HAREKET BOZUKLUKLARI VE DEMANS
Yazanlar: İ. Hakan Gürvit, Aslı Demirtaş-Tatlıdede
Son güncelleme tarihi: 14.10.2020
Lewy cisimcikli demans (LCD), Parkinson hastalığı demansı, Huntington hastalığı, progresif supranükleer paralizi (PSP), kortikobazal dejenerasyon (KBD), multisistem atrofi (MSA) gibi bir grup hastalıkta hiperkinetik, akinetik-rijid, serebellar ya da bunların iç içe geçmesinden oluşan motor bulgular, klinik tabloda zamansal ve ağırlık olarak “subkortikal” tarzda bir zihinsel bozukluğun daha önünde olurlar. Bu bölümde “Hareket Bozuklukları” kısmında ayrıntılı olarak anlatılmış olan Parkinson artı sendromlarının kognitif profillerine yer verilmiş ve LCD, Parkinson hastalığı demansı ve Huntington Hastalığı daha detaylı incelenmiştir.
LEWY CİSİMCİKLİ DEMANS
Birçok seride sıklık açısından nörodejeneratif demanslar içinde Alzheimer hastalığı’ndan (AH) sonra gelen bu kendine özgü demansın, özellikle iyatrojenik bir kötüleşmeye neden olmamak için, tanısı ve ayırıcı tanısının yapılması önemlidir.
Epidemiyoloji
Popülasyon temelli çalışmaların gözden geçirildiği bir metaanalizde LCD prevalansı %0,1-5,0 arasında, demanslar arasında sıklığı ise %1,7-30,5 arasında değişmektedir.
AH bölümünde sözedilen ülkemizde yapılmış TAPS çalışmasında LCD 2. sırada yer almıştır. Otopsi serilerinde de benzer şekilde bu oran %15 ile 25 arasındadır. İnsidans, Cache County çalışmasında izlenen 65 yaş üzeri 5092 birey arasında yılda 6 yeni LCD olgusu ile %0,1 olarak saptanmıştır. 2014 yılında yapılan bir sistematik gözden geçirme ve metaanalizde, tüm yeni demans tanıları içinde LCD insidansı %3,8 olarak bildirilmiştir. Popülasyonda LCD tanısı alan hasta oranı %4,2 iken, ikinci basamak klinik popülasyonda bu oran %7,5’e yükselmekte olup burada bildirilen rakamların LCD için gerçek prevalansı yansıtmadığı ve AH tanısı alan LCD hasta sayısının çok olduğu düşünülmektedir. LCD tanı kriterlerindeki gelişmeler tanı konulan hasta sayısını da yükseltmektedir. 1996 ve 2005 yılları tanı kriterleri karşılaştırıldığında muhtemel LCD tanısı alan hasta sayısında %25 artış olduğu bildirilmiştir; ancak 2017 kriterlerinin tanıya etkisi henüz bilinmemektedir.
Cinsiyetin bir risk faktörü olarak niteliği çok aşikar olmasa da nöropatolojik olarak aynı zamanda kesin AH tanı kriterlerini dolduran ama beyinsapında Lewy cisimcikleri (LCi) saptanan 85 otopsi olgusunda, saf AH’li 192 olgunun tam tersine erkek baskınlığı bildirilmiştir. Şu ana kadar yürütülen 8 prevalans çalışmasının 5 tanesi kadın, 3 tanesi de erkek cinsiyetinin daha fazla olduğu yönündedir. Mevcut tek insidans çalışması ise vakaların %58,6’sının kadın olduğunu bildirmiştir. Yaşın LCD için de bir risk faktörü olduğu, buna karşılık Brezilya ve Sri Lanka ile Japonya kırsal alanından aynı prevalans rakamlarının bildirilmiş olmasına dayanılarak çevresel faktörlerin rolü olmadığı ifade edilmiştir.
Klinik Tablo
LCD AH’ye benzer tarzda, senil dönemde sinsi başlangıçlı, kronik progresif seyirli, genellikle belirgin görsel-mekansal ve yürütücü fonksiyonlarda bozulmanın ön planda olduğu bir demans tablosudur. Son yıllarda edinilen bilgilerden sonra, LCD’nin klinik ve patolojik tanı kriterleri için, McKeith ve arkadaşları tarafından 1996 yılında tanımlanan uzlaşı kriterlerinin kabul edilebilir bir özgüllüğünün olmasına rağmen duyarlılığının suboptimal olması ve bununla beraber temel özelliklerden olan zihinsel işlevlerde dalgalanmanın tanınmasındaki zorluklar ve temel özelliklerin (görsel halüsinasyonlar, seyirde dalgalanma ve parkinsonizm) üçünün bir arada düşük sıklıkta görülüyor olması nedeniyle tanı kriterlerinde revizyonlar yapılması ihtiyacı doğmuştur. McKeith ve arkadaşları tarafından son olarak 2017 yılında yayınlanan yeni uzlaşı kriterleri Tablo 1’de belirtilmiştir.
Revize edilmiş kriterlerde önceki merkezi (zorunlu), çekirdek ve destekleyici özellikler kategorilerine inceleme bulgularından oluşan gösterge niteliğinde biyobelirteçler ve destekleyici biyobelirteçler eklenmiştir. Revize kriterlerdeki en önemli değişikliklerden biri REM uykusu davranış bozukluğunun (RUDB) çekirdek özelliklere dahil edilmesidir.
Zorunlu Özellik
Hastanın sosyal ya da iş hayatındaki fonksiyonelliğini veya günlük rutin aktivitelerini etkileyen ilerleyici bir kognitif bozulma olarak tanımlanan demans LCD tanısı için zorunlu kriterdir. Bu kognitif kayıp erken evrede özellikle karmaşık dikkat, görsel-mekansal ve yürütücü işlevlerde bozulma ile karakterizedir. Hastalığın erken evresinde göreli olarak daha az olan bellek ile ilgili yakınmalar ve muayene bulguları ancak hastalık ilerledikçe aşikar hale gelir. Epizodik bellek ile ilgili testlerdeki performansları (çoktan seçmeli ile hatırlamaları serbest hatırlamadan iyidir) AH hastalarına göre daha iyidir. Ayrıca sözel bellek testlerinde görsel bellek testlerine göre daha iyi performans gösterirler. Erken ve ön planda görsel-mekansal bozukluk LCD için tipiktir. Mini mental durum değerlendirme testinde (MMSE) AH’ye kıyasla göreli yüksek puan alabilen, hatta demans için kesme puanının üzerinde kalan bir hastanın kesişen beşgenlerden puan kaybetmesi özellikle ayırt ettiricidir. Görsel-mekansal bozukluğun şiddeti erken dönemde posterior kortikal atrofide izlenen şiddette bir simultanagnoziye neden olabilir.
Çekirdek Klinik Özellikler
Dalgalanma: Klinik pratikte çekirdek kriterler arasında en zor karakterize edilebileni zihinsel işlevlerdeki deliryum-benzeri dalgalanmalardır. Bu kriter için uyanıklık durumu, dikkat veya kognitif ve işlevsel performansta dakikalar, saatlerden, günler, haftalara kadar değişen sürelerde, hasta yakını ve bazen de hekim için aşikar kayda değer dalgalanmalar geçerli sayılmalıdır. Dalgalanmalar diğer demansların ileri evrelerinde de görülebileceği için özellikle erken dönemde ortaya çıktıklarında LCD’yi öngördürebilir.
Görsel halüsinasyonlar: Hastalığın genellikle erken evrelerinde görülen tekrarlayıcı canlı, renkli, kompleks görsel halüsinasyonlar tanı için çekirdek özelliklerden ikincisidir. Halüsinasyonlar tipik olarak iyi forme insanlar, çocuklar veya hayvanları içerir, geçiş halüsinasyonları ve görsel illüzyonlar da eşlik edebilir. Hastaların halüsinasyonlara tepkisi iç görünün korunmasına ya da halüsinasyonların yarattığı duygusal yanıta göre değişiklik gösterir.
Görsel halüsinasyonları olan LCD’li hastaların (%80), halüsinasyonları olmayan grup ile karşılaştırıldığında görsel-algısal bozulmaları daha belirgindir. SPECT görüntülemelerinde oksipital kortekste tutulum azalmıştır. Görsel halüsinasyonları olan posterior kortikal atrofili (PKA) grubun, olmayan PKA’lılara göre bunların gerçekte LCD olduklarını düşündürecek şekilde daha fazla parkinsonizm ve RUDB’ye sahip oldukları gösterilmiştir. Otopsi çalışmalarında, görsel halüsinasyonları olan vakalarda, görsel kompleks imajların jeneratörü olduğu bilinen anterior ve inferior temporal lob ve amigdalada Lewy cisimciklerinin artmış olduğu bildirilmiştir. Görsel halüsinasyonları olan hastalarda kortikal asetilkolin defisitleri daha belirgindir ve bu durum belki de kolinerjik tedaviye daha iyi yanıtın nedeni olabilir. Görsel halüsinasyonları karakterize ve kantifiye etmek için değerlendirme skalaları mevcuttur.
RUDB: Uykudaki sorunlar demans ve parkinsonizmden çok önce başlamış olabilir. RUDB LCD’ye eşlik eden aşırı gündüz uyuklaması gibi diğer uyku bozukluklarıyla birlikte zihinsel işlevlerdeki dalgalanmalara katkıda bulunuyor olabilir. RUDB öyküsü polisomnografi ile doğrulanabilir.
Parkinsonizm: Spontan parkinsoniyen özellikler LCD’de sıktır (>%85). LCD’de ekstrapiramidal motor bulguların ağırlığı genellikle demansı olsun veya olmasın benzer yaş grubundaki Parkinson hastalarındaki (PH) motor bulguların ağırlığına benzer şekilde olabileceği gibi daha geri planda ve hafif de olabilir. PH’nin ayırdedici asimetrik sunumu belirgin olmayabilir. LCD’de postüral instabilite, yürüyüş bozukluğu, bradimimi gibi aksiyal tutulum, demanssız PH’lilere göre daha belirgindir. İstirahat tremoru daha az görülür. Parkinsonizmin L-Dopa cevabı çok iyi olmayabilir. Miyoklonus, tedavi gerektirmeyecek kadar hafif bir parkinsonizm, tremorun yokluğu, L-Dopa cevapsızlığı özelliklerinden birinin LCD’de PH’ye göre 10 kat daha muhtemel olduğu bildirilmiştir.
Destekleyici Klinik Özellikler
Destekleyici özellikler LCD’de sıklıkla görülür ancak özgüllükleri çekirdek ve işaretleyici özellikler kadar yüksek değildir. Nöroleptik duyarlılık reaksiyonlarıyla birlikte olan yüksek mortalite ve morbidite nedeniyle, D2 reseptör bloke edici ajanlar tanı veya tedavi amacıyla kullanılmamalıdır. LCD’li hastalar ve demansı olsun olmasın PH’liler özellikle tipik nöroleptiklere karşı, demansta kullanılmaya alışılan çok düşük dozlarına dahi aşırı duyarlılık gösterirler ve ağır, akut bir akinetik-rijid tablo geliştirebilirler. Bu durum malign nöroleptik sendrom şiddetinde olabilir ve ketiapin ile dahi bildirilmiştir. Ancak LCD tanısı alan hastaların yaklaşık %50’sinde tipik veya atipik antipsikotik ajanlar almalarına rağmen belirgin yan etki bildirilmemesine dayanarak, nöroleptik duyarlılığının olmaması, LCD tanısını dışlamazken varlığı ise tanıyı kuvvetle destekler.
Hastalığın erken döneminde ortostatik hipotansiyon, üriner inkontinans, nörokardiyovasküler instabilite, konstipasyon, impotans gibi otonomik semptomlar görülebilir. Bazı hastalarda görülebilen tekrarlayan düşmeler, senkop ve geçici tepkisizlik epizodlarının gelişmesine otonomik disfonksiyon da katkıda bulunuyor olabilir. Hezeyanlar içinde misidentifikasyon ve Capgras hezeyanı özellikle sıktır. Sistematize hezeyanlar, apati, anksiyete ve depresyon hastalığın herhangi bir döneminde görülebilir.
Gösterge Niteliğinde Biyobelirteçler
Tek foton emisyon bilgisayarlı tomografisi (SPECT) veya pozitron emisyon tomografisi (PET) görüntülemeleriyle gösterilebilen bazal gangliada düşük dopamin taşıyıcı tutulumu (dopamin taşıyıcı taraması -DAT) görüntülemenin LCD'yi AH'den %78 duyarlılık ve %90 özgüllük ile ayırt edebildiği gösterilmiştir. Yine de otopsi onaylı bazı LCD hastalarında nigral nöronların kısıtlı kaybı, minimal beyinsapı tutulumu veya dopaminin tüm striatumda dengeli bir şekilde kaybedildiği durumlarda normal DAT tutulumu olabileceği bildirilmiştir. Bu görüntülemenin idiopatik PH, multi-sistem atrofi (MSA), KBD ve PSP’de de bozuk olabileceği göz önünde tutulmalıdır.
123Iodine-MIBG miyokard sintigrafisinin bozuk (düşük tutulum) olması: [I-123]-metaiodobenzyl guanidin (MIBG) ile yapılan ve postganglionik sempatik kardiyak innervasyon hakkında bilgi veren sintigrafi LCD’de anormaldir ve AH’den ayırmada özgüllük ve duyarlılığı yüksektir. Başka bir çalışmada bu ayrımın özellikle de belirgin ortostatik hipotansiyonu olan hastalarda parkinsonizm olmaksızın da aşikar olduğu gösterilmiştir.
Atonisiz REM uykusunun polisomnografik konfirmasyonu: Atonisiz REM uykusunun polisomnografi ile konfirmasyonu Lewy patolojisinin oldukça spesifik bir göstergesidir. Demans ve RUDB olan bir kişide polisomnografik inceleme atonisiz REM uykusu olduğunu gösteriyorsa bu durum %90 oranında sinükleinopatiye işaret eder ve diğer çekirdek bulgular veya biyobelirteçler yokluğunda dahi muhtemel LCD tanısı için yeterlidir.
Destekleyici Biyobelirteçler
Beyin tomografi/manyetik rezonans görüntülemede (MRG) hippokampal ve medial temporal lob volümlerinin korunmuş olması, putamen atrofisi ve MRG’de oksipital atrofi olmaksızın SPECT ve PET ile oksipital hipoperfüzyon ve hipometabolizmanın görülmesi tanı için yardımcıdır. FDG-PET’de posterior veya orta singulat bölgede metabolizmanın göreceli olarak korunmuş olduğu ‘singulat ada işareti’ bildirilmektedir. Yukarıda söz edildiği gibi MRG’de PKA tarzı oksipito-parietal atrofi sergileyen hastalar arasında görsel halüsinasyonlar kuvvetle LCD düşündürse de bu bulgu ayırt edici değildir.
Elektroensefalografide (EEG) belirgin posterior yavaş dalga aktivitesi ile birlikte pre-alfa/teta aralığında periyodik dalgalanmalar: EEG’de posterior bölgelerdeki spesifik anormalliklerin LCD biyobelirteci olabileceği yönünde kanıtlar birikmektedir ve bu bulgunun AH ile karşılaştırıldığında tanı için %90’ın üzerinde öngörü sağladığı bildirilmiştir. Bu pre-alfa/teta aralığındaki EEG değişiklikleri bilişsel dalgalanmaların şiddeti ile de pozitif korelasyon gösterir ve hastalığın hafif kognitif bozukluk (MCI) döneminde de görülebilir.
Genetik ve sıvı biyobelirteçleri: Şu anki bilgilerin ışığında, LCD tanısını desteklemek için klinik olarak uygulanabilir genotipik veya beyin-omurilik sıvısı (BOS) işaretleyicisi yoktur.
Tablo 1. Muhtemel ve Mümkün LCD Klinik Tanısı için Gözden Geçirilmiş Yeni Uzlaşı Kriterleri (McKeith ve ark., 2017)
LCD tanısı için demans (normal sosyal ya da mesleki işlevselliği etkileyecek ağırlıkta ilerleyici kognitif bozulma) olması zorunludur. Erken evrelerde bellek bozukluğu baskın ve kalıcı düzeyde olmayabilir, fakat ilerlemeyle birlikte aşikar hale gelir. Dikkat, frontal-subkortikal yetenekler ve vizuo-spasyal işlevleri ölçen testlerdeki bozulmalar ön planda olabilir ve erken ortaya çıkabilir.
Çekirdek klinik özellikler (İlk üçü tipik olarak erken ortaya çıkar ve hastalık sürecinde devam edebilir)
a. Dikkat ve uyanıklıkta ciddi değişikliklerle birlikte giden zihinsel işlevlerde dalgalanma
b. Tipik olarak iyi forme ve ayrıntılı, tekrarlayıcı görsel halüsinasyonlar
c. REM uykusu davranış bozukluğu, kognitif bozulmadan önce görülebilir
d. Parkinsonizmin kardinal özelliklerinin bir veya fazlasının kendiliğinden mevcudiyeti: Bunlar bradikinezi (harekette yavaşlık, amplitüd veya hızda azalma), istirahat tremoru veya rijiditedir.
Destekleyici klinik özellikler
Antipsikotik ajanlara ağır duyarlılık; postüral instabilite; tekrarlayan düşmeler; senkop veya diğer geçici tepkisizlik epizodları; ağır otonomik disfonksiyon, örneğin, konstipasyon, ortostatik hipotansiyon, üriner inkontinans; hipersomni; hipozmi; diğer modalitelerde halüsinasyonlar; sistematize hezeyanlar; apati, anksiyete ve depresyon.
Gösterge niteliğinde biyobelirteçler
SPECT veya PET görüntülemeleriyle gösterilebilen bazal gangliada düşük dopamin taşıyıcı tutulumu
123Iodine-MIBG miyokard sintigrafisinin bozuk (düşük tutulum) olması
Atonisiz REM uykusunun polisomnografik konfirmasyonu
Destekleyici biyobelirteçler
BT/MRG’de medial temporal lob yapılarının göreli olarak korunmuş olması
SPECT ve PET’de oksipital aktivitede azalma ile birlikte genel olarak düşük tutulum ± FDG-PET görüntülemede “”singulat ada” işareti
EEG’de belirgin posterior yavaş dalga aktivitesi ile birlikte pre-alfa/teta aralığında periodik dalgalanmalar
Muhtemel LCD tanısı için:
a. İki veya daha fazla çekirdek klinik belirti vardır, gösterge niteliğinde biyobelirteçler olabilir veya olmayabilir,
veya
b. Sadece bir çekirdek klinik belirtiyle birlikte bir veya daha fazla gösterge niteliğinde biyobelirteç vardır
Muhtemel LCD tanısı tek başına biyobelirteçlere dayanarak konulmamalıdır.
Mümkün LCD tanısı için:
a. Sadece bir LCD çekirdek klinik belirtisi vardır ancak gösterge niteliğinde biyobelirteç kanıtı yoktur,
veya
b. Bir veya daha fazla gösterge niteliğinde biyobelirteç vardır ancak çekirdek klinik belirti yoktur
LCD tanısı ihtimali aşağıdakilerin mevcudiyeti durumunda azalır:
a. Klinik tabloya tamamen ya da kısmen neden olabilecek, serebrovasküler hastalığı da içeren, başka bir fiziksel hastalık veya beyin hastalığının varlığında, yine de bunlar LCD tanısını dışlamaz ve klinik sunuma katkıda bulunan mikst veya multipl patoloji olduğunu gösterebilir,
veya
b. Eğer parkinsonizm özellikleri tek çekirdek klinik özellikse ve ilk kez ağır demans evresinde ortaya çıkarsa
LCD tanısı, demans parkinsonizmden (eğer varsa) önce veya birlikte ortaya çıkar ise düşünülmelidir. Parkinson hastalığı demansı (PHD) terimi, yerleşmiş Parkinson hastalığı bağlamında oluşan demansı tanımlamak için kullanılmalıdır. Pratik uygulamada, klinik durum için en uygun terim kullanılmalıdır ve ‘Lewy cisimcik hastalığı’ gibi genelleyici terimler sıklıkla yardımcıdır. LCD ve PHD ayrımı yapılması gereken araştırma projelerinde ise demansın başlangıcı ile parkinsonizm arasındaki 1 yıl kuralı, LCD tanısı için hala önerilmeye devam edilmektedir.
Prodromal Lewy Cisimcikli Demans
LCD hastalarının demans öncesi evrede tanınması için oluşturulan Prodromal LCD Tanı Çalışma Grubu, bu yıl içinde Prodromal LCD tanısı için araştırma kriterlerini tanımlamıştır. LCD’nin prodromal sendromları (1) hafif kognitif bozukluk (MCI-LC), (2) deliryum başlangıçlı ve (3) psikiyatrik başlangıçlı olmak üzere üç ayrı prototipe ayrılmıştır. Bu üç prodromal LCD sendromu ayrı ayrı tanımlanmış olsa da bu prototiplerin birbirini dışlamadığı ve önemli ölçüde örtüşme olabileceği belirtilmiştir. Muhtemel ve mümkün MCI-LC klinik tanısı için araştırma kriterleri tanımlanmış ancak diğer iki prodromal tip, yani deliryum başlangıçlı ve psikiyatrik başlangıçlı olan sendromların tanısı henüz netleştirilememiş ve bu iki sendrom için spesifik kriterlerin kullanımı henüz konfirme edilememiştir.
Lewy Cisimcikli Hafif Kognitif Bozukluk (MCI-LC) kognitif tutulum paterni olarak LCD’ye benzer ve tipik olarak dikkat, yürütücü işlevler ve görsel işlemlemede daha baskın etkilenmenin olduğu, bellek ve obje isimlendirmenin ise rölatif olarak korunduğu bir profil gösterir. MCI-LC’de kognitif performans profili sıklıkla tek alan non-amnestik MCI ya da çoklu alan non-amnestik MCI ile uyumludur. Muhtemel ve mümkün MCI-LC’nin klinik tanısı için belirlenmiş araştırma kriterleri Tablo 2’de gösterilmiştir.
Deliryum başlangıçlı prodromal LCD'li hastalarda deliryum demans gelişmeden önce ilk yakınma olarak ortaya çıkabilir. LCD'li hastalar deliryuma diğer demanslardan daha duyarlıdır. Deliryum epizodları, ameliyat, enfeksiyonlar, ateş, diğer sistemik hastalıklar veya alkol/psikoaktif ilaçların kullanımı gibi birçok faktör tarafından tetiklenebilir. Deliryum için yeterli provoke edici faktörlerin bulunmadığı, uzun süreli veya tekrarlayan deliryum öyküsü olup kognitif açıdan giderek bozulma gösteren hastalarda özellikle prodromal LCD'den şüphelenilmelidir. Deliryum başlangıçlı prodromal LCD'nin tanısında MCI-LC biyobelirteçlerinin kullanımı yardımcı olabilir, ancak bu prototiplere özel biyobelirteç bulguları hakkında henüz yeterli kanıt bulunmamaktadır.
Psikiyatrik başlangıçlı prodromal LCD tipik olarak geç başlangıçlı majör depresif bozukluğa veya geç başlangıçlı psikoza karşılık gelen baskın psikiyatrik semptomlarla karakterizedir. Görsel ve diğer modalitelerde halüsinasyonlar ve Capgras sendromunu da içerebilen sistematik sanrılar içerir. Aynı zamanda apati, anksiyete ve depresyonla da ortaya çıkabilir ve bulgular hastane yatışı gerektirecek kadar şiddetli olabilir. Bu hastalarda hafif kognitif bozukluk mevcut olabilir, ancak baskın değildir, dalgalanabilir ve psikiyatrik hastalığa bağlı kognitif durumla ilişkili görünebilir. Bradikinezi, depresif bozukluklarda yaygın olarak görülen psikomotor gerilikle karışabilir. Geç başlangıçlı psikiyatrik semptomları baskın olan bu prototipin tanı kriterleri konusunda henüz bir görüş birliği oluşmamıştır. Mortalite ve morbiditeyi artıran ağır antipsikotik duyarlılık reaksiyonları nedeniyle klinisyenlerin bu ihtimalin farkında olmaları önemlidir. Mevcut kanıtlar henüz kesin seviyede olmasa da, şüphe duyulan olgularda tanı için 123I-MIBG sintigrafi gibi önerilen MCI-LC biyobelirteçlerinden destek alınabilir.
Tablo 2. Muhtemel ve Mümkün Lewy Cisimcikli Hafif Kognitif Bozukluk (MCI-LC) Klinik Tanısı için Araştırma Kriterleri (McKeith ve ark., 2020)
MCI-LC tanısı için aşağıdakilerin her birinin varlığıyla tanımlanmış MCI bulunması zorunludur:
a. Hasta, bilgi veren ya da klinisyen tarafından bildirilen kognitif gerilemeye dair endişe
b. Bir veya daha fazla kognitif alanda bozulmaya dair objektif kanıt, kognitif bozukluk herhangi bir alanı içerebilir, ancak daha çok dikkat-yürütücü işlevler ve/veya görsel işlemleme eksiklikleri ön plandadır
c. Fonksiyonel yeteneklerde daha önce elde edilen bağımsız performansın korunmuş olması veya demans kriterlerini karşılamayacak şekilde minimum düzeyde etkilenme
Çekirdek klinik özellikler
a. Dikkat ve uyanıklıkta değişikliklerle birlikte giden zihinsel işlevlerde dalgalanma
b. Tekrarlayıcı görsel halüsinasyonlar
c. REM uykusu davranış bozukluğu
d. Parkinsonizmin kardinal özelliklerinin bir veya fazlasının kendiliğinden mevcudiyeti: Bunlar bradikinezi (harekette yavaşlık, amplitüd veya hızda azalma), istirahat tremoru veya rijiditedir.
Önerilen biyobelirteçler
a. SPECT veya PET görüntülemeleriyle gösterilebilen bazal gangliada düşük dopamin taşıyıcı tutulumu
b. Atonisiz REM uykusunun polisomnografik konfirmasyonu
c. Miyokard sintigrafisinde azalmış meta-iyodobenzilguanidin (MIBG) tutulumu
Muhtemel MCI-LC tanısı için:
a. LCD’nin iki veya daha fazla çekirdek klinik belirtisi mevcuttur, önerilen biyobelirteçler olabilir veya olmayabilir,
veya
b. Sadece bir çekirdek klinik belirtiyle birlikte bir veya daha fazla önerilen biyobelirteç vardır
Muhtemel MCI-LC tanısı, tek başına biyobelirteçlere dayanarak konulmamalıdır.
Mümkün MCI-LC tanısı için:
a. Sadece bir LCD çekirdek klinik belirtisi vardır ancak önerilen biyobelirteçler yoktur,
veya
b. Önerilen biyobelirteçlerden biri veya daha fazlası mevcuttur, ancak çekirdek klinik belirti yoktur
Destekleyici klinik belirtiler
Antipsikotik ajanlara ağır duyarlılık; postüral instabilite; tekrarlayan düşmeler; senkop veya diğer geçici tepkisizlik epizodları; uzamış ya da tekrarlayan deliryum, ağır otonomik disfonksiyon, örn. konstipasyon, ortostatik hipotansiyon, üriner inkontinans; hipersomni; hipozmi; diğer modalitelerde halüsinasyonlar (geçiş ve varlık hissi fenomenleri dahil); sistematize hezeyanlar; apati, anksiyete ve depresyon.
MCI-LC için potansiyel biyobelirteçler
a. Kantitatif EEG’de yavaşlama ve dominant frekansta değişkenlik görülmesi
b. Yapısal görüntülemede medial temporal lob yapılarının göreli olarak korunmuş olması
c. MRG'de insular incelme ve gri cevher hacminde kayıp
d. Perfüzyon/metabolizma görüntülemelerinde düşük oksipital tutulum
MCI artı destekleyici klinik belirtiler veya potansiyel biyobelirteçler, MCI-LC teşhisi için yetersizdir, ancak şüphe uyandırabilir ve biyobelirteç araştırmasını başlatabilir, böylece bir MCI-LC teşhisine ağırlık kazandırabilir.
MCI-LC ihtimali klinik tabloya tamamen ya da kısmen neden olabilecek, serebrovasküler hastalığı da içeren, başka bir fiziksel hastalık veya beyin hastalığı varlığında azalır, yine de bunlar MCI-LC tanısını dışlamaz ve klinik sunuma katkıda bulunan karma veya çoklu patoloji olduğunu gösterebilir.
Nöropatoloji
Nöropatolojik olarak tabloya adını veren intraselüler LCi başlıca özelliktir. LCi’ler, intranöronal sitoplazmik, eozinofilik, sferik, filamantöz inklüzyon cisimcikleridir. LCi’lerin temel bileşeni α-sinüklein isimli proteindir. 4. kromozomda kodlanan ve ağırlıkla akson sonlanmalarında lokalize olan bu proteinin işlevi çok iyi aydınlatılamasa da, aksonal nakilde rol oynadığı ve sinaptik bütünlüğün korunması şeklinde bir plastisite rolü olduğu düşünülmektedir. LCi’ler, α-sinükleinin anormal katlanması ve fosforilasyonu sonrasında oluşurlar. PH’de ağırlıklı olarak substantia nigranın dopaminerjik hücre gövdelerini içeren pars kompaktasına sınırlı olan LCi ve Lewy nöritleri, LCD’de beyinsapına ilave olarak limbik-paralimbik sistem ve neokortekste birikir. Bununla birlikte saf LCD nöropatolojisi tüm LCD klinik fenotipinin küçük bir bölümünden sorumludur. Klinik olarak LCD tanısı alan hastaların postmortem çalışmalarında AH patolojisi de sıklıkla bir arada görülmekte ve hem nomenklatürü (AH’nin Lewy cisimcikli varyantı, yaygın Lewy cisimciği hastalığı gibi) hem de klinik fenotipi bulanıklaştırmaktadır. Örneğin, daha belirgin olarak neokortikal AH tipi patolojisi olan LCD hastalarında, klasik LCD klinik bulguları da daha hafiftir. LCD’li hastalarda en sık görülen Alzheimer patolojisi amiloid plaklar iken nörofibriler yumaklar (NFY) daha seyrektir. NFY’ler görüldüğü takdirde, AH’ye kıyasla daha düşük Braak ve Braak evrelerinde, ağırlıkla limbik ve paralimbik alanlarda, amiloid plaklar (AP) de nöritik de olsalar, genellikle eşleşmiş helikal filamentler (PHF) olmaksızın ve neokortekse sınırlı olma eğilimi taşımaktadırlar. Ayrıca, LCD’deki AP çevresinde mikroglial aktivasyon ve inflamasyona da AH’ye kıyasla çok daha az rastlandığı ileri sürülmektedir. Hippokampal CA2 ve CA3 bölgelerinde görülen ubikütin pozitif nöritik dejenerasyonun da AH’de görülmeyen LCD’ye özgü bir patolojik özellik olduğu ileri sürülmektedir.
LCD nörokimyasal olarak kortikal kolinerjik işaretleyiciler ve nigrostriatal dopamin kaybı ile karakterizedir. Raphe çekirdeği ve locus ceruleus’taki nöron kaybı ve gliozis serotonin ve norepinefrin azalmasına neden olur ve duygudurum, dikkat ve uyanıklıktaki dalgalanmaları açıklayabilir. Meynert çekirdeğindeki kolinerjik nöron kaybı da bellek, dikkat, uyanıklığın derecesi ve belki de dalgalanmaları anlamlı olarak etkileyebilir. Meynert çekirdeğindeki dejenerasyona ek olarak başlıca işlevi diurnal ritmin düzenlenmesi olan talamik kolinerjik innervasyonun kaynağı pedinkulo-pontin çekirdek de AH’den farklı olarak dejenerasyona katılır. Anılan beyinsapı nörotransmitter kayıplarının LCD’ye özgü parasomni, parkinsonizm ve otonom sinir sistemi belirtilerinden sorumlu olduğu düşünülmektedir. Bununla birlikte, nöropatolojik olarak konfirme hastalardan %18’inin hastalık boyunca motor bozukluk geliştirmediği ve LCD hastalarının %10’unda beyinsapı tutulumuna rastlanmadığı bildirilmiştir. Kolin asetiltransferaz (ChAT) aktivitesi ile belirlenen neokortikal kolinerjik kayıp AH’ye göre çok daha belirgindir. Bu kaybın genel olarak demans şiddetiyle, özel olarak temporal lober kaybın ise davranışsal belirtilerle korelasyonu bildirilmiştir.
Çözünemeyip biriken fibriler α-sinükleinin, LCD’nin yanı sıra bir dizi dejeneratif hastalıkta daha temel patolojik işaretleyici olduğunun gösterilmesi sonrasında bu grup α-sinükleopatiler olarak adlandırılmaya başlanmıştır. Bunlar arasında LCD’den ayrı bir antite olup olmadığı tartışmalı olan PH demansı öncelikle sayılabilir. Kısa süre öncesine kadar bir PH-AH örtüşmesi olduğu düşünülen idyopatik PH’de sonradan gelişen demansın, artık beyinsapı tipi LC hastalığının zamanla kortikal tip LC’ye yaygınlaşması sonucu geliştiğine dair bir görüş birliği oluşmaktadır. α-sinüklein fibriler birikimleri MSA’da glial inklüzyonlar şeklinde görülmektedir. Glial α-sinüklein inklüzyonları LCD ve PH’de geç evrelerde görülürken, MSA’da klinik fenotipten sorumlu patolojik değişiklik gibi durmaktadır. Pantotenat kinaz ilişkili nörodejenerasyon (PKAN) hastalığında nöronal ve glial inklüzyonların yanısıra aksonal sferoidler içinde de görülürler. Bir çalışmada otonomik ganglionlarda ilerleyici dejenerasyonla giden saf otonomik yetmezlik de bir α-sinükleinopati olarak tarif edilmiştir.

Şekil 1. Lewy cisimciği. Solda substantia nigra’da çevresinde halosuyla intranöronal eozinofilik inklüzyon izlenirken sağda ise kortekste ubikitin (+), halosu seçilmeyen intranöronal inklüzyon izlenmekte (Kaynak: http://www.path.sunysb.edu/faculty/woz/NPERESS/webclass5.htm)
Sağkalım
LCD’de sağkalım AH’ye kıyasla belirgin olarak daha kısa gibi durmaktadır. Yakın zamanda yapılan 473 hastayı içeren doğal bir kohort çalışmasında klinik olarak LCD tanısı almış hastaların medyan sağkalımı 3,72 yıl ve AH hastalarının sağkalımı ise 6,95 yıl olarak bildirilmiştir. Daha önceki çalışmalar LCD için hastalık başlangıcından itibaren 5,5-7,7 yıl ve tanıdan itibaren 1,9-6,3 yıl sağkalım bildirmişlerdir. AH’ye göre sağkalımın kısalmasının, özellikle motor ve otonom bozukluklar olmak üzere, LCD’nin zengin klinik sunumunun engelliliği çok daha fazla arttırması nedeniyle olduğu ileri sürülebilir. Otopsi doğrulamalı büyük bir çalışmada LCD ve PHD 243 olgunun retrospektif analizinde LCD için medyan sağkalımın 5 yıl olduğu bildirilmiştir. PHD’liler LCD’lilere göre daha uzun sağkalıma sahiptiler. Hastalık başlangıcında aşikar dalgalanma kötü sonlanımı en iyi öngören özellik olarak belirlenmiştir.
Tedavi
LCD hastasını tedavi ederken ilk adım hastanın semptomlarına olumsuz katkıda bulunabilecek ilaçların belirlenmesi ve bu ilaçlardan kaçınılmasıdır. Bu ilaç grupları öncelikle benzodiazepinler, antikolinerjik/antimuskarinik ilaçlar (örneğin, inkontinans tedavisi için), çeşitli antipsikotikler ve trisiklik antidepresanları içerir.
Non-farmakolojik yaklaşım: Hastalar motor semptomlara yönelik fizik tedavi ve genel egzersiz uygulamalarından fayda görebilirler. Görsel halüsinasyonlar ve kognitif kötüleşme, dikkat ve uyanıklığın az olduğu zamanlarda daha ağır olabilir. Bu nedenle sosyal ilişkilerin arttırılması gibi yaklaşımlarla uyanıklığın ve dikkatin arttırılması yararlı olabilir. Non-farmakolojik girişimlerin LCD’deki etkinliği ile ilgili için henüz yeterli veri oluşmamıştır.
Farmakolojik yaklaşım: LCD’de parkinsonizmin L-Dopa cevabının iyi olmaması ve dopaminerjiklerin psikotik belirtileri arttırma riskine karşın hastanın kendisine optimize edilmiş bir antiparkinson tedaviden yararlanıp yararlanmayacağı tetkik edilmelidir. L-dopa düşük dozlarda başlanıp, yavaş doz artışı yapılarak psikiyatrik semptomların artmasını engellemek amacıyla gerekli olan minimum dozda kullanılmalıdır. Diğer antiparkinson ilaçlar hastanın kognitif ve davranışsal semptomlarını kötüleştirebileceği için kullanılmamalı, tremor gibi semptomlar için antikolinerjik kullanımından özellikle kaçınılmalıdır. Rahatsızlık vermeyen halüsinasyonlar tedavi gerektirmeyebilir. Farmakolojik yaklaşımın gerektiği durumlarda atipik antipsikotikler ve kolinesteraz inhibitörleri (ChEİ) düşünülmelidir. Yeni tanı alan bir hastada kognitif tedavi amaçlı başlanan ChEİ, nöroleptik tedaviye karşı gelişebilecek aşırı duyarlılık da göz önüne alınarak görsel halüsinasyonlar ve diğer psikotik belirtilerin tedavisi için bir süre tek başına denenebilir ve cevap olmadığında atipik nöroleptiklerden ketiapin ve klozapin seçilebilir. Tipik antipsikotiklerin kullanımından kaçınılmalıdır. Depresyon ve anksiyete için SSRI veya SNRI’lar kullanılabilir. Antikolinerjik etkileri nedeniyle trisiklik antidepresanlardan kaçınılmalıdır. Apati üzerine etki için kanıtlar anektodaldir; ChEİ ve SSRI/SNRI kullanımı denenebilir, öğlene kadar düşük miktarda kahve kullanımı da önerilmiştir. RUDB için ilk tedavi seçeneği melatonindir ve 3-12 mg arası dozlarla tedavi edilebilir. Etkilidir ve genellikle iyi tolere edilir. Melatoninden yeterli etki alınamadığı durumlarda 0,25 mg ile klonazepam başlanabilir. Doz artışı her zaman yavaş yapılmalı ve yan etkiler açısından dikkatli olunmalıdır. Aşırı gündüz uyuklaması hastalarda zihinsel performansın düşmesine katkıda bulunabilir. Tedavide sabah ve öğlen dozları şeklinde 100-400 mg arası modafinil veya 150-250 mg/gün armodafinil kullanımı ile yarar sağlanabilir.
Kognitif semptomların tedavisinde kolinerjik temelli tedavi AH’ye benzer biçimde yararlı olabilir. Parkinson hastalığı demansı için Amerikan Gıda ve İlaç Dairesi (FDA) onayı olan rivastigminin aksine LCD tedavisi için FDA onayı olan ChEİ henüz bulunmamaktadır. Ancak çeşitli sistematik gözden geçirmeler ve metaanalizler ChEİ kullanımını destekler nitelikte olup bu çalışmalar LCD hastalarında özellikle donepezilin daha iyi tolere edildiğini ve daha iyi bir risk/fayda profili gösterdiğini bildirmektedir. Memantin için kanıt daha az olup genellikle iyi tolere edildiği belirtilmiştir; tek başına ya da diğer ilaçlara ek olarak denenebilir.
PARKİNSON HASTALIĞI DEMANSI
PH uzun yıllar için saf bir motor bozukluk olarak tasarlandıktan sonra zaman içinde non-motor bozukluklar ve bunlar içinde ağırlıklı bir yeri olan kognitif bozukluklar kaydedilir olmuştur. Giderek yeterince uzun sürmüş bir PH’de kognitif bozulmanın belli bir şiddeti olarak demansın nerdeyse kaçınılmaz olduğu gözlenmiştir. Çalışmalarda kortikal LCi’lerin demans şiddetiyle korelasyon gösteren ana patolojik bulgu olduğu saptanmıştır. Braak ve arkadaşlarının anatomo-klinik korelasyon çalışmalarında (halen tartışmalı olsa da) alfa-sinüklein patolojisinin kaudo-rostral bir ilerleyişle beyinsapından limbik ve heteromodal asosiasyon kortekslerine doğru yükseldiğinin gösterilmesiyle PH’de demansın neden geç dönemde ortaya çıktığı sorusunun cevabı da teorik bir temele kavuşmuştur.
Epidemiyoloji
Belirli kriterlere göre seçilmiş̧ 12 prevalans çalışmasını sistematik olarak gözden geçiren bir metaanalizde PHD kesitsel nokta prevalansının %25-30 oranında olduğu bildirilmiştir.
Toplum-tabanlı prospektif bir çalışmada, Aarsland ve arkadaşları başlangıç, 4 yıl ve 8 yıl sonraki PHD kümülatif prevalansını sırasıyla %26, %51, %78 olarak bildirmişlerdir. Diğer çalışmalarda başlangıçta demansı olmayan Parkinson hastalarında 4 yıl sonunda %42, bir diğerinde ise 15 yıl içinde %48 oranında demans ve %36 oranında MCI geliştiği bildirilmiştir. Sekiz merkezin data analizinden elde edilen veriler demansı olmayan Parkinson hastalarının %25,8’inde MCI bulunduğunu bildirmiştir. Yirmi yıldan daha uzun süre yaşayan Parkinson hastalarında PHD prevalansı %83’e ulaşmakta ve ölümden önce hastaların büyük çoğunluğu demans bulgularını göstermektedir. Sağkalımdan etkilenmediği için prevalans ölçümlerine göre daha sağlıklı olan insidans hesaplamalarında üç ila beş yıllık izlem süresinde PH’de insidansın kontrol grubuna göre 4-6 kat daha fazla arttığı izlenmiştir.
PHD için ana risk faktörleri sırasıyla halüsinasyonların varlığı, ileri yaş, ağır motor özürlülük (özellikle bradikinezi-rijidite ve aksiyel semptomlar) ve konuşma bozukluğu olarak bilinmektedir. Değişik çalışmalarda bildirilen diğer risk faktörleri arasında şunlar sayılabilir: PH başlangıcının ileri yaşta olması, bradikinezi şiddeti, ağır Hoehn-Yahr evresi, postüral instabilite gibi aksiyel tutulum bulgularının belirgin olması, düşük eğitim düzeyi, depresyonun varlığı, erkek cinsiyet, hastalıkta ileri evre, akinetik-dominant tipte PH, L-dopa ile tedavi esnasında psikozun erken dönemde ortaya çıkması, hastanın ilk muayenesindeki mini-mental durum muayenesi puanının düşük olması ve yürütücü işlevler (özellikle sözel akıcılık), görsel-mekansal işlevler ve bellek fonksiyonlarında hafif de olsa işlev bozukluklarının varlığı. Özellikle ileri yaş ve ağır motor bulguları olan hastalar genç yaş ve daha hafif motor bulguları olan hastalara kıyasla yaklaşık 10 kat kadar artmış bir demans riski gösterirler. İki yıllık prospektif bir çalışmada postüral instabilite ve yürüme güçlüğü fenotipi bulunan hastalarda tremor-dominant gruba kıyasla PHD ortaya çıkma riskinin daha yüksek ve kognitif bozulma oranının daha hızlı olduğu bulunmuştur. Bir başka çalışmada başlangıçta tremor-dominant bir fenotip gösteren hastaların demans gelişmesinden önce tremor dominansını kaybedip akinetik-rijid belirtilerin ön plana çıktığı saptanmıştır.
Genetik özellikler
Ailevi PH’nin nadir nedenlerinden olan alfa-sinüklein geninde triplikasyon mutasyonu taşıyan ailelerde PH motor semptomlarının yanında, bazen erken dönemden itibaren demans görülebilirken, gen duplikasyonu olan ailelerde bu durum ya hiç olmamakta ya da nadir görülmektedir. PH’de kognitif bozuklukla ilişkili olduğu düşünülen diğer faktörler arasında glukoserebrozidaz (GBA) geninde mutasyonlar, MAPT geninin H1 haplotipi ve APOE4 alleli taşıyıcısı olmak yer alır.
Klinik Özellikler
PHD sinsi başlangıçlı ve yavaş ilerleyici bir demans tablosu ile karakterizedir. Tipik klinik özellikler ön planda bir yürütücü işlev bozukluğuyla birlikte dikkatte ve görsel mekansal işlevlerde erken dönemlerden itibaren bozulma (sıklıkla demansın genel şiddetinden daha ağır ölçüde), genelde tanımanın korunduğu daha hafif bir epizodik bellek bozukluğu, kelime akıcılığı ve kelime bulma zorluğu dışında göreli olarak korunmuş temel linguistik işlevleri içerir. Bazı hastalarda ileri, limbik tipte bellek bozukluğunun ön planda olduğu Alzheimer tipi bir demans tablosunun belirtileri tabloya katılabilir ya da ön plana geçebilir. Bu hastalar büyük olasılıkla her iki tip patolojinin değişik oranlarda bir arada olduğu karma-patolojili tabloları temsil ederler. AH ile kıyaslandığında PHD’de daha fazla apati, daha belirgin kognitif yavaşlama (bradifreni), dikkatte daha ağır ve daha dalgalanan bir bozulma vardır. Bu kognitif profile sıklıkla apati, halüsinasyon ve hezeyanlar gibi davranışsal semptomlar eşlik eder. Sık bir bulgu güniçi uyuklamaların artması, gündüz-gece ritminin tersine dönmeye başlamasıdır. LCD’de görülen dalgalanmalar PHD için de oldukça tipiktirler.
PHD’de davranışsal problemler ve kişilik değişiklikleri oldukça sıktır. Nöropsikiyatrik envanter (NPI) kullanılarak yapılan bir çalışmada PHD olan hastaların %89’unda bir veya daha fazla psikiyatrik semptom bulunduğu saptanmıştır. En sık görülen nöropsikiyatrik semptomlar depresyon, halüsinasyonlar, apati, anksiyete ve hezeyanlardır. Depresyon ya da depresif belirtiler hastaların en az %50’sinde görülüp hastalığın seyri esnasında herhangi bir zamanda ortaya çıkarlar. Halüsinasyonlar genelde görsel nitelikte olup LCD’de görülenlere benzerler. Kapı ya da zil sesi, veya ismin çağrılması şeklinde duysal halüsinasyonlar daha nadirdir; multi-modal halüsinasyonlar ise çok nadir görülürler. İçgörü sıklıkla korunurken bazen bozulur ve halüsinasyonlar korkutucu bir nitelik kazanırlar. Hezeyanlar en sık “phantom boarder” (evde bir yabancının olduğu hissi), hastanın arkasında ya da yanında birisinin durduğu hissi, bir gölgenin geçtiği düşüncesi, aldatma hezeyanları olarak ortaya çıkarlar. Hastalar bazen evlerinin kendi evleri olmadığı, ya da yakınlarının (sıklıkla eşlerin) kendi yakını olmadığı hissine kapılabilir ve “eve” gitmek isteyebilirler. Halüsinasyonlar ve hezeyanlar dopaminerjik ilaçların alımından sonra ortaya çıkabilir ya da belirginleşebilirler. Bu durum PHD olan ya da demans geliştirmekte olan hastalarda daha erken, daha kolay ve daha düşük dozlarda ortaya çıkar.
PHD’de motor fenotip sıklıkla simetrik bradikinezi, rijiidite ve postüral instabilitenin ön planda olduğu akinetik-rijid formdur. Daha önce tremor-dominansı gösteren hastalarda zamanla tremor tamamen ortadan kalkabilir. PHD’de L-dopa cevabının giderek azaldığı düşünülür. Ancak bu gözlem en az kısmen non-dopaminerjik bulguların giderek ön plana geçmesiyle açıklanabilir. LCD hastalarına benzer şekilde ölümle sonlanabilecek nöroleptik duyarlılığı PHD hastalarında da bildirilmiştir. Uyku ile ilgili bozukluklardan artmış gün uykusu yanında REM uykusu davranış bozukluğu da PHD’de sık görülen özelliklerden birisidir. Bu belirti demansı olmayan hastalarda da ortaya çıkabilmekle birlikte, demanslı hastalarda muhtemelen daha sıktır ve bilinen bir Parkinson hastasında yeni olarak ortaya çıkması kognitif bozulmanın ön habercisi olabilir. Kapsamlı ve karşılaştırmalı olarak araştırılmamalarına karşın ortostatik hipotansiyon, senkop, düşmeler, barsak ve mesane bozuklukları, kalp atım hızında azalma, seksüel disfonksiyon gibi otonomik bozuklukların demansı olan hastalarda daha sık görüldüğü düşünülmektedir.
Tanı Kriterleri
“Movement Disorders Society”’nin oluşturduğu ve Çalışma Grubu tarafından hazırlanan PHD klinik tanı kriterleri (Tablo 3), ilk yazarı kliniğimiz hocalarından Murat Emre olacak şekilde 2007 yılında yayınlanmıştır (Tablo 3).
PHD tanısı temel prensipleri itibarıyle herhangi bir hastada demans tanısı koymaktan farklı değildir. İlk aşamada demansı taklit edebilen diğer durumların ekarte edilmesi gerekir. İkinci aşama ayırıcı tanıyı oluşturur. Bu aşamada demans ve parkinsonizm belirtilerinin bir arada olabileceği diğer hastalıklar (PSP, KBD, vasküler demans, normal basınçlı hidrosefali ve LCD gibi) düşünülmeli, PH’ye eklenebilecek subdural hematom, B-12 vitamin eksikliği gibi diğer nedenler de göz ardı edilmemelidir. Ayrıntılı bir öykü, tüm kognitif alanları içeren mental muayene, diğer hastalıkları dışlayan laboratuvar incelemeleri ile yukarıda sözü edilen PHD klinik tanı kriterlerinin dikkatlice uygulanması tanıyı kolaylaştırır.
PHD için özgül bir nörogörüntüleme bulgusu yoktur. BT ve MRG gibi yapısal görüntülemelerde daha ziyade beynin posterior bölgelerinde atrofi dikkat çeker; hippokampal ve mediyal-temporal atrofi eşlik edebilmesine karşın AH’de görülen kadar sık ve ağır değildir. Fonksiyonel görüntülemede SPECT ve PET incelemelerinde frontal ve temporoparietal kortekste hipoperfüzyon bulunur; ancak bu bulgu AH’de de ortaya çıkabildiği için özgül değildir ve tek bir hastanın ayırıcı tanısına katkı sağlamaz. Presinaptik dopaminerjik terminallerin yoğunluğunu gösteren DAT-SPECT incelemesinde PHD ve LCD hastalarında striatal DAT tutulumunda azalma görülürken AH’de bu tutulum normaldir. Böylelikle bu yöntem ekstrapiramidal bulgular geliştiren Alzheimer hastalarının (örneğin, nöroleptik yan etkisine bağlı) PHD ve LCD hastalarından ayırt edilmesinde yardımcı olabilir. Aynı şekilde 123I-MIBG SPECT görüntüleme de PHD ve AH hastalarını ayırt etme potansiyeli göstermektedir.
BOS biyobelirteçleri açısından BOS’ta erken dönemde saptanan düşük amiloid beta (Aβ) seviyeleri ve yüksek tau seviyelerinin gelecekteki bilişsel düşüşü öngördürebileceği düşünülmektedir. Bazı çalışmalar PHD'de BOS α-sinüklein seviyelerinin azaldığını bildirse de, metaanaliz çalışmalarına göre α-sinüklein seviyeleri ile kognitif düşüş arasındaki ilişki henüz belirsizdir.
Tablo 3. Parkinson Hastalığı Demansının Özellikleri. (Emre ve ark. 2007)
I. Temel Özellikler
1. Tanısı “Queen Square Beyin Bankası” Kriterlerine göre konulmuş Parkinson Hastalığının varlığı
2. Sinsi olarak başlayan, yavaş ilerleyen, tanısı öykü, klinik ve mental muayene bulguları ile konan, aşağıdaki şekilde tanımlanmış demans sendromunun varlığı:
· Birden fazla kognitif alanda bozulma
· Bu alanlarda premorbid düzeye göre bir düşüş
· Kognitif bozuklukların günlük hayatı (sosyal, mesleki veya kişisel bakım) etkileyecek ağırlıkta olması (motor veya otonomik bozukluğa atfedilen bozukluktan bağımsız olarak)
II. Eşlik eden klinik özellikler
1. Kognitif özellikler
· Dikkat: Bozulmuş. Spontan ve odaklanmış dikkatte bozulma, dikkat testlerinde kötü performans; performans gün içinde veya günden güne dalgalanabilir.
· Yürütücü işlevler: Bozulmuş. Bozukluk başlama, planlama, konsept oluşturma, kural bulma, kural değiştirme veya koruma testlerinde olabilir; yavaşlamış mental hız (bradifreni)
· Görsel-mekansal işlevler: Bozulmuş. Bozulma görsel-mekansal işlevler, görsel-mekansal oryantasyon, görsel algı veya yapılandırma testlerinde görülebilir
· Bellek: Bozulmuş. Bozulma yakın geçmişteki olayların serbest hatırlanması veya yeni bilgileri öğrenme testlerinde görülebilir; hatırlama genellikle ipucuyla düzelir, tanıma genellikle serbest hatırlamadan daha iyidir
· Dil: Ana fonksiyonlar büyük oranda korunmuş. Kelime bulma güçlükleri ve kompleks cümleleri anlamada bozulma olabilir.
2. Davranışsal özellikler
· Apati: Spontanitenin azalması; motivasyonun, ilginin ve iradi davranışların kaybı
· Kişilikte ve duygu durumda, depresif özellikler ve anksiyeteyi içeren değişiklikler
· Halüsinasyonlar; çoğunlukla görsel, genellikle kompleks, kişi, hayvan veya objelerin iyi şekillenmiş görüntüleri
· Hezeyanlar; genellikle paranoid özellikte, aldatılma veya başka birinin varlığı (evde istenmeyen misafirlerin varlığı) gibi hezeyanlar
· Aşırı gündüz uyuklamaları
III. Parkinson Hastalığı demansını dışlattırmayan, ancak tanıyı kuşkuda bırakan özellikler
· Demansın sebebi oldukları düşünülmeyen, ancak kognitif bozukluğa neden olabilecek diğer anormalliklerin varlığı, örneğin, görüntülemede anlamlı vasküler hastalık varlığı
· Motor ve kognitif semptomların gelişimi arasındaki zaman ilişkisinin bilinmemesi
IV. Mental bozukluğun nedeni olabilecek diğer hastalıkların veya bozuklukların mevcudiyeti, bunların varlığında Parkinson hastalığı demansı tanısını koymak mümkün olmaz
Kognitif ve davranışsal semptomların aşağıdaki nedenler çerçevesinde ortaya çıkması
· Akut konfüzyon
· Sistemik hastalıklar veya anormallikler
· İlaç intoksikasyonu
· DSM IV’e göre majör depresyon
· NINDS-ARIEN kriterlerine göre “Muhtemel Vasküler Demans” ile uyumlu özellikler
Muhtemel Parkinson Hastalığı Demansı
A. Temel Özellik: Her iki temel özellik de bulunmalı
B. Eşlik eden klinik özellikler:
· Dört kognitif alanın en az ikisinde, tipik profil gösteren bozulma (dikkatte dalgalanan bozulma, yürütücü işlevlerde bozulma, görsel-mekansal işlevlerde bozulma ve serbest hatırlama belleğinde bozulma-genellikle ipucu ile düzelme)
· Davranışsal semptomlardan (apati, depresif ve anksiyöz duygudurum, halüsinasyonlar, hezeyanlar, aşırı gündüz uyuklaması) en az birinin varlığı muhtemel Parkinson Hastalığı Demansı tanısını destekler, ancak davranışsal semptomların olmaması tanıyı dışlatmaz
C.
Grup III’de yer alan özelliklerin olmaması
D. Grup IV’de yer alan özelliklerin olmaması
Mümkün Parkinson Hastalığı Demansı
A. Temel özellik: Her iki temel özellik de bulunmalı
B. Eşlik eden klinik özellikler:
Birden fazla alanda
var olan kognitif bozukluğun atipik profili; örneğin, dikkat
korunmuş iken saf depo-hasarı biçiminde amnezi (bellek ipucuyla
veya tanıma görevleri ile düzelmiyor) veya reseptif tipte (akıcı)
afazi.
Davranışsal semptomlar olabilir veya olmayabilir
VEYA
C. Grup III’te yer alan özelliklerden bir veya daha fazlasının
varlığı
D. Grup IV’te yer alan özelliklerin olmaması
Parkinson Hastalığında Hafif Kognitif Bozukluk (PH-MCI)
İlerleyici demanslarda olduğu gibi PH’de kognitif bozulma hafif kognitif bozukluk (PH-MCI) dönemi ile başlayabilmektedir ve bununla ilgili tanı kriterleri Tablo 4’de gösterilmektedir.
Bu kriterlere göre PH-MCI tanısı, PH teşhisi konulan bireylerde kognitif becerilerde hasta veya yakını tarafından bildirilen veya klinisyen tarafından gözlemlenen kademeli düşüş olması, nöropsikolojik testler veya PH için valide edilmiş global bir kognitif fonksiyon skalasında kognitif bozulmanın gösterilmesi ve kognitif bozulmanın fonksiyonelliği önemli ölçüde etkilemek için yeterli düzeyde olmaması ile konabilir. PH-MCI kriterleri hastaların kognitif tutulum alt tipleri açısından değerlendirilmesine de olanak tanımaktadır. PH-MCI hastalarının nöropsikolojik muayenesinde tespit edilen kognitif bozukluklar değişiklik göstermekle birlikte en sık etkilenme yürütücü fonksiyonlar, görsel-mekansal fonksiyonlar, bellek ve dikkat kognitif alanlarında görülmektedir. Beş bilişsel alanın her birinin en az iki testle değerlendirilmesiyle hastalar alt tip açısından tek alan MCI (tek bir kognitif alanda etkilenme) ve çoğul alan MCI (iki veya daha fazla kognitif alanda etkilenme) olarak sınıflandırılabilir. Sekiz farklı kohortun analiz edildiği geniş bir çalışmada hastaların %11,3’ü amnestik olmayan tek alan PH-MCI, %8,9’u amnestik tek alan PH-MCI, %4,8’i amnestik çoğul alan PH-MCI ve %1,3’ü ise amnestik olmayan çoğul alan PH-MCI olarak sınıflanmışlardır.
Parkinson hastalarının yaklaşık %10-20’sinde tanı sırasında MCI bulunduğu bildirilmektedir. Diğer MCI’larda olduğu gibi PH-MCI da ilerleyebilir, stabil kalabilir ya da normal kognisyona geri dönebilir. Bununla birlikte, PH-MCI olan bireylerde PH demansına ilerleme riski artmış olup, bu dönüşüm yaş, hastalık süresi gibi faktörlere bağlı olarak değişiklik gösterebilir. PH hastalarının zamanla demansa ilerlemesinin beklenen bir gelişme olduğu göz önüne alındığında, PH-MCI kriterlerini karşılayan pek çok birey için hem zamansal seyir hem de semptom şiddeti açısından kademeli bir geçiş dönemini yansıtmaktadır.
Parkinson hastalığında MCI varlığı muayene sırasında ileri yaş, hastalığın ileri yaşta başlaması, erkek cinsiyet, depresyon, motor bulguların ağır oluşu ve ileri hastalık evresiyle ilişkili bulunmuştur. Uyku-uyanıklık siklusunda bozulmalar (RUDB, aşırı gündüz uykusu), MRG’de ak madde hiperintensiteleri, BOS’ta düşük Aβ seviyeleri, APOE ε4 durumu, DAT görüntülemede kaudat çekirdekte düşük tutulum PH’de kognitif bozulma ile ilişkili görünmektedir. FDG-PET görüntüleme bulgularının PH’de kognitif gerileme başlamadan önce kortikal hipometabolizmayı saptamada oldukça duyarlı olduğu bildirilmiştir. FDG-PET incelemesi 4 yıl içinde demansa ilerleme riskini %85 sensitivite ve %88 spesifisite ile göstermekte ve özellikle posterior bölgelerde LCD- ve AH-tipi atipik PET metabolizma paternleri gösteren hastaların demansa ilerleyeceğini öngörebilmektedir.
Tablo 4. Parkinson Hastalığı-MCI Tanı Kriterleri (Litvan ve ark. 2012)
I. Dahil etme kriterleri
İngiltere PH Beyin Bankası Kriterlerine göre Parkinson hastalığı teşhisi
Mevcut PH tanısı bağlamında, kognitif becerilerde hasta veya yakını tarafından bildirilen veya klinisyen tarafından gözlemlenen kademeli düşüş
Formal nöropsikolojik testlerde veya global kognitif beceriler skalasında kognitif bozulmanın gösterilmesi (bölüm III'te ayrıntılandırılmıştır)
Kognitif bozulma, fonksiyonel bağımsızlığı önemli ölçüde etkilemek için yeterli düzeyde değildir, ancak karmaşık fonksiyonel görevlerde ılımlı güçlükler olabilir
II. Dışlama kriterleri
“Movement Disorders Society”’ Çalışma Grubu tarafından önerilen kriterlere göre PH demans tanısı
Kognitif bozukluk yapabilecek diğer primer sebeplerin varlığı (örneğin, deliryum, inme, majör depresyon, metabolik anormallikler, ilaç yan etkileri veya kafa travması)
Klinisyenin görüşüne göre kognitif testleri önemli ölçüde etkileyebilecek PH ile ilişkili diğer komorbid durumlar (örneğin, motor bozukluk veya şiddetli anksiyete, depresyon, gündüz aşırı uykululuk veya psikoz)
III. Seviye I ve seviye II PH-MCI kategorileri için özel yönergeler
A. Seviye I (kısaltılmış değerlendirme)
PH’de kullanımı valide edilmiş bir global kognitif fonksiyonlar ölçeğinde (MoCA, PD-CRS, SCOPA-COG, MDRS*) bozulma olması
veya
Sınırlı bir nöropsikolojik test bataryası (yani batarya, beş kognitif alanın her biri için ikiden az test içerir veya beşten daha az kognitif alan değerlendirilir) uygulandığında en az iki testte bozulma olması
B. Seviye II (kapsamlı değerlendirme)
Beş kognitif alanın her bir için iki test içeren nöropsikolojik testler (yani, dikkat ve çalışma belleği, yürütücü, dil, bellek ve görsel-mekansal alanlar)
En az iki nöropsikolojik testte bozulma (tek kognitif alanda bakılan iki testte bozulma veya bir testte iki ayrı kognitif alanda bozulma ile gösterilebilir)
Nöropsikolojik testlerdeki bozulma şu şekilde gösterilebilir:
· Uygun normların yaklaşık 1 ila 2 standart sapma altında performans, veya,
· Arka arkaya yapılan kognitif testlerde gösterilen anlamlı düşüş, veya,
· Hastalık öncesi (premorbid) seviyelere göre önemli azalma
IV. PH-MCI için alt tip sınıflandırması (isteğe bağlı, değerlendirilen beş bilişsel alanın her biri için iki test gerektirir ve araştırma amacı olduğunda şiddetle önerilir)
Tek alan PH-MCI; tek bir kognitif alan içindeki iki testte anormallik olmalıdır (alanı belirtin), diğer alanlar bozulmamıştır, veya,
Çoğul alan PH-MCI; iki veya daha fazla kognitif alanda en az bir testte anormallik olmalıdır (alanları belirtin)
*Kısaltmalar: MoCA: Montreal Kognitif Değerlendirme Ölçeği, PD-CRS: Parkinson Hastalığı Kognitif “Rating” Skalası, SCOPA-COG: Parkinson Hastalığı Sonlanımları için Ölçekler-Kognisyon, MDRS: Mattis Demans Değerlendirme Ölçeği
Nöropatoloji ve Fizyopatoloji
PHD’de asendan modülatör sistemlerin hemen hepsinde hücre ve innervasyon kaybı bildirilmiştir. Değişik çalışmalarda kolinerjik, dopaminerjik, noradrenerjik ve serotoninerjik monoaminerjik sistemlerin etkilendiği bulunmuştur. Kolinerjik kayıplar dışındaki bulgular ve bunların demans ile bağlantısı net olarak gösterilmemiş olmakla birlikte bu kayıpların tümü de demansın değişik belirtilerinin ortaya çıkmasına katkıda bulunabilirler. Örneğin, dopaminerjik kayıp frontostriatal bağlantıların normal çalışmasını bozarak yürütücü işlevlerde ve çalışma belleğinde bozulma yapabilir. Ancak PHD’deki kognitif bozukluğun biyokimyasal kökeni olarak en sistemli şekilde gösterilebilen bulgu kolinerjik innervasyondaki azalmadır. Bu azalma anatomik, biyokimyasal ve fonksiyonel çalışmalarda gösterilmiş olup AH’de görülenden daha fazla orandadır (gözden geçirme için bkz. Emre M, 2007). Yapısal olarak bazal önbeyni innerve eden Meynert’in nükleus bazalisindeki kolinerjik hücrelerin sayısının azaldığı saptanmıştır. Biyokimyasal olarak asetilkolin yapılmasını sağlayan enzim olan kolinasetiltransferaz aktivitesinin PHD olan hastaların korteksinde demansı olmayan Parkinson hastalarına göre daha fazla oranda azaldığı, bu azalmanın özellikle frontal ve temporal kortekslerde daha belirgin olduğu bulunmuştur. Kolinerjik işaretleyiciler ile yapılan fonksiyonel görüntüleme çalışmalarında benzer şekilde kolinerjik aktivitenin PHD olan hastalarda olmayanlara ve AH olanlara kıyasla daha çok azaldığı, bu azalmanın miktarının kognitif tutulumun, özellikle de yürütücü işlevlerdeki bozukluğun miktarı ile orantılı olduğu saptanmıştır. Tüm biyokimyasal bulgular birlikte ele alındığında dopaminerjik eksikliğin yürütücü işlev ve çalışma belleği bozukluğuna, kolinerjik eksikliğin bellek, dikkat ve yürütücü işlevlerde bozukluğa, noradrenerjik eksikliğin dikkat bozukluğuna, serotoninerjik eksikliğin ise afektif bozukluğa katkıda bulunabileceği öne sürülmüştür.
Zamansal gelişim içinde PHD’nin nöropatolojik karşılığı olarak üç tip patoloji sorumlu tutulmuştur. Bunlar a) subkortikal çekirdeklerin, özellikle medial substantia nigranın dejenerasyonu, b) AH-tipi patoloji (amiloid plaklar ve NFY) c) LCi patolojisidir. Her bir patoloji için demanstan sorumlu ana mekanizma olduğunu gösteren farklı çalışmalar vardır. Ancak son yıllarda yapılan, değişik patolojilerin eş zamanlı olarak değerlendirildiği, özellikle de LCi patolojisini saptamak için daha hassas bir yöntem olan alfa-sinüklein immünhistokimyası kullanılan çalışmalarda altta yatan ana patolojinin kortikal ve limbik alanlarda hücre kaybı ve LC-tipinde dejenerasyon olduğu gösterilmiştir. Bir LC hastalığı kohort çalışmasında PH olgularının %80’i McKeith kriterlerine göre limbik geçiş evresinde iken, hem PHD hem de LCD olgularının (sırasıyla %90 ve %97 oranında) neokortikal seviyeye ulaştığı görülmüştür. Bu çalışmalarda LCi patolojisi, kognitif bozuklukla en yakın korelasyon gösteren bulgudur; buna sıklıkla AH-tipi patoloji eşlik etmekte, ancak nadiren patolojik olarak AH tanısını koydurmaya yetecek miktarda olmaktadır. Daha fazla AH-tipi patoloji yükü olan PHD olguları demansa daha hızlı geçiş göstererek LCD’ye benzer bir profil sergilemektedirler.
Sağkalım
PHD hastaları için sağkalım LCD hastalarından daha uzundur. Sidney Çokmerkezli Çalışmasında ortalama 10,9 yıl olarak bildirilmiştir. PH semptomları göstermekte olan PHD hastalarının demans tanısı aldıktan sonra median sağkalım süresi 54 ay olarak bildirilmiştir. Daha uzun hastalık süresi ve motor ilerleme göz önüne alındığında, PHD hastalarının, LCD'li olanlara göre daha fazla motor özürlülüğe, sık motor dalgalanmalara, daha yüksek ilaç yüküne ve invazif tedavi geçmişlerine (örneğin, derin beyin stimülasyonu) sahip oldukları görülmektedir.
Tedavi
PHD düşünülen bir hastada farmakolojik tedaviye başlamadan önce zihinsel bozukluğu tetikleyebilecek diğer faktörler dışlanmalıdır. Bu bağlamda sistemik hastalıklar, özellikle yeni başlanan veya dozu arttırılan ilaçlar gözden geçirilmeli, hasta antikolinerjik, trisiklik antidepresan veya benzodiazepin grubundan bir ilaç kullanıyorsa bu ilaçlar kesilmelidir. Dopaminerjik ilaçlar halüsinasyon, hezeyan gibi psikotik belirtileri ve konfüzyonu arttırabilecekleri için özellikle agonistlerin dozu azaltılmalı ya da bunlar kesilmeli, tedavi mümkün olduğu kadar yeterli dozda yalın levodopa preparatlarına indirgenmelidir.
PHD’de kolinerjik eksikliğin tanımlanmasından sonra hastalığın kognitif semptomlarının tedavisi için kolinesteraz inhibitörleri ile arka arkaya çalışmalar yapılmış ve bu ajanların PHD için faydası birden çok metaanaliz ve sistematik gözden geçirme ile desteklenmiştir. Rivastigmin PH-MCI hastalarında da küçük bir çalışmada değerlendirilmiş, ancak kognitif ölçeklerde düzelme eğilimi görülse de anlamlı klinik etki saptanamamıştır. Diğer asetilkolinesteraz inhibitörlerden donepezil ile PHD’de yapılan büyük çaplı bir çalışmada 10 mg dozunun kognitif ölçeklerde ve klinisyen değişim izleniminde plasebodan üstün olduğu bildirilmiştir. Memantin kullanımının etkinliği üzerine mevcut kanıt daha azdır, iyi tolere edilen bu ajanın asetilkolinesteraz inhibitörleri yanında ek olarak kullanımı düşünülebilir.
Sistematik gözden geçirmeler Parkinson hastalığında egzersiz ve fizik tedavi programlarının hem motor hem de kognitif fonksiyonlar üzerinde faydalarının aktivite sonlandıktan 3-12 ay gibi uzun süreler sonrasında hala devam ettiği üzerine kanıtlar sunmakta ve nöroplastisiteyi arttırdığı düşünülen bu yöntemler giderek daha çok önerilmektedir.
Non-kognitif belirtilerin tedavisi de LCD’ye benzerdir.
HUNTİNGTON HASTALIĞI
Huntington Hastalığı (HH) orta yaşlarda başlayan ve otozomal dominant genetik geçiş gösteren progresif bir nörodejeneratif hastalıktır. Hastalık esas olarak kore, psikiyatrik bozukluklar ve kognitif yıkım triadı ile karakterizedir. Erken dönemlerde striatum ve ilişkili ak madde yolaklarının seçici olarak tutulmasıyla başlayıp ilerleyen aşamalarda daha yaygın bir nörodejenerasyonun ortaya çıkması, HH’nin striatumun kognisyondaki rolünü anlamak için bir model olarak kullanılmasını mümkün kılmıştır.
Bu hastalıkta 4. kromozomda ‘‘Huntingtin proteini’’ni kodlayan ‘‘Huntingtin’’ geninde bir CAG trinükleotid tekrar genişlemesi olduğu belirlenmiştir.
Epidemiyoloji
HH’nin prevalansı 100,000’de 5-10 olup nadir görülen bir nöropsikiyatrik hastalıktır. Hastalık Avrupa kökenli bireyler arasında çok daha yüksek frekanslarda görülürken Japonya, Hong Kong ve Tayvan'da Avrupa'nın yaklaşık onda biri kadardır. HH’nin kökene özgü prevalans oranlarının, HTT lokusundaki genetik farklılıklardan kaynaklandığı düşünülmektedir. Kadın ve erkek cinsiyet eşit etkilenmektedir.
Patogenez
Erken nöropatolojik değişiklikler, ilk olarak striatumda ve özellikle kaudat çekirdekteki orta boy GABAerjik dikenli projeksiyon (MSP) nöronlarının kaybı ile karakterizedir. İstemli hareketleri indirekt olarak kontrol eden ve striatumda enkefalini-baskılayan MSP nöronlarının kaybı, HH koresinin ortaya çıkışında nörobiyolojik temeli oluşturur. Bu nedenle motor semptomların kortikostriatotalamokortikal devrelerde, özellikle kaudat, putamen ve globus pallidustaki nöronal kayba sekonder olarak gelişen fonksiyonel değişikliklere bağlı olduğu kabul edilmektedir. Hastalıkta striatumu frontal loblarla birbirine bağlayan frontostriatal devreler de etkilenir; bu tutulum hastalığın kognitif ve psikiyatrik yönleri ile ilişkilendirilmiştir. Nöronal hücrelerin ölümü korteksin 3, 5 ve 6. katlarında, substantia nigrada ve hippokampusun CA1 bölgesinde yavaş yavaş devam eder.
HH patolojisi esas olarak nörotransmitter işleyişi ve sinaptik haberleşmede bozulma, glutamat-aracılı eksitotoksisite ve beyin kökenli nörotrofik faktörde (BDNF) azalmayı içermektedir. Huntingtin proteininin sitotoksik formları hücre içi taşıma ve sinyalizasyon, sekretuar yolak, endositik geri dönüşüm, mitokondriyal bozukluk, sinaptik işlev bozukluğunu içeren, son derece kompleks bir patojeniteye sebep olmaktadır. Bu hücresel bozukluk, şebekelerde disfonksiyona neden olmaktadır. Farklılaşan nöronal şebeke ve hücre dışı otonom disfonksiyondan kaynaklanan eksitotoksisite HH’nin tüm semptomlarına katkıda bulunmaktadır.
Kognitif Bulgular
HH’de ilk kognitif değişikliklerin hastalığın erken döneminde, motor başlangıçtan yaklaşık 12-15 yıl önce başladığı bilinmektedir. Prodromal dönemdeki ilk değişiklikler, yürütücü işlevlerde (özellikle kognitif esneklik, akıl yürütme, planlama ve sözel akıcılıkta) bozulma ile karakterizedir. Hafif kognitif bozukluk tanımı 2010 yılında HH olan bireylerde de tanımlanmıştır. Genetik testi pozitif olup henüz motor semptomları belirmemiş bireylerde yapılan büyük bir çalışmada, grubun %40’ında işlemleme hızı, yürütücü fonksiyonlar, epizodik bellek ve/veya vizyospasyal algıda hafif bozukluklar olduğu tespit edilmiştir. Tek alan kognitif etkilenme (tek alan MCI) oranı %25,9 ve çoğul alan MCI oranı %13,9 olarak bildirilmiştir. Amnestik olmayan MCI alt tipinin, amnestik alt tipten iki kat daha sık olduğu görülmüştür. Çalışma belleği ve dikkat de erken dönemde bozulmalar gösterir. Karmaşık akıl yürütme, ardışık görevleri yerine getirme ve eşzamanlı ikili işlemleme yeteneği kademeli olarak kötüleşir. Erken dönemde dil özellikleri göreceli olarak korunmuş olup hastalığın başlangıcından sonra cümle karmaşıklığı ve sözel akıcılık azalır. Epizodik bellek bozukluğu, aktif öğrenme zorlukları ile başlar; geri çağırmada bilgi kodlamaya kıyasla daha büyük bir sorun ortaya çıkar. Özellikle negatif emosyonların tanınmasında bozulma başlayabilir. Hastalık ilerledikçe, yürütücü işlevlerdeki bozulma da artar. Soyut düşünme yeteneği, yargılama ve planlama kabiliyeti giderek azalır. Çalışma belleği bozulmaya devam eder. Epizodik ve prospektif bellek kötüleşir, hem yeni bilgileri öğrenme, hem de eski bilgileri geri çağırmada bozukluk görülür. Fonemik ve semantik akıcılık belirgin azalır. Görsel mekansal bozukluklar bu aşamada ortaya çıkar. Son evrede bütün kognitif işlevler ağır şekilde azalır. Bilgiyi harekete geçirememe ve motivasyon eksikliği ile birlikte yaygın demansiyel bir tablo ortaya çıkar. Hastalığın ilerlemesiyle konuşma ve dil yeteneği gittikçe bozulur ve sonunda hasta tamamen sessiz hale gelebilir. Nihai aşamada ağır bir demans hakimdir.
Bradifreni kognitif tablonun en karakteristik özelliklerindendir; örneğin, hastaların cevabını bildiği bir soruya uygun yanıtı vermesi normalde beklenenden çok daha uzun sürebilir. Psikomotor hızda bozulma HH'de sık olarak bildirilmiş ve bilişsel performanstaki tüm farklılıkların motor bozukluğun etkisi ile açıklanamayacağı ve motor performanstaki bozulmadan ayrı olarak gerçekleştiği gösterilmiştir.
Nöropsikiyatrik Bulgular
HH hastaların yaklaşık üçte ikisinde nörolojik bulgularla başlangıç gösterirken, üçte birinde psikiyatrik ya da davranışsal belirtilerle ortaya çıkmaktadır. Psikiyatrik belirtiler %33-76 oranında görülmekte olup anksiyete, depresyon, obsesif kompulsif bozukluk, apati ve psikozu içerir. Bu semptomlar içinde yalnızca apati hastalık evresi ilerledikçe progresif olarak artma eğilimindedir ve nörodejenerasyon ile doğrusal ilişki gösteren tek nöropsikiyatrik semptom olduğu söylenebilir.
İntihar girişimi hastaların %2,3-12 kadarında görülür ve genel popülasyonun 4-5 katı sıklıktadır. Hastalarda sık görülen impulsivite ve öfke kontrol bozukluğu intihar riskini arttırmakta ve intiharı öngörmeyi zorlaştırmaktadır. HH’de anksiyete oldukça sıktır, sosyal anksiyete ya da obsesif kompulsif bozukluk özellikleri görülebilmektedir. Hastalığın tipik olarak geç evrelerinde psikotik bozukluklar ortaya çıkabilir ve sıklıkla kognitif gerilemeye eşlik eder. Klinik olarak psikotik depresyon ya da paranoid ve işitsel halüsinasyonların eşlik ettiği alevli bir tablo gösterebilir. Davranışsal problemler içinde iritabilite, ajitasyon, disinhibisyon, impulsivite, öfke patlamaları sıktır.
Motor Bulgular
HH’de görülen motor bozukluk iki ana bileşen altında incelenebilir: (1) hastalığı karakterize eden istemsiz hareketler ve (2) istemli hareketlerde bozulma (bakınız: Hareket Bozuklukları).
Tanı
Reilmann ve arkadaşları 2014 yılında preklinik dönem, prodromal dönem ve klinik olarak aşikar (manifest) dönem HH terimlerini daha formal olarak tanımlamışlardır. Preklinik dönem kendi içinde iki alt bölümde incelenebilir. Kişilerin subjektif semptomları veya klinik bulguları göstermediği ve bu nedenle "presemptomatik" olarak adlandırılan dönem, genellikle hastalığın başlangıcının 10 ila 15 yıl öncesinde görülür. Kişi bundan sonra "prodromal" döneme girer ve motor başlangıçlı HH tanısı için geçerli kriterleri karşılamayan, ancak motor, bilişsel ve davranışsal değişikliklerin zamanla ortaya çıktığı bir dönem başlar. Klinik olarak aşikar HH için şu anda geçerli kriterler: 1) HH geninde bir CAG-genişlemesi taşımak ya da bir aile öyküsü olmak ve 2) Birleşik Huntington Hastalığı Derecelendirme Ölçeği'nin (UHDRS) Tanısal Güven Düzeyinde (DCL) tanımlandığı şekilde HH’nin kesin motor belirtilerini göstermeyi gerektirmektedir.
Hastalığı karakterize eden semptomlar, daha önce de belirtildiği gibi, motor disfonksiyon, bilişsel bozukluk ve nöropsikiyatrik problemler triadını içerir. HH’nin motor belirtileri UHDRS motor skalası kullanılarak değerlendirilebilir.
Kranial görüntülemede kaudat nukleus başı ve putamende belirgin simetrik atrofi ve buna bağlı olarak frontal boynuzlarda genişleme mevcuttur. Hastalığın ilerlemesi ile kortekste de rölatif olarak yaygın bir atrofi görülür. Bununla birlikte globus pallidus, thalamus, substantia nigra ve amigdala ve özellikle frontal serebral ak maddede volümetrik değişiklikler olduğu gösterilmiştir. Fonksiyonel nörogörüntüleme çalışmaları nörokognitif görev performansı boyunca, kortikostriatal şebekeye dahil bölgeler arasında fonksiyonel konnektivitede bozulma ve bölgesel hiperaktivite olduğuna işaret etmektedir. FDG-PET görüntüleme bazal ganglia ve frontal kortekste hipometabolizmayı kaudat nukleustaki hacim kaybı aşikar hale gelmeden önce gösterebilmektedir.
Ayırıcı Tanı
Klinik olarak HH tanısı alan vakaların yaklaşık %1-3’ünde genetik test tanıyı doğrulamamaktadır. HH’yi taklit eden bu durumlar HH fenokopileri olarak adlandırılır. HH’yi en sık taklit eden hastalık HDL-4 (SCA 17) ve Afrika kökenlilerde HDL-2’dir. Bilinen diğer başlıca HH fenokopileri HDL-1 ve ailesel prion hastalığı, HDL-3, SCA 1,2,3, dentatorubral-pallidoluysian atrofi (DRPLA), nöroferritinopati, beyinde demir birikimi ile giden nörodejenerasyon (NBIA), benign herediter kore ve nöroakantositozu içerir.
Tedavi
HH’de kore tedavisinde FDA onayı olan ilk ilaç dopamin depolarını boşaltan bir ilaç olan tetrabenazindir. Özellikle depresyon öyküsü olan hastalarda depresif semptomlar ve intihar gibi ciddi yan etkilerinin izlenmesi kritik önem taşır ve juvenil HH’de parkinsonizmi arttırabilir. Yine yakın dönemde FDA onayı alan tetrabenazin türevi bir farmakolojik ajan olan deutetrabenazin’in koreiform hareketler üzerine etkilerinin tetrabenazinle karşılaştırılabilir olmasına rağmen, sedasyon gibi bazı tepe dozu yan etkilerinin daha az olduğu ve depresyon skalalarını daha az etkilediği bildirilmiştir. Koreyi azaltmak için ilk sırada tercih edilen diğer ilaçlar arasında dopamin reseptör blokerleri yer alır. Mevcut literatür bu ilaçların koreiform hareketler üzerine faydalarının yanısıra psikiyatrik semptomlarda da düzelme yaptıklarını ve duygudurum, uyku bozuklukları, kilo kaybını önlemede faydalı olduklarını desteklemektedir. Erken başlangıçlı hastalarda ve hastalığın ileri evrelerinde dopaminerjik nörotransmisyonu azaltan bu ilaçlar parkinsonizmi arttırabileceğinden dikkatli olunmalıdır.
Psikiyatrik semptomlar için farmakolojik yaklaşımlar semptomatik açıdan etkilidir. Depresif semptomlar için SSRI, SNRI, bupropion, ya da mirtazapin gibi atipik antidepresanlar kullanılabilir. Anksiyete için yine SSRI ve SNRI’ların yanında olanzapin, ketiapin, risperidon gibi nöroleptikler veya diazepam, alprazolam, klonazepam gibi benzodiazepinler kısa süreli olarak tercih edilebilir. Apatinin tedavisi güçtür, SSRI ve SNRI’lardan fayda görmeyen hastalarda modafinil/armodafinil ya da kontrollü bir şekilde psikostimülanlar denenebilir. İritabilite ve öfke için SSRI ve SNRI’lar yetersiz kaldığında karbamazepin, valproat ya da lamotrijin gibi duygudurum stabilizatörleri veya nöroleptikler uygulanabilir.
Hastalığın yol açtığı kognitif bozukluklar için henüz semptomatik bir tedavi geliştirilmemiştir. Kognitif rehabilitasyon düşünülebilir; 9 ay süreli multidisipliner küçük bir kognitif rehabilitasyon çalışması verbal öğrenme ve bellekte anlamlı artışlar bildirmiştir.
PARKİNSON ARTI SENDROMLARI
Kortikobazal Dejenerasyon
KBD, bazal ganglia, frontal ve parietal korteksleri tutan progresif nörodejeneratif bir hastalıktır. Hastalığın kliniği başlangıçta bir ekstremiteyi etkileyen asimetrik parkinsonizm, apraksi, distoni, miyoklonus, kortikal duyu kaybı ve progresif kognitif bozukluk ile karakterizedir. Armstrong ve arkadaşları tarafından 2013 yılında tanımlanan yeni kriterlere göre KBD 5 farklı klinik sendrom halinde görülebilmektedir. Muhtemel kortikobazal sendrom tanısı için asimetrik prezantasyon gösteren ekstremite rijiditesi veya akinezi, distoni, miyokloni’den biri ve orobukkal/ekstremite apraksisi, kortikal duyusal defisit, yabancı el fenomeni’nden ikisinin bulunması gereklidir. Diğer dört sendrom ise (1) Mümkün kortikobazal sendrom, (2) Frontal-davranışsal-spasyal sendrom, (3) Agramatik varyant progresif afazi ve (4) Progresif supranükleer paralizi olarak belirlenmiştir. Bu beş varyanttan ikisinin kognitif fonksiyon ağırlıklı tanılar olması dikkat çekicidir. Muhtemel frontal-davranışsal-spasyal varyant tanısı için yürütücü disfonksiyon, davranışsal değişiklikler ve vizyospasyal bozukluktan en az ikisi gerekmekte olup bu bulgulara en az bir kortikobazal motor belirtisi eşlik etmelidir. Muhtemel tutuk/agramatik PPA varyantı tanısı için ise tutuk ve agramatik konuşmaya bozulmuş gramer/cümle anlama veya konuşma apraksisinden biri ve en az bir kortikobazal motor belirtisi eşlik etmelidir. Bilişsel veya davranışsal sunumları vurgulayan KBD serilerinde, erken dönemde parkinsonizm oranlarının düşük olduğu belirtilmiştir.
KBD'de görülen zengin kortikal belirtiler arasında apraksi, yabancı uzuv (alien limb) fenomeni, kortikal duyu kaybı, afazi, kognitif bozukluklar ve davranış değişiklikleri bulunur. Bu kitapta “Progresif Apraksi” bölümleri içinde detaylı olarak değinilen apraksi, önceki tüm tanı kriterlerinin temel bulgusu olup derlenen KBD vakalarının % 57'sinde mevcuttur. KBD'de en sık tanımlanan apraksi, ideomotor apraksidir, onu ekstremite kinetik apraksi takip etmektedir. Afazi %52 vakada mevcuttur, çoğunlukla PPA olarak kayıt edilmiştir ve mutizme kadar ilerleyebilir. Yabancı uzuv fenomeni derlenmiş vakaların %30’unda tanımlanmıştır. Kayıtlara göre kortikal duyu kaybı vakaların yaklaşık dörtte birinde görülmektedir. Derlenen vakaların %70’inden fazlasında kognitif bozukluk olduğu bildirilmiştir. Bildirilen seriler hastaların yürütücü fonksiyonlar, bilişsel esneklik, öğrenme, dil, görsel mekansal fonksiyonlar ve sosyal kognisyon alanlarında fonksiyon kaybı yaşadığını ortaya konmuştur. KBD'de bellek bozukluğu gösteren hastalarda altta patoloji olarak atipik AH'nin yatabileceği vurgulanmaktadır.
KBD’de davranışsal değişiklikler sık görülür ve hastanın kliniği başlangıçta davranışsal varyant FTD şeklinde prezante olabilir. Semptomlar apati, garip veya antisosyal davranışlar, kişilik değişiklikleri, ajitasyon, iritabilite, disinhibisyon ve hiperseksüaliteyi içerir. Gözden geçirilen KBD vakalarının %55’inde davranış değişiklikleri olduğu bildirilmiştir. Depresyon oldukça sık (%51-73) görülmektedir.
Nörogörüntüleme KBD'de oldukça yardımcıdır. MRG’de posterior frontal, inferior parietal, superior frontal, thalamus ve striatumda asimetrik atrofi tanımlanabilir ve sıklıkla KBD'yi diğer parkinson sendromlarından ayırmada yardımcı olur. FDG-PET benzer şekilde asimetrik kortikal hipometabolizmayı ortaya çıkarabilir. Dopa-PET alımı striatumda azalır ve kortekste de oldukça asimetriktir. DAT-Scan ve 123I CIT SPECT benzer bir asimetrik striatal bağlanma paterni gösterir, ancak bu bulgu KBD'ye spesifik değildir. KBD'yi diğer ilgili bozukluklardan ayırmada hem tau hem de amiloid ligandlarının kullanımı araştırılmaktadır.
KBD patolojisi tau birikimi ve frontoparietal bölgede oksipital lobların göreceli olarak korunduğu asimetrik kortikal atrofi ile karakterizedir, ancak PSP, AH, Pick hastalığı, FTLD-ubiquitin ve TDP-43+, LCD, FTLD-FUS ve Creutzfeldt- Jakob hastalığı dahil olmak üzere farklı patolojiler de KBD’nın belirti ve semptomlarını taklit edebilir. Altta yatan patoloji KBD’dan farklı olduğunda daha geniş kapsamlı olan kortikobazal sendrom terimi kullanılmalıdır. Bununla birlikte, KBD patolojisinin de birçok başka sendrom görünümünde karşımıza çıkması mümkündür: klasik PSP sendromu, ilerleyici tutuk afazi, davranışsal varyant FTD ve AH benzeri demans olgularında KBD patolojileri bildirilmiştir.
KBD 4R tau birikimi ile karakterize bir taupatidir. Şişmiş vakuolasyonlu nöronlar, atrofik kortikal alanlarda ve daha az derecede etkilenen subkortikal bölgelerde bulunur. Bu balonlanmış nöronlar fosforile nörofilamentler ve bazen tau ve ubikitin içerir. Sadece etkilenen kortekste değil, globus pallidus, putamen, nukleus ruber, thalamus, subtalamik nukleus, substansia nigra, locus coeruleus ve daha az ölçüde dentat çekirdekte yaygın nöron kaybı ve gliozis görülür. Tipik glial bulgular, astrositik plaklar adındaki astrositleri çevreleyen tau içeren birikimlerdir.
Progresif Supranükleer Paralizi
PSP’de aksiyel baskın bir akinetik-rijid bozukluk ve özellikle aşağıya bakış felci şeklinde bir vertikal bakış paralizisi mevcuttur. Provoke olmayan sık düşmeler öykünün başlıca özelliklerindendir. Göz bulgularının yanısıra otonom tutulumun beklenmemesi MSA, LCD ve idyopatik Parkinson hastalığı gibi α-sinükleopatili diğer parkinsonizm sendromlarından ayırmaya yardım eder.
Kognitif tutulum PSP alt tiplerine göre farklılık göstermektedir. Hastalığın ilerlemesi ile kognitif fonksiyonlar da giderek geriler ve tablo demansa doğru ilerler. Demans genel olarak HH’dekine benzeyen tipik bir frontal subkortikal demans tarzındadır. İşlemleme hızı yavaşlamış (bradifreni), verbal akıcılık azalmış ve yürütücü fonksiyonlar bozulmuştur. Bellekte ön planda geri çağırma etkilenmiştir. Otomatik davranışların perseverasyonu sıktır ve alkış işareti pozitiftir. Yakalama, glabellar, “snout“ gibi ilkel refleksler ve taklit davranışları da görülebilir. Konuşma yavaşlamıştır, kekeleme, ekolali, istemsiz vokalizasyonlar ve fonasyon apraksisi görülebilir. Emosyon tanıma bozuklukları sık görülür ve bu bulgunun kognitif bozulmanın şiddeti ile korele olduğu bildirilmiştir.
Başta depresyon olmak üzere duygudurum bozuklukları sıktır. Depresyon olmadan da apati sık görülür ve yürütücü fonksiyonlarla ilişkilendirilmiştir. FTD’ye benzer şekilde disinhibisyon, disfori, anksiyete, iritabilite ve anti-sosyal davranış görülebilir. Emosyonel labilite (emosyonel inkontinans), psödobulber uygunsuz gülme ve ağlama sıktır. Nadir görülen PSP-FTD varyantında klinik tamamen kognitif ve davranış belirtileriyle başlayabilir ve tanı düşmeler, supranükleer bakış paralizisi ve L-dopa’ya cevapsız parkinsonizm gibi diğer tipik semptomların eklenmesiyle hastalığın ileri evrelerinde netleştirilebilir.
MRG’de alışık bir göz özellikle sagital T1 kesitlerde mezensefalon atrofisini ve sinekkuşu ‘hummingbird’ görüntüsünü ayırt edebilir. Superior serebellar pedinküller atrofiktir. Volümetrik olarak saptanan globus pallidus atrofisinin normal kontrollerin yanı sıra PH’liler ve LCD’lilerden de ayırabildiğine ilişkin bir bildirim bulunmaktadır. PSP’de kortikal atrofi paterni FTD'nin davranışsal varyantına benzerlik gösterecek şekilde frontal ve temporal bölgelerde ve thalamus gri cevher hacminde azalma içerebilir. Benzer şekilde PSP-kortikobazal varyantında frontoparietal atrofi görülebilir. SPECT ve PET gibi fonksiyonel görüntüleme yöntemleri ile genellikle beyinsapı, kaudat başı ve thalamus ile birlikte medial prefrontal ve dorsolateral prefrontal alanlarda simetrik hipoperfüzyon/hipometabolizma saptanır. Yakın zamanda AH için geliştirilen tau PET ligandları, 18F-T807/8 ve 18F-THK523, PSP için de ümit verici görünmekle beraber ileri çalışmalar gereklidir.
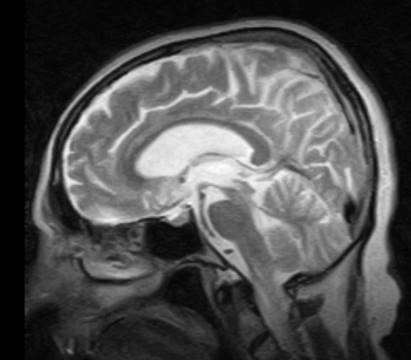
Şekil 2. PSP tanılı bir hastada sagital T2 ağırlıklı orta hattan geçen kesitte mezensefalon atrofisine bağlı sinekkuşu “hummingbird” bulgusu izlenmekte.
Patolojik olarak bir taupati olarak sınıflanabilecek bir nörodejenerasyondur. Taupati kendini intranöronal globoz NFY’ler, nöropil iğcikler şeklinde ortaya koyar. Nöron kaybı ve gliozis eşlik eder. Bu patoloji, beyinsapında superior kollikuli dahil peri-akuaduktal gri madde, pre-tektal alan yanı sıra, substantia nigra pars reticulata, subtalamik çekirdek ve globus pallidusta saptanır. Kortikal tutulum daha seyrek ve hafiftir. Başlıca tau birikimleri ve daha da seyrek olarak nöron kaybı, gliozis ve mikrovakuolizasyon görülür. KBD ve PSP’nin tipik klinik sunumları dahil birbirlerini taklit edebildikleri yapılan otopsi doğrulamalı çalışmalardan bilinmektedir.
MULTİSİSTEM ATROFİ
MSA tabloları, parkinsonizm, serebellar ve piramidal belirtiler ve otonomik disfonksiyonun iç içe geçmiş, ancak birinin ön planda olduğu farklı prezantasyonları ile karakterizedir.
MSA’da kognitif fonksiyon bozuklukları giderek daha çok tanınmakta ve hastalığın erken dönemlerinden itibaren MSA hastalarının %22’sinde kognitif bozukluk olduğu bildirilmektedir. Klinik semptomlara genellikle hafif düzeyde ve demans şiddetine ulaşmayan frontal yürütücü bozukluk şeklinde bir kognitif disfonksiyon eşlik eder. Sıklıkla bellek ve vizyospasyal fonksiyonlar da etkilenebilir. Kognitif bozukluk hastalık süresi ve frontal atrofi ile ilişkili görünmektedir; MSA-Parkinsonizm (MSA-P) hastalarının MSA-Serebellar (MSA-C) hastalarına göre daha ağır kognitif bozukluk gösterdiği bildirilmiştir. Sinükleinopatilerin bilişsel ve nöropsikiyatrik bulgularının değerlendirildiği bir çalışmada MSA hastaları Parkinson hastalarından daha kötü, LCD hastalarından daha iyi kognitif performans göstermişlerdir. MSA’da günlük aktiviteleri bozacak kadar ağır kognitif bozukluk sık ve klasik bir özellik değilse de hastaların yaklaşık %14-16’ünde demans olduğu düşünülmektedir. Yakın zamanda FTD kliniği ile prezente olup MSA patolojisi gösteren vakalar bildirilmiş ve bunlar FTLD-sinüklein olarak adlandırılmıştır.
MSA hastalarında en sık görülen davranışsal belirtiler anksiyete ve depresyon olup bu bulguların şiddeti MSA'nın iki alt tipi arasında farklılık gösterebilir: MSA-P'li hastalarda artmış depresyon ve anksiyete seviyeleri bildirilirken, MSA-C'li hastalarda anksiyete yüksek bulunmuştur. MSA hastalarında bildirilen diğer davranış değişiklikleri arasında panik ataklar, intihar düşünceleri ve özellikle MSA-C tipinde emosyonel inkontinans ve patolojik gülme veya ağlama davranışı yer almaktadır.
Wilson hastalığı, PKAN, Kufs hastalığı metabolik bozukluklara bağlı yirmili yaşların demans hastalıkları arasındadır. Genç başlangıçlı demansların ayırıcı tanısına girerler. İlk iki hastalık tipik olarak hareket bozuklukları ön planda olan sendromlara sahiptir. Genç erişkin demansları arasında olduğu için burada anılan Kufs’a demansın yanı sıra epilepsi eşlik eder. Whipple hastalığı da, bazen PSP’yi taklit eden diğer nadir hareket bozukluğu – demans sendromları arasındadır. Bu bahsedilen demans hastalıklarına kitabın başka bölümlerinde yer verildiğinden burada detayları anlatılmamıştır.
Kaynaklar
1. Duff K ve ark. Mild cognitive impairment in prediagnosed Huntington disease. Neurology. 2010; 75: 500–507.
2. Emre M ve ark. Clinical diagnostic criteria for dementia associated with Parkinson's disease. Mov Disord. 2007; 22: 1689-1707.
3. Emre M. Clinical features, pathophysiology and treatment of dementia associated with Parkinson's disease. Handb Clin Neurol. 2007;83:401-419.
4. Litvan I ve ark. Diagnostic criteria for mild cognitive impairment in Parkinson's disease: Movement Disorder Society Task Force Guidelines. Mov Disord. 2012; 27: 349-356.
5. McKeith IG ve ark. Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology. 2017; 89: 88-100.
6. McKeith IG ve ark. Research criteria for the diagnosis of prodromal dementia with Lewy bodies. Neurology. 2020; 94: 743-755.