmaya yardımcı olur (Şekil 12). Zaaf giderek artar ancak ilerleme DMD’ye göre yavaştır. Üst ekstremite kas gücü uzun süre korunabilir. Hastalığın erken başladığı durumlarda ilerleme DMD’ye yakın ancak yine de ondan daha yavaştır. Becker tipi kas distrofisinde dikkat edilmesi gereken en önemli nokta, kardiyomiyopati olasılığının yüksekliğidir. Ekstremite zaafı ile orantısız olarak kalp kası tutulabilir ve yaşamsal tehlike oluşturabilir. Bu nedenle hastanın yakınması olmasa bile her yıl etraflı kardiyolojik incelemesinin yapılması ve ancak ilk kardiyolojik muayene yapıldıktan sonra fizyoterapi egzersiz programının başlatılmasına karar verilmesi gerekir.
Şekil 12a. Becker tipi kas distrofisinde kuvvetli iliopsoasa karşılık çok zayıf gluteus maksimus.
{kind=link}

Şekil 12b. Spinal müsküler atrofide (SMA) çok zayıf iliopsoasa karşılık) kuvvetli gluteus maksimus.
{kind=link}
Açık distrofinopati taşıyıcılarında sorun hasta allelin aktif durumda olduğu kas liflerinin sayısının, klinik bulgu verecek kadar çok olmasıdır. Bu durumda, normalde asemptomatik olması gereken taşıyıcı kadında kas zaafı ortaya çıkabilir. CK değişik derecelerde yüksek bulunur. Asemptomatik taşıyıcılarda da CK bazen hafif yüksek bulunabilir. Normal CK düzeyi, açık veya gizli distrofinopati taşıyıcılığını dışlamaz. Bazı çalışmalara göre asemptomatik taşıyıcıların büyük bir bölümünde kalp kası, değişik derecelerde tutulmuştur. Tersinden bakıldığında, kardiyomiyopatili bir kadında öncelikle hasta erkek aile bireyleri sorgulanmalı, bunlar yoksa bile CK, EMG ve genetik inceleme yapılmalıdır.
Distrofinopatilerde tanı, anamnez ve klinik bulguların yanı sıra genetik inceleme ile konur. Kas immünhistokimyası distrofinin hiç olmadığını göstermek, yani DMD tanısını kesinleştirmek için kullanılır ve özellikle, genetik incelemede delesyon gösterilemeyen hastalarda değerlidir. Ayrıca, henüz 13 yaşına ulaşmamış ve yürüyebilen, erken başlangıçlı bir distrofinopatide, delesyon gösterilmiş bile olsa, eğer bilinmesi isteniyorsa, hastalığın prognozunu belirlemek amacı ile immünhistokimya gereklidir (Şekil 13). İmmunhistokimyasal olarak distrofinin bulunduğu durumlarda distrofinin kalitesini göstermek amacı ile “Western blotting” yapılması gerekir. Bu inceleme Becker tipi kas distrofisini, diğer benzer seyirli kavşak tipi distrofilerden ayırt etmeye yarar. Son yıllarda çok gelişen genetik yöntemler ile hastalığın tanısını koymak ve ayırıcı tanı yapmak dışında taşıyıcıların tanınması ve prenatal tanı yapılması mümkündür.

Şekil 13. Kas kesitinde distrofin immünhistokimyası A) Normal kasta pozitif boyanma,
B) Erkek distrofinopatili hastada negatif patern, C) Distrofinopati taşıyıcısı olan bir hastada mozaik şeklinde distrofin boyanma paterni
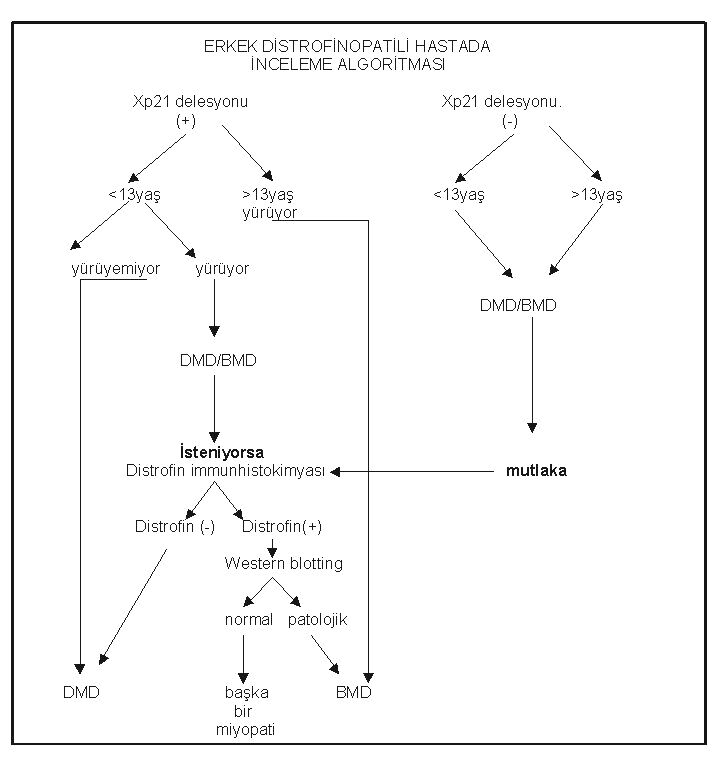
Şekil 13d. Erkek distrofinopatili hastada inceleme şeması
Hastalığın, bugün için bilinen, şifa sağlayıcı bir tedavisi yoktur. Kortikosteroidlerin hastanın yürüme süresini uzattığı, tekerlekli iskemleye bağlanma yaşını geciktirdiği gösterilmiştir. Bu nedenle, 5 yaştan sonra hastalara kortikosteroid başlanması konusunda ortak bir görüş oluşmuştur. Kortikosteroid uygulama protokolleri değişiktir. Kliniğimizde hastalara, genellikle 7-8 yaşından sonra, 0,75mg/kg/gün dozunda prednizolon başlanmakta ve 6 ay sonra doz azaltılarak bu 15 mg/günaşırı olmak üzere idame dozunda yıllarca tutulmaktadır. Kortikosteroidler öncelikle düşmeleri azaltmakta hatta bazen durdurmaktadır. Bu nedenle, düşmeleri çok sık olan çocuklarda, kortikosteroidlere başlama yaşı daha erken olabilmektedir. Yan etkilerin mutlaka izlenmesi gerekmektedir. Son yıllarda, mutasyona özel tedaviler gündeme gelmiştir. Genetik incelemelerde prematür stop kodon oluşan az sayıda olguda Ataluren isimli ilaç kullanılmaktadır. Çalışmaları bittiğinde, ekson atlatma tedavilerinin yakın gelecekte daha geniş bir hasta grubunda kullanılabilir olacağı umulmaktadır. Bunlar dışında, kreatin monohidrat ve albuterol ile yapılan çalışmalar beklenen sonuçları göstermemiştir. Hastalığın tedavisinde ne kullanılırsa kullanılsın, fizyoterapi mutlaka eşlik etmelidir. Çünkü fizyoterapi hem fiziksel kapasiteyi artırmakta, hem de gelişebilecek deformiteleri önlemektedir. Skolyoz, Aşil kontraktürü gibi durumlarda cerrahi girişim düşünülebilir. Bu cerrahi girişimlerin zamanlanması, cerrahi sonrası fizyoterapi ve hastanın çok erken mobilizasyonu büyük önem taşımaktadır.
OTOZOMAL GEÇİŞLİ KAVŞAK TİPİ KAS DİSTROFİLERİ
Otozomal genlerle Mendel yasalarına göre kalıtılan ve kavşak kaslarını tutan kas distrofileridir. Dominant veya resesif olarak aktarılabilirler. Çoğu zaman “Limb girdle muscular dystrophy” veya “LGMD” terimleri ile bu grup kastedilmektedir.
Bulunan genetik anormallikler, kas hücrelerindeki ilgili proteinin yapılamaması veya bozuk yapılması ile ilişkili olarak, fonksiyon bozukluğuna yol açar. Bunlardan en iyi bilinen mekanizma, sarkoglikanopatilerde defektif yapılmış veya hiç yapılamamış olan, distrofinle ilişkili glikoproteinlerin, distrofinle birlikte oluşturdukları kas membranı sağlamlığının bozulması ve kasılmaya dayanıklılığın azalmasıdır. Elbette her proteinin genindeki defekt, o proteinin normaldeki işlevinin ortadan kalkması veya bozulmasına neden olur.
İyi bilinmemekle birlikte, otozomal resesif geçişli olanların akraba evliliklerinin sık olduğu bölgelerde daha sık görüldüğü söylenebilir. Otozomal dominant geçişli kavşak tipi distrofiler, otozomal resesif geçişli olanlara göre daha seyrektir. Her ikisi de hem kadın, hem de erkeklerde görülebilirler. Defektif gen ve genetik geçiş paterni dikkate alınarak bir sınıflama yapılmıştır. Ancak, tanımlanan genlerin fazlalığı, bazı tiplerin yalnızca bir ailede tanımlanmış olması, bazılarında kavşak tipi tutulum dışında bazı özelliklerin ön planda olması nedeni ile yeni bir sınıflama yapılması ihtiyacı doğmuş ve bu konuda görüş birliği oluşmuştur. Yeni sınıflamada, eski sınıflamadaki bazı distrofiler çıkarılmış, olmayan bazıları ise eklenmiştir. Tablo 2’de her iki sınıflama birlikte verilmiştir.
Otozomal geçişli kavşak distrofileri klinik olarak değişik yaşlarda başlayan, başlıca ve öncelikle ekstremite kavşak kaslarında zaaf, buna bağlı olarak da yürüme, merdiven-yokuş çıkma, oturduğu yerden kalkma, kollarını kaldırma işlevlerinde bozulma yapar.
Tanı, klinik bulgular, serum CK değeri, EMG, kas patolojisi, ilgili proteinlerin immünhistokimyasal olarak araştırılması, gereğinde protein incelemesi (Western blotting) ve DNA analizi ile konabilir.
Tablo 2. Otozomal Geçişli Kavşak Tipi Kas Distrofileri.
{kind=link}
|
ESKİ İSİM |
DEFEKTİF GEN |
YENİ İSİM |
|
LGMD 1D |
DNAJB6 |
LGMD D1 DNAJB6-ilişkili |
|
LGMD 1F |
TNP03 |
LGMD D2 TNP03-ilişkil |
|
LGMD 1G |
HNRNPDL |
LGMD D3 HNRNPDL-ilişkili |
|
LGMD 1I |
CAPN3 |
LGMD D4 Kalpain-3 ilişkili |
|
|
COL VI |
LGMD D5 Kollajen 6-ilişkili |
|
LGMD 2A |
CAPN3 |
LGMD R1 Kalpain-3-ilişkili |
|
LGMD 2B |
DYSF |
LGMD R2 Disfelin-ilişkili |
|
LGMD 2C |
SGCG |
LGMD R5 Gama Sarkoglikan-ilişkili |
|
LGMD 2D |
SGCA |
LGMD R3 Alfa Sarkoglikan-ilişkili |
|
LGMD 2E |
SGCB |
LGMD R4 Beta Sarkoglikan-ilişkili |
|
LGMD 2F |
SGCD |
LGMD R6 Delta Sarkoglikan-ilişkili |
|
LGMD 2G |
TCAP |
LGMD R7 Teletonin-ilişkili |
|
LGMD 2H |
TRIM32 |
LGMD R8 Triparite motif içeren protein-32-ilişkili |
|
LGMD 2I |
FKRP |
LGMD R9 Distroglikan-ilişkili |
|
LGMD 2J |
TTN |
LGMD R10 Titin-ilişkili |
|
LGMD 2K |
POMT1 |
LGMD R11 Distroglikan-ilişkili |
|
LGMD 2L |
ANO 5 |
LGMD R12 Anoktamin 5-ilişkili |
|
LGMD 2M |
FKTN |
LGMD R13 Distroglikan-ilişkili |
|
LGMD 2N |
POMT2 |
LGMD R14 Distroglikan-ilişkili |
|
LGMD 2O |
POMGn-T1 |
LGMD R15 Distroglikan-ilişkili |
|
LGMD 2P |
DAG1 |
LGMD R16 Distroglikan-ilişkili |
|
LGMD 2Q |
PLEC |
LGMD R17 Plektin-ilişkili |
|
LGMD 2S |
TRAPPC11 |
LGMD R18 TRAPPC11-ilişkili |
|
LGMD 2T |
GMPPB |
LGMD R19 GDP-mannoz pirofosforilaz-ilişkili |
|
LGMD 2U |
ISPD |
LGMD R20 2-C-metil-D-eritritol 4-fosfat sitidil transferaz benzeri protein-ilişkili |
|
LGMD 2Z |
POGLUT1 |
LGMD R21 POGLU1-ilişkili |
|
|
COL VI |
LGMD R22 Kollajen 6-ilişkili |
|
|
LAMA2 |
LGMD R23 Laminin alfa 2-ilişkili |
|
|
POMGNT2 |
LGMD R24 POMNGT2-ilişkili |
LGMD-1A, -1B, -1C, -1E, -1H, LGMD-2R, -2V, -2W, -2X, -2Y yeni sınıflamada yer almamaktadır.
LGMD-1I, Bethlem miyopati-OD ve -OR, Laminin alfa2-ilişklili kas distrofisi, POMGNT2-ilişkili kas distrofisi sınıflamada yeni yer almışlardır.
ÖZGÜN KAS ZAAFI DAĞILIMI VEYA KLİNİK TABLO GÖSTEREN KAS DİSTROFİLERİ
Fasyoskapulohumeral Distrofi (FSHD)
Yüzün mimik kaslarını, periskapular kaslar ile humerus çevresi kaslarını en çok ve öncelikli olarak tutan bir kas distrofisidir. Erişkin çağın en sık görülen distrofilerindendir. Görülme sıklığı 100.000’de 1-3’dür. Her toplumda görülebilir. Otozomal dominant geçişlidir.
Hastalığın en sık görülen tipi FSHD-1’dir ve “double homeobox protein 4” (DUX4) proteinini kodlayan ve 4q35 bölgesinde D4Z4 tekrarlarında delesyon sonucu gelişir. Bu mutasyonun, başka gen veya fonksiyonları etkileyerek hastalık geliştirdiği düşünülmektedir. 4q35 bölgesinde, mutasyon sonrası, normalden küçük bir tekrar fragmanı kalmakta (kontraksiyon) ve birçok hastada klinik tablo ile kalan bu fragmanın büyüklüğü arasında ters bir orantı gösterilmektedir. Bir başka deyişle daha büyük fragmanlar daha iyi klinik tablo ile korelasyon göstermektedir. Klinik bakımdan benzer olan ve yine otozomal dominant geçen diğer tip, FSHD-2 ise 18p bölgesindeki SMCHD1 geni ile birlikte 4qA allelindeki değişimlerin birlikteliğinde, yani digenik olarak ortaya çıkar.

Şekil 14. Fasyoskapulohumeral distrofili hastada tipik yüz görünümü, “humping” ve skapula alata.
Hastalık her yaşta, en sık olarak ikinci onyılda belirti verir. Buna rağmen sorgulandığında bu hastalardan bazılarında, bebeklikten beri “gözleri açık uyuduğu” anamnezi alınabilir. İsminden de anlaşılacağı gibi zaaf ve atrofinin genellikle ilk başladığı ve en belirgin olduğu kas grupları yüzün mimik kasları, skapula çevresi kaslar ve humerus çevresi kaslardır. Hastalığın bir başka önemli özelliği, kaslardaki tutulumun genellikle asimetrik olarak başlamasıdır. Yüz kaslarının tutulumu nedeni ile hastada göz kapatma veya sıkma zaafı, alt yüz kaslarında atrofi, buna bağlı olarak dudakların belirgin ve dolgun duruşu, ancak buna karşılık orbikülaris oris kasının tutulmasına bağlı olarak üfleme veya ıslık çalma işlevlerinin bozulması söz konusudur. Skapula çevresinde tutulan kaslar, başlıca skapulayı yerinde tutan (fikse eden) gruptur. Bu nedenle omuzun abduksiyonu veya fleksiyonu sırasında mekanik olarak aşağıda tutulması, yerinde kalması gereken skapula burada tutulamaz- ve yukarı kayar, bu hareketler sırasında trapez kası da itilerek yukarı çıkar, omuza pelerin görünümü verir (humping) (Şekil 14). Deltoid kas birçok zaman normal veya çok az zayıf olmasına rağmen bu fiksasyon bozukluğu nedeni ile fonksiyonunu yapamaz. Bir manevra olarak eğer önce skapula mekanik olarak aşağı doğru itildikten sonra deltoid fonksiyonlarına bakılırsa, kasın bu işlevleri daha iyi yerine getirdiği görülür. Deltoid kası en azından başlangıçta, kavşak tipi miyopatilerin tersine, az tutulur veya hiç tutulmaz. Buna karşılık triseps ve biseps, öncelikle ve genellikle asimetrik olarak tutulan kaslardır. Bazen hastalar yalnızca, tek taraflı pektoral kas atrofisine bağlı göğüs kafesindeki asimetri nedeni ile doktora başvururlar. Yıllar içinde kas zaafı üst ekstremite distallerine ve alt ekstremite kaslarına yayılabilir. Alt ekstremitede en sık tutulan kas tibialis anteriordur. Daha sonra alt kavşak kasları da tutulabilir. Hiperlordoz ve bel ağrısı bazen hastayı hekime getiren en belirgin yakınma olabilir ve lomber disk hernisi ile ayırıcı tanı sorunu oluşturabilir. Hastalığın ilerlemesi de hastadan hastaya değişir. Hastalık yaşam süresini etkilemez, bazı hastalar sınırlı bulgularla normal yaşam süresince yaşayabilirler, bazı hastalar ise genç erişkin yaşta tekerlekli iskemleye bağımlı hale gelebilirler. Hastalık genellikle kalp kasını tutmaz. Hastalarda bütün bu bulgular görülebileceği gibi bazı hastalarda bulguların bazılarının, hatta bazı bireylerde yalnızca dolgun görünümlü dudakların bulunması söz konusu olabilir. Hastalık fenotipi aynı aile bireyleri arasında da farklılıklar gösterebilir. Bu nedenle gerek çok hafif tutulumlu bireylerin -saptanabilmesi gerekse otozomal dominant geçişin gösterilebilmesi için tüm aile bireylerinin muayene edilmesi şarttır.
Skolyoz gelişimi enderdir. Pektus ekskavatum deformitesi daha sık görülür. Kardiyomiyopati görülmez. Bazı hastalarda, yalnız odiyometrik testlerle ortaya konabilen, yüksek frekanslı tonlara karşı işitme azalması bulunur. Retinal vasküler hastalıklar ile birlikte görülen FSHD, Coats hastalığı olarak tanımlanmıştır.
Klinik bulgular genellikle tanı koydurucudur. CK normal veya hafif yüksek, EMG miyopatik özellikte, nadiren de normaldir. Kas biyopsisi, yalnızca kuşkulu durumlarda ayırıcı tanı amacıyla kullanılır. Bunun dışında FSHD tanısında kas biyopsisinin yeri yoktur. Kesin tanı ve genetik danışma için genetik inceleme yapılması gerekir.
Diğer distrofilerde olduğu gibi bu hastalıkta da tam iyileştirme, bugün için söz konusu değildir. Beta2-adrenerjik stimülatörlerden Albuterol kullanımı denenmiş, bu ilaç atrofinin azalmasına neden olmuş ancak güçsüzlüğün düzelmesinde başarılı olamamıştır. Fizyoterapi ve kolların rahat kullanılabilmesi için skapulayı fikse eden özel korseler, kısıtlılığı ağır olmayan hastalarda skapulotorasik artrodez uygulanabilir.
Miyotonik Distrofi Tip 1 (DM1)
Genel olarak miyotonik distrofi denildiğinde, DM1 (klasik miyotonik distrofi 1) anlaşılmaktadır. Bu klasik tip dışında, çok daha az görülen konjenital DM1, DM2 (miyotonik distrofi tip 2) / PROMM (proksimal miyotonik miyopati) de mevcuttur.
Klinik olarak kas zaafı ve atrofisine eşlik eden miyotoni ve diğer sistemik bulguların belirleyici olduğu DM1, erişkin çağın en sık görülen kas distrofisidir. Kas distrofisi olarak bilinmekle birlikte, aslında iskelet kası dışında kalp kası, düz kaslar, göz merceği, endokrin sistem ve merkezi sinir sistemini de tutabilen, sistemik bir hastalıktır.
Otozomal dominant kalıtım özelliği gösteren klasik DM1, 19. kromozomda bulunan DMPK (dystrophia myotonica protein kinase) geninin intronlarındaki CTG üçlü nükleotid tekrar artışı ile ortaya çıkar. Bu artış, RNA’da patojenik fonksiyon kazanımına (gain of function) neden olur. Böylece, geniş CUG tekrarı taşıyan RNA’lar çekirdek içinde birikir, agregatlar oluşturur ve birden fazla, çok sayıda düzenleyici alternatif kesim proteininin aktivitesini değiştirir. Hastalığın değişik bulguları, örneğin klor kanalının etkilenmesi ile miyotoni, insülin reseptör geninin etkilenmesi ile artmış diyabet riski gibi birbirinden farklı sistemik bulgular bu mekanizma ile ortaya çıkar. Bu klasik form dışında, CTG tekrar sayısı 1000’in üzerinde başlayan, doğumda veya daha önce başlayan konjenital DM1 ya hayatla bağdaşmaz veya ağır anomalilerle birlikte olur.
Miyotonik distrofi tip 2 (DM2) / PROMM, önceleri farklı iki klinik tablo gibi bildirilmiş olsa da bugün için her iki durum, DM2 adı altında toplanmaktadır. DM2, otozomal dominant olarak kalıtılır, 3q21 bölgesindeki ZNF9 (Zinc finger protein 9) genindeki CCTG dörtlü nükleotid artışı nedeni ile ortaya çıkar. Artan nükleotid tekrarı, diğer genlerin fonksiyonlarını bozmaktadır. Bu tipin konjenital formu yoktur. Kas zaafı, aşağıda belirtileceği gibi, DM1’deki gibi distal değil, proksimaldir. Miyotoni, kardiyak tutulum ve diğer sistemik tutulumlar klinik tabloya eşlik edebilir.
Aşağıda en sık görülen DM1 anlatılacaktır.
DM1’de CTG üçlü tekrarlarındaki artışın düzeyi klinik tablonun ağırlığını belirler. Aynı aile içindeki bireylerde bile tekrar sayısı, dolayısı ile fenotipteki ağırlık, farklılıklar gösterir. Ardışık kuşaklarda tekrar sayısı ve klinik tablonun ağırlığı giderek artabilir (antisipasyon) (Ayrıca bakınız: Klinik Nörogenetik). Konjenital miyotonik distrofi gibi daha ağır tablolar anne tarafından aktarılırlar. Bu nedenle konjenital DM1 saptanan bireylerin anneleri de mutlaka DM1 açısından incelenmelidirler. Genel olarak 36-50 CTG tekrarı premutasyon aralığını oluşturur. 100 ve üstündeki tekrarlar klasik DM1 fenotipine yol açar. 1000-4000 CTG tekrarı (bazı kaynaklara göre 730-4000 tekrar) ise ağır konjenital miyotonik distrofi fenotipinde görülür.
Klasik DM1’in klinik bulguları:
1. Nöromüsküler bulgular:
a. Zaaf ve atrofi: DM1’li hastalarda zaaf ve atrofinin dağılımı yüzde başlıca frontal kas ile temporal, masseter, levator palpebra superior kaslarını içerir. Temporal ve masseter kaslarındaki atrofi oldukça tipik bir yüz görünümüne neden olur. Bazen buna ptoz da eşlik edebilir. Boyunda fleksör kaslar ve sternokleidomastoid kas zayıf ve atrofiktir. Üst ekstremitede başlıca bilek ve parmak derin fleksör kasları, alt ekstremitede ise ayak bileği dorsal fleksör kasları (tibialis anterior), yani distal kaslar tutulur (Şekil 15). Zaman içinde zaaf proksimal kaslara da yayılabilir. Zaaf simetriktir. Bazı hastalarda yutma çok yavaş progresif olarak bozulabilir.
b. Kas zaafı dışında miyotoninin bulunması önemlidir. Miyotonik distrofide klinik miyotoni, iyon kanalı hastalıklarına bağlı konjenital miyotonilerde olduğu kadar ön planda değildir. Özellikle zaafın fazla olduğu durumlarda klinik miyotoni bulmak olanaksız olabilir. Bu nedenle EMG sırasında, bu klinik tanı düşünülüyor ise, miyotonik boşalım mutlaka aranmalıdır. Klinik miyotoni bulunduğu durumda hasta bir süre dinlenmeden sonra sıktığı elini veya gözkapağını açmakta zorlanır (aksiyon miyotonisi). Ayrıca refleks çekici ile bir kas üzerine vurulduğunda o kasta kasılma olur ve bu kasılma bir süre gözle görülebilir halde kalır (perküsyon miyotonisi).
2. Nöromüsküler sistem dışı bulgular:
İskelet kası dışında DM1’de en önemli tutulum kalp iletim sistemi ve kalp kasındadır. Ani ölüm riskinin yüksek olduğu bir hastalık grubunu oluşturur. Tanı konduktan sonra tüm hastaların ve hatta hastalık belirtisi göstermeyen artmış CTG artışı olan aile bireylerinin kardiyolojik muayene ve takipleri yapılmalı, hastalar anestezi almak durumunda kaldıklarında mutlaka dikkat edilmelidir. Diğer sistemik bulgular şöyle özetlenebilir:
a. Frontal kellik
b. Katarakt: Posterior subkapsüler, multipl noktasal
c. Hipogonadizm, FSH artışı, insülin direnci, gebelik komplikasyonları ve düşük
d. Apati, hipersomni, MR’da serebral beyaz madde lezyonları (bu lezyonlar bazen demiyelinizan hastalıklarla karıştırılabilir), bazen hafif duysal nöropati konjenital tipte mental retardasyon,
e. Hipoventilasyon, hiperkapni (solunum fonksiyonları izlenmeli ve gerektiğinde non-invazif ventilasyon uygulanmalıdır)
g. Disfaji (palatal veya özofageal kaynaklı), konstipasyon, megakolon, kolelityazis.

Şekil 15a. Miyotonik distrofide yüz görünümü

Şekil 15b. Atrofik sternokleidomastoid kası

Şekil 15c. Atrofik tibialis anterior kasları

Şekil 15d. Parmak fleksör zaafı
Konjenital tipte ise tüm bu bulgular oldukça ağırdır. Eğer kaybedilmemişse bebek hipotoniktir ve çok sık alt solunum yolu infeksiyonu geçirir. Bunlardan kurtulup yaşamını sürdürebilirse çocuk, miyotonik distrofinin bulguları dışında non-progresif mental retardasyon ile birlikte yaşamak durumunda kalır. Konjenital miyotonik distrofide yüksek damak gibi iskelet anomalileri de mevcuttur.
Genetik inceleme hem hastalığın tanısı için gereklidir, hem de hastalık prognozunu belirler. Prenatal tanı ve hastalık belirtisini henüz göstermeyen bireylerin tanınması da genetik inceleme ile mümkündür ve kalp tutulumunun izlenmesi açısından önemlidir. Genetik inceleme yapılamadığı durumda indeks olgunun klinik bulgularının yanında EMG’de aranarak bulunmuş miyotonik boşalımların varlığı büyük önem taşır. Asemptomatik bireylerde, özellikle konjenital miyotonik distrofi olgularının annelerinde, yapılacak dikkatli bir EMG incelemesi veya genetik inceleme özellikle önemlidir.
Hastalığın kesin çaresi yoktur. Semptomatik tedavi, kataraktın düzeltilmesi, hipersomnide modafinil uygulaması, uyku-apnesi olan hastalarda cihaz, gerektiğinde anti-miyotonik ajanların kullanılmasını içerir. Bazen hipoventilasyon veya hiperkapniyle savaşmak için kullanılan non-invazif ventilasyon hastada aşırı gündüz uyuklamasını düzeltir, günlük yaşam kalitesini yükseltir. Miyotonik distrofide aritmojenik ajanların kullanılmaması, herhangi bir cerrahi girişim sırasında gelişebilecek aritmilere karşı hazırlıklı olmak ve yakın kardiyolojik takip yaşamsal önem taşır.
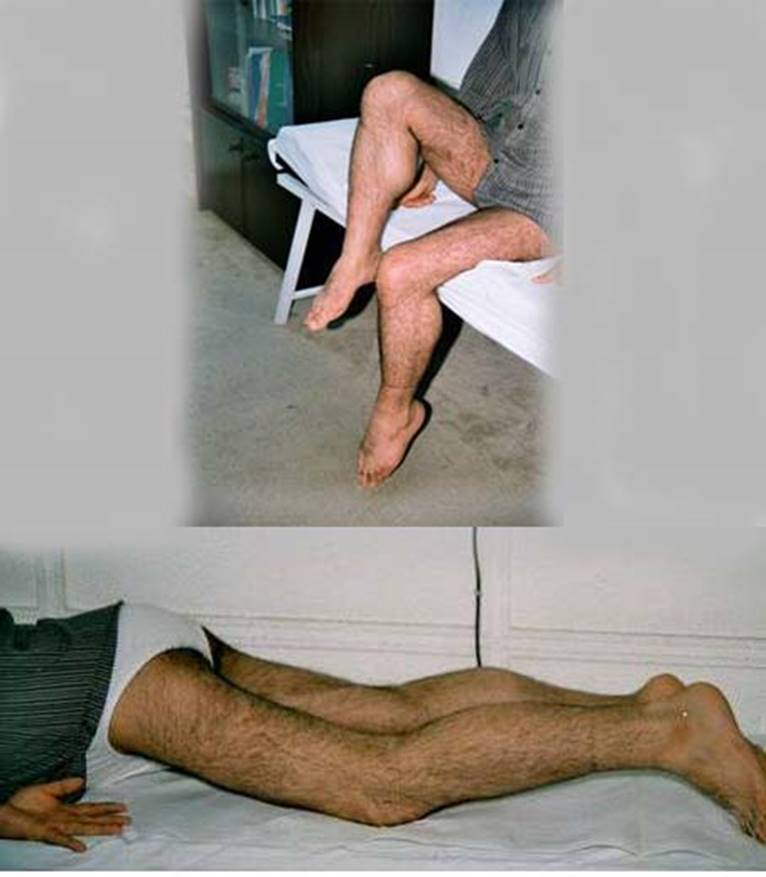
Emery-Dreifuss Kas Distrofisi
Emery-Dreifuss müsküler distrofi (EDMD) bir hastalık antitesi olarak 1960’larda A. Emery ve F. Dreifuss tarafından tanımlanmıştır ve iskelet kası ile birlikte kardiyak ileti sistemi ve kalp kasını en sık tutan miyopatilerdendir. Bu anlamda Kardiyoloji disiplinini de yakından ilgilendirir. Erken çocukluk çağında ortaya çıkan kontraktürler, daha geç gelişen ve yavaş ilerleyen kas zaafı ve atrofisi ile ikinci dekaddan sonra görülen kardiyak tutulum üçlüsü ile kendini gösterir. Başlangıçta XLR tipi tanımlanmış olsa da, daha sonra genetik bilimindeki gelişmeler, aynı klinik tabloya neden olan başka genlerin de tanımlanmasına neden olmuştur. Böylece, hastalığın X’e bağlı resesif (XLR), otozomal dominant (OD) ve otozomal resesif (OR) kalıtılan ve şu an kalıtımsal defekti belirlenerek OMIM’e (Online Mendelian Inheritance in Man) giren 7 tipi ortaya çıkmıştır (EDMD-1, -2, -3, -4, -5, -6, -7) (Tablo 3). Bu yedi genin altısı, hücre çekirdeği zarfının birbiri ile ilişkili proteinlerini kodlayan genlerdir. Birisi ise transmembran bir proteini kodlayan gen olmasına rağmen çekirdek zarfı proteinlerinden bazıları ile iletişimi vardır. Üç ayrı (XLR, OD, OR) genetik geçişli tipin klinik tablosu birbirine benzemekle birlikte daha hafif veya ağır formları görülebilmektedir. Ayrıca, hastalık sınıflamalarında henüz yer almamış olsa da bu fenotip ile ilişkilendirilen üç ayrı gen daha bildirilmektedir (SUN1, SUN2 ve TTN genleri). Burada başlıca EDMD-1’in klinik özelliklerinden söz edilecektir.
EDMD-1: X’e bağlı resesif geçiş gösteren tipi en sık olarak bilinmekle birlikte son yıllarda, LMNA mutasyonu gösteren OD ve OR tiplerinin daha sık olduğunu bildiren yayınlar mevcuttur. Klasik EDMD, yani XLR EDMD durumunda hastalık yalnızca erkeklerde görülür. Ancak sapmış X inaktivasyonu olduğu durumda taşıyıcı kadınlarda da klinik tablo manifest hale geçebilir. Bu durumda aile içinde, erkekten erkeğe geçişin olmadığı, psödo-dominant kalıtım paterni gözlenir. Hastalığa neden olan mutasyon Xq28 bölgesindeki, emerin proteinini kodlayan EMD genindedir. Emerin, nükleer zarfın integral proteinlerinden birisidir.
Tablo 3. Emery-Dreifuss Müsküler Distrofiler.
|
TİP
|
GEN |
PROTEİN |
GEÇİŞ ŞEKLİ |
|
EDMD 1 |
EMD |
Emerin |
XLR |
|
EDMD 2 EDMD 3 |
LMNA |
Lamin A ve C |
OD veya OR |
|
EDMD 4 |
SYNE 1 |
Nesprin 1 |
OD |
|
EDMD 5 |
SYNE 2 |
Nesprin 2 |
OD |
|
EDMD 6 |
FHL1 |
FHL1 protein |
XLR |
|
EDMD 7 |
TMEM43 |
LUMA |
OD |
EDMD: Emery-Dreifuss müsküler distrofi. XLR: X2e bağlı resesif. LMNA: Lamin A. OD: Otozomal dominant. OR: Otozomal resesif. SYNE: Spektrin tekrarı içeren nükleer zarf proteini. FHL1: Dörtbuçuk LİM bölgesi 1. TMEM43: Transmembran protein 43. LUMA: LUMA proteini.
EDMD-2 ve -3: LMNA genindeki mutasyonlara bağlı olarak OD veya OR olarak kalıtılır. Lamin A/C de nükleer zarfın lamina proteinlerindendir. Bu genin mutasyonları ile ayrıca LGMD 1B, dilate kardiyomiyopati ile birlikte kuadriseps miyopatisi, “rigid spine” lı konjenital miyopati, AR-CMT2A, A-V ileti blokları ile birlikte giden ailesel dilate kardiyomiyopati (DCM-CD), erken başlangıçlı atriyal fibrilasyon ve Köbberling-Dunnigan tipi ailesel parsiyel lipodistrofi (FPLD), mandibuloakral distrofi ve prematüre yaşlanma da ortaya çıkmaktadır. Lamin A/C mutasyonları ile ortaya çıkan tüm bu hastalıklar alleliktir ve aynı ailede değişik klinik tabloların birlikteliği bildirilmiştir. EDMD-4: Otozomal dominant geçişlidir ve 6q25’teki sinaptik nükleer zarf proteini-1 (SYNE-1; Nesprin-1) geninde oluşan mutasyonlarla ortaya çıkar. EDMD-5: Otozomal dominant geçişlidir ve 14q13’teki sinaptik nükleer zarf proteini-2 (SYNE-2; Nesprin-2) geninde oluşan mutasyonlarla ortaya çıkar. EDMD-6: Dörtbuçuk LİM bölgesi proteinini kodlayan FHL1 geninin neden olduğu tiptir ve XLR olarak kalıtılır. FHL proteininin de, en azından miyoblastlarda hücre çekirdeğinde olduğu gösterilmiştir, kalp ve iskelet kasında bulunur. EDMD-7: Transmembran protein 43 geni (TMEM43), çekirdek zarfında bulunan ve emerin proteinleri ile ilişkide olan, TMEM43 (veya LUMA) proteinini kodlamaktadır. Genin mutasyonları OD olarak kalıtılmaktadır ve EDMD dışında aritmojenik sağ ventrikül displazisi-5 (ARVC-5)’e de neden olmaktadır.
Emery-Dreifuss müsküler distrofilerde başlangıç neonatal dönem ile 3. onyıl arasındadır. Klinik tablo, aynı aile içinde bile değişkenlik gösterir. Genellikle önce, kas zaafı olmaksızın veya zaafla orantısız kontraktürler başlar. Kontraktürler erken yaşlarda dirsekler ve ayak bileğinde, daha sonra boyun ekstansör kasları, sırt ve bel kaslarında gelişir (Şekil 16). Daha sonra buna eklenen ve çoğunlukla geri planda kalan kas zaafı, hastadaki hareket zorluğunun ana sebebi değildir. Kontraktürler zaaftan daha sakatlayıcıdır. Kas zaafı başlangıçta skapulo-humero-peroneal dağılımdadır ancak daha sonra proksimal kaslara da yayılabilir. Deltoidler genellikle korunur. Üst ekstremitelerde en sık tutulan kaslar biseps ve trisepstir. Alt ekstremitelerde başlıca distal kaslar tutulur. 20-40’lı yaşlarda kalp tutulumu belirir. Kalp tutulumu bu yaşa ulaşmış tüm hastalarda görülür ve en önemli nöromüsküler ani ölüm nedenlerinin başında gelir. Görülen kardiyak patolojiler başlıca taşikardi, bradikardi, atriyal ritim bozuklukları ve paralizileri, A-V ileti defektleri ve/veya kardiyomiyopatidir. Hastaların çoğuna 30’lu yaşlarda “pace-maker” takılmak zorunda kalınır ve bu girişim hayat kurtarıcıdır. Bu nedenle hastalığın tanınması ve hastanın kardiyak ileti defektleri açısından yakından izlenmesi, gerektiğinde “pace-maker” takılması çok önem taşır. Kardiyomiyopati, “pace-maker” takılmasından sonra da gelişebilir. Bu nedenle hastaların kardiyolojik açıdan, “pace-maker” uygulamasından sonra da yakın kardiyolojik izlemde tutulması çok önemlidir. Emerin eksikliğinde manifest taşıyıcılar da kardiyolojik açıdan benzer riskleri taşırlar ve izlenmelidirler.
CK normal veya yüksek bulunur. EMG’de miyojen değişikliklerin yanı sıra nörojen potansiyeller de bulunabilir. Kas biyopsisi miyopati veya distrofi ile uyumludur. İmmünhistokimya ile emerin eksikliği gösterilebilir. Genetik incelemede gösterilen mutasyonlar kesin tanı koydurur. Kardiyolojik inceleme, ekokardiyografi dışında mutlaka EKG ve 24 saatlik Holter de içermeli ve her yıl (bazen daha sık) tekrarlanmalıdır.
Tedavide ‘pace-maker’ takılmasını gerektirebilen kardiyolojik tedavi en önemli yeri tutar. Olası bir anestezi sırasında da durumun bilinmesi hayat kurtarıcı olabilir.
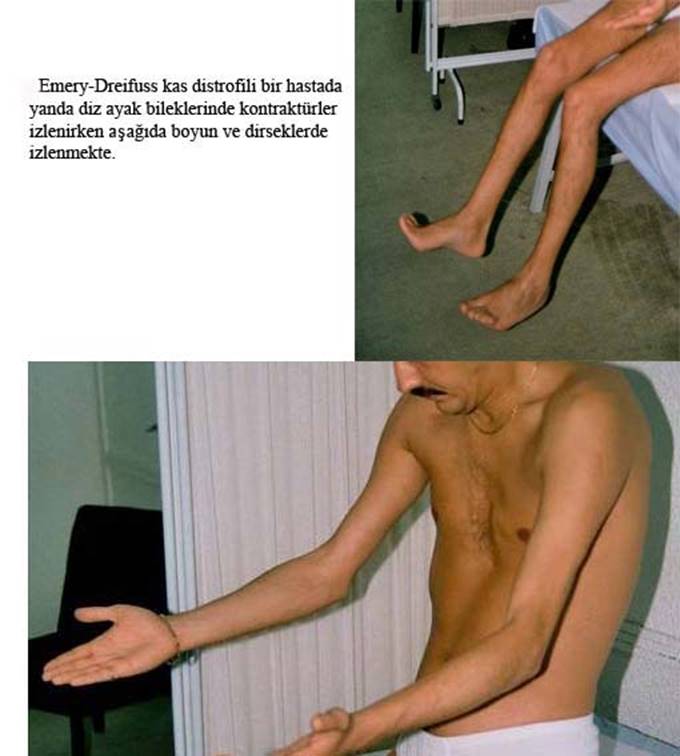
Şekil 16. Emery-Dreifuss kas distrofili bir hastada boyun ve dirsekler ile diz ve ayak bileklerinde kontraktürler.
{kind=link}
Okülofaringeal Kas Distrofisi (OFMD)
Okülofaringeal müsküler distrofi seyrek görülen bir miyopatidir ve ileri erişkin yaş hastalığıdır. Başlangıcı 40 yaş üzeri olarak bilinmektedir, ancak hastaların birçoğunda belirtiler genellikle altıncı onyılda başlar. Otozomal dominant (bazen resesif) olarak geçer. Hastalığa neden olan genetik defekt 14q11.2-q13 bölgesindeki nükleer poly(A) binding protein-1 (PABN-1; PABP2) genindeki GCG tekrar artışlarıdır.
En belirgin klinik özelliği iki yanlı ptoz ve disfajidir. Başlangıç belirtisi her ikisinden biri olabilir. Ptoz başlangıçta asimetrik olabilse de hastalık ilerledikçe simetrik hale gelir, çok belirgindir. Göz hareketleri genellikle kısıtlanmaz, kısıtlandığında da hafiftir ve genellikle yukarı bakışta ortaya çıkar (Şekil 17). Göz hareketlerinin iyi korunmuş olması OFMD’nin, ptozla giden bir başka hastalık grubu olan mitokondriyal hastalıklardan en önemli farkıdır. Diplopi pek gelişmez. Disfaji ilerleyicidir. Klinik tabloya özellikle alt ekstremite proksimal kaslarının zaafı eklenir.
CK hafif yüksektir. EMG miyopatiktir. Kas biyopsisinde kırmızı çerçeveli vakuoller vardır. Elektron mikroskopik olarak kas lifleri içinde ve çekirdeklerinde çok ince filamentler gösterilir ve tanı koydurucudur. Kesin tanı genetik incelemede mutasyonun gösterilmesi ile konur.

Şekil 17. Okülofaringeal kas distrofisinde belirgin ptoz olmasına rağmen göz hareketleri genellikle etkilenmez.
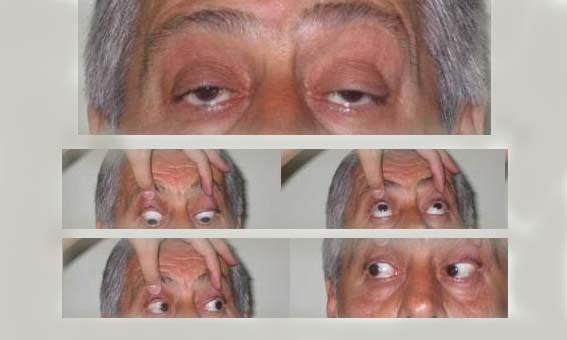{kind=link}
Konjenital Kas (Müsküler) Distrofileri (KMD)
Konjenital müsküler distrofiler (KMD), doğumda veya neonatal dönemde başlayan, klinik ve genetik olarak heterojen bir kas distrofisi grubudur. Yeni nesil genetik incelemelerle alt grupları çok artmıştır (Tablo 4). Bu grup hastalığa neden olan genlerin bazıları, bazı konjenital miyopatilere ve kavşak tipi kas distrofilerine neden olan genler arasında yer almaktadır. Hastalıkların 1-2’si dışında kalıtımsal geçiş OR’tir.
Hastalık doğumda veya ilk yaş içinde başlar. Genel özellikleri, infantil hipotoni (gevşek bebek), genellikle kontraktürler, bazen merkezi sinir sistemi ve/veya göz tutulumu bulguları, kas biyopsisinde ise distrofi bulgularıdır. Çocuk büyürken motor gelişim basamaklarını yakalayamaz veya geç yakalar. Kas zaafı bazılarında aksiyal kaslarda ağırlık taşırken, bazılarında ekstremite zaafı ön plandadır. Distal ağırlıklı zaaf genellikle göstermezler. Bazı hastalar, spinal veya ekstremite kontraktürleri, hiperlaksite gösterebilirler. Bazı hastalarda klinik tabloya mental retardasyon eşlik ederken bazılarında, kranial MR bulgularına (beyaz madde lezyonları, lizensefali, serebellar malformasyonlar) rağmen mental durum normaldir. Özel bir grubunda, göz tutulumu vardır (retinal disgenezi, mikroftalmi, ön kamara malformasyonları). Bazı KMD’lerde, henüz ambulatuvar dönemde iken solunum bozulur, eğer fark edilmezse hasta kaybedilebilir. Kalp tutulumu ve ani ölüm başlıca laminopatilerde ve fukutinle ilişkili protein (FKRP) defektlerinde görülür. Hastalarda CK serum düzeyi orta yükseklikte veya çok yüksektir. Kas biyopsisi bulguları, hafif veya kronik miyopatik değişikliklerden, daha çok açık distrofi bulgularına kadar değişiklik gösterir.
Konjenital müsküler distrofilerin ayırıcı tanısını klinik planda yapmak oldukça zordur. Kasın diğer hastalıklarından konjenital DM1, konjenital miyastenik sendromlar, kendine özgü histopatolojik özellikler gösteren konjenital miyopatiler, metabolik miyopatiler, ayrıca motor nöron hastalıkları (SMA1), periferik sinir hastalıkları (CMT), merkezi sinir sistemi hastalıkları veya non-nöromüsküler hastalıklardan Prader-Willi sendromu’ndan, özellikle de tedavisi mümkün olan infantil Pompe hastalığından bu hastalık grubunu ayırmak, ayrıca konjenital kas distrofilerini de birbirinden ayırmak gerekmektedir. Yeni nesil genetik incelemeler bu ayırıcı tanıyı ve etyolojiyi bulmayı kolaylaştırmış olmakla birlikte, çıkan genetik sonuç ile klinik tablonun uyumuna yine de klinisyenin karar vermesi gerekmektedir.
Tablo 4. Konjenital Müsküler Distrofiler.
|
GRUP |
KISALTMA |
GEN |
|
Ekstraselüler matriks protein defektleri |
||
|
Laminin α2-ilişkili KMD |
MDC1A, Merozin eksikliği KMD, LAMA-2-KMD |
LAMA2 |
|
Kollajen 6 ilişkili KMD |
UCMD, ara formlar, Bethlem |
COL6A1, COL6A2, COL6A3 |
|
İntegrin α7 -ilişkili KMD |
ITGA7 |
ITGA7 |
|
Plektin-ilişkili KMD |
EB-MDC |
PLEC1 |
|
α -Distroglikan ve Glikolizasyonu defektleri |
||
|
α -Distroglikan-ilişkili KMD |
WWS, MEB, FCMD, FKRP, MDC1B, MDC1D, DOLK-CDG |
POMT1, POMTGnT1, FKRP, FKTN, LARGE, ISPD, GTDC2, DAG1, TMEM5, B3GNT1, GMPPB, SGK 196 |
|
İntraselüler ve nükleer protein defektleri |
||
|
SEPN1-ilişkili KMD (Sert omurga) (Rigid spine) sendromu
|
RSMD1 |
SEPN1 |
|
RYR1-ilişkili KMD |
RYR1-KMD |
RYR1 |
|
LMNa-ilişkili KMD |
LMNA |
LMNA |
|
Diğer ender KMD |
Merozin (+), Mitokondriyal, Nesprin |
|
|
Genetik defekti bilinmeyen KMD’ler |
||
*Bonneman CG ve ark. “Diagnostic approach to the congenital muscular dystrophies”. Neuromusc Disord 2014; 24: 289-311’den uyarlanmıştır.
KMD: Konjenital müsküler distrofi. MDC1A, B, C, D: Konjenital müsküler distrofi1A, B, C, D. LAMA2: Laminin α -2. UCMD: Ullrich konjenital müsküler distrofi. COL6A1, A2, A3: Kolajen6A1, A2, A3. ITGA7: İntegrin α 7. EB-MDC: Epidermolysis bullosa konjenital müsküler distrofi. PLEC1: Plektin1. WWS: Walker Warburg sendromu. MEB: Kas Göz Beyin hastalığı. FCMD: Fukuyama konjenital müsküler distrofi. FKRP: Fukutin ilişkili protein. DOLK-CDG: Dolikol kinaz konjenital müsküler distrofi. POMT-1: Protein-O-Mannoziltransferaz 1. Protein-O-Mannozil beta-1-2-N-asetilglikozaminiltransferaz. FKTN: Fukutin. ISPD: Isoprenoid sentaz bölhesi içeren. GTDC2: Glikoziltransferaz benzeri bölge içeren 2. DAG 1: Distroglikan geni. TMEM5: Transmembran protein 5. B3GNT1: α -1-3N-asetilgalaktozaminiltransferaz2. GMPPB: GDP-mannoz pirofosforilaz B. SGK 196: Sugen kinaz 196. SEPN1: Selenoprotein1. RSMD1: Sert omurga (rigid spine) konjenital müsküler distrofi1. RYR1: Ryanodin reseptör 1. LMNA: Laminin A
Performans artırmak ve deformiteleri önlemek amacı ile fizyoterapi, oluşmuş deformitelerin bazılarının düzeltilmesi için cerrahi girişim, solunumsal ve kardiyolojik takip dışında tedavi bugün için söz konusu değildir.
Distal Miyopatiler
Distal kasları da tutan miyopatilerin bir grubu, başka özellikleri nedeni ile bu bölümün başka başlıkları altında irdelenmiş miyopatilerdir. Bu grupta distal kaslar da proksimal kasların yanında tutulur (Tablo 5). Distal miyopati terimi, proksimal bir miyopatinin klinik olarak çok ilerlemiş döneminde zaafın distal kaslara yayılımını anlatmak için kullanılmaz. Aksine distal miyopatilerde klinik tablonun belirleyici özelliği, başlangıçtan veya erken dönemlerden itibaren distal kasların tutulumunun varlığı ve ağırlığıdır (Tablo 6).
Tablo 5. Distal Kas Tutulumu da Gösteren Ancak Asıl Özelliği Bu Olmayan Miyopatiler.
|
|
||||
|
|
Yalnızca, başlıca veya en erken dönemde kol ve bacakların distal kasların tutulumu ile kendini gösteren, heterojen bir grup genetik hastalık özgün “distal miyopati” grubunu oluşturur (Tablo 6). Bu nedenle bu miyopatilerde klinik olarak nöropatilerden ayırt edilme zorluğu yaşanabilir. Klasik olarak polinöropatilerde zaaf ve özellikle atrofi ekstremitelerin en distal (parmaklar, eller, ayaklar) kaslarında varlık veya belirginlik gösterirken distal miyopatiler alt bacak (krus) veya önkol kaslarında atrofi yaparlar, en distal kaslar genellikle korunmuştur (Şekil 18). Bazı özel durumlarda el ve ayakların en distal kasları da tutulabilir.
Histopatolojik olarak bu miyopatilerden bazıları nonspesifik miyopati, bazıları distrofik değişiklikler, bazıları ise miyopati veya distrofi ile birlikte kırmızı çerçeveli vakuoller (RV) gösterirler. Distrofik değişiklikler gösterenler zaman zaman otozomal geçişli kavşak tipi kas distrofileri ile iç içe geçer.
Tablo 6. Herediter ve Özgün Distal Miyopatiler.

CK: Kreatin kinaz. KOMP: Kompartman. MPD1: Gowers-Laing distal miyopati. MYH7: Miyozin ağır zincir 7. WDM: Welander distal miyopati. TMD: Tibial müsküler distrofi. TTN: Titin. NM: Nonaka miyopati (Herediter inklüzyon cisimcikli miyopati). GNE: Üridin difosfat-N-asetilglukozamin 2-epimeraz / N-asetilmannozamin kinaz. MM: Miyoshi miyopati. DYSF: Disferlin

Şekil 18. Distal miyopatide: Zaaf ve atrofi distal bacak kaslarındadır (a). Ayak kaslarında atrofi görülmez, EDB kasının çok belirgin olduğu dikkati çekmektedir (b). Gastroknemius kasının güçsüzlüğünden dolayı hasta parmak ucunda kalkamamaktadır (c). Nöropatide ise zaaf ve atrofi ayak intrensek kaslarındadır (d).
Bu grup hastalıklar içinde en kolay tanınabileceklerden birisi olması nedeni ile Miyoshi Miyopatisi (MM), dikkat edilmesi gereken bir distal miyopatidir. Genç bir hastada kas zaafı ve atrofisi alt bacak arka grup kasları tutmuşsa ve CK çok yüksek ise MM mutlaka akla gelmelidir. Kas biyopsisinde yoğun inflamasyon gösteren distrofi bulguları vardır, disferlin negatiftir. Bazen hastalık polimiyozitle karıştırılır ve hastalara kortikosteroid tedavi başlanır. Bu durumda, bazı hastalarda klinik progresyonun hızlandığı bilinmektedir. Nonaka miyopatisi ise, en azından başlangıçta, alt bacak ön kompartman kaslarını tutar ve düşük ayak belirtisine neden olur. Hastalık daha sonra alt proksimal ve üst distal kaslara da yayılabilir. Serum CK düzeyi hafif yüksektir. Kas biyopsisinde vakuollü miyopati bulunur. Otozomal resesif geçişli olan bu hastalıkta genetik defekt GNE genindedir.
Distal miyopatilerde patogenezin farklılığı serum CK düzeyine de yansımakta, CK normal, yüksek veya çok yüksek bulunabilmektedir. EMG miyopatik değişiklikler gösterir. Kas biyopsisinde nonspesifik miyopatik bulgular, distrofik bulgular veya kırmızı çerçeveli vakuol gösteren miyopati bulguları gösterilebilir.
Performans artırmak ve deformiteleri önlemek amacı ile fizyoterapi, oluşmuş deformitelerin bazılarının düzeltilmesi için cerrahi girişim dışında tedavi bugün için söz konusu değildir. Nonaka miyopatisi açısından bazı tedaviler henüz deneme aşamasındadır.
KONJENİTAL MİYOPATİLER
Konjenital miyopatiler, birbirinden bağımsız bazı yapısal kas histopatolojik değişiklikleri ile kendilerini gösteren bir grup kas hastalığıdır. Konjenital isimlendirmesi aslında “doğuştan beri olan ve yaşamın ilk yılları içinde kendini gösteren” anlamına gelmesine rağmen bu hastalıklar bir yandan konjenital miyasteniler gibi bazı konjenital hastalıkları içermemekte, bir yandan da bu grup içindeki hastalıklardakine benzer veya onlarla aynı patolojik değişiklikleri gösteren, ancak gençlik veya erişkinlik döneminde ortaya çıkan tabloları içine almaktadır. Konjenital miyopatilerin bir diğer özelliği de, bir yandan değişik genlerdeki defektler aynı hastalığa yol açarken, diğer yandan bir gendeki defektin, değişik konjenital miyopatilere veya başka miyopatilere neden olmasıdır (Tablo 7).
Otozomal dominant, otozomal resesif veya X’e bağlı resesif kalıtım özelliği gösteren ve alt gruplarının her biri ayrı hastalıklar olan bu miyopatiler her ırk ve cinste görülebilmektedir.
Konjenital miyopatilerin ortak özelliği, kas hücreleri içinde morfolojik olarak patolojik değişiklikler göstermeleridir. Bu morfolojik değişikliklerin her biri hastalığa özgüdür ve çoğu zaman hastalığın isimlendirilmesinde kullanılır; nemalin (rod/çubuk), santral kor, multi-minikor, sentronükleer ve miyofibriler miyopatiler bu miyopatilerin bazılarıdır. Hastalıkların her biri için ayrı bir etyopatogenetik mekanizmadan söz etmek mümkündür.
Primer olarak kası tutan bu hastalıklarda belirtiler çoğunlukla bebeklikte başlar, yaşamla bağdaşacak hafiflikte olanlar genellikle yavaş ve selim seyirlidir. İlk yaş içinde daha ağır olan zaaf, çocuk büyüdükçe ilk yaşlar içinde kısmen düzelme gösterir. Yani motor gelişim gecikmiştir ancak genellikle çocuk bu basamakları geç de olsa yakalar. Bu miyopatilerden bazıları ekstremite zaafı dışında kranial alan kaslarını, yine bazıları kalp ve/veya solunum kaslarını tutabilir, birçoğuna dismorfik özellikler eşlik eder. Histopatolojik olarak sıklıkla tip-I kas liflerinin belirginliği veya atrofisi gözlenir. İnfantil tiplerin dışında juvenil veya erişkin tipler ve bu genel ve benzer özelliklerin dışına çıkan konjenital miyopatiler veya formlar da mevcuttur.
CK genellikle normal veya hafif yüksek, EMG normal veya miyopatiktir. Bazı konjenital miyopatilerin tanınması rutin histokimyasal ve enzim histokimyasal incelemelerle mümkün olmakla birlikte, çoğunlukla ultrastrüktürel inceleme amacı ile elektron mikroskopisi gerekmektedir. Defektif geni bilinen konjenital miyopatilerde kesin tanı genetik analiz ile kesinleştirilmektedir.
Tablo 7. Konjenital Miyopatiler ve Genetik Defektleri.
|
|
GENETİK DEFEKTLER |
|||||||||||||
|
KLİNİK/ HİSTOPAT. TİP |
ACTA1
|
NEB |
MTM1 |
MYH7 |
DNM2 |
RYR1 |
TPM2 |
TPM3 |
TNNT1 |
CFL2 |
SEPN1 |
BIN1 |
TTN |
|
|
Nemalin |
x |
x |
|
|
|
x |
x |
x |
x |
x |
|
|
|
|
|
Kor miyopati |
|
|
|
x |
|
x |
|
|
|
|
x |
|
|
|
|
Sentronükleer |
|
|
x |
|
x |
x |
|
|
|
|
|
x |
|
|
|
Miyotübüler |
|
|
x |
|
|
|
|
|
|
|
|
|
|
|
|
Konjenital lif tipi oransızlığı |
x |
|
|
x |
|
x |
x |
x |
|
|
x |
|
|
|
|
Miyozin depo miyopati |
|
|
|
x |
|
|
|
|
|
|
|
|
|
|
Histopat: Histopatoloji. ACTA1: Aktin1. NEB: Nebulin. MTM1: Miyotubularin1. MYH7: Miyozin ağır zincir 7. DNM2: Dinamin2. RYR1: Ryanodin reseptör 1. TPM2: Tropomiyozin 2. TPM3: Tropomiyozin3. TNNT1: TroponinT1. CFL2: Kofilin2. SEPN1: Selenoprotein 1. BIN1: Bridging integrator 1. TTN: Titin
Performans artırmak ve deformiteleri önlemek amacı ile fizyoterapi, oluşmuş deformitelerin bazılarının düzeltilmesi için cerrahi girişim dışında tedavi bugün için söz konusu değildir. Gen tedavi çalışmaları sürmekle birlikte henüz patolojik süreci düzeltici bir tedavi bulunmamıştır.
HEREDİTER METABOLİK MİYOPATİLER
Büyük bir bölümü kas dokusunda enerji açığı yaratan, bazıları ise yıkılamayan maddelerin depolanması ile giden ve bu morfolojik veya biyokimyasal bozukluğun yansıması olan klinik bulgularla kendini gösteren miyopatilerdir. Belirtiler çok çeşitli düzeylerde ve lokalizasyonlarda kas zaafı olmakla birlikte, enerji açığı yaratan hastalıklarda egzersiz intoleransı önemli bir bulgudur. Bu gruptaki hastalıkların bazıları yalnızca kas dokusuna özgü patolojik değişiklik oluştururken birçoğu vücudun başka sistemlerinde de kendini gösterir. Bazı metabolik hastalıklar, aşırı enerji gerektiren durumlarda (ağır egzersiz, açlık gibi) rabdomiyoliz ve buna bağlı miyoglobinüri oluşturur. Yaşamı tehdit eden bu sonuç nedeni ile hastalar, hastalıkları hakkında bilinçlendirilmelidir. Kandaki bazal veya egzersiz sonrası laktat, piruvat ve amonyak düzeyleri bu hastalıklar konusunda çok kaba bir ayırıcı tanı yaptırabilir. Bu gruptaki hastalıklar enerji üretmekte kullanılan glikojen ve lipid metabolizmasını ve en yüksek derecede ATP üretiminin yapıldığı mitokondrileri ilgilendirmektedir.
GLİKOJEN DEPO HASTALIKLARI
Lizozomal Tipte Glikojen Depo Hastalıkları
Glikojenin, enerji üretmekte kullanılmayan lizozomal yoldan yıkımının engellenmesi ile ortaya çıkan ve kalıtımsal olan klinik tablolardır. En önemli özelliği, kas hücrelerinde lizozomal otofajik vakuollerin varlığıdır. Pompe hastalığı, bugün artık otofajik vakuoler miyopatiler olarak bilinen bir grup hastalığın ilk tanımlanmış üyesidir. Lizozomal enzimlerden asit maltaz eksikliğine bağlıdır (Tip II glikojenoz). Otofajik vakuoler miyopatiler grubunun diğer üyeleri lizozomla ilişkili membran proteini-2 (LAMP-2) eksikliği olan Danon hastalığı (ilk tanımlandığında asit maltaz eksikliği göstermeyen lizozomal glikojen depo hastalığı olarak bildirilmişti), X’e bağlı geçen artmış otofajili miyopati (XMEA) ve infantil otofajik vakuollü miyopatidir.
Asit maltaz enziminin eksik olduğu Pompe hastalığında klinik tablonun ağırlığı, başlangıç yaşı ve rezidüel asit maltaz miktarına bağlıdır. Kas zaafının nedeni yıkılamayan lizozomal glikojenin lizozom ve sitoplazmada birikerek kas liflerini harabiyete uğratmasıdır. Böylece kalıcı ve ilerleyici kas zaafı oluşur. Kalbi ve karaciğeri de tutabilir. İnfantil tipi doğuştan başlayan yaygın kas zaafı nedeni ile ortaya çıkan hipotoni, ayrıca kardiyomiyopati ve hepatomegali ile seyreder. İlerleyici olan bu tabloyu gösteren bebekler yaklaşık 1,5 yaşında kaybedilirler ancak enzim replasman tedavisinin kullanılması ile birlikte bu kayıp yaşı çok ileri taşınmıştır. Juvenil ve erişkin tipleri biraz daha selim olmakla birlikte, bunlarda birincil sorun solunum kaslarının tutulumudur. Diyafragma ve diğer solunum kaslarının tutulmasına bağlı olarak ortaya çıkan solunum bozukluğu, kas zaafından bağımsız olarak, hasta henüz ambulatuvar iken ortaya çıkmaktadır. Tanının erken konması, solunum fonksiyonlarının izlenmesi ve gerektiğinde non-invazif ventilasyonun başlanarak hastanın yaşam süresinin uzatılması açısından büyük önem taşımaktadır.
Serum CK düzeyi, az yüksek veya distrofilerde olduğu gibi çok yüksek düzeyde olabilir. Bu nedenle, çok yüksek CK düzeyi gösteren hastalarda distrofilerle birlikte Pompe hastalığı da düşünülmelidir. EMG, fibrilasyon, pozitif diken ve bazen miyotonik boşalımların eşlik ettiği miyopati gösterir. Hastalığın tanısı, basit histokimyasal incelemelerle rahatlıkla konabilir. Ancak kesin tanı için dokuda biyokimyasal olarak enzim düzeyi tayini ve genetik inceleme yapılmalıdır. Sistemik bulgular açısından belli aralıklarla kalp ve solunum sistemi fonksiyonları izlenmelidir.
Rekombinant enzimin (alglukozidaz alfa) ömür boyu sistemik olarak kullanılması infantil başlangıçlı hastalarda yaşam süresini uzatmakta, erişkin hastaların bazılarında ise iyileşme sağlamaktadır. Tedaviye, hastalığın yıkıcı etkilerinin en az olduğu başlangıç döneminde başlanması en akılcı yol gibi görünmektedir. Bu nedenle hastalığın erken tanısı önem taşımaktadır. Bunun dışında solunum yetersizliği ve kardiyomiyopatinin destek tedavisi birincil yaklaşım olmalıdır.
Non-Lizozomal Tipte Glikojen Depo Hastalıkları
Glikojenin enerji üreten, yapım ve yıkım yollarındaki enzim defektleri nedeni ile ortaya çıkan miyopatilerdir. Bazıları sistemiktir. Tümü kalıtımsal hastalıklardır. Miyopati ortaya çıkaran glikojenozlar “debranching” enzim, “branching” enzim, miyofosforilaz, fosfofrüktokinaz, fosforilaz-b-kinaz, fosfogliserat kinaz, fosfogliserat mutaz, laktat dehidrogenaz, aldolaz eksiklikleridir.
Bu tipteki miyopatilerde kas liflerinde yıkılamayan glikojenin birikimi olmakla birlikte, geri plandadır. Bazılarında glikojen birikimi hemen hiç olmamaktadır. Dokularda yalnızca glikojen değil, bloğa uğratan enzimin fonksiyonuna göre, başka polisakkaridler de birikebilir. Daha önemli sorun enerji elde edilememesidir.
Başlıca sorunun enerji elde edilememesi olması nedeniyle egzersizle ortaya çıkan ağrı, yorgunluk gibi belirtiler (egzersiz intoleransı), kas zaafına göre daha belirgindir. Bazen hiç kas zaafı olmamasına rağmen yalnızca egzersiz intoleransı görülebilir. Yıllar içinde bazen zaaf gelişebilir. Karaciğer ve periferik sinirler de tutulabilir. Bazı durumlarda hipoglisemi atakları, ensefalopati tabloları görülebilir. Rabdomiyoliz gelişebilir.
Serum CK düzeyi normal veya hafif yüksek olabilir, rabdomiyoliz dönemlerinde geçici olarak çok yükselebilir. EMG normal veya miyopatik olabilir. Miyofosforilaz eksikliğinde (Mc Ardle hastalığı) bazen tek bulgu, iskemik egzersizle ortaya çıkan ve EMG’de sessiz kalan kontraktür olabilir. Bu hastalıktan kuşku duyulduğunda bu bulgunun özellikle aranması gerekir. İskemik egzersiz testi sırasında laktatın bazal değere göre hiç yükselmemesi değerli bir bulgudur, ancak bulunamayabilir. Bu grup hastalıkta biyokimyasal incelemeler, kas biyopsisi ve genetik incelemeler değer taşımaktadır.
Bu grup hastalığın kesin tedavisi bugün için bilinmemektedir. Hastanın, hastalığı konusunda bilinçlendirilmesi, diyetin enerjiyi glikojenden farklı bir maddeden elde edecek şekilde düzenlenmesi, rabdomiyolize yol açacak aktivitelerden kaçınılmasının sağlanması, kardiyovasküler performansı artırıcı hafif ve düzenli egzersiz verilmesi önemlidir.
LİPİD DEPO HASTALIKLARI
Kas kasılmasının bazı aşamalarında gerekli olan lipid aktarım veya yıkım yolu bozukluklarına bağlı olarak gelişen, egzersiz intoleransı veya kas zaafı, bazen eşlik eden sistemik bulgularla kendini gösteren bir grup metabolik hastalıktır.
Lipid metabolizması bazı hareketlerimiz ve hareketlerimizin bazı aşamalarında gerekli olan bir enerji yoludur. Bu yolun, kas lifleri içindeki nötral lipidlerin aktarımı veya yıkımı sırasında bloke olması hem enerji açığı ortaya çıkarır, hem de yıkılamayan lipidlerin hücreler içinde birikmesine (bazı tiplerinde) neden olur. Belirtiler de bu iki faktöre bağlı olarak ortaya çıkar. Kas veya sistemik karnitin eksikliği, karnitin palmitil transferaz I ve II (CPT I ve II) eksikliği, karnitin-acilkarnitin translokaz eksikliği, kısa-, orta-, uzun- ve çok uzun- zincirli acil-koenzimA dehidrogenaz eksiklikleri, elektron transfer flavoprotein (ETF) defektleri, koenzim-Q (CoQ10) eksikliği, miyopati yapan lipid (veya lipid-mitokondri) depo hastalıklarıdır.
Yoğun egzersiz ve özellikle açlık durumlarında rabdomiyoliz gelişebilir. Bazen ilerleyici kas zaafı olabilir. Kas tutulumu dışında bazı tiplerde karaciğer tutulumu, bazı durumlarda ise ensefalopati (Reye sendromuna benzer tablo) ve sıklıkla kalp tutulumu görülebilir. Özellikle infantil dönemde başlayan bazı tiplerde hipoketotik hipoglisemi de birlikte olabilir.
Serum CK düzeyi normal veya hafif yüksektir, rabdomiyoliz dönemlerinde çok yükselebilir. EMG’de miyopati bulguları vardır. Kas biyopsisinde kas lifleri içinde lipid birikiminin gösterilmesi bu grup hastalığa tanı koydurur. Bazı tiplerde lipid birikimi görülmez. Birikim gösterilebilse bile bu yolun hangi düzeyde bloke olduğunu anlamak için dokuda biyokimyasal olarak enzim düzeyi saptamak gereklidir. Bazı tiplerde genetik inceleme yapılabilir.
Bugün için bilinen kesin bir tedavi yoktur. Karnitin eksikliğinde 3gm/gün L-karnitin kullanılabilir. Elektron transfer flavoprotein (ETF) defektleri riboflavin (B2 vitamini), koenzim-Q (CoQ10) eksikliği ise CoQ10 tedavisine dramatik yanıt verir. Ancak, başlıca kardiyomiyopatinin, ayrıca gelişebilecek ensefalopatinin iyileştirilmesi en önemli yaklaşımlardan biridir. Bunun yanında hastanın hastalık hakkında bilinçlendirilmesi, diyetin düzenlenmesi, rabdomiyolize yol açan açlık, oruç, aşırı egzersiz gibi aktivitelerden kaçınmasının sağlanması önemlidir.
MİTOKONDRİYAL HASTALIKLAR
Mitokondriyal hastalıklar genellikle yalnızca kas dokusunu ilgilendirmeyen multisistemik ve mitokondri disfonksiyonu ile giden hastalıklardır. Bu nedenle “mitokondriyal sitopatiler” olarak da adlandırılmaktadırlar. Kalıtımsal veya edinsel olabilirler. Kalıtımsal olanlarda genetik geçiş Mendel kanunlarına göre, yani nükleer gen defektlerine bağlı olarak veya mitokondriyal DNA defektlerinin maternal yoldan aktarımı ile olur. Nükleer-mitokondriyal intergenomik iletişim bozukluğu da mitokondriyal hastalık ortaya çıkarabilir. Bazı olgular sporadiktir.
Defektif mitokondrilerin oranı, her dokunun eşik değerine bağlı olarak, patolojik bulgu oluşmasına neden olur veya bulgunun derecesini belirler. Mitokondrinin, hücrenin en önemli enerji deposu olması nedeni ile enerji açığı ortaya çıkar. Bu nedenle en sık etkilenen dokular, enerjiye en çok gereksinimi olan iskelet kası, düz kas ve kalp kası ile retina ve beyin dokularıdır.
İyi belirlenmiş sendromik mitokondriyal hastalıklar:
MELAS: Mitokondriyal ensefalomiyopati, laktik asidoz, inme benzeri epizodlar MERRF: Miyoklonik epilepsi, “ragged red” lifler CPEO: Kronik progresif eksternal oftalmopleji MNGIE: Miyo-nöro-gastrointestinal sendrom, ensefalopati NARP: Nöropati, ataksi, retinitis pigmentosa, Leigh sendromu, Kearns Sayre sendromu FRDA: Friedreich ataksisi
İyi belirlenmiş bu mitokondriyal sendromlar dışında, her biri bulunan mutasyona veya oluşmuş klinik bulguya göre adlandırılmış çok sayıda mitokondriyal hastalık olduğu bilinmektedir. Tanımlandıkça bu sayının çok artması kaçınılmazdır. En iyi tanımlanmış mitokondriyal hastalıklarda ptoz ve oftalmopleji sık görülen bulgulardır (Şekil 19). Kas zaafı değişik derecelerde olabilir. Egzersizle yorulma, geri planda da olsa bulunabilir. Diğer sistem tutulumları da kendilerine özgü bulguları gösterirler. Bazen rabdomiyoliz gelişebilir.
Serum CK düzeyi çoğunlukla normal, bazen hafif yüksektir. EMG normal veya miyopatik olabilir. Kas biyopsisinde “ragged red” liflerin (RRF) bulunması, enzim histokimya ile mitokondriyal agregatların varlığı veya sitokrom oksidaz üretiminin olmadığının gösterilmesi genel olarak bu grup hastalığın tanınmasına yardımcı olur. Ancak bazı durumlarda bu histopatolojik bulguların bulunmayacağının bilinmesi önemlidir. İskemik egzersiz testinde serum laktat düzeyi normalden çok daha fazla yükselir. MELAS’ta beyin omurilik sıvısında laktat düzeyi artmıştır, bazen laktat/piruvat oranı değişmiştir. Ancak alt grupların belirlenmesi için biyokimyasal inceleme ve dokuda genetik inceleme gereklidir. Diğer sistem tutulumları da kendilerine özgü incelemelerle saptanmalıdır.
Bugün için hiçbir alt grupta kesin tedavi söz konusu değildir. Ancak, aerobik egzersiz, rabdomiyolizden korunma, diklorasetat (DCA) (laktik asidozda), riboflavin (kompleks-1 eksikliğinde), folik asit, L-karnitin, CoQ-10, L-karnitin + CoQ-10, E vitamini, C vitamini, K3vitamini, diğer sistem tutulumlarının tedavisi, nöbet kontrolü (valproata dikkat edilmelidir, karnitin eksikliği yapabilir), katarakt cerrahisi, ptoz cerrahisi, transfüzyon (sideroblastik anemi), büyüme hormonu (endokrin sistem tutulmuşsa), “pace-maker”, gerekirse kardiyak transplantasyon, nazogastrik besleme, periton diyalizi (MNGIE), işitme cihazı, koklea transplantasyonu, hidrasyon ve elektrolit balansının düzenlenmesi, diyaliz (renal tutulumda), nikotinamid ribozid (NR), bezafibrat, ve bazı deneysel ilaçlar, ilgili durumlarda ve gerektiğinde uygulanmalıdır.
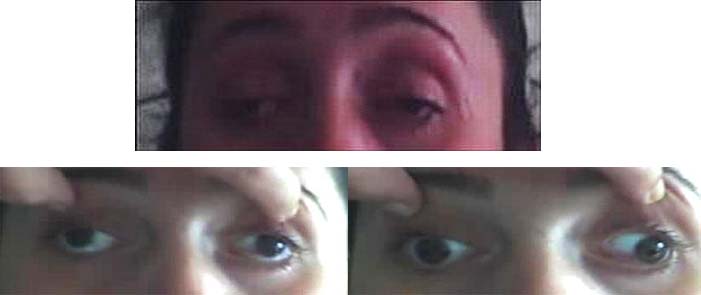
Şekil 19. Mitokondriyal miyopatide ptoza göz hareketlerinde belirgin bozulma eşlik eder
İSKELET KASI İYON KANALLARININ HEREDİTER HASTALIKLARI
Kas iyon kanalı hastalıkları, kas klor, sodyum, kalsiyum ve potasyum kanallarını kodlayan genlerdeki bozukluklara bağlı olarak ortaya çıkan nadir kalıtsal hastalıklardır (Tablo 8). Prevalansları tüm dünyada yaklaşık 1/100,000 civarıdır.
Aynı gen içindeki farklı mutasyonlar aynı fenotipe neden olabileceği gibi farklı fenotiplere de yol açabilir. Örneğin, sodyum kanalındaki mutasyonlar, paramiyotonia konjenita, potasyumun artırdığı miyotoni, hiperkalemik periyodik paraliziye veya hipokalemik periyodik paralizi tip 2’ye yol açabilir (Tablo 8).
Kas iyon kanalı hastalıklarının en önemli özelliği belirtilerin paroksizmal olmasıdır. İki ana belirtileri vardır: Miyotoni ve epizodik güçsüzlük. İyon kanalının hafif bir bozukluğu kas membranında depolarizasyon yaratarak hipereksitabiliteye yol açar ve klinik olarak miyotoni görülür. İyon kanalının daha ağır bozukluğu ise depolarizasyonun uzamasına neden olarak kas membranı hipoeksitabilitesine yol açar ve klinik olarak güçsüzlük görülür.
Kas iyon kanalı hastalıklarını bir spektrum halinde düşünmek mümkündür: Bu hastalıkların bazılarında sadece miyotoni, bazılarında sadece güçsüzlük, bazılarında ise biri ön planda olmak üzere her iki belirti bulunur (Tablo 9). Spektrumun bir ucunda, sadece miyotoniyle seyreden potasyumun arttırdığı miyotoni ve dominant miyotonia konjenita bulunur. Spektrumun diğer ucunda ise sadece güçsüzlük atakları ile seyreden, miyotoninin olmadığı hipokalemik periyodik paralizi yer alır. Diğer kas iyon kanalı hastalıklarında, değişik oranlarda miyotoni ve güçsüzlük atakları vardır: Resesif miyotonia konjenita’da ön planda miyotoni olmasına rağmen genellikle eklenen geçici zaaf, paramiyotonia konjenita’da ise benzer oranlarda miyotoni ve soğuk ve hareketle provoke olan güçsüzlük atakları görülür. Hiperkalemik periyodik paralizi ve Andersen-Tawil Sendromu’nda epizodik güçsüzlük ön plandadır, ancak klinik ve/veya elektrofizyolojik olarak çok hafif miyotoni görülebilir.
Seyrek görülen bu hastalıklar, otozomal resesif olarak kalıtılan miyotonia konjenita dışında otozomal dominant tipte bir geçiş gösterir.
Bu hastalıkların malign hipertermi ile ilişkisi net değildir, ancak nadir olgular tanımlanmış olduğundan anestezi kullanımında dikkatli olunmalıdır.
Tablo 8. İskelet kası iyon kanallarının herediter hastalıkları ve sorumlu genler

NON-DİSTROFİK MİYOTONİLER
Miyotoni, kas hipereksitabilitesinin neden olduğu gecikmiş relaksasyon sonucunda ortaya çıkan bir bulgudur. Dışarıdan mekanik uyarımla (perküsyon miyotonisi) ya da istemli kası ile (aksiyon miyotonisi) ortaya çıkabilir. Tekrarlayan hareketler ile örneğin hastanın elini açıp kapatmasıyla, miyotoni azalır (ısınma fenomeni). Miyotoninin görüldüğü iki hastalık grubu, kas iyon kanalı hastalıklarından non-distrofik miyotoniler ve müsküler distrofilerden miyotonik distrofidir. Miyotoni, non-distrofik miyotonilerin en önemli belirtisidir. Miyotonik distrofide ise belirtilerinden sadece biridir, hatta zaafın belirginleştiği dönemlerde saptanmayabilir.
Miyotonia Konjenita
Miyotonia konjenitanın başlıca belirtisi miyotonidir. Miyotoni, ani korku, ani hareket ya da sadece uzun süre istirahatten sonra harekete geçme ile tetiklenebilir. Açlık, soğuk ve yorgunluk da miyotoniyi arttırabilir. Bu hastalarda, kaslardaki sürekli hafif istemsiz gevşeme güçlüğü nedeniyle kol ve bacak kasları hipertrofik görünümde olabilir (Şekil 20).
Serum CK değerleri normal veya hafif yüksektir. İğne EMG’sinde incelenen hemen her kasta miyotonik boşalımlar vardır. Rutin olarak yapılmayan kas biyopsisinde, hipertrofik lifler, santral nükleuslar ve tip 2 liflerinde belirgin azalma, hatta kaybolma görülebilir. Kesin tanı moleküler genetik testler ile konur.
Miyotonia konjenitada, kasın ana klor kanalı geni CLCN1’de bozukluk saptanır. CLCN1 geninde hastalığa neden olacak 250’den fazla mutasyon tanımlanmıştır, bunların çoğu nokta mutasyonudur, ancak nadiren delesyon ve duplikasyon da tanımlanmıştır. CLC-1, CLCN1 geni tarafından kodlanan voltaj bağımlı bir klor kanalı proteindir ve çoğunlukla sarkolemmada eksprese olur. Bu proteinin temel görevi kas lifi membranının eksitabilitesi düzenlemek ve istirahat potansiyelini stabilize etmektir. Normal şartlarda klor iyonu, kas lifinden dışarı atılan potasyum iyonunun transvers tübüllerde birikmesine engel olur. Klor iletisi azaldığında transvers tübüllerde artan potasyum konsantrasyonu yeniden depolarizasyona neden olur ve böylece ortaya çıkan repetitif elektrik deşarjlar klinik olarak miyotoninin görülmesine neden olur. Klor kanalını sorumlu bulan ve birbirini tamamlayan bu fizyolojik ve genetik çalışmalara rağmen klor kanalındaki bozukluğun tek başına miyotonia konjenitadaki hipereksitabiliteyi açıklamaya yetmediği, bu hastalarda sodyum kanalında da bazı bozuklukların olduğu ve mekanizmanın henüz tam olarak anlaşılamadığı düşünülmektedir.
Miyotonia konjenitanın otozomal resesif olarak
kalıtılan Becker ve otozomal dominant olarak kalıtılan Thomsen olmak üzere iki
allelik tipi vardır. Aile içi evlenmelerin yüksek oranda olduğu ülkemizde
resesif miyotonia konjenita kas iyon kanalı hastalıkları arasında en sık
görülenidir. Dünyada da resesif tip, dominanta göre daha sık görülür. Bu iki
tip klinik olarak birbirine benzer ancak bazı farkları vardır.
Otozomal Resesif Miyotonia Konjenita (Becker Tipi)
Hastalığın başlıca belirtisi olan miyotoni çocukluk çağında (4 ile 12 yaş arası) başlar. Daha çok erkeklerde görülür. Miyotoni çok belirgin ve çoğunlukla oldukça ağırdır. Tüm çizgili kasları etkileyebilmesine rağmen en çok ekstremitelerde kendini gösterir, göz kasları genelde etkilenmez. Miyotoni önce bacaklarda, bundan birkaç yıl sonra ellerde, kollardan aylar veya yıllar sonra da yüz ve boyunda fark edilir, yani asandan (yukarı doğru) bir seyir söz konusudur. Miyotonik semptomların şiddeti yaşla azalabilir. Bu azalmanın bir nedeni de hastaların belirtileri daha iyi idare edebilir hale gelmesidir.
Birçok hastada ‘geçici zaaf’ diye adlandırılan özgün bir fenomen görülür. Bir hareketin ilk kontraksiyonu normal iken güç ikinci kontraksiyonda aniden azalır, birkaç kontraksiyon boyunca azalmış olarak kalır, hareket sürdürüldükçe güçsüzlük giderek düzelir ve normale döner. Birçok kasta muayene ile ortaya çıkarılabilen bu fenomeni hastalar daha çok ellerde ağır bir yük kaldırırken aniden boşalma ve elden düşürme şeklinde ifade ederler. Miyotoninin ortaya çıktığı erken yaşlarda görülmeyebilir, birkaç yıl sonra ortaya çıkabilir. Geçici zaaf, hastaya bazen miyotoniden daha çok rahatsızlık verir ve miyotoniden ayırdedilmesi gerekir.

Şekil 20. Miyotonia konjenitalı hastada kasların hipertrofik görünümü
{kind=link}
Otozomal Dominant Miyotonia Konjenita (Thomsen Tipi)
Bebeklik veya erken çocukluk çağında kendini gösterir. Baştan itibaren miyotoni yaygındır, yani asandan seyir yoktur. Geçici zaaf görülmez. Resesife göre daha hafiftir, belirtiler aylar-yıllar içinde fluktuasyon gösterebilir. Şikayet yaratmayacak ölçüde silik belirtileri olan olgular da vardır.
Bir klor kanalı hastalığı olan ‘dominant miyotonia konjenita’ ile aşağıda anlatılan bir sodyum kanal hastalığı olan ‘potasyumun artırdığı miyotoni’ birbirine çok benzeyebilir, ancak genetik incelemelerle ayırt edilmeleri mümkün olabilir.
Resesif ve dominant miyotonia konjenitayı birbirinden ayırmak her zaman kolay değildir. Ender olarak başlangıç yaşı, asandan seyir olup olmaması, miyotoninin ağırlığı, geçici zaaf olup olmaması gibi karakteristiklerde yukarıda anlatılan klasik bilgilerin tam tersi, yani biri için anlatılan diğeri için geçerli olabilir. Ayırım başlıca aile ağacında resesif veya dominant geçişe göre yapılır.
Miyotonia konjenitadan ayırt edilmesi gereken en önemli hastalık miyotonik distrofidir (DM). Miyotonia konjenitada miyotoni çok belirgin iken DM’de genellikle geri plandadır. DM’de okülobulber kaslarda, boyun ve distal ekstremite kaslarında güçsüzlük vardır; frontal kellik ve temporal atrofiyle birlikte tipik bir yüz görünümü dikkati çeker; kas dışı organ tutulumu bulunur. Serum CK düzeyi, DM’de genellikle daha yüksektir.
Miyotoni hafif olduğu zaman tedavi gerekmez. Tedavide en sık meksiletin kullanılır, karbamazepin ve fenitoin sodyum diğer seçeneklerdir. Karbonik anhidraz inhibitörleri (asetazolamid) de yararlı olabilen ilaçlardandır. Bu tedavilerin her biri tek başına, düşük dozda başlanarak verilir; semptomlar yeterince kontrol altına alınamamışsa doz yükseltilir. Beklenen cevap, miyotoninin tamamen ortadan kalkması değil, mümkün olabileceği kadar azalmasıdır. Özellikle meksiletin ve fenitoin sodyum kullanımından önce hastaya kardiyolojik inceleme yaparak bunların kullanımında bir sakınca olmadığının belirlenmesi gerekir.
Paramiyotonia Konjenita (VON EULENBURG)
Paramiyotonia konjenita, voltaj bağımlı sodyum kanalının alfa alt ünitesini kodlayan SCN4A genindeki mutasyonlarla gelişir. Otozomal dominant geçişlidir. Çok nadir olan bu hastalığın prevalansının 1/250.000 civarında olduğu bildirilmiştir.
Miyotoni ve güçsüzlüğün birlikte görüldüğü bu hastalık bebeklikte başlar. Her iki belirti de en çok yüz ve ellerde belirgindir. Miyotoni diğer hastalıklarda görülenin aksine hareketle azalacağına artar (paradoks miyotoni, paramiyotoni). Göz kapağı paramiyotonisi çok tipiktir ve neredeyse patognomonik olduğu düşünülür. Hastaya gözünü üst üste sıkıca kapaması söylenir, hasta bir süre sonra gözünü açamaz hale gelir. Hareketle miyotoni arttığı gibi kuvvetli ve tekrarlanan hareket güçsüzlük de oluşturabilir. Hastalığın en tipik özelliği güçsüzlüğün soğukta ortaya çıkmasıdır.
Hastalar birçok defa belirtilerini ağrı, katılık, şişlik gibi müphem sözlerle ifade ettiklerinden tanı koymak için güçsüzlüğü ortaya çıkaracak provokatif testlerin yapılması gerekebilir. El soğuk suya kısa bir süre sokulursa hasta elini hareket ettiremez hale gelir, bu ağır zaaf (kontraktür) el sudan çıkarıldıktan saatler sonra düzelir.
CK normal veya hafif yüksektir. El soğuk sudayken EMG ile kasın elektriksel olarak sessiz hale geldiği gösterilebilir. Ayrıca, EMG’de miyotonik boşalımlar görülür. Kesin tanı moleküler genetik testler ile konur.
Tedavisinde, hafif olgularda soğuktan sakınmak yeterlidir. Meksiletin gibi sodyum kanal blokerleri ya da tiazid/asetazolamid gibi ilaçlar işe yarayabilir.
Potasyumun Arttırdığı Miyotoni (Potassium Aggravated Myotonia, Myotonia Fluctuans)
Potasyum alımı ile provoke olan miyotoni atakları ile karakterize OD geçişli nadir bir sodyum kanalı hastalığıdır. Çocukluk ve gençlik çağlarında ortaya çıkar. Güçsüzlük görülmez. Paramiyotonia konjenitadan farklı olarak soğukla kötüleşme yoktur. Bazen egzersiz miyotoniyi provoke edebilir. Miyotoni ağrılı olabilir. Miyotonia fluktuans, miyotonia permanens ve asetazolamide yanıtlı miyotoni gibi formları içerir. Ayırıcı tanıda dominant miyotonia konjenitayı düşünmek gerektiğini yukarıda belirtmiştik. Tedavisinde, meksiletin veya asetazolamid kullanılabilir.
Tablo 9. Non-distrofik miyotoniler ve periyodik paralizilerin klinik, elektrofizyolojik özellikleri ve tedavi seçenekleri
|
|
HASTALIK |
BAŞLANGIÇ YAŞI |
MİYOTONİ/EMG’DE MİYOTONİK BOŞALIM |
GÜÇSÜZLÜK |
PROVOKE EDEN FAKTÖRLER |
TEDAVİ |
|
NON-DİSTROFİK MİYOTONİLER |
Resesif Miyotonia Konjenita (Becker) |
Çocukluk çağı |
+ |
+ Tekrarlayan hareketle geçici güçsüzlük (belirgin) |
Uzun istirahat, ani hareket, ani korku, açlık, soğuk |
Meksiletin Karbamazepin Fenitoin Asetazolamid |
|
Dominant Miyotonia Konjenita (Thomsen) |
Bebeklik ve erken çocukluk çağı |
+ |
- (Tekrarlayan hareketle hafif geçici güçsüzlük olabilir) |
Uzun istirahat, ani hareket, ani korku, açlık, soğuk |
Meksiletin Karbamazepin Fenitoin Asetazolamid |
|
|
Paramiyotonia Konjenita (von Eulenburg)
|
Bebeklik |
+ (paradoks) |
+ Soğuk ve hareket sırasında güçsüzlük |
Soğuk, uzun süre hareket |
Meksiletin Karbamazepin Asetazolamid |
|
|
Potasyum ile kötüleşen miyotoni |
Genç erişkinlik çağı |
+ |
- |
Potasyum içeren yiyecekler |
Meksiletin Asetazolamid |
|
|
PERİYODİK PARALİZİLER |
Hipokalemik Periyodik Paralizi
|
Genç erişkinlik çağı |
- |
+ Uzun süren güçsüzlük atakları (saatler-günler), Kalıcı güçsüzlük gelişebilir |
Aşırı egzersiz, fazla karbonhidrat tüketimi |
Atak: Hafif egzersiz, Potasyum Önlem: Karbonhidrattan fakir diyet Önlem: Asetazolamid, Sprinolakton, Eplerenon |
|
Hiperkalemik Periyodik Paralizi |
Çocukluk ve genç erişkinlik çağı |
+/- |
+ Kısa süren güçsüzlük atakları (1-2 saat) Kalıcı güçsüzlük gelişebilir |
Aşırı egzersiz, açlık |
Atak: Hafif egzersiz, karbonhidrat alımı Önlem: Hidroklorotiazid, Asetazolamid Diklorfenamid |
|
|
Andersen-Tawil sendromu
|
Bebeklik ve çocukluk çağı |
+/- |
+ |
Uzun istirahat, aşırı egzersiz |
Atak: Serum potasyumunun düzenlenmesi Önlem: Aşırı egzersiz ve uzun istirahatten kaçınma Aritmi tedavisi Asetazolamid |
PERİYODİK PARALİZİLER
Periyodik paraliziler (PP), ağır egzersiz, açlık, yüksek karbonhidrat alımı gibi nedenler ile tetiklenebilen ağrısız kas güçsüzlüğü atakları ile karakterize kalıtsal kas iyon kanalı hastalıklarıdır. Bazı hastalarda kalıcı güçsüzlük de gelişebilir. Atakların sıklığı değişkendir, bazı hastalar hayatlarında bir defa paralitik atak geçirirken bazılarında her gün atak görülebilir. Paralizi, kas liflerinin geçici olarak uyarılabilirliğinin azalmasından kaynaklanır.
Ailesel Hipokalemik Periyodik Paralizi
Hipokalemik PP, PP’ler içinde en sık görülenidir, ancak yine de oldukça nadir bir hastalıktır, prevalansı 100,000’de bir civarındadır. İki farklı gendeki bozukluklar bu hastalığa neden olabilir. Hastaların yaklaşık %70’inde 1. kromozomda yer alan ve kasın L-tipi kalsiyum kanalını Cav1.1’i kodlayan CACNA1S’de nokta mutasyonları (Tip 1) saptanırken, nadiren 17q23’de yer alan SCN4A geninde kas sodyum kanalı Nav 1.4’i kodlayan nokta mutasyonları (Tip 2) bulunabilir. Bu mutasyonların hastalığa nasıl neden olduğu tam olarak açıklanmış değildir.
Otozomal dominant geçişli olan bu hastalığın penetransı değişkendir. Güçsüzlük atakları genellikle puberte sıralarında başlar. Tipik bir atakta hasta sabah güçsüzlükle uyanır ya da sabah uyanınca bacaklarında ağırlık hisseder, bir saat içinde kol ve bacaklarında güçsüzlük başlar. Güçsüzlük ilerledikçe derin tendon refleksleri hipoaktif hale gelir. En ağır şeklinde hasta hiçbir ekstremitesini hareket ettiremez hale gelir. Çoğu zaman solunum ve yüz kasları etkilenmez, ancak çok nadiren solunum kaslarının da tutulup ölüme yol açtığı çok ağır ataklar olabilir. Atak sırasında duysal yakınma olmaz. Ataklar birkaç saatten bir güne kadar sürebilir ve başladığı gibi hızlıca sona erer. Ataklar arasında hasta tamamen normaldir, fakat hafif bir kalıcı zaafın sürdüğü olgular da vardır. Atak sıklığı çok değişkenlik gösterir, yılda birkaç kez veya daha az olabildiği gibi haftada birkaç kez olabilir. İleri yaşlarda atakların seyrekleştiği gözlenir, ancak çoğu hastada ileri yaşlarda ekstremitelerin proksimallerinde baskın ilerleyici ve kalıcı bir zaaf geliştiği gözlenir. Ataklar ağır karbonhidratlı yiyecekler ve ağır egzersiz sonrası dinlenme ile provoke olabilir.
Ataklar sırasında serum potasyumu genellikle düşük (3 mEq/L veya daha aşağıda) bulunur. Bu durum potasyumun böbrekten atılımı ile ilgili değildir, çünkü idrar potasyumu da azalmıştır. Mesele potasyumun kas lifi içine girmesidir. Ağır karbonhidrat alımı ve insülinin bu hastalığı provoke etmesi, insülinin Na/K pompasını stimüle ederek potasyumun hücre içine girmesini sağlayan fizyolojik etkisine bağlıdır. Tanının belirsiz olduğu durumlarda glikoz ve insülin infüzyonu yapılarak güçsüzlük provoke edilebilir. Bu test, düşük potasyumun kardiyak etkilerinin çok ağır olabileceği göz önünde tutularak, ancak çok gerektiği zaman, sıkı kontrol altında ve acil girişimin mümkün olduğu şartlarda yapılmalıdır.
Serum CK, atak sırasında veya sonrasında yüksek olabilir. EMG’de güçsüzlük derecesine bağlı olarak bileşik kas aksiyon potansiyelinin amplitüdünün düştüğü, motor ünite potansiyellerinin azaldığı görülür; hasta hareket edemez hale geldiğinde kas ineksitabldır. Miyotonik boşalımların olmaması dikkat çekicidir. Kas biyopsisi normal olabilir veya miyopatik değişiklikler gösterebilir. Özellikle kalıcı zaafın olduğu hastalarda kas liflerinde vakuoller veya tübüler agregatlar bulunur. Atak sırasında potasyumun düşük olmasına bağlı olarak tipik EKG değişiklikleri (bradikardi, PR ve QT aralıklarının uzaması, T dalgalarının düzleşmesi ve belirgin U dalgaları) ortaya çıkar. Kesin tanı genetik inceleme ile konur.
Hasta dört ekstremitede de güçsüzlükle geldiğinde ayırıcı tanıda servikal miyelopati ya da vertebrobaziler sistemi tutan serebrovasküler hastalık, Guillain-Barré sendromu gibi kuadriparezi yapan nedenler akla gelmelidir. Familyal hipokalemik PP’yi sekonder PP’den ayırmak çok önemlidir. Güçsüzlük atakları üriner veya gastrointestinal sistemden potasyum kaybı sonucu ortaya çıkıyorsa (sekonder PP), familyal PP’nin aksine, ataklar arasında da potasyum düşüktür. Ayırıcı tanıda düşünülmesi gereken bir diğer hastalık da tirotoksikoza bağlı PP’dir. Tirotoksikozun tedavisi ile ataklar da kaybolur.
Paralizi atakları 5-10 gram oral potasyum ile tedavi edilebilir, genellikle intravenöz potasyuma gerek kalmaz. Atakların düzelmesi dakikalar ya da saatler sürebilir. Kardiyak monitorizasyon gereklidir. Koruyucu tedavide düşük karbonhidratlı ve düşük sodyumlu diyet ile ataklar önlenebilir, hastalar aşırı egzersizden kaçınmalıdır. Ataklar arası koruyucu tedavi karbonik anhidraz inhibitörü olan asetazolamid veya diklorfenamid ile yapılabilir. Triamteren, sprinolakton ve eplerenon da denenebilir.
Hiperkalemik Periyodik Paralizi
Hiperkalemik PP, oldukça nadir bir hastalıktır; prevalansının 200,000’de bir olduğu tahmin edilmektedir. Otozomal dominant geçişli bu hastalığa kasın sodyum kanalında yer alan SCN4A proteinini kodlayan SCN4A genindeki bozukluklar neden olmaktadır.
Hastalık her iki cinste de görülür. Paralizi atakları bebeklikte fark edilebilir ama daha çok çocukların uzun süre oturmak zorunda kaldıkları ilkokul çağlarında dikkati çekmeye başlar. Nadiren de olsa erken erişkinlik dönemine kadar hastalık sessiz kalabilir. Güçsüzlük atakları kol ve bacakları tutar; yüz ve solunum kasları etkilenmez. Güçsüzlük çoğu zaman dört ekstremiteyi de etkiler, ancak tek ekstremitenin etkilendiği ataklar da vardır. Hiperkalemik PP’deki atağın hipokalemik PP’dekinden bazı farkları vardır: Gün içinde, daha çok egzersiz sonrasındaki istirahat sırasında ortaya çıkar, maksimum zaafa daha çabuk ulaşılır, kısa sürer (çoğu bir saatten az), daha hafiftir ve daha sıktır. Yine hipokalemik PP’den farklı olarak karbonhidratlı yiyecekler atakları provoke edeceğine atağın gelişmesini engelleyebilir. Soğuğa maruziyet, anestezi, açlık, potasyum alımının atağı tetiklediği bildirilmiştir. Atakların bazen daha uzun sürdüğü, daha ağır olduğu, bazı hastalarda ise hafif bir kalıcı zaaf zemininde geliştiği gözlenir. Bazı hastalarda miyotoni görülebilir.
Ataklar sırasında genellikle potasyum hafifçe yüksek veya normal bulunabilir. Potasyum verilerek yapılan provokasyon testleri ciddi bir atağı tetikleyebileceğinden artık önerilmemektedir, sadece genetik testin sonuç vermediği durumlarda dikkatli monitorizasyon altında denenebilir. Egzersiz testi (30 dakika koşma) daha az tehlikeli bir alternatif olabilir. Serum CK düzeyi atak sırasında veya sonrasında yüksek bulunabilir. EMG’de hipokalemik PP’de görülmeyen miyotonik boşalımlar görülebilir. Miyotonik boşalımların gözlenmesi hipokalemik PP olasılığını dışlar. Atak sırasındaki güçsüzlüğün ağırlığına bağlı olarak bileşik kas aksiyon potansiyelinde düşme ve motor ünit potansiyellerinde azalma saptanır. Kas biyopsisinde tübüler agregatlar olabilir, vakuoller görülebilir. Potasyumun yüksek bulunmasına uygun olarak EKG değişiklikleri görülebilir. Kesin tanı genetik inceleme ile konur.
Paralizi atakları genellikle hafif olduğundan tedavi gerektirmez. Bol karbonhidratlı, düşük potasyumlu diyet ve sık aralıklı öğünlerle ataklar önlenebilir. Gerekirse kalsiyum glukonat veya glikoz ve insülin verilebilir. Ataklar arası koruyucu tedavi olarak asetazolamid veya diklorfenamid kullanılabilir. Diüretikler atakları azaltabilir.
Andersen-Tawil Sendromu
Otozomal dominant veya sporadik geçişli bu sendromda paralizi ataklarına kardiyak disritmi ve dismorfik belirtiler (düşük kulak, hipotelorizm, mandibula agenezisi, sindaktili) eklenir. Hastalığa potasyum kanalı genlerindeki (KCNJ2 ve nadiren KCNJ5) bozukluklar neden olur. PP, hipo-, normo- veya hiperkalemik olabilir. Uzamış QT aralığı tipik bir bulgudur. Hafif miyotoni de semptomlara eşlik edebilir. Tanınması, “pace-maker” takılmasını gerektirebilecek ciddi kardiyak komplikasyonların önlenmesi açısından önem taşır.
MALİGN HİPERTERMİ
Malign hipertermi (MH), hassasiyeti olan
kişilerde halotan gibi uçucu anestezikler
veya depolarizan kas gevşeticisi süksinilkolin ile tetiklenen,
hipermetabolik kriz olarak kendini gösterir. Nadiren aşırı egzersiz ve sıcağın
da atağı tetiklediği gösterilmiştir. Genel popülasyonda, anestezi uygulanan
kişilerde 1:100,000 oranında geliştiği tahmin edilmektedir, ancak hafif
reaksiyonlar atlanabildiğinden bu oran gerçekte daha yüksek olabilir.
MH’nin patofizyolojisinde, çeşitli anestetikler ile tetiklenen kas hücresi sitoplazmasında aşırı kalsiyum birikmesine yol açan genetik bozukluklar rol oynar. Artmış sitozolik kalsiyum, kasta hiperaktivite ve hipermetabolizmayı tetikler. MH, çoğunlukla OD kalıtılır, ancak sporadik hastalar da vardır. Genetik bozukluğu saptanabilen hastaların büyük çoğunluğunda ryanodin reseptörü RYR1 geninde mutasyon bulunmuştur. RYR1 geninde bugüne kadar 350’den fazla patolojik mutasyon bildirilmiştir. Bunlardan 30 kadarı MH’den sorumludur. MH ile allelik ve nadir bir konjenital miyopati olan santral kor miyopatisinde de MH eğilimi görülür. MH ile allelik ve nadir bir konjenital miyopati olan santral kor miyopatisinde de MH eğilimi görülür. RYR1 genindeki mutasyonlar ayırca nadir görülen sentronükleer miyopati ve sert omurga sendromuna da neden olabilir. Az sayıda ailede, CACNA1S ve SCN4A genlerinde MH ilişkili bozukluklar gösterilmiştir. Hastaların %50 kadarında ise MH ile ilişkili olduğu gösterilmiş genlerde bozukluk saptanmaz.
Çocukluk ve genç erişkinlikte görülme sıklığı daha fazladır. Hastaların birçoğunda daha önce sorunsuz bir şekilde anestezi alma öyküsü vardır ve çoğu hasta MH öncesinde asemptomatiktir, bazı hastalarda CK hafif yüksek saptanabilir. MH krizi, çoğunlukla anestezinin verildiği ilk 1 saat içinde başlar ancak bu sürenin anesteziden 24 saatten sonraya kadar uzadığı olgular bildirilmiştir. Hastada, ventilasyona dirençli açıklanamayan hiperkapni gelişmesi MH’nin önemli bir erken işaretidir. Taşikardi, yine hipermetabolizmayı gösteren erken bir bulgudur. Klasik MH bulguları ise, trismus, iskelet kası rijiditesi, rabdomiyoliz ve hipertermidir. Hipertermi bazen geç ortaya çıkabilir. Hastalarda, rabdomiyolize ikincil kaslarda ağır ödem ve böbrek yetersizliği gelişebilir. Ağır ataklar, özellikle tanı geç konulduğunda ölümcül seyredebilir. Kriz atlatıldığında özellikle rabdomiyoliz ağır ise hastanın kas gücünün normale dönmesi haftalar alabilir, hatta düzelme tam olmayabilir.
Kriz geliştiği anda tetikleyen anestetik madde hemen kesilmelidir. Hastaya vakit kaybetmeden, sarkoplazik retikulumdan aşırı kalsiyum salınım eğilimini önlediği ya da inhibe ettiği düşünülen “dantrolen” uygulanmalıdır. Bu arada hasta, olası elektrolit bozuklukları, asidoz, kardiyak aritmiler ve böbrek yetersizliği açısından takip edilmelidir. Hasta atak sonrasında MH ve tetikleyicileri açısından bilgilendirilmeli, OD kalıtım paterni göz önünde bulundurularak diğer aile bireyleri de MH konusunda uyarılmalıdır.
EDİNSEL SİSTEMİK- METABOLİK- ENDOKRİN MİYOPATİLER
Çok çeşitli sistemik, metabolik ve endokrin hastalıklar kası da tutabilirler. Bunlar arasında en sık görülenler inflamatuvar veya granülomatöz hastalıkların ortaya çıkardığı miyopatiler (lupus eritematozus, sarkoidoz gibi), infeksiyon hastalıkları, tiroid hastalıkları ile birlikte görülenler (hipotiroidi, tirotoksik miyopati, tiroid oftalmopatisi, miyastenia gravis, tirotoksik periyodik paralizi) ve glikokortikoidlerle ilgili (Cushing) olanlardır. Ayrıca hiperparatiroidi, üremi, hipo- veya hiperkalemi ve osteomalazi de miyopatiye neden olabilir. Bu durumlarda endokrin anormalliğin düzeltilmesi çoğu kez miyopatinin düzelmesine neden olur. Aşağıda örnek olarak, yoğun bakım koşullarında ortaya çıkan miyopatilerin bir özeti verilmiştir:
YOĞUN BAKIM MİYOPATİLERİ (Critical illness myopathies)
Bazı durumlarda yoğun bakım koşullarında, intübasyon uygulanmış hastaların, beklendiği sürede spontan solunuma başlayamamaları sonucu dikkati çekmiş olan bir klinik tablodur. Uzun süre yoğun bakım nöropatisi olarak değerlendirilmiş ancak yakın yıllarda ayrı bir antite olarak kabul edilmiştir.
Tanınmaya başladığından bu yana tahmin edilenden daha sık olduğu düşünülen bu miyopatiler ancak, söylenen durumdaki koşullarda bulunan ve spontan solunuma başlatılamayan hastalarda kuşkulanılması durumunda tanınabilir. Yüksek doz kortikosteroid kullanımı (%80 olguda), organ transplantasyonu, sepsis, diyaliz, ağır hastalık durumu, kalıcı zaaf, status asthmaticus için yapılan paralitik tedaviler, nondepolarizan kas gevşeticiler gibi faktörlerin herhangi birisi veya birkaçı söz konusu tabloya neden olabilir. Hastalar tümüyle kuadriplejiktir. Bazen klinik tabloya ekstraoküler kasların zaafı da eklenir. Başlangıçta CK değeri yüksek olsa da sonraları normale dönebilir.
Yoğun bakım miyopatilerinin en sık görüldüğü durum, özellikle yüksek doz kortikosteroidlerle non-depolarizan kas gevşetici ajanların birlikte kullanıldığı durumlardır. Ancak her iki ajanın yalnız başına kullanıldığı durumlarda da ortaya çıktığı gözlenmiştir. Bu faktörler dışında ortaya çıkan miyopatide sepsis ve sepsisin yarattığı multisistemik inflamatuvar yanıtın rolü üzerinde durulmaktadır. Bugüne kadar yoğun bakım koşullarında ortaya çıktığı bildirilen üç grup miyopatiden söz edilmektedir:
1. En sık görülen tip, akut miyozin kaybı miyopatisidir. Özellikle yüksek doz kortikosteroidlerin non-depolarizan kas gevşeticilerle birlikte kullanıldığı durumlarda görülmektedir. Akut, kuadripleji yapan bir miyopatidir. CK değeri çoğunlukla normaldir. EMG normal, nöropatik veya miyopatik olabilir. Kas patolojisinde Tip-2 atrofisi ve miyozin kaybı saptanır.
2. Non-nekrotizan miyopati
3. Yaygın nekrozlu miyopati
Her üç durum da altta yatan nedenin düzeltilmesi ile haftalar içinde geriler. Bu hastalardaki ölüm miyopatiden çok, genellikle altta yatan neden veya komplikasyonlarla ile ilişkilidir.
TOKSİK VE İLACA BAĞLI MİYOPATİLER
Çok çeşitli toksinler ve ilaçlar miyopati yapmaktadırlar. Bu nedenle, ilaç ve özellikle çoklu ilaç kullanımının daha yoğun olduğu geç erişkinlik ve yaşlılık döneminde gelişen miyopatilerde ilk akla gelmesi gereken olasılık, ilaca bağlı miyopati olasılığıdır. Bu ajanlardan bazıları sessiz CK yükselmesine neden olurken bazıları nöromüsküler kavşak hastalığı, açık miyopati (özgün olmayan miyopati, mitokondriyal miyopati, miyofibril kaybı miyopatisi, vakuollü miyopati, inflamatuvar miyopati, rabdomiyoliz, gibi), veya kas ağrısı, krampları, miyotoni ile kendini gösterir. Bu nedenle, miyopatiye neden olduğu bilinen ilaçların kullanımı sırasında mutlaka hastanın miyopati açısından da aralıklı kontrolü, gerektiğinde miyopati tanısı için gerekli incelemelerin yapılması, miyopati saptandığında ise ajanın dozunun azaltılması veya tümüyle kesilmesi gerekir. Aşağıda miyopati yaptığı bilinen ilaçlar ve toksinlerden bazıları liste olarak verilmiştir.
Tablo 10. Miyopatiye Neden Olan İlaçlar ve Toksinlerden Bazıları
|
· Kortikosteroidler · Lipid düşürücü ajanlar o Klofibrat, o Bezafibrat o HMG-CoA redüktaz inhibitörleri (statinler) · e-Aminokaproik asit · Emetin · Zidovudin (AZT) · Non-depolarizan blokan ajanlar: o Pankuronyum o Vekuronyum o Atrakuronyum · İnterferon a-2a ve 2b · Siklosporin-A |
· Vinka alkaloidleri o Vinkristin o Vinblastin, vs. · Doksorubisin · Metronidazol · Na-Valproat · Kolşisin · Klorokin · D-Penisilamin · Altın · L-Triptofan · Amiodaron · Ranitidin · Baryum · Propofol · Germanium |
· Prokainamid · Hidralazin · Klozapin · Haloperidol · Risperidon · Olanzapin · Fosfolipaz A2 inhibitörleri · Vitamin E · Pentaboran · Alkol · Eroin · Kokain · Toluen · Organofosfatlar · Yılan zehirleri
|
|
|
MİYOGLOBİNÜRİ-RABDOMİYOLİZ
Bazı faktörlerin etkisi ile kas liflerinde çok yaygın nekroz (rabdomiyoliz) ve bu süreç sonunda açığa çıkan miyoglobinin idrarla atılması (miyoglobinüri) ile belirlenen durumdur. Kaslarda güçsüzlük, miyalji, bazen kaslarda şişlik, koyu idrar rengi (hafif durumlarda idrar rengindeki değişiklik fark edilmeyebilir) başlıca klinik bulgularıdır. CK ileri derecede yükselir. CK yüksekliği bazen kas zaafından önce ortaya çıkabilir. İdrarda yüksek oranda miyoglobin saptanır. Hasta idrar renginin koyu olduğunu ifade eder (hematüri ile karıştırılmamalıdır). Rabdomiyolize yol açan nedenler çok çeşitlidir. Miyoglobinüri akut böbrek yetersizliğine yol açabilir ve yaşamı tehdit edebilir. Ayrıca hiperkalemi, hiperfosfatemi, hipo- veya hiperkalsemi, hiperürisemi geliştirebilir, kardiyak aritmilere yol açabilir. Nörolojik, nefrolojik ve kardiyolojik acil durumlardandır. Acil tedavide böbrek ve kalp fonksiyonlarının izlenmesi ve gerektiğinde düzeltilmesi büyük önem taşır. Bu dönemi atlatan hastalarda düzelme genellikle tamdır. Ayırıcı tanıda hemoglobinüri nedenleri ve akut intermittan porfiri önemlidir.
Miyoglobinüriye neden olan ana hastalığı araştırmak amacıyla gereken kas biyopsisi, bu acil tablonun gelişiminden en az 1 ay sonra yapılmalıdır. Biyopsi eğer rabdomiyoliz sırasında yapılmışsa daha sonra mutlaka yinelenmelidir. Aşağıda rabdomiyoliz-miyoglobinüriye en sık neden olan durumların listesi verilmiştir:
Tablo 11. Rabdomiyoliz ve Miyoglobinüriye Neden Olan Durumlar
|
Aşırı fiziksel aktivite (istemli, status epileptikus, delirium tremens vs.) Travma İnfeksiyonlar Viral Bakteriyel İskemi Vasküler oklüzyon (fokal) Orak hücreli anemi Hipokalemi Hipertiroidi, Hipotiroidi Hiperparatiroidi
|
İlaçlar ve toksinler Lipid düşürücü ajanlar Laksatifler (aşırı kullanım) Kolşisin Emetin Monensin e-Amino kaproik asit Valproik asit Lamotrijin Nöroleptikler Serotonin sendromu Eroin, Kokain, Amfetamin Alkol Pentaboran Hidrokarbonlar (toluen gibi) |
İnflamatuvar kas hastalıkları Polimiyozit Dermatomiyozit Distrofiler DMD BMD Bazı mitokondriyal miyopatiler Bazı konjenital miyopatiler Bazı herediter metabolik hastalıklar Çok uzun zincirli yağ asitleri defektleri CPT2 Defekti Glikojen depo hastalıkları Miyoadenilat deaminaz defekti Malign hipertermi Familyal rekürran rabdomiyoliz |
İNFLAMATUVAR MİYOPATİLER
İnflamatuvar miyopatiler kas dokusunun edinsel miyopatilerindendir. Diğer birçok miyopatinin tersine subakut seyirlidirler. Kas ve çevre dokularda inflamasyona yol açan nedenler çok çeşitlidir. Aşağıdaki tabloda bu değişik nedenler görülebilir. Bu bölümde yalnızca idyopatik inflamatuvar miyopatiler konusu derinlemesine işlenecektir.
Tablo 12. İnfeksiyöz ve İnflamatuvar Miyopatiler
{kind=link}

İDYOPATİK İNFLAMATUVAR MİYOPATİLER
Polimiyozit (PM), dermatomiyozit (DM), otoimmün nekrotizan miyopati (ONM) ve inklüzyon cisimciği miyoziti (İCM) bu grup içinde yer almaktadır. Polimiyozit, dermatomiyozit ve otoimmün nekrotizan miyopatinin ortak özellikleri subakut (haftalar veya aylar içinde gelişen) proksimal kas zaafı, boyun fleksiyon veya ekstansiyon zaafı ve yüksek serum CK değeri ile seyretmeleridir. Dermatomiyozitte ek olarak deri bulguları da vardır. Bu benzerliğe rağmen iki miyopatinin patogenezleri birbirinden tümüyle farklıdır ve ayrı ayrı anlatılacaktır. Sporadik inklüzyon cisimcikli miyozit (s-İCM) ise, ileri yaşta başlaması, distal kas zaafı ile birlikte olması, inflamasyon dışındaki bazı histopatolojik özellikleri ve immünsupresiflere yanıtsızlığı açısından, tümüyle farklı bir hastalıktır.
İlk üç miyozit tablosunun bazı özelliklerinin iç içe geçmesi nedeni ile önce ortak özellikleri bildirmek daha yararlı olacaktır.
Ortak klinik ve laboratuvar özellikler (PM, DM, ONM):
Kas zaafı proksimaldir ve kişiyi, haftalar, aylar içinde özürlülüğe ulaştırır. Bu anlamda, çok yavaş ilerleyen ve özürlülüğe ulaştırma süresi yıllar, hatta on-yıllar alan kalıtımsal kas hastalıklarından, özellikle de kas distrofilerinden farklı bir seyir izler. Proksimal zaafa çoğunlukla boyun fleksiyon zaafı ve yutma güçlüğü de eşlik eder.
Serum CK değeri, hastalığın aktif dönemlerinde yüksek veya çok yüksektir. Tedavi ile CK düzeyi düşer. Bu yükseliş ve düşüşler, klinik tablonun öncesi veya sonrasında olabilse de, genel olarak klinik ağırlık/CK düzeyi ilişkisi mevcuttur.
Kas MR’ında, aktif dönemde kas dokusu içinde yama şeklinde kontrast madde tutulumu, inflamasyona işaret eder.
Kas biyopsisinde PM ve DM’de inflamasyon mevcuttur ancak ONM, başlıca nekroz ile giden bir miyozit tablosudur.
Kas dışı organ tutulumları (PM, DM, ONM):
Otoimmün/inflamatuvar miyopatiler, sistemik hastalıklardır ve kas dışındaki organları da tutabilirler.
Deri: Deri bulgularından en eski ve iyi bilineni DM’te deri tutulumudur. Göz kapaklarındaki heliotrop ödem, boyundaki V belirtisi, eklemlerin ekstansör yüzlerindeki makülopapüler lezyonlardan oluşan Gottron belirtisi, periungual değişiklikler, özellikle juvenil DM’deki kalsinozis bunların başlıcalarıdır. Anti-sentetaz hastalıklarında da Raynaud fenomeni ve işçi eli görünümü dikkat çekicidir. Overlap miyozitlerde ise skleroderma (Scl) ve sistemik lupus eritematozusa (SLE) ait deri lezyonları görülebilir.
Solunum sistemi: Miyozitlerde akciğer tutulumu; pulmoner hipertansiyon, serozit, ama en sık olarak da interstisyel akciğer hastalığı (İAH) şeklinde kendini gösterir. Antisentetaz sendromunun ana solunumsal tablosu olan İAH ayrıca anti-SRP, anti-RNP, anti-PM/Scl, anti-Ku ve anti-MDA5 antikorları ile birlikte olan miyozitlerde de görülebilir. İnterstisyel akciğer hastalığının ana belirtileri kuru öksürük ve nefes darlığıdır, yüksek çözünürlüklü göğüs bilgisayarlı tomografisi ile tanınır. Malignite dışında mortalitesi en yüksek miyozitle ilişkili organ tutulumu olması nedeni ile hastalarda mutlaka aranması gereken bir tablodur ve kendine özgü bir tedavi düzenlemesi gerektirir.
Eklemler: Eklemlerde şişlik, ağrı ve tutuklukla giden artralji tablosu antisentetaz sendromunun ana bulgularından birisidir. Küçük el, bilek, kalça, diz ve ayak bileği eklemleri tutulabilir. Antisentetaz sendromu dışında, overlap sendromlarda da görülebilir.
Kardiyovasküler sistem: Aslında seyrek olmamasına rağmen kardiyak tutulum miyozit ve overlap miyozit hastalarında gözden kaçan bir bulgudur. Kardiyomiyopati, perikardit, aritmi şeklinde kendini gösterir ve mortaliteyi artıran bir tutulumdur. Hastada eforla dispne ve aritmi ana semptomlardır. Bu nedenle, kardiyak tutulum olasılığı miyozitli hastalarda düşünülmeli, incelemeler 24 saatlik Holter ve ekokardiyografiyi içermelidir. Kalp tutulumu olduğunda kardiyoloji izlemi ve tedavisi yanında immünsupresif tedavi dozu da artırılmalıdır.
Gastrointestinal sistem: Yutma güçlüğü miyozitlerde iyi bilinen bir semptomdur ve farinks ve üst özofagus kaslarının tutulumuna bağlıdır. Aspirasyon riski taşımaktadır. Alt gastrointestinal sistem kanamaları ise juvenil DM’de görülebilir.
Otoantikorlar (PM, DM, ONM, s-İCM):
İnflamatuvar miyopatilerde, diğer bağ dokusu hastalıklarında da görülen miyozitle ilişkili antikorlar (“myositis associated antibodies” - MAA) ve yalnızca miyozitte saptanan, miyozite özgü antikorlar (“myositis specific antibodies” - MSA) görülebilir. Tanımlandıkları ilk yıllarda, özellikle MSA’ların belli bir klinik tabloyu işaret etmekte yetersiz kaldıkları düşünülmüş ancak yıllar içindeki deneyim, bazı MSA’ların belli klinik tablolarla ilişkisini kesinleştirmiştir. Böylece hem bu klinik tablolar daha iyi tanınmaya başlamış, hem de sınıflamalara girmiştir. Örneğin Jo1 MSA taşıyan bireylerdeki anti-sentetaz sendromu ve bazı başkaları, klinik bulgular, prognoz ve tedaviye yanıt açısından ayrı birer hastalık olarak yerlerini almışlardır. (Tablo 13). Bugüne kadar 17 MSA tanımlanmıştır ve hastaların %70-80’inde, gösterilebilir bir MSA mevcuttur. Geriye kalan MSA negatif %20-30’luk hasta grubunda ise muhtemelen henüz saptanmamış MSA’lar mevcuttur.
Tablo 13. Miyozite özgü antikorlar (MSA) ve ilişkili olduğu önemli klinik tablolar
|
OTOANTİKOR |
KLİNİK TABLO |
İLİŞKİLİ OLDUĞU KLİNİK ÖZELLİK |
|
Anti-sentetazlar:
(-Jo1, -PL-7, -PL-12,-EJ, -OJ, -KS, -Z, -YRS/HA) |
PM ve DM (OM) |
En sık anti-Jo1 (%15-20) ve serum düzeyi hastalık aktivitesi ile ilişkili. Anti-sentetazların hepsi interstisyel akciğer hastalığı (İAH) ile ilişkili. En sık anti-Jo1 (Hastaların %50-75’inde) anti-PL-7, anti-PL-12, anti-OJ ve anti-KS pozitif hastalarda İAH miyozitten daha ön planda veya tek başına olabilir
|
|
anti-Mi-2 |
DM |
Klasik DM tablosu Tedaviye iyi yanıtlı
|
|
anti-MDA5 (anti-CADM140) |
DM |
A-Hipomiyopatik DM tablosu yapar. Deri lezyonları ağır ve atipik olabilir, İAH olabilir
|
|
anti-TIF1 g/a (155-140) |
DM |
Juvenil DM. Kanser ile ilişkili a/hipomiyopatik erişkin DM
|
|
anti-TIF1b (140) |
DM |
Juvenil DM Kanser ile ilişkili erişkin DM
|
|
anti-NXP2 (anti-MJ) |
DM |
Juvenil DM. Kalsinozis yoğun Kanser ile ilişkili erişkin DM
|
|
anti-SAE |
DM |
Deri lezyonları ve disfaji ağır olabilir Kanserle ilişkili olabilir
|
|
anti-SRP |
ONM |
Çok akut başlangıç (genellikle sonbaharda),
|
|
anti-HMGCR (200/100) |
ONM |
Statin kullanan veya kullanmayan hastalarda görülür
|
|
anti-cN-1A |
s-İCM |
Henüz standardize edilmemiş
|
Tetikleyici faktörler (PM, DM, ONM):
Otoimmün/inflamatuvar miyopatilerden PM, DM ve ONM’de MSA’ların varlığı, dolayısı ile öz antijenlere karşı toleransın kırıldığı otoimmün bir yanıt söz konusudur. Sporadik-İCM’de bu yanıtın başka özellikleri vardır ve patogenez henüz aydınlatılamamıştır. Otoimmün olduğu gösterilmiş inflamatuvar miyopatilerde öz toleransın kırılmasını tetikleyen faktörlerin neler olduğu iyi bilinmemekle birlikte, bazı klinik tabloların özelliklerinden yola çıkılarak bu toleransın kırılmasına neden olan bazı faktörlerin olduğuna kanısına varılmaktadır
Viral tetikleyiciler:
Bazı virüslerin (Coxackie B, parvovirüs, enterovirüs, HTLV-1, HIV) kasa yönelimi olduğu bilinmektedir. Ayrıca bazı miyozitlerin mevsimsel kümelenme özelliği, miyozitli bazı çocuk ve erişkin hasta popülasyonlarında hastalarda öncül bir infeksiyon varlığı da miyozit oluşumunda viral tetikleyicilerin olabileceğini düşündürmektedir. Viral etmenler, birkaç yoldan bu öz toleransın kırılmasına neden olabilir: Öncelikle, var olan öz proteinlerde bir değişim oluşturabilirler. Veya hücre içi proteinlerin yapısını veya konformasyonunu değiştirebilirler. Ayrıca moleküler benzerlik (çapraz reaksiyon), immün toleransın kırılmasına yol açabilirler.
Tetikleyici faktör olarak kanser:
Kanser ile erişkin DM arasındaki ilişki iyi bilinmektedir. Üstelik erişkin DM, kanser başlamadan 2 yıla kadar önce de görülebilir. Miyozite özgü antikorlar konusundaki deneyimin birikmesi ile birlikte yakın yıllarda, erişkin DM’de görülen bazı MSA’larla kanser ilişkisinin kuvvetli birlikteliği, her DM hastasının kanser adayı olmadığını göstermiştir. Örneğin, TIF-1g/a, TIF-1b, NXP-2 ve çok daha seyrek görülen, SAE antikorlarının var olduğu erişkin DM’de kanser olasılığı çok yüksek iken diğer MSA’larda bu ilişki gösterilememektedir (Tablo 13). Ayrıca, HMGCR antikorları da kanserle ilişkili olabilmektedir. Kanserle miyozit ilişkisi günümüzde şöyle açıklanabilmektedir: Kanser hücrelerindeki, somatik mutasyonlarla oluşmuş neoantijenler çok immünojeniktirler ve yoğun bir immün yanıt oluşturabilirler ki bu da konak proteinleri ile çapraz reaksiyon oluşturabilir (Örneğin, kanserle ilişkili erişkin DM’li TIF-1b pozitif hastalarda var olan tümörlerin çoğunun TIF-1b mutasyonu barındırdığı görülmüştür). Diğer yandan, tümör hücreleri üzerinde bulunan proteinlerin normal konak proteinleri olması durumunda, vücutta oluşan anti-tümör immünitesi doğrudan konak proteinlerine de yönelebilir. Kanserle ilişkili miyozitlerde sıklıkla kanser hücresinde de, rejenere kas hücrelerinde de miyozitle ilişkili proteinler eksprese edilmektedir ve bu ortaklık da otoimmüniteyi kasa yönlendirebilir. Yine de unutulmamalıdır ki, kanserle ilişkili antijen taşıyan tüm hastalarda malignite görülmemektedir. Bu, ya miyozitin kanserden önce teşhis edilmesi, ya da başka mekanizmaların varlığını düşündürebilir.
Tetikleyici ilaçlar:
Çok sayıda olan miyozit tetikleyici ilaçlar arasında en iyi bilineni statinlerdir. Statin kullanımı sırasında ortaya çıkan nekrotizan miyozitte, çoğu hastada HMGCR antikorlarının gösterilmesi ile otoimmün bir zemin olduğu ortaya çıkmış ve hastalık statinle ilişkili ONM adını almıştır. Statinler kas dokusunda HMGCR’nin konformasyonunu değiştirmekte veya miktarını artırmaktadır. Statin dışında immün denetim noktası baskılayıcı (immune check-point inhibitors) ilaçlar, bu tetiklemeyi yapan başka bir ilaç grubudur. Bu ilaçlar, kontrolsüz immün yanıttan koruyan mekanizmaları bozarak bir yandan tümör immünoterapisi yapmakta ama diğer yandan da, değişik organlarda kontrolsüz immün yanıt oluşturmaktadır. İlginçtir ki bu yan etkiyi oluşturan kanser immünoterapileri kanser üzerinde daha etkili olmaktadır.
Polimiyozit
Her iki cinste ve her yaşta görülebilen, ancak kadın baskınlığı olan PM’in en sık görüldüğü yaş grubu 45-60 yaş arasıdır. Kadın baskınlığı 15-44 yaş arası en belirgindir. Çocukluk çağında çok ender görülür.
Kas zaafı genellikle alt ekstremitelerin kök kaslarından başlar ve hem aksiyal kaslara, hem de omuz kavşağı kaslarına yayılır. Tedavi edilmediğinde hastalığın varacağı nokta hastanın yatağa bağımlı hale gelmesi ve kasların atrofiye uğramasıdır. Polimiyozitte en ağır etkilenen kas grupları kalça fleksör, ekstansör ve abduktörleri, omuz abduktörleri ve boyun fleksörleri olmakla birlikte, tüm bu kas gruplarının DM’e göre daha ağır etkilenmesi ve ek olarak diz ekstansörleri ile ayak plantar fleksiyonunda güçsüzlük saptanması da dikkat çekicidir. Genel olarak ağrısız bir hastalık olmasına rağmen PM’de, olguların %20’sinde kas ağrıları görülebilir. Tüm bulgular subakut yerleşebileceği gibi ender olarak akut yerleşen, yaygın kas nekrozu ile seyreden bir klinik tablo da sergileyebilir. Yutma güçlüğü genellikle, özofagusun 1/3 proksimal bölümündeki çizgili kasların tutulumuna ikincildir. Kanserle miyozit birlikteliği gösteren hastalardaki MSA’lar DM ve ONM tablolarına yol açmaktadır. Dolayısı ile PM’de kanser ile artmış ilişki görülmez.
Kas biyopsisindeki tanı koydurucu tipik özellik, nekrotik olmayan kas liflerinin çevresindeki CD8(+) sitotoksik lenfosit infiltrasyonu ve tüm kas liflerinin sarkolemmasında majör doku uygunluğu kompleksinin (MHC-1) pozitifliğidir (Şekil 21). Bu birliktelik ve henüz bozulmamış kas liflerinde MHC-1 pozitifliği, öncelikle kas hücresinde antijen prezentasyonu olduğunu ve sonra hücresel immün yanıtın oluştuğunu, sitotoksik bir hücre hasarına uğradığını göstermektedir.
Anti-Sentetaz sendromu:
Miyozit, İAH, inflamatuvar artrit, işçi eli görünümü ve Raynaud fenomeninden oluşan klinik bulgular ile anti-sentetazların herhangi birinin birlikteliği, anti-sentetaz sendromunu (ASS) oluşturur. Bu klinik tablo ile en sık birlikteliği olan, aynı zamanda ilk tanımlanmış olan anti-sentetaz, anti-Jo1’dir. Anti-Jo1 pozitif hastalar daha çok miyozitin ön planda olduğu klinik tablo sergilerken diğer anti-sentetazlar, ön planda İAH ile seyrederler. Anti-Jo1 pozitif hastalarda kas biyopsisi de klasik polimiyozitten bazı farklılıklar gösterir. Bu hastalar kas biyopsisinde endomizyal değil, perimizyal inflamasyon, perifasiküler nekroz ve perimizyal bağ dokusunda fragmantasyon gösterirler. Bu anlamda, deri bulguları uymamakla birlikte, histopatolojik olarak DM ile karıştırılabilirler.
Overlap miyozit: Sınıflamalarda giderek yerini almış olan overlap miyozit, miyozit tabloları içinde en geniş yeri tutmaktadır. Miyozit tablosu ile, en sık Sjögren (SjS), sistemik skleroz (SSc) ve SLE olmak üzere, bir bağ dokusu hastalığı birlikte görülür. Miyozit klinik tablosu ve CK düzeyi, polimiyozitte olduğu gibi seyreder. Bazen bağ dokusu hastalığı daha geç ortaya çıkar ve klinik tablo PM ile karışabilir. Overlap sendromlar ASS ile birlikte de olabilirler. Overlap miyozitlerde anti-sentetaz antikorları dışında anti-SS-a, -SS-B, -Ro52, Ro-60, -U-snRNP, -Ku ve SSc ile birliktelikte -PM/Scl antikorları da görülür.
Hastalığın tedavisinde immünsupresyon yer alır. Birinci basamak tedavi, 1-1,5 mg/kg başlangıç dozunda prednizolondur. Ağır olgularda tedaviye 1000 mg/gün metilprednizolon ile başlayıp sonra oral prednizolona geçmek daha yararlı olabilir. Yanıt almak için iki, bazen üç ay beklemek gerekebilir. Hastalığa yanıt almadan ilaç dozunu azaltmamak gerekir. Düzelmeye klinik bulguların gerilemesi, ayrıca başlangıçtaki CK düzeyinin düşmesi ile karar verilir. Ancak sonraki izlemlerde CK değerini sürekli gözlemek ve tedaviyi buna göre yönlendirmek yanlıştır. Genel olarak önemli olan kas zaafının düzelmesidir. Klinik yanıtın başlamasından sonra tedavi dozu giderek ama yavaş olarak azaltılır ve bir idame dozuna inildiğinde bu düzey çok uzun süre (bazen yıllarca) korunur. Bir diğer olasılık, başlangıçtan itibaren, klinik tabloda düzelme olmazsa veya kortikosteroid dozunu azaltmak gerekirse ek olarak öncelikle metotreksat 20 mg/hafta veya azatioprin 2,5-3mg/kg/gün başlanabilir ve zaman içinde bir idame dozuna inilir. Bu ilaçların eklenmesi, toksik etkilerin yakından izlenmesini gerektirir. İVİg kullanımı da polimiyozitte yararlı olabilir. Sirolimus ve mikofenolat mofetil yeni tedavilerdendir. Diğer tedavilere yanıtsız PM’te rituksimab kullanımı önerilmektedir. Ayrıca infliksimab, takrolimus gibi yeni nesil immünsupresif ilaçlar ile ilişkili çalışmalar sürmektedir. Unutulmamalıdır ki ilaç tedavisine miyozitlerde baştan itibaren yoğun fizyoterapi eşlik etmelidir.
Şekil 21. Polimiyozit ve dermatomiyozitte histopatolojik görünüm.

Şekil 21a. Polimiyozitte endomizyal (a) ve non-nekrotik lifler çevresinde inflamasyon (b)
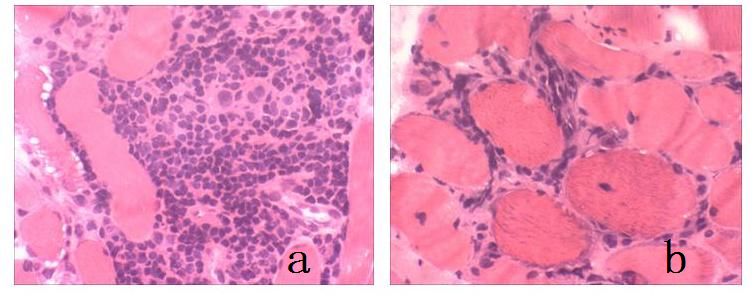{kind=link}

Şekil 21b. Dermatomiyozitte perimizyal inflamasyon (a), perifasiküler atrofi (b)
{kind=link}
{kind=link}
Dermatomiyozit
Dermatomiyozit, özgün deri döküntülerinin veya buna ilerleyici kas güçsüzlüğünün eşlik ettiği, kolay tanınabilen bir inflamatuvar kas hastalığıdır. Her yaşta görülebilmesine rağmen, çocukluk çağında ve 40 yaş sonrası olmak üzere, iki yaş grubunda görülme sıklığı yüksektir. Her iki yaş grubunda da kadınlarda daha sıktır. Juvenil DM çocukluk çağının en sık görülen inflamatuvar kas hastalığıdır. Kas zaafının dağılımı ve ilerlemesi PM’dekine benzerdir ancak DM’deki zaaf görece daha hafif sayılabilir.
Dermatomiyozitin en önemli klinik özelliği deri bulgularının varlığıdır (Şekil 10 ve 22). Deri bulguları göz kapaklarındaki leylak rengi ödem (heliotrop ödem), yüzde eritemli döküntü (rash) (Şekil 10), V-belirtisi, eklemlerin ekstansör yüzlerinde makülopapüler döküntü (Gottron papülleri) ve tırnak diplerindeki değişikliklerden oluşur. Hastalarda bu belirtiler değişik birlikteliklerde bulunurlar. Çoğunlukla deri bulguları miyopatiden önce başlar. Genellikle yüzdeki döküntü ilk belirtidir. Klinik miyopatinin eklenmediği veya geri planda olduğu, yalnızca deri belirtilerinin bulunduğu tablo a-hipomiyopatik DM, deri belirtisi olmaksızın miyozit tablosu ve DM’e uygun histopatolojik özellikler “DM sine miyozit” adını alır. A/hipomiyopatik DM’de de klasik döküntülerden 1-2 sene sonra miyozit gelişebilmektedir. Ancak bu dönemde de diğer sistemik tutulumların, özellikle interstisyel akciğer hastalığının gelişebileceği unutulmamalıdır. Yukarda anlatılan bu nedenlerle DM, miyozit ve deri bulgularının çeşitli ağırlıklarda olabileceği geniş bir spektrum olarak kabul edilir.
Kas yıkımının olduğu dönemde serum CK değeri de yine normalin 10-20 katına çıkar. Ancak CK değerlerinin PM’e göre daha düşük olduğu ve zaman zaman, özellikle a/hipomiyopatik DM’de normal sınırlar içinde olabileceği de akılda tutulmalıdır.
Kas biyopsisindeki en karakteristik özellik, perimizyal dokudaki inflamasyondur. Bu inflamasyon endomizyal bölgeye de yayılabilir. Histopatolojik olarak başlıca, kas dokusu içindeki damarlarda inflamasyon, daralma ve tıkanmalara bağlı olarak kas liflerinde dejenerasyondan nekroza kadar değişik yelpazede letal ve sub-letal değişiklikler gözlenir (Şekil 21b). Endomizyal damarların sayısında ciddi oranda azalma gözlenir. İnflamasyonu oluşturan hücreler başlıca B lenfositleri, az miktarda CD4(+) T lenfositleri ve çok geri planda CD8(+) T lenfositlerden oluşur. İmmünhistokimyasal çalışmalar perimizyum ve endomizyumdaki damarlarda C5b9 membran atak kompleksi (MAC) birikimi gösterir. Tüm bu bulguların birlikteliği DM’te, komplemana bağımlı humoral bir immün yanıtın birincil hedefinin kas ve derideki damar endoteli olduğunu, damarlarda non-nekrotizan bir vaskülitin, buna ikincil olarak da kas hücreleri dejenerasyonu ve nekrozunun geliştiğini göstermektedir. Kronikleşmiş olgularda fasiküllerin kenar kısımlarında (perifasiküler) gözlenen kas lifleri atrofisi çok tipik ve tanı koydurucudur (Şekil 21b).
Geç dönemdeki erişkin DM kanser ile birlikte bulunabilir. Dermatomiyozitli hastalarda kanser riskinin benzer yaş ve cinsiyetteki genel popülasyona göre altı kat daha fazla olduğu gösterilmiştir. Anti-TIF1b ve NXP2 antikorlarının varlığının DM/kanser birlikteliğine işaret ettiği gösterilmiştir. Bunlardan anti-TIF1b, juvenil DM’de de görülen ve kanserle ilişkili olmayan bir antikor iken 40 yaş üzeri DM’li hastalarda kanser riski oluşturmaktadır. Bu hastalar daha çok a/hipo-miyopatik DM tablosu gösterirler, İAH gibi sistemik bulgu geliştirmezler, bazen, klasik DM deri bulguları dışında palmar döküntü gösterirler. En sık görülen kanserler over, akciğer ve meme solid kanserleridir. Kanser-DM birlikteliğinde görülen diğer antikor NXP-2’dir. Bu antikor da kalsinozisli juvenil DM’de görülmekte ve kanser ile birliktelik göstermemekle birlikte, erişkin DM’de, özellikle erkek hastalarda, kanser riskine işaret etmektedir. Negatif sonuçlanan bir kanser taramasından sonra, taramanın tekrarlanması konusunda genel kanı, bu hastaların hassas bir şekilde senelik fizik muayenelerinin ve rutin laboratuvar testlerinin tekrarlanması yönündedir. Ancak over kanserinin DM’li kadınlarda uzun yıllar sonra da saptanabilmesi, senelik serum Ca-125 kontrolü ve pelvik muayenenin tekrarının gerekliliğini düşündürmektedir.
Juvenil DM, erişkin DM’e benzemekle birlikte sistemik bulguların daha ön planda olduğu bir durumdur, kas güçsüzlüğü diğer inflamatuvar miyopatiler ile karşılaştırıldığında daha hafiftir ve benzer dağılım gösterir. Etkilenmiş çocuklar huzursuz ve yorgundur. Yüzde kırmızı raş belirgindir. Ayak bileğindeki fleksiyon kontraktürü nedeniyle parmak ucunda yürüme sıktır. Çocukluk çağı DM’nin diğer önemli özelliği, vaskülitin ön planda olması, kontraktürler ve kas içi (ektopik) kalsifikasyonlarla seyretmesidir. Kalsifikasyonlara karşı etkili bir tedavi bulunmamaktadır. Ancak kontraktürlerin oluşması, zaten bu hastaların alması gereken fizyoterapi uygulamalarının ayrıca kontraktürlere yönelik olarak da uyarlanması ile önlenebilir.
Tedavi, PM’deki prensipleri içerir.
Şekil 22. Dermatomiyozitte deri belirtileri

Şekil 22a. Dirsekte ve elde Gottron belirtisi
{kind=link}

Şekil 22b. Periungual değişiklikler
{kind=link}

Otoimmün Nekrotizan Miyopati
Geçmişte PM olduğu düşünülen ancak kas biyopsilerinde inflamasyon gösterilemeyen, kas nekrozu gösterilen hastaların bir bölümü, doksanlı yılların başlarındaki sınıflamalarda, özel histopatolojileri, eşlik ettiği klinik durumlar (otoimmün hastalıklar ya da kanser) ve tedaviye yanıtları ile ayrı bir grup hastalık olarak değerlendirilmeye başlanmış, daha sonraki sınıflamalarda ONM olarak yerini almıştır. Klinik olarak PM gibi subakut ilerleyici proksimal zaaf, yutma güçlüğü ve yüksek CK düzeyi göstermektedir. Hastaların bir bölümünün miyozitin geliştiği sırada veya öncesinde statin kullanmış olmaları, statin ile ilişkili miyozit tanımına da neden olmuştur.
Hastalık kendini akut/subakut yerleşen, proksimal kaslarda belirgin orta veya ağır derecede güçsüzlük, yüksek CK ve EMG incelemelerinde bol iritatif miyopatik bulguların eşlik ettiği bir miyozit olarak gösterir. Klinik tablo PM’e çok benzerdir. Kas histopatolojisinde, kas lifi nekrozu gözlenir, buna zaman zaman, geri planda inflamasyon da eşlik edebilir. Polimiyozitten farklı olarak T hücre infiltratları gözlenmez ve MHC-1 pozitifliği olmayabilir, temel patojenik hücre grubu makrofajlardır.
Otoimmün nekrotizan miyopati, MSA’lardan anti-SRP ve anti-HMGCR antikorları ile birlikte de görülmektedir. Son ENMC kriterlerine göre ONM, anti-SRP pozitif, -antiHMGCR pozitif veya seronegatif olarak ayrılmaktadır.
Hastalık, her iki antikorun varlığında da, hem çocuklarda hem de erişkinlerde görülmektedir. Çocuklukta kas histopatolojisinde nekrozlu bir miyopati görülmesi nedeniyle kas distrofileri ile karıştırılabilir. Bu nedenle, kas distrofisi düşünülen ancak genetik incelemede sorumlu mutasyonu bulunamayan çocuk hastalarda anti-SRP ve -HMGCR antikorları mutlaka aranmalıdır. Erişkinlerde anti-SRP pozitif hastalar görece daha genç popülasyonda görülürken, anti-HMGCR pozitif hastalık daha yaşlılarda kendini göstermektedir. Kanserle ilişki açısından değerlendirildiğinde, anti-SRP pozitif hastaların kanserle ilişkili olmadığı, anti-HMGCR pozitif hastaların bağının zayıf olduğu, seronegatif hastaların kanserle birlikteliğinin ise sık olduğu bildirilmektedir. Hastalarda statin maruziyeti sıkça gözlenmektedir. Hastalık statin kullanımına başlandıktan yıllar sonra (ortalama 3 yıl) hatta ilaç kullanımı bırakıldıktan sonra bile ortaya çıkabilmektedir. Dolayısı ile statin kullanımı sırasında veya sonrasında gelişen proksimal kas zaafı ve düşmeyen CK düzeyi görüldüğünde hasta, diğer olasılıklar yanında, ONM olasılığı açısından da değerlendirilmelidir.
Yeni tanımlanan bir hastalık grubu olması nedeni ile ONM’de tedavi konusunda randomize kontrollü çalışmalar bulunmamaktadır. Ancak genel kanı, bu hasta grubunun daha güçlü ve daha uzun süreli immünsupresan tedaviye ihtiyacı olduğu yönündedir. Fizyoterapi yine, immünsupresif tedavinin ayrılmaz bir parçası olmalıdır.
Sporadik İnklüzyon Cisimcikli Miyozit (s-ICM)
Geleneksel olarak idyopatik inflamatuvar miyopatiler arasında sayılan inklüzyon cisimciği miyoziti, klinik olarak bu iki hastalıktan bazı farklar gösterir: Öncelikle hastalığın başlangıç yaşı ileri (50-60) olup erkeklerde daha sık görülür. Kas zaafının dağılımında distal kasların tutulumu erken ve belirgindir. En sık tutulan kaslar kuadriseps femoris ve el parmak fleksörleridir (Şekil 23). Böylece hastalık sık düşmeler veya ellerin ince hareketlerinde bozulma ile kendini gösterir. Bazen tutulum asimetriktir. Zaafa paralel gelişen atrofi önkol kaslarında belirgin olabilir. Yutma güçlüğü %30 hastada görülür. Bu klinik bulgular hastalığın sıklıkla amiyotrofik lateral skleroz (ALS) ile karıştırılmasına neden olur. Hastalık seyri, ancak 10 yılda tekerlekli iskemleye bağımlılık geliştirecek kadar yavaştır. Zaafa paralel gelişen atrofi önkol kaslarında belirgin olabilir. Yutma güçlüğü önemli bir sorun yaratabilir.
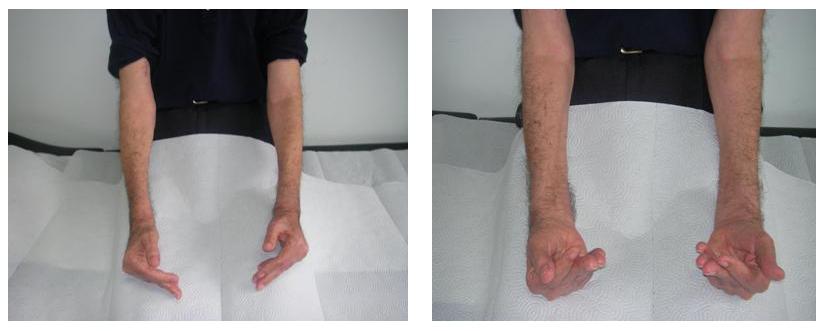
Şekil 23. Sporadik inklüzyon cisimcikli miyozitli bir hastada önkoldaki uzun parmak fleksör kaslarının zaaf ve atrofisi (hasta el parmaklarını kapatamamaktadır).
{kind=link}
Serum CK değeri en çok normalin 10 katına kadar yükselir. Elektromiyografi miyopatik, bazen buna eşlik eden çok belirgin nöropatik özellikler gösterebilir. Kas biyopsisi, polimiyozittekine benzer özelliklerin yanında dağınık atrofik lifler ve kırmızı kenarlı, b-amiloid (+) vakuoller gösterir. Bu vakuollerin gösterilemediği durumlarda tanı koymak mümkün olmaz, bazen birkaç kez kas biyopsisi yapmak gerekebilir. Elektron mikroskopik olarak kas lifleri içinde ve çekirdekte çift sarmallı, elektron-mikroskopik olarak “tübülofilamentöz” 16nm çaplı inklüzyonların birincil belirleyici olduğu bir inflamatuvar miyopatidir.
Patolojik görüntüleri ve soygeçmiş özelliklerini dikkate alarak İCM’ni üç alt gruba ayırmak mümkündür: a) Sporadik İCM, b) Familyal inflamatuvar İCM (ailede aynı hastalık var), c) Herediter inklüzyon cismi miyopatileri (inflamasyon yok).
Hastalık hiçbir immünsupresif tedaviye yanıt vermez. İVİg ve kortikosteroid uygulamalarına yanıt veren olgular anektodal olarak bildirilmiştir.
MİYASTENİA GRAVİS
“Myasthenia gravis”, Yunanca kas güçsüzlüğü anlamına gelen “myasthenia” ve Latince ağır, vahim anlamına gelen “gravis” kelimelerinden oluşur. MG, hareketle artan kas güçsüzlüğü (bitkinliği) ile karakterize, öncelikle okülobulber kasları tutan, çoğunlukla postsinaptik yerleşimli nikotinik asetilkolin reseptörlerinin (AChR) hedef alındığı otoimmün kökenli bir hastalıktır. Doğal seyrinde solunum krizi nedeniyle mortalitesi çok yüksek olan bu hastalık, uygun tedavi ile hastalar tamamen normal bir yaşam sürebileceklerinden, nöromüsküler hastalıklar arasında, hatta tüm nöroloji pratiğinde özel bir öneme sahiptir. Tedavi, hastalığın prognozunu tamamen değiştirdiğinden MG artık gravis ekini hak etmemektedir.
KISA TARİHÇE
Oxford’dan Thomas Willis tarafından 1672 yılında tarif edilen bir hastanın büyük olasılıkla MG olduğu 1903’de anlaşıldı. MG hakkında daha kapsamlı yazılar ancak 200 yıl kadar sonra yazılmaya başlandı. On dokuzuncu yüzyıl sonlarına gelindiğinde artık hastalığın tüm özellikleri ayrıntılı olarak açıklanmış, okülobulber belirtilerin ön planda olduğu, fluktuasyon ve remisyonların varlığı, çok ağır seyredebildiği ve mortalitenin yüksek olduğu kaydedilmişti. Okülobulber belirtileri nedeniyle beyinsapı tutulumu düşündüren MG’de yapılan otopsilerde beyinsapında lezyon olmadığı anlaşıldı. Çizgili kasa kısa aralıklarla verilen faradik stimülasyon sırasında kas yanıtının hacminin giderek azaldığı gösterildi ve ardışık sinir uyarımının temelleri atılmış oldu. Hastalığa ‘myasthenia gravis’ ismi 1899 yılında verildi.
Bin dokuz yüz otuzlu yılların başında antikolinesterazların dramatik bir düzelme sağladığı fark edildi, bu ilaç hastalığın tedavisinde önemli bir yer tuttu. İlk timektomi 1936 yılında timomalı bir hastada yapıldı. Daha sonraki yıllarda timektomi ile ilgili birçok çalışma, timektominin görece genç hastalarda erken yapıldığında yararlı olduğunu gösterdi. İki binli yılların başından itibaren videotorakoskopik yöntemle yapılan timektomi, ameliyatla ilgili mortalite ve morbiditenin çok azalmasını sağladı. Miyasteni prognozunu değiştiren en önemli ilerleme 1960’ların başlarında pozitif basınç ve hacim kontrollü asiste ventilasyon kullanılması, bronşiyal bakım ve pnömoninin iyi tedavisi oldu. Bin dokuz yüz yetmişlerde kortikosteroidlerin kullanılmaya başlanması da eklenince mortalite çok düşük seviyelere indi. Yine bu yıllarda azatioprin gerektiğinde verilmek üzere tedaviye eklendi. Plazma değişimi 1970’lerin sonlarında, İVİg ise 1980’lerde kullanılmaya başlandı.
Bindokuz yüz yetmişte AChR proteini izole edilip tanımlandıktan sonra ardarda gelen keşifler sayesinde 1976’de serumda anti-AChR antikoru saptandı. MG hastalarından alınan İgG’nin enjekte edildiği farelerde güçsüzlük gelişmesi ve postsinaptik AChR sayısında azalma olması, plazma değişimi yapılan hastaların düzelmesi ve MG hastalarının nöromüsküler kavşaklarında (NMK) İgG ve kompleman bulunması ile bu antikorların patolojik olduğu anlaşıldı. Anti-MuSK antikorlarının 2001 yılında saptanması önemli bir aşamaydı.
Türkiye’de MG’ye ilgi İstanbul Üniversitesi İstanbul Tıp Fakültesi emekli öğretim üyesi Prof. Dr. Coşkun Özdemir öncülüğünde 1950’li yıllarda Haseki Hastanesi Nöroloji Kliniği’nde başladı, 1960’ların sonlarından itibaren İstanbul Üniversitesi İstanbul Tıp Fakültesi MG tanı ve tedavisinin yerleşmesinde önemli bir rol oynadı.
EPİDEMİYOLOJİ
Birçok Avrupa ülkesi ve A.B.D.’de yapılan epidemiyolojik çalışmalarda elde edilen farklı değerler, MG’nin insidansının 3-30/milyon/yıl, en yüksek prevalansının ise 140-200/milyon arasında değiştiğini göstermektedir. Hastalığın prevalansının giderek arttığı dikkati çekmiştir, bunda hastalığın daha iyi tanınmasının, gelişen tanı/tedavi yöntemlerinin ve yoğun bakım şartları ile hastaların daha uzun yaşatılmasının etkisi mevcuttur. MG daha çok genç hastalığı olarak bilinirken yeni epidemiyolojik çalışmalar MG insidansının 50 yaş üzerinde de artmakta olduğunu ortaya çıkarmıştır. Buradaki artışın, genç ve çocuklardakine göre çok daha yüksek olmasının nedeni tartışmalı olmakla birlikte yaşlılarda da hastalığın daha iyi tanınmasının ya da çevresel faktörlerin etkisinin olabileceği düşünülmektedir.
Hastalığın en sık ortaya çıkış yaşı kadınlarda 20-30 yaş arası ve 50 yaş üstü olmak üzere bimodal, erkeklerde ise 50 yaş üstündedir. Başka bir deyişle, genç kadınlarda erkeklere göre daha sık, geç yaşlarda ise kadın ve erkekte aynı oranda görülür. MG’nin 1 yaşın altında görülmesi beklenmez, 1-10 yaşları arasında da nadirdir. MG tüm etnik grupları etkilemekle birlikte, prepubertal olguların oranının Uzak Doğu’da yüksekliği dikkati çekmektedir. Anti-MuSK pozitifliğinin, siyahi ırkta, özellikle genç kadınlarda, fazla olduğu bildirilmiştir. Anti-MuSK pozitifliği oranı Kuzey Avrupa ve Uzakdoğu’da daha düşüktür, ekvatora yaklaştıkça artmaktadır.
ETYOPATOGENEZ
MG, nöromüsküler kavşaktaki (NMK) proteinleri hedef alan antikorlar aracılığıyla oluşan otoimmün bir hastalıktır. Normal bir NMK’de iletim, presinaptik sinir terminalinden salınan asetilkolinin postsinaptik yerleşimli asetilkolin reseptörü (AChR) ile birleşmesi sonucu sağlanır. Antikorlar, bu iletimin gerçekleşmesini engelleyerek hastalığa neden olur.
MG’de en sık görülen ve en iyi bilinen anti-AChR antikorunu ele alırsak, antikor aracılı hastalıkları tanımlamada kullanılan tüm kriterlerin burada mevcut olduğunu görürüz: Hastaların birçoğunda antikor bulunur ve antikorun AChR ile birleştiği gösterilebilir. Hastalık pasif transfer ile hayvanlara geçirilebilir, antijen ile immünizasyon sonucu hayvanlarda hastalık oluşur ve serumda antikorların azaltılması (plazmaferez) hastalığın belirtilerini azaltır.
Anti-AChR antikorları çizgili kaslarda postsinaptik olarak yerleşmiş olan nikotinik AChR’ye karşı oluşur. Anti-AChR antikorları birkaç farklı yoldan reseptör sayısının azalmasına neden olur. Bunlardan en önemli iki yol kompleman aracılığıyla postsinaptik membran harabiyeti ve antikorların birleştirdiği (cross-linking) reseptörlerin daha hızlı harap olmasıdır. ACh birleşme yerlerinin fonksiyonel olarak blokajı söz konusu ise de bu daha az önemlidir.
Anti-AChR antikorları B lenfositleri tarafından yapılır, ancak otoimmün yanıtının oluşmasında yardımcı T lenfositlerinin de katkısı gerekmektedir (T hücresine bağımlı humoral immün yanıt). Patogenez hakkında ayrıntılı bilgi edinilmiş olmasına rağmen otoimmün yanıtı neyin tetiklediği tam olarak bilinmemektedir. MG’li hastaların yaklaşık %10-15’inde timoma, %70 kadarında timus hiperplazisi bulunması ve hastaların önemli bir kısmının timektomiden yararlanması dikkatleri timus üzerine çekmiştir. İmmünolojik olarak kendi antijenlerine yanıt veren “zararlı” T hücrelerinin elimine edildiği yerin timus olması, burada yüzeylerinde AChR taşıyan ‘miyoid’ hücrelerin bulunması, timik lenfositlerin anti-AChR antikoru sentez edebilmeleri, timomalarda AChR veya bazı subünitelerine benzeyen bir protein ekspresyonu olması gibi özellikler timusun önemli bir rolü olabileceğini düşündürmektedir. Ancak timusun olayın başlangıcından mı sorumlu olduğu yoksa immünolojik sürecin daha sonraki aşamalarında mı devreye girdiği sorusu halen yanıtlanamamıştır.
Son yıllarda MG’de anti-AChR antikoru dışında başka antikorlar da daha önce seronegatif olarak bilinen hastalarda saptanmıştır. Bunlar, kasa spesifik tirozin kinaz reseptörü (muscle specific kinase, MuSK), low density lipoprotein 4 (LRP4), agrin ve asetilkolinesterazın (AChE) bir komponenti olan kollajen Q (ColQ) proteinlerine karşı olan antikorlardır.
Bunların hemen hepsi agrin-MuSK yolağında bulunduğundan bu yolağı kısaca gözden geçirmekte yarar vardır. Agrin, presinaptik sinir ucundan salınır ve postsinaptik LRP4’e bağlanır, LRP4 de MuSK’a bağlanıp onu fosforilize ederek aktifleştirir. Docking protein 7 (DOK7) de MuSK’u aktive eden bir proteindir. Fosforile MuSK, rapsin aracılığıyla AChR’yi etkileyerek AChR’nin NMK’ta postsinaptik yarıklarda kümelenmesini sağlar. Bu kümelenmenin nöromüsküler bileşkedeki iletinin sağlıklı şekilde sürdürülmesi için gerekli olduğu bilinmektedir. ColQ proteininin de MuSK’u sinapsa bağlı tutarak MuSK’un fonksiyonu ile ilişkili olduğu düşünülmektedir.
Bu antikorlar arasında MG açısından en önemli olan anti-MuSK antikorlarıdır. Anti-MuSK MG’nin patogenezi henüz aydınlatılabilmiş değildir. Anti-AChR pozitif hastalarda antikorlar İgG1 ve İgG3 iken, buradaki antikorların esas olarak İgG4 olduğu görülmüştür. Anti-MuSK antikoru pozitif hastalarda anti-AChR pozitiflerden farklı olarak timus patolojilerinin gözlenmesi oldukça nadirdir.
Herhangi bir antikorun gösterilemediği seronegatif hastalarda, fare deneylerinde pasif transfer ile hastalığın geçirilebilmesi, seronegatif MG’li annelerin bebeklerinde nadir de olsa neonatal MG gözlenmesi ve plazmafereze yanıt olması otoantikorların varlığına kanıttır. Seronegatif hastalarda da, anti-AChR antikor pozitiflere göre daha az oranda olmakla birlikte, timusta hiperplazi bulunur. Timoma ise tek tük bildiri niteliğinde olup son derece nadirdir.
MG’de genetik bir yatkınlığın varlığını düşündüren gözlemler vardır. MG’li bir kişinin aile bireylerinde MG gelişme olasılığı diğer kişilere göre biraz daha yüksek bulunmuştur. Bizim 2000 üzerinde olan hastalarımız arasında anti-AChR pozitif MG olan çok az sayıda kız kardeşler ve ebeveyn-çocuk bulunmaktadır. HLA ile ilişki, genetik bir predispozisyonun söz konusu olduğunu desteklemektedir. Timomasız erken başlangıçlı MG’de, özellikle kadınlarda, A1, B8, DR3 ile ilişki saptanmıştır. Timomasız geç başlangıçlı MG’de ise daha zayıf bir ilişki B7 ve DR2 ile bulunmuştur. Hollanda, İtalya ve Türkiye’den yapılan çalışmalar, anti-MuSK pozitif MG ile HLA-DR14-DQ5 arasında kuvvetli bir ilişki olduğunu göstermiştir.
KLİNİK
Belirtiler
Hastalığın en önemli özelliği yorulmakla artan ve dinlenmekle en azından kısmen düzelen kas güçsüzlüğüdür. Hastalar sabahları düzeldiklerini, belirtilerin akşama doğru ya da yorulunca arttığını ifade ederler (Şekil 24). Hastalık remisyon ve alevlenmelerle seyreder. Remisyonlar birkaç günden birkaç yıla kadar sürebilir.
Hastalık çoğu zaman oküler belirtilerle, en sık da asimetrik ptozla başlar. Buna çift görme eşlik eder ya da birkaç gün veya hafta içinde eklenir. Hastaların çoğunda oküler belirtilere kısa zamanda bulber (orofarengeal) kaslara ve ekstremite kaslarına ait belirtiler eklenir. Bulber belirtiler konuşma, yutma ve çiğneme güçlüğü, hastalığın en ağır halinde de solunum zorluğudur. Ekstremite kaslarındaki belirtiler yokuş-merdiven çıkarken zorlanma, kollarını yukarı kaldırma zorluğu ya da bir iki el parmağını bir süre kaldıramama şeklinde kendini gösterir. Hastalık oküler kaslarda başlayabildiği gibi bulber kaslarda da başlayabilir. Ekstremite kaslarına ait belirtilerle başlaması nadirdir ve daha çok gençlerde görülür. Yine aynı şekilde diğer kaslara ait belirtiler başlangıç belirtilerine kısa zamanda eklenir.

Şekil 24. Miyastenia gravis’li hastada sağdaki ptozun dinlendikten sonra düzelmesi
Bulgular
Ptoz unilateral veya bilateral olabilir, bilateral olduğu zaman asimetrik olması dikkati çeker. Ekstraoküler kas tutulumu genellikle bilateraldir. Ekstraoküler kas tutulumu belli bir paterne uymaz, yani güçsüz kasların dağılımının belli bir sinir innervasyonunu düşündürmesi, ya da beyinsapı hastalıklarında görülen internükleer oftalmopleji veya konjuge bakış kusuru tarzında olması olağan değildir. Ancak nadiren bu tarzda tutulmalar da görüldüğünden her türlü göz hareket bozukluğunda, özellikle fluktuasyon varsa, MG’yi akla getirmek gerekir. İç rektuslar sık etkilenen kaslardandır (Şekil 25). MG’de pupillalar tutulmaz.

Şekil 25. Miyastenik bir hastada sağ göz kapağında ptoz, göz hareketlerinde içe-aşağıya-yukarıya bakış parezisi (sağ 3. sinir parezisi gibi, ancak solda da ptoz var) izlenmekte (sol taraftaki resimler). Sağdaki resimlerde ise antikolinesteraz sonrası düzelme göze çarpmakta.
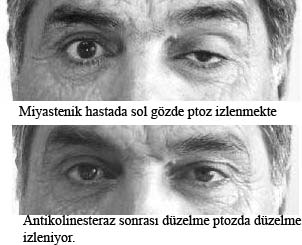{kind=link}
Orbikularis okuli kasının güçsüzlüğü MG’de en sabit bulgulardan biridir. Remisyonda olan hastalarda dahi tek muayene bulgusu bu olabilir. Bu kasta güçsüzlük olmayan jeneralize miyastenik hasta çok nadir görüldüğünden orbikularis okulilerin kuvvetli olması MG tanısından şüphe doğurabilir. Genellikle bilateral olan yüz kaslarının güçsüzlüğü hastalarda ağzın horizontal yerine vertikal yönde kayması şeklinde tipik bir gülüşe neden olur (Şekil 26). Bulber tutulumu olanlarda dil, yumuşak damak, masseter ve boyun kaslarında güçsüzlük görülür. Nazone konuşma dikkat çekicidir.

Şekil 26. Resmin solunda izlenen miyastenik gülüşün (vertikal gülüş) resmin sağ tarafında antikolinesteraz sonrasında düzeldiği izlenmekte
Ekstremite kaslarındaki güçsüzlük kollarda proksimal ve distal, bacaklarda ise daha çok proksimal kasların tutulması şeklindedir. En çok güçsüzlük bulunan kaslar triseps, el parmak ekstansörleri ve iliopsoastır. Deltoid ve hamstringler de sık tutulan kaslardandır. Kuadriseps femoris ve tibialis anterior kasları genellikle normal bulunur. Ancak iliopsoasların çok zayıflamış olduğu durumlarda tibialis anterior kasında güçsüzlük saptanabilir. Tibialis anteriorun ön planda etkilendiği olgular son derece nadirdir.
Kaslardaki güçsüzlüğü ortaya çıkarmak için hastalara yukarıda anlatılan bitkinliği ortaya çıkarıcı testler (bir süre yukarıya bakmak ya da kolları havada tutmak gibi) yaptırmak gerekebilir. Muayene bulguları motor alana sınırlıdır. Kemik veter refleksleri normaldir, hatta canlı bulunabilir. Duyu muayenesi normaldir.
Seyir
Hastalığın seyri genellikle ilerleyicidir. Hastalığın başlangıcında kısa süreli, nadiren uzun süreli remisyonlar olabilir, ancak hastalık çoğunlukla yeniden kendini gösterir. İlk yıllar miyasteninin en ağır seyrettiği dönemdir. Ağır olgularda yemekleri yutmak olanaksız hale gelir, içilen sular burundan gelir, ağzı kapamak için hastalar çeneyi elleriyle desteklerler, beslenme ancak nazogastrik sonda yoluyla olur (Şekil 27). Hastaların bir kısmında, genellikle ilk 3 yıl içinde, ağır solunum yetmezliğiyle giden kriz görülür. Yıllar içinde miyastenik yakınmalar sönerek azalır ama hastalık dalgalanmalarla sürer. İnfeksiyonlar ve ağır stres hastalığın gidişini genellikle olumsuz yönde etkiler. Gebelik ve loğusa dönemleri hastalığın seyrini değiştiren faktörlerdir, ancak bu değişimin hangi yönde olacağını önceden kestirebilmek mümkün değildir. Menstruasyon dönemlerinde de miyastenik yakınmalar artabilir.

Şekil 27. Çenesini kapalı tutamayan ve nazogastrik sonda ile beslenen miyastenik bir hasta.
MG ALT TİPLERİ
MG’yi farklı alt tipleri olan bir hastalık olarak düşünmek, patogenez, tanı ve tedaviye yaklaşım açısından yararlıdır (Tablo 11). En klasik şekliyle MG, erken başlangıçlı, jeneralize, anti-AChR pozitif ve timomasızdır.
Tablo 11. Miyastenia gravis’in alt tipleri.
|
Tutulan kaslara göre · Jeneralize · Oküler |
|
Başlangıç yaşına göre · Erken (50 yaş öncesi) · Geç (50 yaş sonrası) |
|
Antikora göre · Anti-AChR pozitif · Anti-MuSK pozitif · Seronegatif |
|
Timoma varlığına göre · Timomasız · Timomalı |
Tutulan kaslara göre alt tipler
Oküler belirtiler olsun veya olmasın, bulber ve ekstremite kaslarında güçsüzlük var olduğunda “jeneralize MG”den söz edilir. MG hastalarının büyük bir kısmında hastalık jeneralize olarak seyreder. Jeneralize MG’ler arasında son derece nadir olan bir tip ise sadece ekstremite kaslarının tutulduğu “kavşak tipi MG”dir.
MG hastalarının yaklaşık üçte ikisinde başlangıç ptoz ve çift görme gibi oküler belirtilerledir, bunların küçük bir bölümünde (yaklaşık %10-20) hastalık oküler kaslara sınırlı kalır. İki yıl sadece oküler belirtilerle seyreden MG’nin artık oküler dışı kaslarda belirti verme olasılığı azdır ve “oküler MG”den söz edilir. Oküler MG, jeneralize MG’ye göre daha hafiftir, ancak ptoz ve çift görme günlük hayatı çok etkileyebilir. Prepubertal hastalarda oküler MG sıktır. Oküler MG’nin sık görüldüğü diğer bir grup ise yaşlı erkeklerdir.
Başlangıç yaşına göre alt tipler
Hastalık 40 veya 50 yaşından önce başlarsa “erken başlangıçlı MG” (early-onset MG, EOMG), daha sonra başlarsa “geç başlangıçlı MG”den (late-onset MG, LOMG) söz edilir. Artık sınır 50 yaş olarak kabul edilmektedir.
EOMG’de kadın oranı daha yüksektir. Timik hiperplazi görülme olasılığı daha yüksektir. EOMG içinde puberteden önce başlayan ‘prepubertal MG’ kendine has özellikleri ile ayrılabilir. Bu grup beyaz ırkta tüm MG hastalarının %10’undan azdır, Uzak Doğu’da ise oldukça yüksektir. Bu hastalarda uzun spontan remisyonlar gözlenebilir, hastalık oküler kalabilir veya jeneralizasyon yıllar sonra olabilir. Prepubertal MG’de kız-erkek çocuğu oranı birbirine eşittir.
LOMG ise kadın ve erkeklerde eşit oranda görülür veya erkeklerde hafifçe daha yüksektir. Timus çoğunda involüsyona uğramıştır, ancak timoma oranı daha yüksektir.
Antikora göre alt tipler
Alt tipler, klinik olarak anti-AChR antikor pozitif MG (AChR MG), anti-MuSK antikor pozitif MG (MuSK MG) ve bu iki antikor tipini taşımayan seronegatif MG olarak ayrılır. Diğer antikorları taşıyan hastalarda bu antikorlar birçok defa tek başına bulunmadığından ve belli bir fenotip göstermediklerinden bunların bir alt tip oluşturduğu söylenemez.
AChR MG
Jeneralize MG’li hastaların %80 kadarının serumunda anti-AChR antikorları bulunur. Oküler MG’de pozitiflik oranı sadece %50’dir.
MuSK MG
MG hastalarının %7-8’inde anti-MuSK antikorları bulunur. Anti-MuSK ve anti-AChR antikorlarının birlikteliği çok nadirdir. MuSK MG, daha çok kadınlarda görülür, çocuklarda ve çok ileri yaşlarda enderdir. Hastaların neredeyse hepsi jeneralize seyreder, bulber kaslar ve boyun ekstansörleri sıklıkla erken tutulur. Bazı hastalarda miyasteni oküler kaslarda güçsüzlükle başlayabilirse de oküler kaslara uzun süre sınırlı kalması çok nadirdir. Genellikle miyastenik kriz oranının yüksek olduğu ağır okülobulber belirtilerle seyreder, ancak küçük bir bölümü hafif belirtilerle seyredebilir. Ağır seyreder ve tedavi etkin olmazsa dilde atrofi gelişebilir, ancak dil atrofisi MuSK MG’ye özgü değildir. Kol ve bacaklarda güçsüzlük hiç olmayabilir ya da geri plandadır. Timus ile ilişkisi gösterilememiştir.
Seronegatif MG
Hiçbir antikor saptanmamış olan seronegatif MG, hafif belirtilerle seyreden hastaları olduğu gibi kriz gelişen ağır hastaları da içerebilir. Oküler MG’de jeneralizeye göre seronegatiflik oranı yüksektir.
Diğer antikorlar
Yakın zamanda seronegatif hastalarda LRP4, agrin, COLQ antikorları saptanmıştır, ancak bu antikorların klinik önemi hala tartışmalıdır. Anti-LRP4 antikorları, hem anti-MuSK hem de anti-AChR pozitif hastalarda görülebildiği gibi ALS gibi başka hastalıklarda da saptanmıştır. Fenotipin aydınlatılması için daha geniş çalışmalara gerek olmakla birlikte, oküler veya hafif jeneralize hastalarda görüldüğü bildirilmiştir.
Timoma varlığına göre alt tipler
MG’li hastaların %10-15’inde timoma bulunur. Timomalı hastaların ise yalnızca %30 kadarında MG gelişir, bazılarında anti-AChR antikor pozitif olsa da MG olmayabilir. Çoğu selim olan bu tümör nadiren habis olur ve çevreye yayılır, ancak uzağa metastaz çok nadiren görülür. Hastaların çoğunda belirtiler jeneralizedir ve MG ağır seyredebilir. Oküler MG’de timoma nadirdir. Timomalı hastalarda MG, bildirilmiş tek tük olgular dışında hemen daima anti-AChR antikoru pozitiftir. Anti-AChR antikoru negatif bir hastada timoma tanısı koyarken çok dikkatli davranmalı, yanılgı olabileceği göz önünde bulundurulmalıdır. Timomalı hastaların bir bölümünde titin ve ryanodin reseptör antikorları gözlenebilir ancak bunlar timomaya özgü değildir, timomasız yaşlı hastaların bir kısmında da pozitif olabilirler.
TANI
Tanıda klinik bulgular ön plandadır. Okülobulber belirtilerle başlayan ve gün içinde fluktuasyon gösteren bir klinik tablo kuvvetle MG’yi düşündürür. Öyküde spontan remisyonların varlığı bu tanıyı destekler. Tanıya yardımcı olabilecek klinik ve laboratuvar testler aşağıda sıralanmıştır.
Yorma Testi
Ptozu olan hastalarda, hastayı 1 dakika boyunca yukarı baktırıp yorarak ptozun artıp artmadığı gözlenir. Hasta 100’e kadar yüksek sesle saydırılıp sesin nazone hale gelip gelmediği izlenebilir.
Buz Testi
Yine ptozu olan hastalara ptozun olduğu tarafa buz torbası uygulanır, eğer ptozda belirgin bir düzelme olursa test pozitif kabul edilir. Ptozdaki bu geçici düzelme AChE enziminin düşük ısı ile yavaşlamasına ve kavşaktaki asetilkolin miktarının artmasına bağlıdır.
Antikolinesterazlara yanıt
Antikolinesterazlar (AKE) ile kas gücünde objektif ve net düzelme gözlenerek tanı konur (Şekil 25, 26, 28, 29). Tanıda, kısa etkili edrofonyum klorid (Tensilon) veya daha uzun etkili neostigmin bromid (Prostigmin) kullanılır. Edrofonyum’un etkisi birkaç saniyede başlar ve birkaç dakikada sona erer. 10 mg’lık ampulleri olan edrofonyum klorid’in önce 2 mg’ını intravenöz olarak verip 1 dakika cevabı beklemek, yanıt alınmazsa gerisini vermek gerekir. Kas dışı müskarinik etkileri gerektiğinde antagonize etmek için atropin hazır bulundurulur. Neostigmin’in etkisi ise 15-20 dakika kadar sonra başlar ve 2 saat kadar sürer. İki ampul (1 mg) neostigmin bromid, 0,4 mg atropin ile birlikte intramüsküler olarak yapılır. Bu testlerin pozitif sayılabilmesi için cevabın çok iyi olması gerekir, belli-belirsiz bir düzelme önemli değildir. Motor nöron hastalığı ve poliyomiyelit gibi bazı hastalıklarda da bunlar hafif pozitif olabilir ama genellikle yanıt MG’de olduğu gibi net ve tekrarlanabilir değildir. Özellikle yaşlı hastalarda parenteral AKE yerine 2 draje (120 mg) piridostigmin bromid (Mestinon) vererek daha geç çıkacak olan cevabı beklemek daha tedbirli bir tutum olabilir.
Şekil 28. Miyastenik bir hastada dil güçsüzlüğü ve antikolinesteraz sonrası düzelme. Hasta dilini ancak ağız komisürüne kadar götürebilmekte, antikolinesteraz sonrası diliyle yanağını şişirebilmektedir.

Şekil 29. Miyastenik bir hastada kitabı kaldıramayacak kadar kol güçsüzlüğü ve antikolinesteraz sonrası düzelme.
Antikorlar
Serolojik testler tanıda çok yardımcı ve genelde güvenilirdir. MG şüphesi olan bir hastada anti-AChR veya anti-MuSK antikorlarının bulunması tanı koydurucudur. Bazı hastalarda antikor bir kez negatif olduğu halde daha sonra pozitif bulunabilir. Özellikle prepubertal başlangıçlı hastalarda serokonversiyon bildirilmiştir. Bu nedenle testi tekrarlamak gerekebilir. Sonucun sınırda veya çok hafif pozitif olduğu hastalarda da MG tanısı koyarken dikkatli olunmalıdır, kliniğin MG ile uyumlu olup olmadığı gözden geçirilmelidir.
Ardışık sinir uyarım testleri ve tek lif elektromiyografisi
Sinire supramaksimal olarak uygulanan düşük frekanslı (2 Hz veya 3 Hz) ardışık uyarım ile birbirini izleyen bileşik kas aksiyon potansiyellerinin amplitüdünde tipik bir düşme (miyastenik dekrement) kaydedilir.
Klasik olarak 9-10 kez üst üste yapılan stimülasyon sırasında dördüncü potansiyel amplitüdünde ilk potansiyel amplitüdüne göre %10’dan fazla olduğunda anlamlı olarak kabul edilen bu düşüşün MG’de belli birkaç paterni vardır (Ayrıca bakınız: Elektromiyografi). Amplitüd düşmesinin, ikinci potansiyelde, ender olarak da üçüncü potansiyelde ortaya çıkması gerekir. İkinci potansiyelde düştükten sonra amplitüd ya aynı kalır ya da dördüncü veya beşinci potansiyele kadar düşmeye devam eder. Artık beşinci potansiyelden sonra düşme beklenmez, hatta giderek amplitüd artabilir. Beşinci potansiyelden sonra da devam eden sürekli düşüş MG’de görülen bir patern değildir. Bu testi distal kaslarda yapmak daha kolaydır, ancak proksimal kaslar ve yüz kaslarında miyastenik dekrementi ortaya çıkarma olasılığı daha fazladır. En verimli seçim, güçsüzlüğün olduğu alanda bir kasın (örneğin, ekstremite güçsüzlüğü olanda m. abductor digiti minimi veya trapezius, bulber güçsüzlüğü olanda m. nasalis gibi) kullanılmasıdır. Ayrıntılı bir inceleme ile remisyonda olmayan ve çok hafif olmayan jeneralize MG’lilerin % 95 kadarında dekrement bulunabilir. Ardışık sinir uyarım testlerinde en büyük tehlike, artefakt olarak ortaya çıkan dekrementi gerçek sanmak ve yanlış olarak MG tanısı koymaktır. Bu tanı bir kez konulduğunda sonra değiştirmek zor olabilir. Bu bakımdan incelenecek kası çok iyi stabilize etmek, dekrementi bir değil birkaç kez elde ederek emin olmak ve dekrement paternine dikkat etmek çok önemlidir.
Tek lif EMG ile ‘jitter’ ölçümü ve blok aranması MG tanısında kullanılan bütün testlerin en duyarlı olanıdır. Hemen bütün miyasteniklerde artmış ‘jitter’ bulunur, hele güçsüz bir kasta ‘jitter’in normal bulunması MG tanısını neredeyse dışlar. Tek lif EMG, ardışık sinir uyarımı ile dekrement bulma olasılığının oldukça düşük olduğu oküler MG ve çok az bulguları olan hafif jeneralize olgularda özellikle çok yararlıdır. Tek lif EMG’nin duyarlılığı çok yüksek iken özgünlüğü ardışık sinir uyarımına göre daha düşüktür.
MG tanısında çok önemli bir yeri olan elektrofizyolojik incelemeler, antikor öncesi dönemlere göre değerini biraz kaybetmiştir. Anti-AChR veya anti-MuSK antikor sonucu pozitif ise tanı konmuş olur. Seronegatif hastalarda ise elektrofizyolojik testler tanıda çok yardımcıdır, NMK’ta bir patoloji olduğunu destekleyen yegane testler bunlar olduğundan mutlaka dikkatle yapılmaları gerekir.
Tanıda en büyük sorun hiçbir antikor saptanmayan seronegatif oküler MG’li hastalardadır. Bu hastalarda bazen ptoz ve göz hareket bozukluğu AKE’ye yanıt vermez, elektrofizyolojik incelemeler de yeterince destekleyici olmayabilir. Öykü, MG’yi kuvvetle düşündürüyorsa bu tanıdan kesinlikle vazgeçmemek gerekir. Başka olası tanılar elendikten sonra hala MG düşünülüyorsa kortikosteroid verilerek alınan yanıt ile MG tanısını desteklemek mümkündür.
Timusun radyolojik incelemesi
Timusun radyolojik incelemesi MG tanısı konduktan sonra timoma olup olmadığını araştırmak üzere yapılır. Mediastenin bilgisayarlı tomografisi ile hemen tüm timomalar saptanır (Şekil 30). Kontrast maddeyi, MG’li hastayı kötüleştirme olasılığını göz önüne alarak, özellikle bulber tutulumu olan hastalarda vermekten kaçınmak gerekir. Genellikle manyetik rezonans görüntülemesi yapılmasına gerek kalmaz.

Şekil 30. Mediasten bilgisayarlı tomografisi: Normal timus loju (A) ve timoma (B).
Tablo 12. MG’de tanı ve incelemeler.
|
Tanı |
Öykü (okülobulber güçsüzlük, fluktuasyon, remisyon) Antikolinesterazlara (parenteral veya oral) net olumlu yanıt
|
|
Tanıyı kesinleştiren veya destekleyen incelemeler
|
Anti-AChR antikor tayini Anti-AChR antikoru negatifse anti-MuSK antikor tayini (özellikle jeneralize olgularda) Ardışık sinir uyarımı ve/veya tek lif EMG (özellikle seronegatif olgularda)
|
AYIRICI TANI
Okülobulber yakınma ve bulguların birlikte olduğu durumlarda MG ile en çok karışabilecek olan beyinsapını tutan hastalıklardır (tümörler, vasküler ve demiyelinizan hastalıklar). Beyinsapı hastalıkları fluktuasyon olmaması, birinci motor nöron bulguları ve duysal bulguların diğerlerine eşlik etmesi ile MG’den ayırt edilir.
Sadece ptoz olduğunda Horner sendromu ve blefarospazm akla gelir (Tablo 13). Horner sendromunda ptoz unilateral ve hafiftir, genellikle miyoz vardır, pupilla karanlıkta genişleyemez. Blefarospazmda gözkapaklarında kasılma hissi olması tipiktir, bu kasılmayla birlikte gözler kısılır ve bazen bilateral olabilen ptotik bir görünüm verebilir (Şekil 31).
Şekil 31. Blefarospazm (ptoz ile karışabilir).
Ptoza göz hareket bozukluğu eklendiğinde en çok nöromüsküler hastalıklar ile ayırıcı tanı gündeme gelir ve göz hareket bozukluğunun ön planda ya da geri planda olmasına göre farklı hastalıklar düşünülür. Göz hareket bozukluğunun geri planda olduğu miyopatilerden miyotonik distrofi, otozomal dominant geçiş, tipik klinik özellikleri ve EMG bulgularıyla kolayca ayırt edilir, genetik incelemelerle tanı kesinleştirilir. Geç yaşlarda ortaya çıkan okülofaringeal müsküler distrofide genellikle sadece bilateral ptoz vardır, yutma güçlüğü başlangıçta görülmez, göz hareket bozukluğu ise geç dönemde ortaya çıkabilir. Otozomal dominant olması, MG’nin klinik ve laboratuvar özelliklerinin bulunmaması ve genetik incelemeler ile tanı konur. Konjenital miyopatiler genellikle bebeklikte başlar, kas biyopsisi ve genetik incelemeler tanı koydurucudur. Tüm bu miyopatilerde ptoz genellikle bilateral ve oldukça simetriktir. Miyastenide ptoz simetrik hale gelebilir ama öyküdeki asimetrik başlangıç dikkat çekicidir. Nöromüsküler hastalıklar dışında unutulmaması gereken bir ayırıcı tanı da Graves oftalmopatisi ile yapılmalıdır.
Bilateral ptoza ek olarak gözler orta hattan çok az oynayacak derecede ağır ve bilateral göz hareket bozukluğu olan hastada ayırıcı tanıya konjenital miyastenik sendromlar (KMS) ve mitokondriyal hastalıklar (progresif eksternal oftalmopleji) girer. Bunlarda da ptoz bilateraldir. Bu hastalıklarda MG’nin aksine çift görme yoktur veya KMS’lerde olduğu gibi çok azdır. KMS’ler genellikle bebeklik döneminde başlar, MG ise ilk yıldan sonra başlar. Öykü ve genetik incelemelerle tanı konulabilir. Mitokondriyal miyopatilerde kas biyopsisi ve genetik incelemeler ile kesin tanı konur. Nöropatiler de ayırıcı tanıda düşünülmelidir. Akut dönemde Miller Fisher sendromu ve oküler bulguların nadir olarak ön planda olduğu Guillain-Barré sendromu akla gelmelidir. Kemik veter reflekslerinin kaybolması ve EMG bulguları bu hastalıkların tanısında yardımcı olur.
Oküler kaslara sınırlı olan MG, güçsüzlüğün belli bir paterne uymayışı ile izole kranial sinir felçlerinden ayırt edilir. İzole üçüncü sinir parezisine görünürde tıpatıp uyan bir hastamızda (bir gözde ptoz ve içe-yukarı-aşağı bakış parezisi), pupillanın normal olması ve diğer gözde hafif bir ptoz olması ile MG’den şüphelenildi ve oküler MG tanısı konuldu (Şekil 25a). Pupillanın tutulmadığı diyabetik 3. sinir parezisi unilateral ve ağrılıdır. Yaşlı hastalarda iskemik 3. sinir parezisi akla gelmelidir.
Sadece bulber yakınma ve bulgular olduğunda düşünülmesi gereken bir hastalık motor nöron hastalığının bulber formudur. Motor nöron hastalığındaki anartriye kadar varabilen ve fluktuasyon göstermeyen, birinci ve ikinci motor nöronun birlikte tutulumu sonucu ortaya çıkan tipik konuşma şekli MG’nin nazone konuşmasından çok farklıdır. Motor nöron hastalığında bazen hafif bir fluktuasyon öyküsü alınabilir. MuSK MG’deki konuşma bozukluğu bazen motor nöron hastalığındakini andırabilir, bu bakımdan çok dikkatli olunmalıdır.
Sadece ekstremite güçsüzlüğü ile giden MG son derece nadirdir. Ekstremite güçsüzlüğüyle başlangıç daha çok genç hastalarda görüldüğünden müsküler distrofiler ile karışır ve MG hastaları yanlış tanı alabilir. Kas hastalığı düşünülen her olguda fluktuasyon ve remisyona ait ipuçları yakalamaya çalışılmalı ve tedavi edilebilen bu hastalık gözden kaçırılmamalıdır.
Her akut yerleşen miyastenik tabloda botulizm akla gelmelidir. Bacaklarda güçsüzlüğün ön planda olduğu hastalarda Lambert Eaton miyastenik sendromu düşünülmelidir.
MG ile en sık karışan hastalık depresyondur. MG’li hastaların öyküsünden, birçoğunun yıllarca depresyon tanısıyla izlendiği anlaşılır. MG’li hastaların basit bir yorgunluktan yakınmadıkları, bunun gerçek bir güçsüzlük olduğu, sabah-akşam farkı gibi çok net özellikleri olduğu unutulmamalıdır.
Tablo 13. Ptoz ve/veya oftalmoparezi belirtileri olan miyastenia gravis ayırıcı tanı
|
Oftalmoparezisiz ptozun olduğu hastalıklar/sendromlar · Horner sendromu* · Blefarospazm · Konjenital ptoz |
|
Ptozun ön planda, oftalmoparezinin geri planda olduğu hastalıklar/sendromlar · Nöromüsküler hastalıklar o Miyotonik distrofi o Okülofaringeal distrofi o Konjenital miyopati o Lambert-Eaton miyastenik sendromu · Diğer o Graves oftalmopatisi
|
|
Ptozla birlikte belirgin oftalmoparezinin olduğu hastalıklar/sendromlar · Nöromüsküler hastalıklar o Konjenital miyastenik sendromlar o Botulizm* o Mitokondriyal miyopati o Miller-Fisher sendromu* o Guillain-Barré sendromu* · Diğer o Diyabetik 3. sinir parezisi o İskemik 3. sinir parezisi o İntrakranial yer kaplayıcı lezyonlar (tümör, anevrizma)*
|
*Pupilla anormallikleri eşlik eder ya da edebilir.
TEDAVİ
Antikolinesterazlar, immünsupresif/immünmodülatör tedavi ve pozitif basınçlı solunum desteği, eskiden ağır mortalitesi olan MG’nin prognozunu tamamen değiştirmiş, hastalara normal bir yaşam sürme şansını vermiştir. İlaçlardan AKE’ler semptomatik yarar sağlar, diğerlerinin ise hastalığın immünolojik kökenine yönelik olduklarından tedavi edici etkileri vardır.
Hastalığın fluktuasyon ve spontan remisyonlarla seyretmesi ve ağırlık derecesinin her hastada farklı olması tedavi sonuçlarını değerlendirmeyi güçleştirmektedir. Kontrollü klinik çalışmaların zorluğu ve azlığı da göz önüne alınırsa, ilaçların hangi kombinasyon halinde ve hangi dozda kullanılacağı hakkında kesin formüller öne sürmenin mümkün olmadığı kolayca anlaşılır. Tedavinin doktora ve hastaya göre bazı değişiklikler göstermesi kaçınılmazdır. Ancak uyulması gereken bazı prensiplerin olduğunu da unutmamak gerekir.
Antikolinesterazlar
AKE, asetilkolinin yıkılmasını engelleyerek sinaptik aralıkta daha uzun süre kalmasını sağlar. Günlük tedavide en sık kullanılan piridostigmin bromid’in (Mestinon) 60 mg’lık oral formudur. Etkisi 15-30 dakikada başlar, 1-2 saatte maksimuma erişir ve 3-4 saat veya daha fazla sürer. Oral olarak kullanılabilen ve piridostigmin bromid’den çok az farkları olan diğer preparatlar Türkiye’de bulunmayan neostigmin bromid (Prostigmin) ve ambenonium kloriddir (Mytelase).
AKE dozunu ayarlamak oldukça güçtür. Hastaya AKE tedavisinden neler beklenildiğinin anlatılması ve hastayla çok yakın bir işbirliğinin sağlanması şarttır. AKE’yi belirtiler ortaya çıkmaya başladığı zaman almak gerekir. Beklenen, ilacı aldıktan 1-2 saat sonra belirtilerin azalması veya kaybolması, tekrar ilaç saati geldiğinde belirtilerin hafiften başlamış olmasıdır. Bazı hastalarda bir grup semptom kaybolurken başka bir grup hiç etkilenmeyebilir. Bu durumda hastaya hayati fonksiyonları etkileyecek semptomlara (bulber) dikkat etmesi öğretilmelidir. Hastalar göz bulgularına çok önem verdiklerinden sıklıkla göz düzelmiyorsa ilacın fazla işe yaramadığı fikrine varırlar. Hastaya şimdilik gözle ilgili yakınmalara aldırmamaları söylenmeli, bunların er ya da geç düzeleceğini anlatarak hasta rahatlatılmalıdır.
Başlangıçta günde 2-4 kez 60 mg’lık drajelerden (120-240 mg) verilir. Hastalığın ağırlığına göre 3 saatte bir 60 mg’a kadar (480 mg) artırmak gerekebilir. Ağır hastalarda bu doz dikkatle arttırılabilir. Günde sekizden fazla alması gereken hastaları hastane şartlarında izlemek daha uygundur. Yüksek dozlarda aynen miyasteniye benzeyen kolinerjik bir tablo ortaya çıkabilir. Hastalığın ağır dönemlerinde AKE’yi olabildiğince 24 saat içine eşit aralıklara yaymak gerekir. Hastalık kontrol altına alındığında sadece gündüz verilebilir ve aralıklar da daha serbest olarak ayarlanabilir. İmmünsupresif başlanmışsa amaç, immünsupresif ajan etkisini gösterdikçe AKE’yi azaltmak ve hasta remisyona girdiğinde kesmektir. Hasta bozulduğunda AKE’ye yeniden başlanabilir. Görüldüğü gibi, AKE dozunun hastalığın ağırlığına, günlük aktivite derecesine, kullanılan diğer ilaçlara bağlı olarak dinamik olarak değiştirilmesi gerekmektedir.
MuSK MG’li hastaların çoğunda piridostigmin işe yaramadığı gibi aşırı fasikülasyon ve hipersalivasyondan dolayı tolere edilemeyebilir. Bu hastalarda piridostigmin sürekli kullanımında işe yaramadığı halde test dozu verildiğinde ileri bir düzelme görülebilir. Bazı hastalarda piridostigminin düşük dozlarda yarayabileceğini de gözden kaçırmamalıdır.
Timektomi
Timomalı hastalarda mutlaka timomektomi yapılmalıdır. Timomalar çabuk yayılabildiğinden ve yayılma hızını belirlemek mümkün olmadığından olabildiğince erken çıkarılmalıdır. Timoma çıkarıldıktan sonra miyasteninin düzelmesi beklenmez.
Timektomi, timomasız hastalarda miyasteninin düzelmesini sağlayabileceğinden çok önemli bir yer tutar. Jeneralize AChR MG’li genç hastada tedavinin temel basamaklarından biridir. Seronegatif hastalarda da timektomi işe yarayabilir. Elli yaşın biraz üzerindeki hastalarda ise karar, ameliyatın riskleri ve timektominin o yaştaki etkisi tartılarak verilmelidir. İleri yaşlarda ve MuSK MG’de timektomi yapılması yarar sağlamamaktadır. Pür oküler miyasteniklerde timektomi genellikle önerilmez.
Timektominin yararlı olduğu 1950’lerden beri bilinmektedir. Timektomiden sonra hastaların üçte bir kadarının remisyona girdiği, en az üçte birinde miyasteninin minimal belirtilerle veya daha iyi gittiği, %10 kadarında ise miyasteninin aynı kaldığı gözlenmiştir. Ancak iyi yapılmamış çalışmalar, ya da timomalı hastaların da serilere eklenmesi akılları karıştırmıştır. Anti-AChR pozitif hastalarda, özellikle erken dönemde, timektominin yararı yakın zamanda yapılmış bir bilimsel çalışma ile de gösterilmiştir. Bu yarar hemen ortaya çıkmamakta, birkaç ay-bir yıl, hatta üç yıl içinde kendini göstermektedir. Hastalığın erken dönemlerinde yapılan timektomiden yararlanma şansının daha yüksek olduğu bildirilmiştir.
Timektomi hiçbir zaman acil yapılması gereken bir ameliyat değildir. Ameliyat, hastalar iyi duruma getirildikten sonra yapılır. Cerrahi yaklaşım klasik olarak transsternal veya transservikaldi. Artık birçok merkez yeni bir yöntem olan videotorakoskopik timektomi uygulamaktadır. Bu sayede timektominin morbiditesi çok azalmıştır. Bizde de bu yöntem 2003’den beri timoması olmayan ya da küçük timomalı hastalarda uygulanmaktadır.
Kortikosteroidler
Kortikosteroidler hastaların büyük çoğunluğunu tamamen düzeltir (Şekil 32). Bu düzelme 4-6 hafta içinde görülür. Daha kısa zamanda düzelme ortaya çıkabildiği gibi düzelme süreci daha da uzayabilir. En çok prednizolon (PRD) kullanılır.
Hastaların bir bölümünde PRD başlanıldıktan bir hafta-on gün sonra kas güçsüzlüğü geçici olarak artabilir. Özellikle bulber güçsüzlüğü olan hastada bu nedenle denge tümüyle bozulabilir ve solunum güçlüğü belirebilir. Bu bakımdan PRD’yi düşük dozda başlayıp birkaç günde bir yükselterek istenilen düzeye çıkmak, özellikle ilaca hasta hastaneye yatırılarak başlanmıyorsa, daha emniyetli bir yoldur. Hastanede yatan hastada, solunumun korunabileceği şartlar sağlanabiliyorsa, doğrudan yüksek doz başlanabilir, bu yöntem istenen cevabın daha çabuk alınması bakımından avantajlıdır. Amaçlanan doz 1 mg/kg/gün civarındadır. Birçok hastada 0,75 mg/kg/gün gibi düşük bir doz ile istenen yanıt alınabilir. Pür oküler ya da hafif jeneralize miyasteniklerde daha da düşük dozlar yetebilir, her gün yerine günaşırı uygulama denenebilir.
PRD başlandıktan sonra hastada bariz bir iyileşme elde edilince ilaç dozu düşürülmeye başlanır. Yüksek dozları uzun süre sürdürmemeye çok dikkat etmelidir. Doz düşürme ilacı günaşırı vermeye yönelik olarak yapılır ve doz azaldıkça düşürme temposu da yavaşlatılır. Timektomi yapılmamış hastalarda PRD’yi günaşırı 10-20 mg gibi bir idame dozunda sürekli olarak tutmak gerekebilir. PRD tümüyle kesilirse hastalık büyük bir olasılıkla alevleneceğinden ilacı ancak hastayla bu risk tartışıldıktan sonra, hastanın isteği doğrultusunda kesmek uygundur. Timektomi yapılmış genç hastalarda azaltıp kesmeye gayret etmek gerekir. Ek bir immünsupresif alan hastada PRD’yi azaltıp kesmek düşünülebilir. PRD düşürülürken ya da kestikten sonra alevlenme olursa yeniden yüksekçe bir doza çıkarak vermek ve yeni bir immünsupresif eklemeyi düşünmek gerekir.
Kortikosteroidlerin yan etkileri arasında kozmetik bozuklukların yanı sıra uygun tedaviyle kontrol altına alınabilen gastrik yan etkiler, hipertansiyon, hiperglisemi ve hiperkolesterolemiyi sayabiliriz. En büyük problem ise, özellikle menopozdaki kadınlarda kalsiyum ve D vitamini tedavisine rağmen önlenmesi çok zor olan osteoporozun hızlanması ve buna bağlı fraktürlerdir. Birçok hastada bifosfonat kullanmak gerekebilir. Katarakt oluşumu da sıkça rastlanan yan etkilerdendir.

Şekil 32. Miyastenik bir hastada sağda ptoz ve kortikosteroid sonrası düzelme.
Diğer immünsupresif ilaçlar
Azatioprin, kortikosteroid alamayan hastalarda, kortikosteroid dozu düşürülürken alevlenme olduğunda, kortikosteroid dozu hızlıca düşürülmek istendiğinde tedaviye eklenebilir. Bazı durumlarda (özellikle yaşlılarda) azatioprine PRD ile eşzamanlı olarak ya da tek başına başlamanın avantajları olabilmektedir. Fertil dönemdeki hastalarda temkinli kullanılmalıdır. Doz 2-2,5 mg/kg/gün olarak hesaplanır. Etkisi geç (en az 3-6 ay sonra) çıktığından verildiği ilk aylarda yarar beklenmemelidir. Mide bulantısı, kusma, ishal, saç dökülmesi görülebilecek yan etkiler arasındadır. Karaciğer ve lökositler üzerine toksik etki gösterebildiğinden karaciğer fonksiyon testleri ve kan sayımı ile izlemek gerekir. İnfeksiyona yatkınlık görülebilir. Alerjik reaksiyon gelişirse ilaç kesilir. Azatioprin dozu hastalık düzeldikten sonra yavaşça düşürülür, küçük doz azaltmalarının 6-8 aydan kısa sürelerde yapılmamasına dikkat etmek gerekir.
Mikofenolat mofetil, azatioprini tolere edemeyen hastalarda sıklıkla kullanılmaya başlanmıştır. Günde 2 gram iki doza bölünerek verilir. Mide bulantısı, kusma, ishal, nadir olarak da artralji ve karaciğer toksisitesi gibi yan etkiler gözlenebilir. Lökopeni yapabileceğinden kan sayımı ile izlenmelidir. Alerjik reaksiyon nadirdir. Fırsatçı infeksiyonlarda artış bildirilmiştir.
Azatioprine göre daha az kullanılan siklosporin-A, etkisi daha çabuk ortaya çıkan, ancak hipertansiyon ve renal toksisite şeklinde yan etkileri olan bir immünsupresiftir. Lökopeni, hemorajik sistit ve infertilite gibi yan etkileri olan siklofosfamid ancak tedaviye çok dirençli hastalarda kullanılır.
Rituksimab
Rituksimab insan İgG1 sabit bölgeleri ve sırasıyla değişken mürin hafif zincir ve ağır zincir içeren bir glikolize immünoglobulin sunan, genetik mühendisliği ile üretilen kimerik fare/insan monoklonal antikorudur. Yakın zamanda yapılan çalışmalarda dirençli MG hastalarında özellikle anti-MUSK pozitif hastalarda oldukça olumlu sonuçlar elde edilmiştir. Anti-MUSK pozitif hastalarda etkinin uzun süre sürdüğü dikkati çekmiştir.
Farklı doz uygulamaları bulunmakla birlikte erişkin hastalarda 375 mg/m2 dozunu 4 hafta boyunca haftada bir uygulamak ya da 1 ay boyunca iki haftada bir 1000 mg dozunda uygulamak en sık kullanılan iki yöntemdir. Uzun süreli takipte tedavinin nasıl devam ettirileceği tartışmalıdır, ancak 6 aydan daha erken olmayacak şekilde miyastenik bulgularda alevlenme görüldüğünde doz yenilemek önerilebilir.
Rituksimab genel olarak iyi tolere edilir. İnfüzyona bağlı ateş, üşüme, titreme, mide bulantısı, halsizlik, döküntü gibi yan etkiler erken dönemde görülür. İnfeksiyona yatkınlık, hipogamaglobulunemi, hipermakrofaji sendromu ve progresif multifokal lökoensefalopati (PML) nadir ama mortalitesi yüksek yan etkileridir.
İntravenöz immünglobulin ve plazmaferez
İntravenöz immünglobulin (İVİg) çok sayıda donörden toplanan serumlardan ayrılan yüksek miktarda immünglobulin G içeren bir kan ürünüdür. Pek çok otoimmün hastalıkta bağışıklık sistemini düzenleyici bir ajan olarak kullanılmaktadır. Plazmaferez de yine pek çok otoimmün hastalığın özellikle akut dönemde tedavisinde kullanılan kandaki antikorları çeşitli yöntemler ile filtreleyerek temizlemeyi hedefleyen bir yöntemdir.
Bunlar, ani bozulmalarda, timektomi öncesi, gerektiğinde gebelik sırasında, yeni başlanan kortikosteroidlerin etkisi beklenirken kısa süreli olarak kullanılabilen tedavi şekilleridir. Miyastenik kriz öncesinde veya kriz sırasında en çok yararlanılan ajanlardır. Plazma değişiminin olumlu etkisi birkaç değişimde başlar, İVİg’in olumlu etkisi ise birkaç gün içinde başlar, her iki durumda da etki birkaç haftada sona erer. Birinin diğerine üstünlüğü kanıtlanmamış olmakla birlikte, İVİg’in etkili olmayıp plazmaferezin etkili olduğu olgular (özellikle MuSK MG) bildirilmiştir. Diğer taraftan plazmaferezin yan etkileri (infeksiyon, hipovolemi) İVİg’e göre daha çoktur. Hastane şartlarına ve edinilmiş tecrübeye göre seçim yapılır.
İVİg bazen tek başına verilmekte ve çok uzun süre kullanılmaktadır. Birkaç aydan uzun süre kullanım önerilmez. Uzun süre kullanıma ihtiyaç duyuluyorsa mutlaka bir immünsupresif eklemek ve İVİg’i kesmeye çalışmak gerekir. Tek başına kullanım ancak timektomiye hazırlık sırasında, timektomiden sonra düzelmenin beklendiği birkaç ay süresince ve gebelik sırasında uygundur. Prepubertal MG’de de bir süre kullanılabilir. Miyastenik kriz nedeniyle entübe edilmiş ve İVİg sayesinde toparlamış bir hasta katiyen sadece İVİg ile izlenmemeli, tedaviye mutlaka bir immünsupresif eklenmelidir.
MG’DE TEDAVİ ŞEMALARI
Erken başlangıçlı jeneralize MG
Piridostigmin ile MG kısmen de olsa kontrol altına alındıktan sonra hastanın durumunun timektomiye uygun olup olmadığı değerlendirilir. Hastalık hafifse sadece piridostigmin ya da İVİg veya plazmaferez eklenerek timektomi yapılır. Ağır durumlarda timektomi öncesi kortikosteroid başlamak gerekebilir. Timektomi optimal durumda yapılır. İmmünsupresif ilaçlar daha önce başlanmışsa ameliyat döneminde immünsupresif tedavide büyük değişiklikler yapmamak gerekir, ameliyattan sonra da en az bir süre ilaçları sürdürmek uygun olur. Timektomiden önce veya sonra kortikosteroid başlanmış olan genç hastalarda, timektomi sonrası kortikosteroidler düşürülürken hastalık alevlenmiyorsa ilaçları zaman içinde tamamen kesmeye çalışılmalıdır, aksi durumda yani sık alevlenmeler oluyorsa ek bir immünsupresif vermek düşünülebilir. Timektominin etkisi bir yıldan önce ortaya çıkmayabileceğinden yeni bir immünsupresif başlamakta acele etmemek yerinde olur. Bazen ayda bir verilen İVİg tedavisiyle zaman kazanılabilir. MG puberte öncesi başlamış ise mümkünse kortikosteroidlerden puberte sonrasına kadar uzak durmak iyi olur. Timektomi değerlendirilebilir. İVİg ve plazmaferez işe yarar.
Geç başlangıçlı MG
Önce yeterli dozda piridostigmin verilerek hastalık kısmen kontrol altına alınır. Bu yaş grubunda, özellikle 50-55 yaşın üzerinde, timoma yoksa timektominin etkisi tartışmalıdır. Timektomi yapılmayacaksa hastaları sadece piridostigmin ile idare etmeye çalışmak risklidir, bu durumda kortikosteroid başlamak gerekir. Bazı yaşlı hastalarda düşük doz kortikosteroid yeterli olabilir. Gerektiğinde ek olarak İVİg veya plazmaferezden yararlanılabilir. Kortikosteroidlere azatioprin eklenmesi iyi sonuçlar verir. Hastanın MG ağırlık derecesi azatioprinin geç yanıtını beklemeye uygunsa tek başına azatioprin başlamak da bir yoldur. Yukarıda anlatıldığı şekilde ilaçlar yıllar içinde düşük bir seviyeye indirilir ve o seviyelerde süresiz tutulur. Hastanın yaşı ve bu yaşlarda miyastenik krizin daha yüksek olan mortalitesi düşünülürse ilaçları tamamen kesmek doğru olmayabilir.
Anti-MuSK pozitif MG
Buradaki önemli farklılıklar, AKE’lerin işe yaramayabileceği ve timektominin önerilmediğidir. Kortikosteroidler ile hastaların hemen hepsi düzelir, ancak bu düzelme yüksek doz kortikosteroidlerlerin olağandan daha uzun bir zaman kullanımını gerektirebilir. Ek immünsupresif gerekliliği daha çabuk gündeme gelebilir. İVİg veya plazmaferez de bu hastalarda daha fazla kullanılır. İVİg fazla işe yaramayıp plazmaferezin daha iyi geldiği hastalar vardır. MuSK MG’li hastalarda erken dönemde rituksimab tedavisinin verilmesinin etkin olduğu ve etkinin uzun sürdüğü yakın zamanda yapılan çalışmalarda gösterilmiştir. Bazı hastalarda dilde atrofi geliştiğinde nazone konuşma tüm tedavi seçenekleri kullanıldığı halde kalıcı olabilir. MuSK MG’nin her zaman tedaviye dirençli olmadığı, içlerinden küçük bir grubun, aynı AChR MG gibi kortikosteroidlerle kolayca tedavi edilebildiği de dikkatle değerlendirilmelidir.
Oküler MG
Belirtiler hayati tehlike oluşturmasa da birçok hasta sadece AKE ile günlük faaliyetlerini rahat sürdüremez ve immünsupresif kullanmak zorunda kalır. Tipik olarak düşük doz ve günaşırı verilen PRD’den (günaşırı 30-40 mg gibi) yararlanırlar. PRD’ye ek veya tek başına düşük doz azatioprinin de yeterince etkili olabildiği bildirilmiştir, özellikle yaşlı hastalarda denenebilir. Timektomi genellikle yapılmaz.
Timomalı MG
Timomektomi mutlaka yapılmalıdır. Tümör çoğunlukla kapsüllü ve selim niteliktedir. Ancak bir bölümü de habis özellikler taşır ve çevre dokuya invazyon yapar. Bu durumlarda ek radyoterapi ve gerektiğinde kemoterapi uygulanır. Uzak metastazlar çok nadirdir. Timomalar tekrarlayabileceğinden muntazam aralıklarla radyolojik kontrolün yapılması gerekir. MG belirtileri, timomektomi yapılmasından etkilenmeyebilir. MG ağır seyredebildiğinden yüksek doz kortikosteroidler ve erken başlanan ek immünsupresifler, İVİg veya plazmaferez gerekebilir.
Gebelikte MG
Genç kadınlara gebe kalmadan önce miyasteninin kontrol altına alınması ve ilaçların mümkün olabildiğince düşük bir seviyeye getirilmesi gerektiği anlatılır. Gebelik sırasında AKE ihtiyacı değişebilir ve ayarlanması gerekir. Hasta kortikosteroid alıyorsa aynen sürdürülür. Biz gebelik sırasında azatioprin vermiyoruz, ilacı gebelikten 6 ay önce kesiyoruz, zararı olmadığını savunanların yanında kesin bilgilerin olmadığını vurgulayanlar da bulunmaktadır. Alevlenme olursa İVİg verilebilir. Doğum normal yolla yapılabilir, anestezi gerekiyorsa mümkünse genel anestezinden kaçınıp epidural anestezi tercih edilir. Nadir de olsa bebekte neonatal MG gelişebileceğinden ve annede miyastenik alevlenme gelişebileceğinden doğumun, riskli gebeliklerin izlenebileceği hem neonatal hem erişkin YBÜ olan merkezlerde gerçekleştirilmesi önerilmelidir. Loğusa döneminde dikkatli olmak gerekir.
Miyastenik kriz
Gelişen solunum güçlüğü mekanik ventilasyon kullanmayı gerektiren düzeye vardığında miyastenik krizden söz edilir. Miyastenik kriz, özellikle hastalığın ilk iki yılında tedavi henüz sağlanamadan ortaya çıkar. Tarihi bilgi hastaların %40’ında miyastenik kriz geliştiğidir, ancak günümüzdeki daha etkin tedavi seçenekleri ile bu oran düşmüştür. MuSK pozitif ve timomalı hastalarda kriz geçirme olasılığı daha fazladır. Özellikle ağır bulber tutulumu olan hastalarda her an beklenilmesi ve acil müdahale edilmesi gereken bir durumdur. Konuşma, yutma ve çiğnemesi çok iyi olan bir hastada solunum güçlüğü gelişirse bunun miyasteniye bağlı olmadığını, başka bir sebep aramak gerektiğini düşünmek daha doğrudur.
Solunum güçlüğü gelişmeye başlayan hasta genellikle piridostigmin dozunu artırarak iyileşmeye çalışır. Bazen piridostigmin o kadar çok alınır ki miyastenik yerine kolinerjik semptomlar ön plana çıkar ve kolinerjik kriz söz konusu olabilir. İşin zor tarafı, fazla asetilkolinin çizgili kas üzerindeki etkisinin (nikotinik etki) miyasteniye benzer bir tablo yaratmasındadır. Asetilkolinin çizgili kas dışı etkileri (müskarinik etkiler - mide bulantısı, barsak hareketlerinin artması, terleme, hipersalivasyon) ile nikotinik etkiler birlikte gitmediğinden müskarinik etkilerin olması ya da olmaması da ayırıcı tanıda yararlı değildir. Solunum güçlüğü içindeki hastada miyastenik ve kolinerjik krizleri birbirinden ayırmak neredeyse olanaksız olduğundan yapılacak en doğru davranış hastanın solunumunu güvenceye almak ve mekanik ventilasyonun yapılabileceği şartları sağlamaktır. Yine de miyastenik krizin kolinerjikten çok daha sık olduğunu bilmekte yarar vardır; hasta az piridostigmin kullanmaktaysa reanimasyon şartları sağlanana kadar piridostigmin verilmelidir.
Tablo 14. Miyastenide kullanılabilecek tedavi şemaları.
{kind=link}
|
ERKEN BAŞLANGIÇLI JENERALİZE MG (sırayla) · Uygun dozda AKE verilir · Gerekirse İVİg verilir veya plazmaferez uygulanır · Hastalık ağırsa kortikosteroid verilir · Optimal durumda timektomi yapılır · Timektomi sonrasında olabildiğince (en az bir yıl) beklemeye çalışılır, gerektiğinde aylık İVİg verilir ve çok gerekirse kortikosteroid başlanır · Kortikosteroid timektomiden önce veya sonra başlanmışsa zaman içinde yavaşça doz düşürülür ve kesmeye çalışılır · Kortikosteroidler düşürülürken sık alevlenmeler oluyorsa hastanın durumuna göre düşük bir dozda sürekli tutulur · Kortikosteroidler düşük doza düşürülemiyorsa azatioprin eklenir · AKE’ler daima ihtiyaca göre verilir, hasta düzeldikçe azaltılır ve kesilir |
|
MuSK MG · AKE’ler fayda etmeyebilir hatta bozabilir · Timektomi önerilmez · Yüksek doz kortikosteroid verilmelidir · Bağışıklık sistemini baskılayıcı ek ilaçlar erken eklenmelidir · Gereğinde İVİg ya da plazmaferez tedavisi eklenmelidir · Sık alevlenmeler oluyorsa rituksimab tedavisi düşünülmedir |
|
TİMOMALI MG · Uygun dozda AKE verilir · Hastalık ağırsa (veya transsternal girişim gerekiyorsa) kortikosteroid verilir · Gerekirse İVİg veya plazmaferez eklenir · Optimal durumda timomektomi yapılır · Gereğinde timoma tedavisi için ek radyoterapi veya kemoterapi yapılır · Timoma açısından aralıklı takip gerekir |
|
GEÇ BAŞLANGIÇLI JENERALİZE MG’DE FARKLAR · Timoma varsa timomektomi mutlaka yapılır, yoksa birçok merkezde timektomi endikasyonu konmaz (özellikle >50 yaş) · Çoğunlukla kortikosteroid başlamak gerekir · Azatioprin daha çabuk eklenebilir, hatta ağır olmayan olgularda azatioprini tek başına vermek düşünülür · Kortikosteroid düşük bir doza getirip sürdürülür, kesilmesi planlanmaz · Azatioprin de düşük bir doza düşürülür |
|
OKÜLER MG’DE FARKLAR · Timektomi genellikle düşünülmez · Kortikosteroidleri düşük dozda, hatta günaşırı vermek genellikle yeterlidir |
ÖZEL DURUMLAR
Neonatal miyasteni
Miyastenik annelerin %10-15’inin çocuğunda doğumdan sonraki 3 gün içinde miyastenik semptomlar görülür. Belirtiler annedeki antikorların pasif olarak çocuğa geçmesine bağlıdır. Belirtiler 2-3 haftada kaybolur, ancak bu süre içinde AKE ile tedavi gerekebilir. Ağır olgularda İVİg kullanılabilir.
MG ve diğer otoimmün hastalıklar
MG ile birlikte olabilen otoimmün hastalıkların başında tiroid hastalıkları gelir. Tiroid hastalıklarının tedavisi miyasteniyi olumlu yönde etkileyebilir. Romatoid artrit, psoriasis, pernisiyöz anemi ve otoimmün aplastik anemi gibi hastalıklar da MG’ye eşlik edebilir.
MG ve infeksiyonlar
MG’de vücudun bağışıklığı genel olarak düşük değildir. Yukarıda anlatıldığı gibi, bağışıklık bozukluğu sadece tek bir proteine karşı oluşan antikorlar nedeniyle ortaya çıkar. Ancak, özellikle solunum yolunun infeksiyöz hastalıkları miyastenik hastalarda daha ağır geçebilir. Akciğer çabuk etkilenebilir, pnömoni gelişebilir, buna miyastenik belirtilerin artması eklenebilir. Kullandıkları immünsupresif ilaçların da olumsuz etkisi olabilir. Bu bakımdan MG’li hastaların bulaştırma potansiyeli olan hastalardan uzak durmaları ve kendilerine özel itina göstermeleri çok önemlidir. İmmünsupresif ilaçların infeksiyon sırasında azaltılması doğru değildir. Hastaların bu sırada ilaçları aynı dozda alması, gerektiğinde de danışarak artırması önerilir. Gripal infeksiyonlardan korunmak için her yıl grip aşısını, pnömoniden korunmak için daha uzun süreli etkili pnömoni aşılarını olmaları önerilir.
MG’de ilaç kullanımı (Tablo 15)
Nöromüsküler kavşağı etkileyen birçok ilaç miyastenik hastanın durumunu bozabilir, hatta MG’nin ortaya çıkmasına neden olabilir. MG iyi kontrol altındayken kötü etkilenme olasılığı görece olarak azdır. Bunların arasında en çok dikkat edilmesi gereken ilaçlar antibiyotiklerdir. Telitromisin, aminoglikozid grubu, flurokinolon grubu, linkomisin, makrolid grubu ve sulfametoksazol + trimetoprim gibi antibiyotikler miyasteniklere verilmemelidir, mutlaka verilmesi gerekirse ciddi kötüleşme ve mekanik ventilasyon olasılığı göz önünde tutulmalıdır. Yine NMK’ta iletimi bozan botulinum toksini enjeksiyonu miyastenik hastalara uygulanmamalıdır. Antiaritmikler (prokainamid, kinidin) ve beta-adrenerjik blokerler miyasteniyi bozabilir. Depolarizan ilaçlar (süksinilkolin) dikkatle kullanılmalıdır. Magnezyum ve BT yaparken kullanılan iyotlu kontrast madde güçsüzlüğü artırabilir. D-penisilamin MG ortaya çıkarabilir.
Bu hastalarda, ilaçlar miyasteniyi bozacak korkusuyla, MG dışı hastalıkların yetersiz tedavi edilmesi de ciddi bir sorun yaratır. Miyasteninin yanında başka hastalıkları da olan hastalarda ilaç kullanımında karar, yarar/zarar oranına göre verilmelidir. Örneğin, ağır infeksiyon veya sepsis gibi durumlarda, mutlaka gerekiyorsa, sakıncalı bir ilaç kullanılabilir, ancak hasta gözlem altında dikkatle izlenmelidir. Ayırca, çoğu ilacın yan etki profilinin büyük MG hasta gruplarında değerlendirilmediği de akılda tutulmalıdır.
Miyastenik belirtileri artırmayan ilaçları bilmek büyük önem taşır. Daha rahat kullanılabilecek ilaçlar arasında sefalosporin ve penisilin grubu antibiyotikleri, asetilsalisilik asit ve parasetamolu, SSRI grubu antidepresanları ve alprazolamı sayabiliriz. Ancak sakınılacak ilaç listesinde yer almasa bile her yeni ilaç kullanımı sırasında hastayı yakından izlemek, gerektiğinde ilacı kesmek gerekir. İlaç konusunda MG için yapılacak uyarılar aşağıda anlatılacak olan Lambert Eaton miyastenik sendromu ve konjenital miyastenik sendromlar için de geçerlidir.
Tablo
15. Miyastenide ilaç
kullanımı.
|
KULLANILMAMASI GEREKEN İLAÇLAR
|
Bazı antibiyotikler · Aminoglikozidler (göz damlaları dahil) · Fluorokinolon · Makrolidler · Linkomisin · Sulfonamid · Sulfametoksazol + trimetoprim · Tetrasiklin · Polimiksin Beta blokerler Kalsiyum kanal blokerleri Kinin, kinidin (tonik dahil) Bazı antiepileptikler (Fenitoin, Gabapentin) Kürar ve türevleri Morfin ve diğer narkotik analjezikler Trankilizan ve barbitüratlar Amitriptilin Lityum Magnezyum içeren ilaçlar (laksatifler ve antiasidler dahil) D-penisilamin Östrojen içeren preparatlar Bazı göz damlaları (Timolol) Botulinum toksini |
|
KULLANILMASINDA GENELLİKLE SAKINCA OLMAYAN İLAÇLAR
|
Bazı antibiyotikler · Sefalosporinler · Penisilinler (ampisilin dışında) Analjezikler · Parasetamol · Asetilsalisilik asit Antidepresanlar (SSRI, SNRI) Diyabet ilaçları Tiroid ilaçları Proton pompa inhibitörleri Yüksek doz magnezyum içermeyen vitaminler |
LAMBERT EATON MİYASTENİK SENDROMU
Lambert Eaton miyastenik sendrom (LEMS) ön planda bacaklarda güçsüzlük ile karakterize, tümörle ilişkili (paraneoplastik) olabildiği gibi tümör olmadan da ortaya çıkabilen, motor ve otonomik sinir terminallerindeki voltaja bağımlı kalsiyum kanallarının hedef alındığı otoimmün kökenli bir hastalıktır. Ender rastlanan bir hastalık olan LEMS daha çok 40 yaşın üzerinde başlarsa da çocuklarda bile görüldüğü bildirilmiştir. Eskiden erkeklerde daha sık olduğu bilinirken artık kadınlarda ve erkeklerde eşit olarak görülmektedir.
Klinik olarak, kas güçsüzlüğü, azalmış kemik veter refleksleri ve otonomik fonksiyon bozukluğu görülür. Hastalık genellikle subakut olarak gelişen alt ekstremite güçsüzlüğü ile başlar. Bu güçsüzlüğün muayene ile ortaya çıkarılabilen bir özelliği vardır: Kasın ilk kontraksiyonu zayıfken hareket tekrarlandıkça ikinci kontraksiyondan itibaren kas geçici olarak kuvvetlenir (fasilitasyon), sonra yine zayıflar. Nörolojik muayenede hafif güçsüzlüğü olan bir hastanın bu güçsüzlükten beklenmeyecek ölçüde ağır yürüme güçlüğü olduğu dikkati çeker. Güçsüz kas, ağrılı ve hassas olabilir. Kollardaki güçsüzlük daha hafiftir. Ekstremitelerdeki güçsüzlüğe ptoz ve çift görme gibi oküler bulgular, yutma-konuşma-çiğneme güçlüğü gibi bulber belirtiler eklenebilir, ancak bunlar MG’nin aksine çoğu zaman geri plandadır ve geç başlar. Hastalığın belli başlı otonomik belirtileri ağız kuruluğu, kabızlık ve erkeklerde impotansdır, ancak ortostatik hipotansiyon, pupil anomalileri ve terleme azlığı da görülebilir.
LEMS, antikorlar aracılığıyla oluşan otoimmün bir hastalıktır. Voltaja bağlı kalsiyum kanallarına (VGCC) karşı antikorlar (en çok P/Q tipi) LEMS’li hastaların % 90’ında gösterilebilir. VGCC, motor ve otonomik sinir terminallerinde ACh’nin presinaptik membrana füzyonunu ve salınımını sağlar. İşte bu kanalların otoimmün saldırı sonucu azalması ACh salgılanmasının azalmasına neden olur.
Hastaların yarısından fazlasında kanser saptanır, bunların da büyük çoğunluğu küçük hücreli akciğer kanseridir. LEMS’in bulguları çoğunlukla kanserin bulgularının çıkmasından önce ortaya çıkar. Çocuklarda lenfoproliferatif tümörler ile ilişkili olabilir. Nöroektodermal kökenli bu tümörde bol miktarda VGCC bulunur. Kanserle ilişkili olan ve olmayan LEMS klinik olarak birbirinden farklı değildir, ancak kanserle ilişkili olanda belirtilerin daha hızlı ilerlediğini gösteren çalışmalar vardır. Bulber bulguların varlığı, otonomik tutulum, kilo kaybı, sigara kullanımı ve 50 yaşın üstünde olma kanser olasılığını arttırmaktadır. Duyu kaybı ve serebellar bulgular beklenen bozukluklar değildir, ancak varlıkları küçük hücreli akciğer kanseri zemininde gelişen ikinci bir paraneoplastik sendromu destekler. Kanser olmayan hastalarda başka otoimmün hastalıklar ya da otoantikorlar bulunabilir.
Tanıda en yararlı laboratuvar incelemesi EMG’dir. İstirahat halinde bileşik kas aksiyon potansiyellerinin (BKAP) amplitüdü düşüktür. Yüksek frekanslı ardışık sinir uyarımından sonra elde edilen BKAP amplitüdünün istirahat halindekinin iki veya daha fazla katı olduğu görülür (fasilitasyon). Yüksek frekans vermek yerine kasa 10 saniye istemli kontraksiyon (15-20 Hz’e tekabül eder) yaptırmak tercih edilir. Yüksek frekanslı ardışık sinir uyarımı, kalsiyumun sinir terminali dışına çıkmasını önler ve daha çok ACh salgılanmasını sağlayarak nöromüsküler geçişi düzeltir. Elektrofizyolojik (ve klinik) olarak gözlenen fasilitasyon bu şekilde açıklanır. Düşük frekanslı ardışık sinir uyarımı ile ise MG’de olduğu gibi dekrement görülür, ancak dekrement paterni MG’den bazı farklılıklar gösterebilir (Ayrıca bakınız: Elektromiyografi). Tek lif EMG ile de NMK’ta iletimin bozulmuş olduğu saptanabilir. Ancak MG ile LEMS ayırımı motor ileti incelemeleri ve ardışık sinir uyarımı ile yapılabilirken tek lif EMG ile bu ayırım pratik olarak yapılamaz.
Presinaptik bir patolojiyi düşündüren bu elektrofizyolojik bulgular klinik ile birleştirilerek tanı konur. Hastaların çoğunda serumda anti-VGCC antikorları gösterilebilir. Kanserli hastalarda antikor bulma olasılığı daha yüksektir. Tanı konduktan sonra akciğer kanseri ve diğer kanserler yönünden ısrarla araştırma yapmak gerekir. Tümör LEMS tanısı ile aynı zamanda saptanmayabilir, özellikle ilk iki yıl içinde olmak üzere beş yıla kadar ortaya çıkma olasılığı vardır. Bu bakımdan hastaya belli aralarla toraks bilgisayarlı tomografisi yapmak gerekir.
Ayırıcı tanıda, ekstremitelerde subakut güçsüzlük ile başlayan polimiyozit gibi hastalıklar, okülobulber belirtiler de eklendiği zaman MG düşünülmelidir. Yine presinaptik bir patoloji sonucu ortaya çıkan botulizmin kliniği ise çok farklıdır.
Tümörün tedavisi LEMS bulgularının da gerilemesine neden olabilir. Hastalar bir potasyum kanal inhibitörü olan 3,4 diaminopiridinden çok yararlanırlar. Bu ilaç tek başına veya piridostigmin ile kombine edilerek kullanılabilir. Guanidin hidroklorid yan etkileri bakımından tercih edilmez. Birçok hastada ancak immünolojik tedavinin eklenmesi ile hastalık kontrol altına alınabilir. İmmünolojik tedavide MG’de anlatıldığı gibi kortikosteroidler ve/veya azatioprin, kısa vadede yarar için de plazmaferez ya da İVİg kullanılır.
BOTULİZM
Anaerobik bir bakteri olan Clostridium botulinum toksini ile oluşan bir hastalıktır. (Ayrıca bakınız: Sinir Sistemi İnfeksiyonları) Toksin, motor ve otonomik sinir terminallerinden ACh’in salınımını engelleyerek presinaptik bir patoloji yaratır. Evde yapılmış konserve başta olmak üzere toksin içeren gıdaların yenmesiyle, nadiren de yarada toksin üremesiyle oluşur. Son zamanlarda botulinum toksin enjeksiyonu sonrası botulizm gelişen olgular bulunmaktadır.
Belirtiler, kontamine gıdanın yenmesinden 12-36 saat sonra bulanık görme, ptoz ve diplopi ile başlar. Bu sırada hastalarda mide bulantısı ve kusma da vardır. Üç-dört gün içinde bulber ve ekstremite kaslarında güçsüzlük eklenir. Ağız kuruluğu, kabızlık, idrar retansiyonu, midriazis ve pupilla yanıtsızlığı gibi otonomik belirti ve bulgular dikkati çeker. Ağır mortalitesi olan bu hastalıkta genellikle çok kısa zaman içinde solunum yetmezliği belirir ve mekanik ventilasyon yapmak gerekir.
Miyastenik tablonun akut yerleşmesi, mide bulantısı, kusmanın olması, otonomik belirtilerin eşlik etmesi, evde yapılmış konserve yeme öyküsü, birden çok kişide benzer belirtilerin görülmesi ile tanıdan şüphelenilir. Yakın zamanda botulinum toksin enjeksiyonu yapılıp yapılmadığı ayrıntılı olarak sorgulanmalıdır. Presinaptik nöromüsküler hastalıklarda görülenlerle uyumlu olan EMG ile (LEMS için anlatılanlarla bakınız) tanı konur.
Çok erken verilen antitoksin yararlı olur, ancak tedavinin esası mekanik ventilasyonun sağlanmasıdır. Düzelme çok yavaştır, birkaç ay sürebilir.
KONJENİTAL MİYASTENİK SENDROMLAR
Konjenital miyastenik sendromlar (KMS’ler), nöromüsküler kavşağın nadir kalıtsal bir grup hastalığıdır. KMS’ler immünolojik olmadığından serumda antikor bulunmaz ve immünolojik tedaviye yanıt yoktur. Aile öyküsünün varlığı önemli bir ipucudur. Bu hastalıklar genellikle ilk yaş içinde oküler, bulber ve ekstremite kaslarında güçsüzlük ile kendini gösterir. Nadiren daha geç yaşlarda başladığında tanı koymak zorlaşır. Ardışık sinir uyarımı ve tek lif EMG bulguları MG’de görülenlere benzerdir. Tedavide piridostigmin önemli bir yer tutar, ancak piridostigmin ile kötüleşen KMS tipleri olduğunu bilmek gerekir.
Konjenital miyastenik sendromlar, presinaptik, sinaptik veya postsinaptik bir patolojiye bağlı olabilir (Tablo 16). Klinik olarak çoğu zaman mümkün olmayan bu ayırım, dünyada çok az merkezde gerçekleştirilebilen morfolojik incelemeler ve artık bunların yerini alan genetik testlerle yapılabilir hale gelmiştir.
Tablo 16. Konjenital miyastenik sendromlar (çok nadir olanlar listeye alınmamıştır).
|
Presinaptik · Kolin asetiltransferaz eksikliği |
|
Sinaptik · Son plak AChE eksikliği |
|
Postsinaptik · AChR eksikliği · AChR kinetik anormallikleri (yavaş kanal ve hızlı kanal KMS) · Rapsin eksikliği · DOK7 eksikliği
|
|
Glikozilasyon defektleri · GFPT 1 miyastenisi
|
Ülkemizde de, dünyadakine benzer şekilde bozukluğun en sık postsinaptik olduğu ve mutasyonların çoğunun da AChR geni ε subünitesinde yoğunlaştığı gösterilmiştir. KMS’ler arasında en sık görülen AChR eksikliği bebeklikte başlar, ön planda oftalmoparezi ile seyreder ve iyi prognozludur. Genellikle ilk 3 ay içinde çocuğun kısık sesle ağladığı ve iyi ememediği dikkati çeker, daha sonra ptoz fark edilir. Zaman içinde bulber belirtiler geriler ve göz belirtileriyle kol ve bacaklardaki yorgunluk süregelir. Nörolojik muayenede bilateral ptoz olduğu ve gözlerin çok az hareket ettiği, neredeyse orta hatta sabit olduğu görülür (Şekil 33). Bu göz hareket bozukluğu kronik ve büyük ölçüde simetrik olduğundan çift görme yok denecek kadar azdır. Belirtiler gün içinde fluktuasyon gösterir ve AKE’ye kısmen de olsa yanıt verir.

Şekil 33. Konjenital miyastenik sendromlu bir hastada bilateral ptoz ve göz hareket bozukluğu
{kind=link}
Rapsin eksikliği de görece sık görülen KMS’lerden biridir. Rapsinin, NMK’ta AChR’nin kümelenmesinde rolü vardır. Ağır, konjenital malformasyonların gözlendiği erken başlangıçlı tipi olduğu gibi göz hareketlerinin korunduğu ptoza, jeneralize veya proksimal kas zaafının eşlik ettiği erişkin formu da bulunmaktadır. Genelde AKE’ye kısmen de olsa yanıt verir.
Farklı tedavisi olan iki KMS tipinden söz etmekte yarar vardır: Son plak AChE eksikliği ve AChR’nin uzun süre açık kalmasına bağlı yavaş kanal KMS. Bu iki tipi birbirinden ayırmak zordur. Her ikisinde de diğer belirtilere ek olarak kol ve elin ekstansör kaslarında güçsüzlük ve atrofi gelişebilir. Her ikisinde de oküler bulgular geri planda iken, bulber ve solunumla ilgili problemler ön planda olabilir. EMG bulguları da benzer: Motor ileti incelemelerinde, tekrarlayan BKAP dikkati çeker, yani normal BKAP’ı izleyen daha düşük amplitüdlü BKAP’lar görülür. Her KMS olgusunda bu tanıları kaçırmamak için tekrarlayan BKAP’ların aranması önemlidir. Bu iki tip KMS piridostigmine yanıt vermez, hatta piridostigminle kötüleşebilir. Son plak AChE eksikliğinde efedrin veya albuterol, yavaş kanal KMS’de ise kinidin ve fluoksetin yararlı olabilir. Gerektiğinde BiPAP ile solunumun düzeltilmesi de çok önemlidir.
Son zamanlarda, yüz tutulumunun geri planda olduğu ve proksimal kas zaafının dikkati çektiği DOK7 ve GFPT1 miyasteni gibi KMS’lerin de bu sendromlar arasında önemli bir yer tuttuğu anlaşılmıştır. DOK7 miyastenide piridostigmin hastayı kötüleştirdiğinden verilmemelidir. GFPT1 miyastenisinde ise piridostigmin işe yarar. Bu iki grupta olan hastalar da efedrinden yararlanabilirler.
Miyastenik yakınmalar çok silik olduğu halde aniden solunum krizine girildiği ‘epizodik apne’ adı verilen tablonun kolin asetiltransferaz enzimi eksikliğine bağlı olduğu anlaşılmıştır. Bu bebeklerde profilaktik olarak antikolinesterazlar verilmeli ve hastalar çok yakından izlenmelidir.
Sonuç olarak, KMS’lerde farklı tedavilerin varlığı bunların ayırdedilmesinin ne kadar önemli olduğunu göstermektedir. Günümüzde genetik incelemelerle mutlaka bu ayırımın yapılmasına çalışılmalıdır. Ayrıca, seronegatif MG tanısı almış ve immünsupresiflere yanıt vermeyen erişkin hastalarda da KMS olasılığını akla getirerek gerekli incelemeleri yapmak önem taşımaktadır.
KAYNAKLAR
KİTAPLAR
· Amato A, Russell JA. Neuromuscular Disorders. New York: McGraw-Hill; 2016.
· Aminoff MJ, ed. Neurology and General Medicine (5. Baskı). Philadelphia: Churchill Livingstone Elsevier; 2014. (Bu kitap 2010 yılında Türkçeye çevrilmiştir).
· Bradley WG, Daroff RB, Fenichel GM, Jankovic J. Neurology in Clinical Practice. The Neurological Disorders (7. baskı). Boston: Butterworth-Heinemann; 2015. (Bu kitap 2008 yılında Türkçeye çevrilmiştir).
· Carpenter S, Carpati G. Pathology of Skeletal Muscle (2. Baskı). Oxford: Oxford University Press; 2001.
· Darras BT, Jones HR, Ryan MM, De Vivo DC, eds. Neuromuscular Disorders of Infancy Childhood and Adolescence (2. Baskı). Philadelphia: Academic Press; 2015.
· Deymeer F, ed. Neuromuscular Diseases: From Basic Mechanisms to Clinical Management. Basel: Karger; 2000.
· Emery AEH. Diagnostic Criteria for Neuromuscular Disorders (2.Baskı). London: Royal Society of Medicine Press; 1997.
· Emery AEH, ed. Neuromuscular Disorders: Clinical and Molecular Genetics. Chicester: John Wiley & Sons; 1998.
· Engel AG, Franzini-Armstrong C, eds. Myology (3. Baskı). New York: McGraw-Hill; 2004.
· Engel AG, ed. Myasthenia Gravis and Myasthenic Disorders (2. Baskı). Oxford: Oxford Univeristy Press; 2012.
· Kandel ER, Schwartz JH, Jessell TM. Principles of Neural Science. (5. Baskı) New York: McGraw-Hill; 2013.
· Lane RJM, ed. Handbook of Muscle Disease. New York: Marcel Dekker; 1996.
· Mastaglia FL, Hilton-Jones D, eds. Handbook of Clinical Neurology (Myopathies and Muscle Diseases) (1. Baskı). Amsterdam; Elsevier; 2007.
· Ropper AH, Brown RJ, eds. Adams and Victor’s Principles of Neurology. (11. Baskı) New York: McGraw-Hill; 2019. (Bu kitap 20011 yılında Türkçeye çevrilmiştir).
· Schapira AHV, DiMauro S, eds. Mitochondrial Disorders in Neurology. Oxford: Butterworth-Heinemann; 1994.
· Vassalio J, ed. Metabolic myopathies (Neurologic Clinics 18;1) Misafir Ed: R Pourmand. Philadelphia: W.B. Saunders; 2000.
GENEL NÖROLOJİ DERGİLERİ DIŞINDA KAS HASTALIKLARINA ÖZELLİKLE YOĞUNLAŞAN SÜRELİ YAYINLAR (DERGİLER)
· Neuromuscular disorders
· Muscle and Nerve
· American Academy of Neurology Yıllık Eğitim Kursu Materyelleri: Neuromuscular Disease
· Acta Myologica (Myopathies and Cardiomyopathies)
KAS HASTALIKLARINA YOĞUNLAŞAN İNTERNET ADRESLERİ
· www.neuro.wustl.edu / neuromuscular
· www.dmd.nl
· www.mdausa.org