ALZHEIMER HASTALIĞI
Yazanlar: İ. Hakan Gürvit, Zerrin Yıldırım
Son güncelleme tarihi: 13.12.2020
Bu bölüm, sayfa sayısı kısıtlaması nedeni ile kitabın basılı halinde kısaltılmıştır. Daha ayrıntılı metin için bölümün uzun versiyonuna başvurulabilir.
Kazanılmış entellektüel becerilerin yitirilmesi olarak demans antik çağlardan beri bilinen bir durumdur. Yaşlılık gerek Antik Yunan’da, gerekse de Roma’da bir yandan bilgelikle, diğer yandan da kaçınılmaz kognitif bozulmayla eşlenmiştir. Skolastik Çağ’ın sonlanması ve Modernite ile birlikte ampirik bilimlerin öne çıkmasıyla, tıp alanında da deneysel yöntem yeniden itibar kazanacak ve yaşamları içinde zihinsel bozukluk sergileyen hastaların otopsilerinde 17. yüzyıldan itibaren beyin atrofisi bulgusu kaydedilecektir. Ancak, AH’nin ilk olgusu olma onuru bayan Auguste D.’ye aittir ve bu tek olgunun Dr. Alois Alzheimer tarafından bildirilmesi tıp tarihinde demans hastalıklarının modern miladı sayılır.
Alzheimer, beynin nöropatolojik incelemesinde serebral korteksin normale göre incelmiş olduğunu gördü. Ayrıca, daha sonra senil plaklar (SP) ve içerdikleri materyel anlaşıldıktan sonra amiloid plaklar (AP) olarak adlandırılacak miliyer odakları ve yine daha sonra nörofibriler yumaklar (NFY) adını alacak intraselüler fibril birikintilerini tanımladı. Hocası ve çalışma arkadaşı, Emil Kraepelin, Psychiatrie: Ein Lehrbuch für Studierende und Aerzte adlı döneminin ünlü psikiyatri ders kitabının 1910’daki 8. baskısında Alzheimer’in adını anarak ilk kez “Alzheimer hastalığı” terimini kullanmıştır. Alois Alzheimer, 1911 yılında ise, üç yıl boyunca Kraepelin’in kliniğinde yattıktan sonra 56 yaşında ölen Johann F. olgusunu bildirmiştir. Enteresan biçimde yumaklar olmaksızın sadece plakların görüldüğü (“sadece-plak demans”), ailesinde çok sayıda 30 ila 60 yaşında hastalık başlangıçlı demans olgusu mevcut bir hasta olmasından dolayı Johann F. bildirilmiş ilk ailesel Alzheimer hastalığı olgusudur.
KLİNİK TABLO
Terminoloji
AH tipik olarak 65 yaş üzeri bir bireyde, sinsi başlangıçlı, yavaş ilerleyici seyirli, en baştaki sübjektif bellek yakınmalarının sonra objektif olarak saptanabilen izole progresif bellek bozukluğuna dönüştüğü, zaman içinde diğer kognitif alanlardaki bozuklukların da eklendiği ve nihayet zihinsel bozukluğun şiddetinin günlük yaşam aktivitelerindeki (GYAlar) bağımsızlığı da engelleyerek işlevselliği bozar hale geldiği bir klinik tablodur. İşlevsel bozukluk eşiği aşıldığında, kendi içinde hafif, orta ve ağır olarak alt evrelere ayrılan AH demansı (AHD) aşamasına geçilmiş olur. Bu eşiğe kadar olan seyir hafif kognitif bozukluk (“mild cognitive impairment” – MCI) veya prodromal AH olarak adlandırılır. Prodromal AH’nin tipik özelliği olan izole progresif bellek bozukluğu amnestik MCI (aMCI) olarak adlandırılır. AH klinik belirtilerini vermeden önce uzun yıllar süren ve bu sırada sadece hastalığın patolojik özelliklerinin evrildiği ve pre-klinik AH adı verilen bir başlangıç evresine sahiptir. Sübjektif bellek yakınmaları halen pre-klinik evrenin son aşamaları olarak değerlendirilmekte ve sübjektif kognitif bozukluk (“subjective cognitive impairment” – SCI) olarak adlandırılmaktadır. Pre-klinik ve klinik evreler patolojik AH (AH-P) ve klinik AH (AH-K) olarak da adlandırılmaktadır. Tipik hastada aile öyküsü negatiftir, yani hastalık sporadik olarak ortaya çıkar. Tipik AH tüm AH olgularının yaklaşık %90’ından sorumludur. Bu tanımla uyumsuz erken başlangıç (65 yaş altı erken), hızlı seyir, bellek dışı klinik sunumlar, pozitif aile öyküsü atipik olarak değerlendirilir. Erken başlangıçlı sunum, EOAD (“early-onset Alzheimer’s disease”) olarak adlandırılır ve ağırlıkla genetik nedenselliğe sahiptir. Bu nedenle EOAD sınıfında aile öyküsü pozitif ailesel AH (FAH) olguları sıktır. Tipik geç başlangıçlı sunum ise LOAD (“late-onset Alzheimer’s disease”) olarak adlandırılır ve yukarda söz edildiği gibi bu sınıf başlıca sporadik AH’lilerden oluşur. AH seyri bazen Creutzfeldt-Jakob hastalığını taklit edecek kadar hızlı olabilir. Böyle olgular hızlı progresif AH (hpAH) olarak sınıflanır. Bir hasta başlangıç yaşı, seyir hızı ve ailevi özellik açılarından “atipik” olabilse de atipik AH terimi başlıca bellek dışı klinik sunumlar için kullanılır.
Tanı Kriterleri
Alzheimer Hastalığı tanısı için 2011 yılına kadar Dünya Sağlık Örgütü’nün Hastalıkların Uluslararası Sınıflaması dizisinin (ICDler) yanısıra 1984’ün NINCDS-ADRDA (National Institute of Neurological and Communicative Disorders and Stroke-Alzheimer’s Disease and Related Disorders Association) ve Amerikan Psikiyatri Birliği’nin (APA) DSM’ler dizisi (Diagnostic and Statistical Manual of Mental Disorders) tanı kriterleri kullanılmaktaydı.
1948 tarihli DSM-I ve 1966 tarihli DSM-II’deki “kronik organik beyin sendromu” tanımı 1980 tarihli DSM-III’te “primer dejeneratif demans”, 1987’nin DSM-III-R’ında (revize DSM-III) “Alzheimer tipi primer dejeneratif demans” haline dönüştükten sonra bu terim 1994’ün DSM-IV’ünde yaygın kabul görecek “Alzheimer tipi demans” (“dementia of Alzheimer-type” – DAT) oldu ve aynı terim 2000’in DSM-IV-TR’ında da korundu. Her iki tanı kriterleri dizisi de biri bellek olmak üzere en az iki kognitif alanda sinsi başlangıçlı, yavaş seyirli bir bozulmayı Alzheimer demansı tanısı için zorunlu tutmaktadır. GYAların bozulması DSM-IV için zorunlu kriterken, NINCDS-ADRDA için destekleyici bir kriterdir. Böyle bir tabloya yol açabilecek sekonder nedenlerin (sistemik, nörolojik, psikiyatrik) ekarte edilebiliyor olması da her ikisi için zorunlu dışlama kriteridir. Bu koşulların gerçekleştirildiği tipik tabloya, yukarda söz edildiği gibi DSM-IV ve DSM-IV-TR DAT adını verirken, NINCDS-ADRDA için ise aynı tablonun adı “muhtemel Alzheimer hastalığı” (“probable Alzheimer’s disease” - PRAD) olmaktadır. Sekonder nedenlerin varlığı veya bellek dışı sunum gibi atipik özelliklerin mevcudiyetinde ise “mümkün Alzheimer hastalığı” (“possible Alzheimer’s disease – PosAD) tanısı konulmaktaydı. Özellikle 1990’lardan itibaren Alzheimer-dışı demansların tanı kriterlerinin de devreye sokulmaya başlanmasıyla öncesindeki düşük özgüllük de bir ölçüde kompanse edilmiştir. Her iki sisteme göre de kliniko-patolojik serilerdeki tanı duyarlılığı %66-98, özgüllüğü %54-88 arasında değişmektedir. Görüntüleme, EEG ve lomber ponksiyon gibi laboratuvar yöntemleri sadece dışlama tanısı için gerekmekteydi. “Kesin Alzheimer hastalığı” tanısı ancak biyopsi ya da otopsiyle histopatolojik olarak bir nöropatolog tarafından konulabilmekteydi.
AH-P klinik olarak demans gelişmeden, hatta AH-K’dan yıllar önce ortaya çıkmakta, demans bu patoloji için ancak son dönemi temsil etmektedir. AH patofizyolojisi sadece tipik bellek sunumuna değil atipik klinik sunumlara da yol açabilir. Ayrıca geçen sürede AH dışı demansların da daha iyi anlaşılması AH’nin daha iyi ayrılmasını sağladı. Bu kavrayışlarla birlikte tanı için hastalığa özgü nörogörüntüleme bulgularının tanımlanması ve beyin-omurilik sıvısında AH’ye spesifik protein düzeyi ölçümlerinin kullanılmaya başlanmasıyla laboratuvar artık bir dışlama yöntemi olmaktan çıktı ve AH hem klinik hem de laboratuvar özellikleriyle (AH Bİ’leri) tanımlanabilir hale geldi. Bu nedenle yeni tanı kriterleri geliştirilme ihtiyacı doğdu.
İlk müdahale 2007 yılında NINCDS-ADRDA kriterlerinin Uluslararası Çalışma Grubu (“International Working Group” - IWG) tarafından revize edilmesiyle yapıldı. Revize kriterler sadece PRAD tanısı içindi ve PosAD dahil edilmemişti. PosAD’ın dahil edilmeme tercihi nedeniyle NINCDS-ADRDA’de PosAD tanısı alabilecek bütün durumlar (atipik seyir veya klinik sunum, karma durumların varlığı) IWG’de otomatik olarak dışlama kriterleri olmaktaydı. IWG çekirdek klinik kriterler ve destekleyici özelliklerden oluşmaktaydı. Çekirdek kriterler izole veya başka bozukluklarla bir arada olabilecek progresif epizodik bellek bozukluğunu içermekteydi. Epizodik bellek bozukluğunun ipucuyla düzelmeyen limbik tarzda olması gerekliliğine vurgu yapılmaktaydı. Manyetik rezonans görüntüleme (MRG) ile görsel değerlendirme ölçeği kullanarak gösterilen mediyal temporal lob atrofisi, FDG-PET ile temporo-parietal hipometabolizma, BOS’ta düşük Aβ42 veya artmış total tau veya artmış fosfo-tau düzeyleri veya her üçü birden PRAD tanısı için destekleyici özellikler olarak eklenmişti. Kesin AH tanısı için histopatolojik doğrulama geçerliliğini korumaktaydı. Bununla birlikte bir yenilik olarak PRAD tanı kriterlerini dolduran bir hastada Mendelyen mutasyonlardan birinin (APP, PSEN1 veya PSEN2) varlığı tanıyı kesin AH yapmaktaydı. AH’nin demansla değil de pozitif Bİ’ler destekli ilerleyici bellek bozukluğuyla eşlenmesi ve mutasyon taşıyıcı tipik kliniğe sahip hastalarda kesin tanıya izin vermesi IWG kriterlerinin getirdiği en büyük yeniliklerdir. Bununla birlikte atipik sunumların dışlanması, pre-klinik evrenin unutulmuş olması da en büyük eksiklikleridir diyebiliriz.
2011 yılında NINCDS’in yerini alan Ulusal Yaşlılık Enstitüsü (National Institute on Aging-NIA) ve adını kısaltmış olan Amerikan Alzheimer Derneği (Alzheimer’s Association-AA) NIA-AA çalışma grubu olarak yeniden bir araya geldiler. NIA-AA çalışma grubu üç alt gruba ayrılarak AH-K evreleri olan MCI ve demans ve asemptomatik AH-P veya pre-klinik AH için üç ayrı tanı kriterleri seti önerdiler (Tablo 1A, 1B, 1C ve 1D). Böylelikle AH’yi demans ile eşleyen paradigmaya kesin olarak son verilmiş oldu ve hastalık pre-klinik evreyi de kapsayan bir süreklilik olarak yeniden kavramsallaştırılmış oldu. Bu süreklilikte evreler arası geçişler biyolojik nitelik değişikliklerini değil fakat klinik gözleme dayanan ister istemez konulmuş keyfi eşiklerin aşılmasını temsil etmektedir (pre-klinik evreden MCI evresine geçişte objektif bellek bozukluğu, MCI evresinden demans evresine geçişte işlevsel bozukluk, bu bozuklukların tanımlanma biçimine [örn., kaç standard sapma düşüş bozukluğa karşılık gelir?] ve bozukluğun saptanmasında kullanılan enstrümanların seçimine göre değişecektir). Bu kriterlerde AH-K çekirdek kriterleri genel pratikte de kullanım için ayrılmışken AH-P ve AH-K iki evresi için Bİ’lerin kullanımı araştırma amaçlı olarak ayrılmıştır. MCI için çekirdek kriterler bizzat hastanın kendisi, hasta yakını veya yakından izleyen hekim tarafından ifade edilen bozulmuş kognitif yetenekler ile beş kognitif alanda (yani, bellek, yürütücü işlevler, dikkat, dil ve görsel-mekansal yetenekler) bir veya daha fazla, işlevsel bozukluğa yol açacak şiddette olmayan objektif bozukluğu içermektedir. Demans için çekirdek kriterlerde ise dördü kognitif, biri davranışsal toplam beş alanın en az ikisinde işlevsel bozukluğa da yol açmış bozulma bulguları demans sendromu tanısı için gereklidir. Bu şekilde demans tespit edilmiş bir hastada sinsi başlamış ve aşikar kötüleşmenin ifade edildiği bir seyir ile klinik olarak tipik amnestik sunum yanısıra bu kez dil, görsel-mekansal veya yürütücü işlev bozukluğu gibi atipik non-amnestik sunumlar da NINCDS-ADRDA’den farklı olarak, PRAD tanısına olanak vermektedir. Demans tanı kriterlerinde PosAD tanısı, atipik seyire veya karma durumların (serebrovasküler hastalık, LCD özellikleri gibi) varlığına sınırlandırılmıştır. Bİ’ler iki ana gruba ayrılırlar: 1. Amiloid-beta (Aβ) birikimi işaretleyicileri, 2. Nöronal hasar işaretleyicileri. 1. grup beyin-omurilik sıvısında (BOS) düşük Aβ42 düzeyi ve/veya pozitron emisyon tomografisi (PET) ile amiloid görüntülemede beyinde amiloid birikimi delillerini içerir. 2. grup içinde BOS’ta artmış total tau ve fosfo-tau düzeyleri (kriterlerin yayınlandığı 2011’de PET ile tau görüntüleme henüz mümkün değildi), MRG ile kortikal kalınlık ölçümleri ile spesifik anatomik dağılımda gri madde kaybı (yani, lateral ve mediyal parietal, posterior singulat, ve lateral temporal korteksler) ve/veya volümetri ile hippokampal atrofi, FDG-PET ile spesifik anatomik dağılımda metabolik değişiklikler (precuneus ve temporo-parietal hipometabolizma) bulunur. AH-P zamansal seyri içinde 1. grubun daha önce pozitifleştiği bilinmektedir. AH-K evrelerinin her ikisi için de çekirdek kriterleri dolduran bir hastada her iki grubun da pozitivitesi MCI veya demansın AH ilintili olmasını yüksek ihtimalli, sadece birinin pozitivitesi orta ihtimalli, ikisinin de negativitesi ihtimal dışı kılmaktadır. Pre-klinik evrede AH-P sürekliliği üç alt evreye bölünmüştür. Evre 1. Asemptomatik serebral amiloidozis (sadece amiloid pozitif). Evre 2. Amiloid pozitivitesi + sinaptik disfonksiyon ve/veya erken nörodejenerasyon: Amiloid pozitivitesi + nöronal hasar göstergelerinden bir veya daha fazlasının pozitifliği (FDG-PET veya fMRG ile gösterilen sinaptik disfonksiyonun volümetrik kayıptan daha önce başladığına dair deliller vardır). Evre 3. Amiloid pozitivitesi + nörodejenerasyon delilleri + belli belirsiz kognitif bozulma: henüz MCI şiddetine ulaşmamış, normal değerler arasında kalan kognitif performansa rağmen kendi bireysel standartlarının altına düşme (Tablo 1D). Bu tabloya sübjektif kognitif bozulma (SCI) adı da verilir. İlerde biraz daha ayrıntılı tartışıldığı gibi, bu bireylerdeki yakınmalar mevcut nöropsikolojik bellek enstrümanlarıyla objektif olarak saptanamadığı için halen “sübjektif” olarak adlandırılmaktalar. Eğer bu bireylerin bellek yakınmaları tahmin edildiği gibi yenilik işleme ve örüntü ayırt etme bozukluğundan kaynaklanıyorsa ve bunu örüntüyü tamamlamayla (aşinalıkla) kompanse ediyorlarsa yenilik işlemeyi ve örüntü ayırt etmeyi değerlendirebileceğimiz yeni bellek testlerinin geliştirilmesiyle, SCI’lı bireylerin en azından bir kısmının AH-P’den AH-K’ya en erken evre olarak kaydırılmaları mümkün olacaktır. Bu üç kriter setinin yayınlanmasından sonra azımsanmayacak sayıda pre-klinik bireyin amiloid negatif+tau pozitif statüde oldukları görüldü. Bu bireylerin uzunlamasına izlemlerinde genellikle AH dışı klinik tablolar geliştirdikleri saptandı ve bunlara “kuşkulu non-Alzheimer patoloji” (suspected non-Alzheimer’s pathology – SNAP) adı verildi. Bu bireylerin bir kısmı demansa ilerlemeyen aMCI klinik bulguları geliştirirler. Bu hastalarda otopside de AP’ler olmaksızın sadece limbik-paralimbik alanlara sınırlı, AH’de görülenlerden farksız NFY’ler saptanabilmektedir. Eskiden “sadece yumak demansı” adı da verilen bu tabloya aslında bir demans değil de çok büyük sıklıkla bir non-progresif aMCI olması ve çok ileri yaşlarda görülmesi dolayısıyla “primer yaşla-ilintili taupati” (primary age-related tauopathy – PART) adı verilmiştir. Hem amiloid, hem de tau negatif olan ileri yaştaki MCI’lı bir bireyde “arjirofilik tahıl hastalığı” (argyrophilic grain disease – AGD) olabilir.
2014 yılında ise IWG yeniden toplandı ve Bİ kategorilerini ve AH klinik fenotiplerini tekrar gözden geçirerek IWG-2 kriterlerini yayınlandı. Bu kriterlerin öncekinden önemli iki farkı vardır. Öncelikle AH’nin tipik ve atipik klinik sunumları için ve karma tipte AH için ayrı ayrı tanı kriterlerinin önerilmiş olmasıdır. Diğeri ise Bİ’ler “destekleyici özellikler” olmaktan çıkarılmış ve klinik kriterlerle birlikte tanı için zorunlu haline getirilmiştir. PRAD terimi terk edilmiş ve AH-K için üç ayrı terim benimsenmiştir. 1. Tipik AH: Hippokampal tipte ilerleyici bellek bozukluğu izole olabileceği gibi ikinci bir kognitif veya davranışsal bozukluk (MCI veya demans şiddetinde olabilir) da eşlik edebilir. 2. Atipik AH: Üç ayrı atipik klinik sunum (posterior varyant AH, logopenik varyant AH, frontal varyant AH) ve Down Sendromu sunumu dahil edilmiştir. 3. Karma AH: Tipik AH’ye özgü bellek bozukluğuna eşlik eden serebrovasküler hastalık veya LCD’nin klinik ve görüntüleme özellikleri. Bu tanıların konulabilmesi için spesifik klinik fenotipe ek olarak Alzheimer patolojisinin in vivo delillerini gösterecek üç laboratuvar göstergesinden en az birinin varlığı gerekmektedir. 1. BOS’ta azalmış Aβ ile birlikte artmış total tau veya fosfo-tau düzeyi. 2. Amiloid PET ile amiloid birikimi. 3. AH otozomal dominant (OD) mutasyonlarından biri. IWG-2, IWG için yukarda sıralanan eksiklikleri kapatmış gibi görünse de kolay erişilir olmayan Bİ’leri tanı için zorunlu hale getirmesi bu kriterlerin rutin klinik pratikte kullanılabilmesini çok zorlaştırmıştır. Diğer yandan, 1. maddede BOS Bİ’lerinin her ikisinin birden pozitivitesini talep ederken, 2. maddede amiloid PET pozitivitesini ilk maddenin alternatifi olarak kabullenilmesi doğru bir yaklaşım olarak kabul edilemez. Bunun yerine NIA-AA 2011 kriterlerinin yaklaşımı olan serebral amiloidozun iki göstergesi BOS Aβ42 pozitifliği ile amiloid PET’te amiloid yükünün saptanmasının eşdeğer görülmesi doğru olurdu. Artık tau-PET de kabul görmüş bir yöntem olduğuna göre 2. madde “amiloid-PET ve tau-PET ile artmış ligand tutulumu” olarak değiştirilebilir.
Tablo 1A. Bütün demanslar için çekirdek klinik kriterler NIA-AA 2011 (McKhann ve ark., 2011)
|
Bütün demanslar için çekirdek klinik kriterler
1. İş ya da olağan aktiviteler sırasındaki işlevselliğin etkilenmesi, 2. İşlevsellik ve performansın daha önceki düzeylerine göre düşmüş olması, 3. Delirium ya da majör bir psikiyatrik bozuklukla açıklanmaması, 4. Kognitif bozulmanın (1) hasta ve yakınından alınan öykü ve (2) yatak başı mental durum muayenesi ya da nöropsikolojik inceleme gibi objektif bir kognitif değerlendirmeyle gösterilmesi. 5. Kognitif ya da davranışsal bozulma aşağıdaki alanlardan en az ikisini kapsamalıdır: 1. Yeni bilgi edinme ya da hatırlamanın bozulması. 2. Muhakemenin ve kompleks görevlerin yürütülmesinde bozulma, yargılamanın zayıflaması. 3. Görsel-mekansal yeteneklerin bozulması 4. Dil fonksiyonlarında bozulma. 5. Kişilik, davranış ve tutumlarda değişiklikler. |
Tablo 1B. Alzheimer hastalığı
demansı için biyoişaretleyicileri içeren NIA-AA kriterleri (McKhann
ve ark., 2011)
Tanı kategorisi
AH etiyolojisinin biyoişaretleyici olasılığı
Aβ
(PET ya da BOS)Nöronal hasar belirteçleri
(tau, FDG, yapısal MRG)
Muhtemel AH Demansı
Klinik kriterlere göre
Bilgi vermez
Test edilmemiş/ çelişkili/ belirsiz
Test edilmemiş/ çelişkili/ belirsiz
AH patofizyolojik süreci kanıtıyla
Orta
Test edilmemiş/ belirsiz
Pozitif
Orta
Pozitif
Test edilmemiş/ belirsiz
Yüksek
Pozitif
Pozitif
Mümkün AH Demansı (Atipik Klinik Sunum)
Klinik kriterlere göre
Bilgi vermez
Test edilmemiş/ çelişkili/ belirsiz
Test edilmemiş/ çelişkili/ belirsiz
AH patofizyolojik süreci kanıtıyla
Yüksek ama ikinci bir etiyolojiyi dışlamaz.
Pozitif
Pozitif
AH’ye Bağlı Olmayan Demans
---
En düşük
Negatif
Negatif
AH: Alzheimer hastalığı, Aβ: Amiloid beta, BOS: Beyin-omurilik sıvısı, PET: Pozitron emisyon tomografisi, FDG: Florodeoksi glikoz, MRG: Manyetik rezonans görüntüleme.
Tablo 1C. Alzheimer hastalığı -MCI için
biyoişaretleyicileri içeren NIA-AA kriterleri (Albert ve ark., 2011)
|
Tanı kategorisi |
Aβ |
Nöronal hasar belirteçleri (tau, FDG, yapısal MRG) |
|
MCI Klinik tanı (Petersen kriterleriyle aynı) |
Test edilmemiş |
Test edilmemiş |
|
MCI orta ihtimalle AH’ye bağlı |
Pozitif |
Test edilmemiş |
|
Test edilmemiş |
Pozitif |
|
|
MCI yüksek ihtimalle AH’ye bağlı |
Pozitif |
Pozitif |
|
MCI- olasılıkla AH’ye bağlı değil |
Negatif |
Negatif |
Aβ: Amiloid beta, BOS: Beyin-omurilik sıvısı, PET: Pozitron emisyon tomografisi, FDG: Florodeoksi glikoz, MRG: Manyetik rezonans görüntüleme, MCI: hafif kognitif bozukluk.
Tablo 1D. Alzheimer
hastalığı preklinik evreleri için biyoişaretleyicileri
içeren NIA-AA kriterleri (Sperling ve ark., 2011)
Kognitif olarak normal (risk altındaki) birey + Biyoişaretleyici pozitifliği
• Evre I : Aβ
• Evre II : Aβ + tau
• Evre III : Aβ + tau + silik kognitif/davranışsal bozulma
Hastalığın tanısını olabildiğince erken
koyabilmek arzusu klinik çalışmaların odağını
risk altında fakat kognitif yakınması olmayan ya da kognitif
yakınması olan fakat nöropsikolojik testlerle objektif kognitif
bozukluk saptanmayan bireylere yönlendirdi. Böylelikle araştırmalarda
Bİ kullanımı yaygınlaştı ve AH’nin
kliniko-patolojik ilişkisinin yerini klinik-biyoişaretleyici
ilişkisi aldı. AH artık demansla karakterize bir sendrom
olmaktan ziyade zemininde yavaş ilerleyen amiloidoz ve ardından
nörodejenerasyonla karakterize bir nöropatolojinin bulunduğu ve klinik
olarak asemptomatik evreden demans evresine kadar ilerleyen semptomatik
evrelerden oluşan bir hastalık sürekliliği olarak görülmeye
başlandı.
Böylelikle 2018 yılına gelindiğinde NIA-AA çalışma grubu özellikle araştırmalarda kullanılacak bir çerçeve çizmek üzere AH için kliniği geri planda tutan biyolojik ağırlıklı bir tanımlama girişiminde bulundu. AH biyoişaretleyicileri amiloid birikimi belirteçleri (A), tau birikimi belirteçleri (T) ve nörodejenerasyon belirteçleri (N) olmak üzere 3 gruba ayrıldı ve AT(N) Bİ’leri olarak tanımlandılar.
BOS’ta Aβ42 pozitifliği ya da amiloid PET ile amiloid birikiminin gösterilmesi A pozitifliği, BOS’ta fosfo-tau pozitifliği ya da tau PET ile tau birikimin gösterilmesi T pozitifliği, anatomik MRG’de atrofi ya da FDG-PET’te hipometabolizma ya da BOS’ta total tau pozitifliğinin gösterilmesi (N) pozitifliği anlamına gelmektedir. Bu üç biyoişaretleyici grubuna göre 8 tane biyoişaretleyici profili önerilmiştir (Tablo 2). Her üçünün de negatif olması normal biyoişaretleyici profili (NBİP) anlamına gelirken, diğer ikisinden bağımsız olarak sadece A pozitifliği kişiyi Alzheimer sürekliliği (AS) kategorisine sokmaktadır. A negatif iken diğerlerinden en az birinin pozitifliği ise kişiyi non-AH patolojik değişiklik (NAPD) kategorisine sokar. AS içinde A+T-(N)- durum Alzheimer patolojik değişikliği (APD) adını alır. A+T+(N)+/- iki durum AH olarak adlandırılır. A+T-(N)+ durum ise APD+NAPD olarak karma duruma karşılık gelir. Bu kriterlerde klinik, biyoişaretleyicilerden farklı olarak kognitif evrelemeyle tariflenir ve (C) ile gösterilir. A ve T AH’yi tanımlayan spesifik nöropatolojik değişikliklere işaret etmektedir, (N) ve (C) ise AH’ye spesifik değildir ve bu yüzden parantez içerisinde gösterilmiştir. Kognitif yakınması olsun ya da olmasın objektif kognitif bozukluk saptanmamış bireyler kognitif olarak etkilenmemiş (“cognitively unimpaired” - CU), demans eşiğini aşmamış fakat objektif olarak kognitif bozulma saptanan bireyler MCI ve GYA’ları etkiler düzeyde kognitif bozulma saptanan bireyler ise demans (D) evresinde olarak tanımlanmıştır. AT(N) statüleri ile (C) statüleri arasında mütekabiliyetler saptanabilir. Buna göre AS, NAPD ve NBİP statülerinin her biri için CU, MCI ve D durumları mümkündür. AS kendi içinde klinik bozulma şiddetine göre altı evreye ayrılır. 1. Evre: CU. 2. Evre: SCI-CU. 3. Evre: MCI. 4. Evre: Hafif demans. 5. Evre: Orta demans. 6. Evre: Ağır demans.
Tablo 2. Tanımlayıcı terminoloji: Biyoişaretleyicilerle kombine şekilde sendromal kognitif evreleme (Jack ve ark., 2018).
Biyoişaretleyici kategorisi
AT(N) profili
Kognitif evre
Kognitif Olarak Etkilenmemiş
Hafif Kognitif Bozukluk (HKB)
Demans
Biyoişaretleyici Profili
Normal AH biyoişaretleyicileri
A-T-(N)-
Normal AH biyoişaretleyicileri, kognitif olarak etkilenmemiş
Normal AH biyoişaretleyicileri ile birlikte MCI
Normal AH biyoişaretleyicileri ile birlikte demans
Alzheimer sürekliliği
A+T-(N)-
Preklinik Alzheimer patolojik değişikliği
Alzheimer patolojik değişikliği ile birlikte MCI
Alzheimer patolojik değişikliği ile birlikte demans
A+T+(N)-
Preklinik Alzheimer hastalığı
Alzheimer hastalığı ile birlikte HKB
Alzheimer hastalığı ile birlikte demans
A+T+(N)+
A+T-(N)+
Alzheimer ve eşlik eden şüpheli non-Alzheimer patolojik değişikliği, kognitif olarak etkilenmemiş
Alzheimer ve eşlik eden şüpheli non-Alzheimer patolojik değişikliği ile birlikte HKB
Alzheimer ve eşlik eden şüpheli non-Alzheimer patolojik değişikliği ile birlikte demans
Non-Alzheimer patolojik değişiklik
A-T+(N)-
Non-Alzheimer patolojik değişiklik, kognitif olarak etkilenmemiş
Non-Alzheimer patolojik değişiklik ile birlikte HKB
Non-Alzheimer patolojik değişiklik ile birlikte demans
A-T-(N)+
A-T+(N)+
Tanımladığımız “AH’nin geç modern dönemi” büyük ölçüde
Bİ’lerin klinik tanıya katkısı ile belirlenmektedir. Bu
nedenle bu dönem içinde devreye girmiş olsalar da kriterleri içine
Bİ’leri dahil etmeyen DSM-V (2013) ve ICD-11 (2019) gibi tanı
kriterleri setleri burada tartışılmamıştır.
Alzheimer Hastalığının Evreleri- Alzheimer Hastalığı Sürekliliği
İlerleyici hastalıklar patogenetik süreçleri içinde belli bir eşiği geçtikleri aşamada klinik olarak saptanabilirler. Bu eşik de hemen daima normal kabul edilemeyecek bir belirtidir. Örneğin, glioblastoma yeterince büyüyüp kafaiçi basıncını arttırarak başağrısına neden olana kadar subklinik olarak ilerler. Başağrısı kadar aşikar bir belirti olmayan unutkanlık durumunda subklinik dönemle klinik dönem daha kalın çizgilerle içiçe girer. Unutkanlığın klasik olarak yaşlılıkta sıklıkla duyulmaya alışılmış, gayet normal kabul edilen bir yakınma olması da “normal” olan ile “anormal” olanın ayrımını daha da önemli kılmaktadır. Bellek de dahil olmak üzere bazı kognitif yeteneklerde yaşlanmayla birlikte beklenen “normal” azalma nöropsikolojik standardizasyonda eğitimle birlikte göz önüne alınması gereken temel özelliklerdendir. Bir yakın bellek testinde yetmişli yaşların normal değeri yirmili yaşların normalinin yarısı olabilir. Normal yaşlanma kavramı bellek de dahil kognitif kapasitelerini hiç yitirmeyen küçük bir azınlığı (“süper yaşlılar”) içerse de, ağırlıkla nöropsikolojik testlerde yaşa göre uyarlanmış düzeylerde bir performansa karşılık gelir. Sosyal yaşamda başka açılardan sağlıklı bir yaşlının özel isimleri unutuyor, özel eşyalarını kolaylıkla bulamıyor olması anormal kabul edilmez. Aynı soruları tekrarlar olması yakınları arasında kuşku uyandırmaya başlar. İşini sürdüremez olması, ya da yabancı bir mekanda kaybolması ise artık normal olarak kabul edilemez olur. Normal yaşlanma için kabul edilebilir bellek bozukluğunu tanımlamak amacıyla “Selim Yaşlılık Unutkanlığı”, “Yaşla İlintili Bellek Bozukluğu (AAMI)” kavramları önerilmiş ve 1986 yılında Amerikan Ulusal Zihinsel Sağlık Enstitüsü (NIMH) tarafından araştırma amaçlı tanı kriterleri yayınlanmıştır. Bu tanı kriterlerinde AAMI tanımına uygun düşen yakınmaları olan bir yaşlı günlük yaşamında tümüyle bağımsızdır, nöropsikolojik muayenesinde ise özellikle yakın bellek testlerinde yaşa göre normal sınırlarda, fakat genç erişkinlere göre ortalama değerlerin 1 standart sapma (SD) altında yer aldığı saptanır. Fakat bu ayrım hangi yaşlının “selim” yaşlılık değişikliklerini gösterdiği, hangisinin ise AH’nin en erken izlerini taşımakta olduğu sorularının cevabını verememekteydi. Günümüzde artık SCI kavramı yaygın kabul görmüştür.
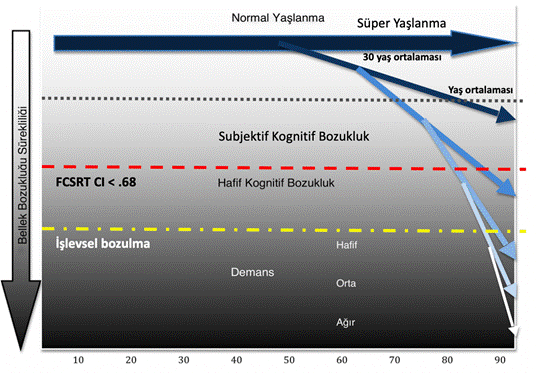
Şekil 1. Normal yaşlanmadan ağır demansa bellek bozukluğu sürekliliği.
Yaşlılıkta pre-demans kognitif bozukluğu tanımlama gayretiyle bir dizi daha kavram ve kriterler dizisi ileri sürülmüş olsa da tümü demansa ilerleyen bir bellek bozukluğu kavrayışından ziyade demans kadar şiddetli olmayan bir “hafif kognitif bozukluk”u çeşitli isimlerle tanımlamışlard. Bunlar arasında 1990’lar boyunca tanımlanan ICD-10’un “minimal kognitif bozukluk – MCD”, DSM-IV’ün “hafif nöro-kognitif bozukluk – MNCD”, Uluslararası Psikogeriyatri Birliği’nin “Yaşla İlintili Kognitif Bozukluk – AACD” ve Kanada Sağlıklı Yaşlanma Çalışması (CSHA) çerçevesinde kullanılan “demans olmayan kognitif bozukluk – CIND” kavramları sayılabilir. Tüm bu tanımlamalar, belli nüanslarına rağmen, tanımladıkları demans şiddetinde olmayan kognitif bozukluk durumunu esas olarak statik özerk antiteler olarak görmektedirler; öyle ki, bunlar arasından bir kısım hasta düzelip normale dönebileceği gibi, bir kısmı ise ilerleyip demans geliştirdiğinde nitelik değiştirip başka bir antiteye dönüşmüş gibi kabul edilmektedir. İlk kez 1997’de Mayo’dan Ron Petersen ve aralarında Türkiye kökenli Emre Kökmen’in de bulunduğu arkadaşları tarafından tanımlanan, MCI’da kişi anılan bellek problemlerinin yarattığı, ancak listeyle alışverişe çıkar olmak gibi genellikle üstesinden gelebildiği güçlükler dışında, günlük yaşamında halen bağımsızdır. Nöropsikolojik muayenesinde ağırlıkla bazen de tek başına bellek alanında, anlamlı düzeyde düşük performans gösterir. Orijinal biçimiyle bu tanım, prodromal AH olması muhtemel, ATD’ye dönüşme riski taşıyan bir alt grubu araştırma ve mümkünse tedavi hedefi yapmak üzere belirlenmişse de sonradan revize edilen biçimiyle amnestik olmayan demanssız kognitif bozukluklar da dahil edilerek prodromal AH’ye olan vurgu çabası kapsayıcılık adına zayıflamış görünmektedir. Sınıflama tümüyle bellek bozukluğunun bulunup bulunmamasına ve mevcut kognitif bozukluğun tek veya çoğul olma durumuna göredir. Bu farklı tabloların ilerledikleri takdirde en mutad olarak belli demans hastalıklarına dönüşecekleri beklenir. İlk bakışta bu sınıflama yukarda tanımlanan “çekirdek sendromlar” yaklaşımının bir benzeri gibi düşünülebilirse de buradaki “mevcut MCI tablosu-ilerdeki demans sendromu” ilişkisi çok daha indirgemeci bir akıl yürütmedir. “Çekirdek sendromların anatomik adreslerinden bu anatomik adrese yatkınlık sergileyen en muhtemel proteinopati” tarzı bir akıl yürütme esnekliği sergileyemez. Örneğin, progresif simultanagnozi-simetrik dorsal parietal alanlar-amiloidopati + taupati silsilesi progresif simultanagnozinin bu sınıflamadaki karşılığı olan izole non-amnestik MCI ile, bu MCI alt sınıfının AH’ye dönüşmesi beklenmediğinden sağlanamaz. Dahası, çekirdek sendrom yaklaşımıyla, ilerde demansa dönüşüp dönüşmeyeceğine bakılmaksızın anatomik yatkınlık ve muhtemel proteinopatiden yola çıkarak hastalık tanısına varmak mümkünken, MCI tanımı ile ancak demans aşamasında bir hastalıktan söz edilebilecek durumla normallik arasında kalmış bir geçiş aşaması tasarlanmış olmaktadır.
Uzunlamasına izlenen büyük serilerde bu kriterlerle tanımlanmış MCI’ın demansa dönüşme hızı yıllık %8-14’ken 5 yıllık izlemlerde %50 kadardır. Oysaki, normal popülasyonda demans insidansı %1-2’dir.
Yukarıda yapılmış olan tanımlar Alzheimer hastalığını bir süreklilik olarak kabul etmekten ziyade her bir kognitif basamağı ayrı bir antite olarak kabul etmekteydi. Bu da hastalığın nöropatolojik ilerleyişini yansıtmıyordu ve pre-klinik/asemptomatik evredeki hasta grubunu tanımlayamıyordu. Genetik açıdan risk taşıyan bireyler ve sağlıklı yaşlılarda yapılan çalışmalar hastalık patofizyolojisinin yıllar, hatta onyıllar (bazı hesaplamalara göre 15-20 yıl) önce başladığını ortaya koydu. Biyoişaretleyicilerin kullanımını sağlayan gelişmeler sayesinde de bu patofizyolojik süreci in vivo izleme şansı elde etmiş olduk. Gözlenen ilk patolojik değişikliğin Aβ birikimi olduğu, fakat kognitif bozulmanın ancak tau birikimi ve nöron kaybı gelişmesiyle ortaya çıktığı görüşü önem kazandı.
2011 NIA-AA kriterleriyle hastalığın pre-klinik evresi artık asemptomatik ve “sübjektif/silik kognitif bozukluk” evresi olarak ikiye ayrılırken klinik evrelerine ise henüz demans eşiğine ulaşmamış objektif kognitif bozuklukla karakterize “Alzheimer Hastalığına bağlı hafif kognitif bozukluk” (AH-MCI) ve ardından “Alzheimer Hastalığı demansı” dahil olmaktadır (Şekil 1).
Alzheimer Hastalığı Sürekliliği, Pre-klinik Evre
Giderek hastalığın patolojik ilerleyişinin daha iyi anlaşılmasıyla hastalığın erken evrelerde henüz asemptomatik iken tanısının konulabilmesi önem kazanmıştır. Hastalığın pre-klinik evresiyle ilgili bilgiler özellikle genetik açıdan riskli gruplarla yapılan çalışmalardan elde edilmiştir. Pre-klinik evre klinik olarak asemptomatik (AH-AS) ve SCI (AH-SCI) olarak ikiye ayrılmaktadır. Akılda tutulması gereken, bir bireyin AH sürekliliği-pre-klinik evresinde değerlendirilebilmesi için zeminde AH patolojisine dair delil bulunması gerekliliğidir. Fakat bu delil kişide açıkça AH demansı gelişeceğinin kanıtı olarak kabul edilemez. Biyoişaretleyici pozitif fakat kognitif olarak normal kişiler “AH demansı için risk altında asemptomatik bireyler” olarak tanımlanmaktadırlar.
Tüm bu tanımlar “asemptomatik bir birey neden AH açısından değerlendirilmekte ve AH sürekliliği içine dahil edilmektedir?” sorusunu akla getirebilir. Bu tanımlara özellikle AH için genetik risk taşıyan gruplarda yapılan çalışmalarla ya da çalışmalarda kontrol grubu olarak incelenmiş bireylerin sonuçlarıyla ulaşılmıştır. Kognitif yakınması olmayan ve nöropsikolojik olarak da kognitif olarak normal bulunan bireylerde de BOS veya PET ile amiloid birikiminin delilleri gösterilmiştir. Bu delilin ne anlama geldiğine dair detaylı bilgiye biyoişaretleyiciler başlığında tekrar değinilecektir.
Bir bireye SCI diyebilmemiz için kişinin kendi normaline göre kognitif/davranışsal bozulma yakınması olması, bu yakınmanın günlük yaşam aktivitelerini etkilememesi ve nöropsikolojik testlerdeki performansının normatif aralıkta olması gerekir. Yukarda da değinildiği gibi yenilik işlemlemeyle ilgili bellek testlerinin rutin nöropsikolojik değerlendirmeye sokulması, muhtemelen SCI’lı bireylerin bir kısmını AH-P’den AH-K’ya sokacaktır. Kişi kendi kognitif durumundaki değişikliği fark ettiği ve ilerde demansa dönüşme riski konusunda endişe taşıdığı için başvurmuş olabilir. Özellikle AH için genetik açıdan riskli grupta olan bireyler bu aşamada bize başvurabilirler.
NIA-AA kriterlerinde bu pre-klinik evreler biyoişaretleyici pozitifliğine göre sınıflandırılmış ve pre-klinik AH kendi içinde 3 evreye ayrılmıştır (bkz. Tablo 1D).
Bu sınıflamalar araştırmalar için bir çerçeve oluşturmak amacı taşırlar. Pre-klinik evrede AH patolojisi için biyoişaretleyici kanıtı olan bireyler kognitif ve davranışsal bozulma ve ardından demans gelişimi için risk altındadırlar. Fakat AH-P pozitifliği olup yaşam boyu semptom geliştirmeyen bireyler de bulunmaktadır. Otopsi çalışmaları da bunu desteklemektedir. Kognitif açıdan normal kişilerin otopsileri, tümüyle normal olabileceği gibi 50’li yaşların başlarından itibaren neokortekste gevşek nitelikli amiloid plaklar ve bazen bunlara eşlik eden entorhinal kortekste NFY’lerin görülebileceğini ortaya koymaktadır. Sağduyu bu 2. gruba ait beyinlerde AH nörodejenerasyonun başladığı fakat henüz klinik belirti eşiğini geçmediğini düşündürmelidir. Başka bir deyişle, henüz tam kesin bir bilgi olarak yerleşmemiş bir şekilde, 2. grup değişiklikleri göstermeye başlayan beynin sahibi yeterince uzun yaşasaydı, ya yakınmaları zaman içinde görünür olup sonunda AH klinik kriterlerini doldurabilirdi veya savunma mekanizmalarının direnci sayesinde nöropatoloji sınırlanabilir ve yaşam boyu asemptomatik kalabilirdi. Bu varsayımların ikisi de bir arada kısmen doğru olabilir.
Alzheimer Hastalığı Sürekliliği, Klinik Evre (AH-K)
Hastalığın klinik evreleri henüz demans eşiğine ulaşmamış objektif kognitif bozuklukla karakterize AH-MCI ve ardından AHD’dir. Yukarda değinildiği gibi AH-K klinik sunum, başlangıç yaşı, seyir hızına göre tipik ve atipik olarak sınıflanır. Klinik sunum yukarda Çekirdek Sunumlar başlığında ayrıntıyla tartışılmıştır. Tipik klinik sunumun (%90) ilerleyici bellek bozukluğu olması beklenir ve çekirdek sendromlardan “progresif amnestik disfonksiyon”a karşılık gelir. Atipik sunumlar (%10) diğer tüm çekirdek sendromlarla olabilir. Bu sunumlar arasında görsel, dilsel ve frontal varyantlar IWG-2 ve NIA-AA gibi formel tanı kriterlerine de dahil edilmiştir. Formel kriterler içinde anılmamakla birlikte motor varyant ile de azımsanmayacak bir sıklıkta sunulabileceği unutulmamalıdır. Görsel varyant-AH çekirdek sendromlardan “progresif görsel-mekansal bozukluk”a karşılık gelir ve geleneksel olarak “posterior kortikal atrofi” olarak adlandırılır. Dilsel varyant-AH çekirdek sendromlardan “progresif afazi”ye karşılık gelir ve primer progresif afaziler altında sınıflanan logopenik varyant PPA ile ilintilidir; agramatik/tutuk ve semantik varyant PPA’larda AH patolojisi beklenmez. Frontal varyant-AH çekirdek sendromlardan “progresif davranışsal/yürütücü bozukluk”a karşılık gelir ve en seyrek atipik AH sunumu olsa da dvFTD fenotipinin göreli yaşlı sunumlarında akla getirilmelidir. Motor varyant-AH çekirdek sendromlardan “progresif apraksi”ye karşılık gelir ve kortiko-bazal sendrom alt tipleri arasında temporo-parietal varyant KBS ile ilintilidir; frontal varyant KBS’de AH patolojisi beklenmez. Tipik başlangıç yaşının 65+ olması beklenir (LOAD). Terminoloji başlığında değinildiği gibi, 65 yaş altı “genç” veya “pre-senil” başlangıç (EOAD) atipik kabul edilir ve genetik faktörleri düşündürür (otozomal-dominant mutasyonlar veya APOE ε4 taşıyıcılığı). AH seyri tipik olarak yıllar içinde yavaş ilerleyicidir. Ancak, yine Terminoloji başlığında değinildiği gibi Creutzfeldt-Jakob hastalığına benzeyen, hızlı ilerleyici bir atipik alt tipi de vardır (hpAH). hpAH hastaları genel olarak MMSE’de yılda 6 veya daha fazla puan kaybeden AH’li bireyler olarak tanımlanır. Bu hastalarda fokal nörolojik bulgular erken dönemde çoğul olarak belirir. BOS Bİ’ler sıklıkla çok düşük Aβ, çok yüksek fosfo ve total tau düzeyleri şeklindedir. Sağkalım 3 yılın altındadır. Nöropatolojik olarak tipik AH bulgularına orta-ağır düzeyde serebral amiloid anjiyopati (CAA) eşlik etmesi de sık bir bulgudur.
AH-MCI
MCI tipik olarak amnestik ya da atipik olarak non-amnestik olabilir. Amnestik MCI’ın (aMCI) AHD’ye dönüşme olasılığı daha yüksektir. NIA-AA kriterlerinde AH-MCI için klinik ve araştırma kriterleri ayrı ayrı tanımlanmıştır. Klinik tanı kriterleri Petersen-Mayo kriterlerinden farksızdır. Hasta veya yakını tarafından ifade edilen ya da klinisyen tarafından gözlenen kognitif bozulmanın varlığı, bir ya da daha fazla kognitif alanda bozulmanın objektif olarak gösterilmesi ve günlük yaşam aktivitelerinin etkilenmemiş olması gerekmektedir. Kognitif yıkıma neden olabilecek başka faktörler dışlanmalıdır.
Klinik olarak MCI tanısı almış bireylerde Bİ’ler kullanılarak altta yatan etyolojiye dair fikir edinilebilir. İlerleyen yıllarda tedavi ajanları kullanıma girdiğinde doğru tedavinin bu şekilde seçilmesi mümkün olabilecektir. Bİ kullanımı kliniğin nasıl seyredeceğine dair de fikir verebilir. Örneğin klinik olarak MCI tanısı almış bir bireyde, NIA-AA kriterlerine göre, amiloid birikimine işaret eden Bİ pozitifliği veya nöronal hasara işaret eden Bİ pozitifliğinden biri mevcut ise, bu bireyin AH-MCI olma olasılığının orta düzeyde olduğunu, her ikisinin birden pozitifliği ise yüksek düzeyde olduğuna işaret etmektedir. Her ikisi de negatif ise bu MCI kliniğinin AH zemininde olma ihtimali zayıftır. MCI otopsilerinde normalden hafif AH patolojilerine kadar değişen bir spektrum izlenmekle birlikte, NFY’nin halen limbik-paralimbik alanlara sınırlı ancak sayısal olarak normallere göre anlamlı düzeyde artmış olması en tutarlı bulgu gibi görünmektedir. NIA-AA kriterlerindeki temkinli söylem günümüze MCI klinik tanısının A+T+(N)+ durumda AH-MCI, özellikle de ileri yaşlı MCI’lı bireylerde A-T+(N)+ durumda PART, özellikle de MCI tablosuna psikotik bulguların da eşlik etmesi durumunda ileri yaşın bir başka taupatisi olan “arjirofilik tahıl hastalığı” (AGD), A-T-(N)+ durumda “limbik-baskın yaşla-ilintili TDP-43 ensefalopati (LATE) patolojik tanısına karşılık gelmesi şeklinde uyarlanabilir.
Alzheimer Hastalığı Dermansı (AHD)
Aynı klinik özellikler belli bir eşiği aştığında artık kişinin günlük yaşam aktiviteleri de bozulmaya başlayacak ve AHD’den bahsedilecektir. Burada da tanı kişinin kendisi, yakını veya hekim tarafından farkedilen kognitif yıkımın varlığı, objektif araçlarla en az 2 kognitif alandaki bozulmanın gösterilmesi ve kognitif yıkımın kişinin günlük yaşam aktivitelerini etkileyecek düzeyde olmasıyla konulur. Bu duruma karşılık gelen otopsilerde NFY’ler artık neokortekse yayılmış, gevşek biçimden nöritik biçime değişmiş AP’ler limbik sistemde görülür durumdadır.
İlerleyici bir hastalıktan söz ettiğimize göre klinik tabloyu statik bir profille tarif edemeyiz. Yine sağduyu ile amatör bir yaklaşım dahi ilerleyen bir tablonun hafif, orta ve ağır olarak sınıflandırılması gerektiğini öngörebilir. Formel evreleme ölçeklerinin de taklit ettiği yöntem bundan farklı değildir. Zihinsel yıkımın demans düzeyine ulaşması günlük aktivitelerde bozulma ile belirlendiğine göre, evrelemeyi de GYA’lara göre kabaca yapabiliriz: SCI’yı sübjektif bellek yakınmaları, MCI’yı GYA’ların korunduğu ama yakın kişilerin farkında olduğu bir bellek bozulması olarak tarif etmiştik; bu durumda devamlılık ölçütünü göz önünde tutarak hafif demansı işte ve ev dışında bağımsızlığın bozulmaya başladığı, orta demansı, bunlar tümüyle bağımlılaşırken ev yaşamı ve kendine bakımda sorunların başladığı, ağır demansı ise sürekli bakım gereken, hastanın tümüyle bağımlı olduğu bir durum olarak tarif edebiliriz.
Bugün yaygın olarak kullanılan iki evreleme sisteminden biri olan “Global Bozulma Ölçeği” (Global Deterioration Scale- GDS)’nin de geliştiricisi olan Reisberg, AH’deki ilerleyici yıkım sürecini, bebeklik-erken ve geç çocukluk ve ergenlik şeklindeki insanın ilerleyici bireyselleşme-bağımsızlaşma gelişimsel sürecinin tam tersine çevrilmesi olduğunu ileri sürer ve bu ilerleyici yıkımı retrogenez olarak adlandırır. Buna göre MCI’lı yaşlı büyük ölçüde bağımsız olsa da bazı kararları için erişkin gözetimine gerek duyan ergene, hafif demanslı, evinde ve ev dışında tanıdık mekanlarda belli bir bağımsızlığı kazanmış, ancak sosyal ilişkiler, muhakeme gerektiren karmaşık işlevlerde halen denetim gereksinen 7-12 yaşlarındaki ilkokul çocuğunu andırır. Orta demanslı ise kabaca, ev yaşamı ve giyinme, yıkanma, yemek yeme gibi temel GYA’larda henüz gözetim gereken 2-6 yaşları arasındaki okul öncesi çocuğu gibidir. Ağır demanslı, yaşamını sürdürmek için 24 saat ana-babaya (bakıcıya) tümüyle bağımlı 0-2 yaş bebeğine benzer. Ağır evrenin kendisi de, yani giderek tüm motor ve verbal yeteneklerin kaybedildiği yatağa tam bağımlı nihai döneme doğru yıkım, oturma, yürüme, konuşma, sfinkter kontrolünün geliştiği bebekliğin dinamik gelişiminin tam tersi olarak kavranıp alt evrelere ayrılabilir.
GDS söz edildiği gibi AH’yi evrelemeye yarayan ölçeklerden birisidir. AH dışı demanslarda AH’ye özgüllüğü dolayısıyla kullanılamaz. GDS evreleri 1 ila 7 arasında değişir. Kabaca, GDS1 hiç yakınması ve bulgusu olmayan normal yaşlıya karşılık gelirken GDS2’ye SCI, GDS3’e ise MCI evreleri denilebilir. GDS 4-5-6-7 ise hafif, orta, ağır ve çok ağır olmak üzere AH’nin klinik evreleridir. Demansın Klinik Evrelendirilmesi (Clinical Dementia Rating Scale-CDR) ise yaygın olarak kullanılan 2. ölçektir. CDR’da bellek yine merkezi önemde olmakla birlikte, çok eksenli tasarımıyla diğer demansların evrelenmesinde de kullanılabilir. CDR evreleri 0-0,5-1-2-3 olarak sıralanırlar. CDR 0, SCI’yı da içerecek şekilde normal yaşlılığa karşılık gelir. CDR 0,5 büyük ölçüde MCI’ya, bazen de çok erken demansa karşılık gelir ve “kuşkulu demans” evresi adını alır. CDR 1, 2 ve 3 sırasıyla hafif, orta ve ağır evrelerdir.
Hafif evredeki demanslı hasta halen çalışmaktaysa artık işinde verimliliğini yitirmiştir. Yaratıcılık gerektirmeyen tekdüze işler başlangıçta sürdürülebilse de, iş arkadaşları performans düşüklüğünün farkındadırlar ve kısa süre içinde emeklilik kaçınılmaz olur. Yakın geçmişe ait olayların hatırlanmasındaki güçlük, aynı soruların tekrarlanması, kelime bulma güçlükleri yakınların dikkatini çeken başlıca özelliklerdir. Halen bildik mekanlarda dolaşabilse ve yolculuk yapabilse de, yabancı mekanlarda kaybolabilir. Araba kullanırken sinyalizasyona dikkatsizlik, tepkilerde yavaşlama, yönleri karıştırma gibi güçlükler başlamıştır. Banka işleri, fatura ödemeleri gibi mali işlerde hatalar olmaktadır. Banka kartı, cep telefonu gibi yenilikleri öğrenip kullanmayı başaramaz. Hobiler (dikiş-nakış, bahçecilik, sanatsal uğraşılar, yetenek oyunları, vb.) sürdürülemez olmuştur. Yemek lezzetinde bozulmalar gibi mutfak işlerinde güçlükler başlamıştır. Çamaşır, bulaşık gibi ev işlerini sürdürebilse de, bunlarda eski özenini bir ölçüde yitirmiştir. Okumak ve gazete-TV aracılığıyla aktüaliteye ilgi azalmıştır. Giyinmek, yıkanmak, sofra alışkanlıkları ve temel hijyende henüz bir sorun yoktur. İritabilite, duygulanımda küntleşme ve inkar eğilimi ile kendiliğindenliğin azalması dışında davranışsal belirtiler yoktur ve sosyal uygunluk iyi korunmuştur. Uyku kalitesi bozulmaya başlar. Cinsel ilgi ve iştah bozulur. Eksikliklerin farkedilmesinin de katkısıyla bazı olgularda depresyon belirtileri ön planda olabilir. Ancak depresyon sıklıkla keder ifadesi gibi afektif belirtilerden çok, isteksizlik gibi motivasyonel belirtilerle kendini gösterir. Muayenede yakın bellek ön planda olmak üzere, görsel-mekansal bozukluk, uzak bellekte bozulmalar, adlandırma güçlükleri, dikkat ve soyutlama-planlamada bozulmalar saptanır. Praksis muayenesinde “beden-parçası-nesne-gibi” cevaplar alınır. Henüz gnostik bir kusur saptanmaz. Somatik nörolojik muayene normaldir. MMSE skoru kabaca 20-26 arasında olabilir. Sıklıkla GDS 4, CDR 1 olarak evrelenirler. Bu hastaların otopsilerinde heteromodal kortekste NFY’ler, limbik sistemde nöritik SP’ler saptanır.
Orta demans evresine ulaşıldığında, hasta ev dışındaki bağımsızlığını artık tümüyle yitirmiştir. Gözetimle sokağa çıkabilse de, yalnız kaldığı takdirde yolunu bulamaz. Başkalarının evinde odaları karıştırabilir. Yeni öğrenme artık hemen hiç mümkün olamamaktadır. Anlama, okuma ve yazma giderek bozulur; evrenin sonlarına doğru imzası tanınmaz olabilir. Birinci derece yakınları hakkındaki bilgiyi genellikle korusa da, torunlarının sayısı, isimleri, okulları gibi bilgileri karıştırmaktadır. Evdeki işlevselliği son derece yüzeyselleşmiştir. Ancak sofrayı toplamaya veya sebze doğramaya yardım düzeyinde olabilir. Giyinme sırasında mevsime ya da günün saatine uygun giysiyi seçmede zorlanma, giysilerin sırasını karıştırma (gömleğin üzerine iç çamaşırı gibi), düğmeleri yanlış ilikleme gibi güçlükler başlar. Sofrada öncelikle bıçağı kullanamaz olduğunda yemeklerinin önceden kesilmesi gerekir. Giderek döküp saçarak yemek belirginleşir. Çatal-bıçağı karıştırmak, sıvıları çatalla almaya çalışmak gibi hatalar görülebilir. Yıkanmakta öncelikle sıcağı soğuğu ayarlamakla başlayan yardım gereksinimi ortaya çıkar. Henüz sfinkter kontrolü seyrek gece kaçırmaları dışında sorunsuzdur. Tuvalet mekaniği, elini yüzünü yıkamak gibi işlevleri kendi başına yapabilir. Davranışsal belirtiler artık vurgulanmaya başlamıştır. Hırsızlık, terkedilme ve sadakatsizlik hezeyanları olabilir. Yalnız kalmaktan ürker ve yakınını (eşi, çocuğu) sürekli gözünün önünde ister. Hekim vizitleri gibi yaklaşan randevular aşikar bir beklenti anksiyetesine yol açabilir. Uyku-uyanıklık ritminde bozulma artık belirginleşmiştir. Gece sık uyanmalar ve gündüz sık uyuklamalarla geçer. Muayenede hafif evre bulguları biraz daha ağırlaşmıştır. Dilsel işlevlere ait bulguların ağırlaşması dile dayanan testlerin yapılamaz olmasına neden olabilir. Praksis bozukluğu taraf apraksisi düzeyine ulaşabilir. Gnostik bozukluklar, özellikle sofrada göz önündeki nesneyi diğerleri arasından ayırıp bulamamak (simultanagnozi) şeklinde olabilir. Temel nörolojik muayenede hafif parkinsonyen değişiklikler saptanabilir. MMSE skoru 10-19 arasında değişir. GDS 5, CDR 2 olarak evrelenirler. Bu hastaların otopsilerinde NFY’ler heteromodal alanları tümüyle istila etmiş ve unimodal asosiasyon kortekslerine de yayılmış olabilir.
Ağır demans evresinde bellekte artık sadece parçacıklar söz konusudur. Yakınını (eşi, çocuğu) ana-babasıyla karıştırabilir, aynadaki kendi yüzünü tanıyamayabilir. Giyinmek, yıkanmak, yemek gibi temel GYA’larda artık tam bir gözetim gerekmektedir. Evrenin sonlarında yutma güçlüğü de ortaya çıkar. Kelime hazinesi son derece fakirleşmiştir. Evrenin sonlarında tüm verbal yetenekler yitirilir. Ambulasyon giderek zorlaşır ve sonlara doğru giderek oturmak dahi mümkün olmaz hale gelir. Televizyondaki kişileri ev içindeymiş gibi sanıp konuşmak, aynadaki kendi hayaliyle yabancıymış gibi konuşmak gözlenebilir. Ambulasyonun korunduğu sırada amaçsız dolaşma, istifçilik, amaçsız tekrarlayıcı hareketler izlenebilir. Tuvalet mekaniğinde bozulmalar (idrar ya da gaita sonrası uygun biçimde temizlenme, sifonu çekme sorunları), idrar kaçırma giderek belirginleşir. Epileptik nöbetler ortaya çıkabilir. Ağır evredeki hastaların formel muayenesi son derece güçtür ve mümkün olamayabilir. Muayenede global bir yıkım saptanır. Temel nörolojik muayenede tonus değişiklikleri, yürüyüş bozuklukları şeklinde parkinsonyen bulgular biraz daha ortaya çıkmıştır. MMSE 0-9 arasındadır. GDS 6-7, CDR 3 olarak evrelenirler. Şekil 2’de AH’nin klinik ve bir dizi laboratuvar ölçütünün pre-klinik evreden başlayarak ağır demans evresine kadar gösterdiği değişkenlik gösterilmektedir.

Şekil 2. Alzheimer hastalığının evreleri boyunca klinik ve laboratuvar göstergelerinin değişimi. BOS Aβ düzeyi daha preklinik evrede PIB-PET ile amiloid yükünün saptanabilmesinden önce düşmeye başlar; klinik evrelerden itibaren PIB-PET ile amiloid yükü fazlaca değişmez. SCI’dan itibaren kognisyon bozulmaya, BOS tau düzeyleri artmaya başlar. MCI evresinde FDG-PET ile bölgesel metabolizma azalması izlenir olur ve sonrasında demans evreleri boyunca yaygınlaşarak şiddetlenir. Hippokampus hacmi MCI’dan itibaren küçülmeye başlar. İşlevsel bozukluk (GYA’lar) tanı gereği demans aşamasından itibaren başlar ve ilerleyici olarak kötüleşir.
BOS: beyin-omurilik sıvısı; FDG-PET: florodeoksiglikoz pozitron emisyon tomografisi; GYA’lar: günlük yaşam aktiviteleri; MCI: hafif kognitif bozukluk; PIB: Pittsburgh bileşeni; SCI: sübjektif kognitif bozukluk
ALZHEIMER HASTALIĞI TANISINDA LABORATUVAR
Yukarıda da bahsedildiği üzere güncel olarak kullandığımız 2011 NIA-AA AH tanı kriterleri tanıya laboratuvarı da dahil etmiş ve biyoişaretleyici (Bİ) olarak adlandırdığı çeşitli laboratuvar yöntemlerini kullanarak hastalığın daha pre-klinik evrede tanınmasına olanak vermiştir. Bİ’ler, biyokimyasal Bİ’ler ve nörogörüntüleme Bİ’leri olarak sınıflanabilir. Yerleşmiş biyokimyasal Bİ’ler şimdilik sadece BOS Bİ’leri olsa da, başta kan ve plazma olmak üzere (diğerleri, oral, oküler ve nazal sıvılar olarak sıralanabilir) erişimi daha kolay olan vücut sıvıları da aday Bİ’ler açısından yoğun bir şekilde çalışılmaktadır. Nörogörüntüleme Bİ’leri MRG ile yapısal görüntüleme ve PET ile metabolik görüntüleme olarak sıralanabilir. Fonksiyonel MRG yöntemlerini de aday nörogörüntüleme Bİ’leri olarak sayabiliriz.
2011 NIA-AA tanı kriterlerinde Bİ’ler 2 gruba ayrılmıştır: (1) Aβ birikimine işaret edenler ve (2) nörodejenerasyona işaret edenler. Birinci grupta amiloid PET görüntülemede anormal tutulum ve BOS’ta düşük Aβ42 düzeyleri yer alırken, ikinci grupta artmış BOS total ve fosforile tau düzeyleri, FDG-PET’te spesifik topografik tutulum gösteren azalmış florodeoksiglikoz tutulumu (hipometabolizma), yapısal MR görüntülemede spesifik topografide atrofi yer almaktadır. 2018 yılı NIA-AA tanı kriterlerinde ise Bİ’ler 3 gruba ayrılmıştır. AT(N) sınıflaması olarak da bilinen bu sistemde amiloid birikimi belirteçleri (A), fosfo-tau (p-T) birikimi belirteçleri (T) ve nörodejenerasyon belirteçleri (N) olarak ifade edilmiştir (Yukarıya bakınız).
Beyin Omurilik Sıvısı Biyoişaretleyicileri
BOS Bİ’leri pratik olarak Aβ42, t-T ve p-T düzeylerine karşılık gelir. AH’de BOS Aβ42 düzeyi düşerken, t-tau ve p-tau düzeyleri yükselmektedir. Alzheimer hastalarında kontrol grubuna göre BOS Aβ42 düzeyinin yaklaşık %50 oranında azaldığı, t-T ve p-T düzeylerinin ise %300 oranında arttığı çeşitli çalışmalarda gösterilmiştir. Semptomatik AH hastalarında BOS’ta Aβ42 düzeyinde azalmayla beraber t-tau ve p-tau düzeylerinde artış hastalık tanısında %80 düzeylerinde bir sensitivite ve spesifiteye sahiptir. Biz Cerrahpaşa Tıp Fakültesi, Tıbbi Biyoloji Anabilim Dalı, Beyin ve Nörodejeneratif Hastalıklar Araştırma Laboratuvarı’nda analiz ettiğimiz BOS’larda eşik değerler olarak Aβ42<813pg/ml, p-T>52pg/ml, t-T>375pg/ml kullanıyoruz.
AH’de beyin parenkiminde birikmesi nedeniyle BOS Aβ42 düzeyi düşmektedir. Amiloid PET görüntülemeyle saptanan serebral amiloid yüküyle yüksek negatif korelasyona sahiptir. Amiloid pozitifliği AT(N) terminolojisinde, klinik belirtilerin mevcut olup olmamasından bağımsız bir şekilde “Alzheimer sürekliliği”ni başlatır. AH-P süresince BOS düzeyi progresif olarak azalır, erken AH-K evrelerinden itibaren plato çizmeye başlar. BOS Aβ42 AH’yi kontrol grubundan ve diğer nörodejeneratif hastalıklarından ayırmada yüksek doğruluğa sahip bir Bİ’dir.
BOS Aβ42 düzeyi kullanımında kişiler arası farklılıklar nedeniyle kısıtlılıklar ortaya çıkmaktadır. Bu sorun Aβ42/Aβ40 oranı hesaplanarak Aβ42’nin normalizasyonu yoluyla kısmen çözülebilmiştir. Aβ40 AH’de değişmemektedir bu nedenle toplam Aβ düzeyini yansıtan bir gösterge gibi kullanılmaktadır. Aβ38 düzeyi de AH ve kontroller arasında değişmediğinden BOS Aβ42/Aβ38 oranı da kullanılabilmektedir.
Bu işaretleyicilerin kan, plazma ve diğer vücut sıvılarında ölçülebilmesine yönelik çeşitli çalışmalar sürse de metodolojik farklılıklar nedeniyle henüz tutarlı sonuçlar elde edilememiştir.
AH’de BOS t-T ve p-T düzeyleri yükselmektedir. p-T serebral NFY/NT yükünü temsil eder ve tau PET’te saptanan tau yükü ile yüksek pozitif korelasyona sahiptir. AT(N) terminolojisinde, Alzheimer sürekliliğinde olan bir bireyde p-T pozitifliği klinik belirtilerin mevcut olup olmamasından bağımsız bir şekilde “Alzheimer hastalığı” tanısı koydurur.
En iyi araştırılmış p-T epitopu threonine 181 (p-tau181)’dir. Ancak p-tau231 ve p-tau199 da p-tau181 ile yüksek derecede koreledir. Her üçü de benzer tanısal doğruluğa sahiptir. Yukarda da değinildiği gibi A-T+ durumlar amiloidozsuz taupatileri, özellikle de ileri yaştaki hastalarda PART ve AGD gibi izole taupatileri düşündürmelidir. t-T nöronal hasar sonucu salındığı için BOS t-T artışı nöron kaybının derecesini ve nörodejenerasyonu yansıtan non-spesifik bir bulgudur. AH dışında LCD, FTD ya da prion hastalıkları gibi birçok nörodejeneratif hastalıkta artmış BOS t-T düzeyleri gösterilmiştir. Travmatik beyin hasarı, inme ve otoimmün ansefalitlerde BOS t-T düzeyinde nöronal hasarın derecesiyle korele olarak geçici ama belirgin bir artış görülebilir fakat p-T düzeyi normaldir. Creutzfeldt-Jakob hastalığında hızlı bir nörodejenerasyon mevcuttur fakat PHF tau birikimi görülmez. BOS t-T düzeyi AH’ye oranla 10-20 kat daha fazla artarken, p-T düzeyi normaldir ya da minör değişiklik gösterir. BOS t-T ve p-T düzeyleri AH’de kognitif bozulmayla Aβ’ye oranla çok daha güçlü bir korelasyona sahiptir, Alzheimer sürekliliğinde sıklıkla AH-K’nın başlangıcını işaret eder ve AH-K boyunca düzeyleri artmaya devam eder.
Nöro-Görüntüleme Biyoişaretleyicileri
Yapısal Manyetik Rezonans Görüntüleme
Alzheimer hastalığında yapısal manyetik rezonans görüntüleme daha önceden sadece ayırıcı tanı amacıyla kullanılırken, hastalığa özgü atrofi örüntüsünün tanımlanmasıyla doğrudan hastalığın tanısında kullanılabilir olmaya başladı. Tipik amnestik AH’de yapısal MRG’de mediyal, bazal ve lateral temporal loblarda ve mediyal parietal kortekste atrofi görülmektedir.
Hippokampus hacmi ve bunun zaman içerisindeki değişimi koronal, T1-ağırlıklı görüntülerle değerlendirilebilmektedir. Sağlıklı yaşlanma, MCI ve AHD gruplarıyla yapılan çalışmalar her üç grupta da zamanla hippokampal hacim kaybı olduğunu fakat AH ve MCI gruplarında bu azalmanın daha hızlı gerçekleştiğini göstermiştir. AH’de saptanan hippokampal atrofi kognitif bozulmayla koreledir. Klinik pratikte hippokampal atrofi görsel derecelendirmeyle değerlendirilebilir. Scheltens ve arkadaşları tarafından önerilen mediyal temporal lob atrofisi (MTA) görsel skorlamasında koronal, T1 ağırlıklı görüntülerde hippokampal atrofi 1-4 arası derecelendirilmektedir. Kantitatif yöntemler ile daha hassas bilgiler edinilebilir. Ayrıca ileri görüntüleme yöntemleri ile de (diffüzyon tensör görüntüleme, nöromelanine duyarlı yöntemler) hastalık süreçleri daha iyi anlaşılmaya başlamıştır.
Pozitron Emisyon Tomografisi
[18F]FDG-PET
[18F]-2-Fluoro-2-deoxy-D-glucose (FDG) yapay bir glikoz analoğudur ve dokunun glikoz metabolizması hakkında bize fikir vermektedir. Nörolojide özellikle nörodejeneratif hastalıkların tanısında kullanılmaktadır. Bölgesel olarak azalmış FDG tutulumu sinaptik ve nöronal hasarı yansıtmaktadır.
AH’de gözle görülür atrofi ortaya çıkmadan önce precuneus, mediyal temporal korteksler ve temporo-parietal simetrik bir hipometabolizma görülebilir. MCI’dan AHD’ye dönüşümü %76 sensitivite ve %82 spesiviteyle öngördüğü bildirilmiştir. Atipik bir AH formu olan posterior kortikal atrofide oksipital, logopenik varyant PPA’da sol parietal hipometabolizma görülür. AH’nin tipik ve atipik formalarını birbirinden ayırabildiği gibi diğer nörodejeneratif hastalıklardan da ayırmada kullanılır.
FDG-PET’teki bölgesel hipometabolizma AT(N) sınıflamasında (N) kategorisine alınmıştır ve nörodejenerasyon belirteci olarak yorumlanmaktadır.
Amiloid-β PET
Karbon (C-11) işaretli Pittsburgh bileşeni B (PiB), ilk kullanılan amiloid ligandıdır ve AP’lere yüksek afiniteye sahiptir. Fakat karbon işaretli ligandların yarı ömrünün çok kısa olması (20 dk) kullanımını oldukça sınırlandırmaktadır. Bu nedenle daha sonra daha uzun ömürlü olan (yaklaşık 110 dk) flor (F-18) işaretli [18F]flutemetamol, [18F]florbetapir ve [18F]florbetaben gibi bileşenler kullanılmaya başlanmıştır. Amiloid PET ile saptanan serebral amiloid yükü ile otopside nöropatolojik olarak saptanan amiloidozun çok yüksek bir uyum gösterdikleri bulunmuştur.
AH tanısı almış kişilerde yapılan amiloid-PET çalışmalarında neredeyse tüm hastalarda amiloid birikimi gösterilmiştir. Bu yüksek tekabül özellikle amiloid görüntülemede kullanılan PiB ile fibriler Aβ arasındadır. Bu bileşen fibriler Aβ’ya bağlanırken, çözünebilir Aβ ya da difüz plaklara bağlanmamaktadır. Çözünebilir formlar daha önce değinildiği gibi sinaptik disfonksiyona neden olan formlardır. Dolayısıyla, amiloid PET görüntülemesi negatif bireylerde dahi nöral ağların fonksiyonel görüntülemesinde özellikle APOE-ε4 taşıyıcıları gibi riskli bireylerde bağlantısallık azalması saptanabilir.
Amiloid birikiminin en erken görüldüğü bölgeler posterior singulat korteks ve retrosplenial korteksi de içine alan precuneustur. Bu mediyal parietal bölgeler tau birikiminin en erken başladığı MTL yapılarıyla ventral cingulum aracılığıyla karşılıklı bağlantılıdır.
Amiloid birikimi kognitif bozulmayla ne yükün şiddeti ne de topografisi açılarından korele eder. Bununla birlikte, tipik ve atipik olgularda aynı birikim topografisi klinik sunumdan bağımsız olarak AH tanısı koydurur. Diğer yandan, pre-klinik evrenin bir işaretidir. Çalışmalar kognitif olarak normal olarak katılan bireyler arasında gerçekten sağlıklı kontrolleri ve pre-klinik AH’li bireyleri birbirinden ayırır. Dolayısıyla, asemptomatik iken gerçek anlamda koruyucu “AH-aşısı” klinik çalışmalarına katılımının sağlanabilmesi açısından önemli katkı sağlamaktadır.
Tau PET
Tau PET görüntüleme, ancak otopside saptanabilen tau yayılımın in vivo olarak gösterilmesine olanak sağlamıştır. Böylelikle, geçen yüzyılda klinisyenlerin elinden alınıp, nöropatologlara teslim edilmiş gibi görünen AH’nin kesin tanısı itibarı henüz yaşamakta olan hastada da klinisyenler tarafından da konulabilir hale gelmiştir. Tau-PET ligandları tau’nun PHF formuna afinite göstermektedirler ve 3R/4R izoformunu tanımaktadırlar. Bu izoform diğer taupatilerde de görülebileceğinden hastalıklar arası ayırım yapabilmek için ligandın patolojiye spesifik anatomik dağılımına göre yorum yapılması gerekmektedir.
İlk jenerasyon ligandlar 11C]PBB3, [18F]THK523, [18F]THK5105, [18F]THK5351 ve [18F]flortaucipir iken, daha sonra hedef-dışı bağlanmaları daha düşük olan ikinci jenerasyon ligandlar [18F]RO948, [18F]GTP1, [18F]PI2620, [18F]PM-PBB3 ve [18F]MK6240 üretilmiştir.
Tau-PET ile saptanan patolojik tau birikiminin hem kognitif bozulma şiddeti hem de topografisi ile korelasyonun yüksek olduğu gösterilmiştir. Topografik farklılıklar tipik ve atipik klinik sunumları birbirlerinden net bir şekilde ayırt eder.
AT(N) sınıflamasında PET ile tau birikiminin gösterilmesi T pozitifliği anlamına gelmektedir.
Fonksiyonel Manyetik Rezonans Görüntüleme
Fonksiyonel MRG (fMRG) kan oksijenlenme seviyesine bağlı (“blood oxygen level dependent”-BOLD-) sinyalleri ölçen bir görüntüleme yöntemidir. BOLD sinyali nöral aktivitenin dolaylı bir yansımasıdır. BOLD sinyalleri görev bağımsız biçimde entrensek yavaş salınımlar göstermektedirler. İstirahat durumu fMRG (rs-fMRG) birden fazla anatomik bölgeye ait yavaş BOLD salınımlarının zamansal korelasyonunun bu bölgelerin birbirleriyle fonksiyonel olarak etkileştiği varsayımından yola çıkmaktadır. Böylece beyinde dinlenim durumu ağları olarak tanımlanan geniş boyutlu, görev-bağımsız ağlar tanımlanmıştır. Nörodejeneratif hastalıklarda bu ağların bağlantısallığının nasıl etkilendiği üzerine giderek artan sayıda çalışma yürütülmektedir.
Bu ağlardan AH için en çok dikkat çekeni, yukarda değinildiği gibi, olağan durum ağı (“default mode network”-DMN) olmuştur. DMN görüntüleme esnasında katılımcıya bir görev verildiğinde aktivitesini istirahat durumuna göre azaltan bir kognitif şebekedir. Ödevden bağımsız, “istirahat” halinde iken en aktif durumdadır.
Yaklaşık 20 yıldır nörodejeneratif hastalıklarda çalışılmakta olan rs-fMRG veya ICN görüntülemesi çalışmaları, özellikle AH’de daha en erken evrelerden başlayan DMN’deki progresif bağlantısallık azalmasını ortaya koymuştur. Son yıllarda, farklı nörodejenerasyonları tek tek ICN yatkınlıklarıyla anlamaktan çok, bunların neden olduğu gerek tek tek ağ-içi değişiklikler, ama aynı zamanda ağlar-arasındaki etkileşim değişiklikleri üzerinde durulmaktadır.
Bununla birlikte, bulunan farklılıklar grup karşılaştırmalarındaki istatistik anlamlılıklardır ve tek tek bireylerde tutarlı bir biçimde gösterilmeleri henüz mümkün olmamıştır. Bu yüzden bu görüntüleme teknikleri henüz bir nörogörüntüleme Bİ’si olarak değerlendirilemez.
EPİDEMİYOLOJİ
Prevalans ve İnsidans
Dünyada şimdiye kadar yapılan prevalans (belli bir zaman kesitinde bulunan tüm olguların nüfusa oranı) çalışmalarının metaanalizi 65-70 yaşları arasında %4-5 kadar olan AH’nin her 5 yılda bir katlanarak artıp 90’lı yaşlarda %50’ye kadar ulaştığını ortaya koymaktadır.
Türkiye’den ilk çalışma diyebileceğimiz Istanbul, Kadıköy’de gerçekleştirilen “Türkiye Alzheimer Prevalans Çalışması” (TAPS) sonuçları Tablo 3’te gösterilmiştir. Doksanlı yaşlardan sonra artışın devam etmeyip bu oranın plato çizdiği (“AH yaşla ilintilidir”) görüşü ile sürekli yükseldiği (“AH yaşlanmayla ilintilidir) görüşü arasındaki geçen yüzyıldaki tartışma ilki lehine çözümlenmiş gibi durmaktadır. Bu ileri yaşlarda, PART, AGD ve LATE gibi yeni nörodejenerasyon tipleri belirmekte ve yaşla birlikte giderek artmaktadır.
Tablo 3. TAPS Çalışması’nda yaşa ve cinsiyete göre katmanlandırılmış prevelans oranları (Gürvit, 2008)
|
|
PRAD |
Toplam AH |
Toplam Demans |
|
Yaş grupları bir arada Tümü Erkek Kadın |
%11 %9 %13 |
%16 %13 %17 |
%20 %17 %22 |
|
Cinsiyet grupları bir arada 70-74 75-79 80+ |
%9 %14 %13 |
%14 %19 %17 |
%18 %22 %23 |
|
Erkek 70-74 75-79 80+ |
%8 %9 %11 |
%12 %12 %16 |
%16 %15 %20 |
|
Kadın 70-74 75-79 80+ |
%10 %18 %15 |
%15 %23 %20 |
%19 %26 %25 |
PRAD: muhtemel Alzheimer hastalığı; toplam AH: PRAD artı mümkün AH; toplam demans: toplam AH artı non-AH demanslar.
2019 yılı verilerine göre dünya çapında demans tanısı almış 50 milyon kişi yaşamaktadır. Bu sayının 2030 yılında 82 milyon, 2050 yılında ise 152 milyon kişiye ulaşacağı varsayılmaktadır. Alzheimer hastalığı (AH) ise tüm demans tipleri arasında %60-80 görülme sıklığına sahiptir.
Yaşa-özgül insidans, ya da belli bir zaman aralığında ortaya çıkan yeni olguların sayısı da yaş aralıkları arttıkça hızla artar. 1998 tarihli bir meta-analizlerinde 65-69 ile başlayıp 85-89 ile sonlanan 5’er yıllık 5 yaş katmanında insidans %1.6, %3.5, %7.8, %14.8 ve %26.0 şeklinde yine katlanarak artmaktadır.
Risk Faktörleri ve Koruyucu Faktörler
Birçok hastalıkta olduğu gibi AH’de de risk faktörleri değiştirilebilir ve değiştirilemeyen risk faktörleri olarak ikiye ayrılmaktadır. AH için değiştirilemeyen risk faktörleri yaş, genetik ve cinsiyettir. Değiştirilebilir risk faktörlerine ise tedavi edilebilir tıbbi durumlar ve yaşam tarzı dahildir.
Günümüzde yaşam beklentisindeki dramatik artış global bir fenomendir ve yaşlı nüfusun artması ile AH daha fazla bir toplumsal sorun olmaktadır. Hastalığın toplumsal maliyetinin demans şiddetiyle doğru orantılı olduğunu gösteren çalışmalar mevcuttur.
Bir risk faktörü olarak cinsiyet tartışmalıdır. Birçok çalışma AH prevalansının kadınlarda erkeklere göre daha fazla olduğunu göstermektedir. Ancak bu prevalans farklılığı genellikle kadınlarda yaşam beklentisinin daha uzun olmasıyla açıklanmaktadır. Bununla birlikte östrojen beyinde bir nörotrofik faktör olarak işlev görmekte, erkeklerde ömür boyu mevcut olan testosteron beyinde östrojene çevrilip işlevini sürdürürken, kadınlar menopoz sonrası östrojensiz kalmaktadırlar. Bu durum yaşamın ikinci yarısında bir nörotrofik faktörden yoksun kalan kadınların neden demans için daha fazla risk taşıdıklarının açıklamalarından biri olabilir. Nitekim, epidemiyolojik çalışmalarda post-menopozal dönemde östrojen replasmanı kullanan kadınlarda kullanmayanlara göre demans prevalansının daha düşük olması bu varsayımı destekler niteliktedir ama jinekolojik kanser riski artışı nedeni ile bu yöntem koruyucu bir tedavi olarak kullanılamamaktadır.
Dominant geçişli ailesel AH olguları harici, ailede demans öyküsü AH için kendi başına bir risk faktörüdür. AH’li hastaların kardeşlerinde yaşam boyu hastalık riski beklentisi ikiye katlanarak %23’ten %48’e çıkar. Monozigotik ikizlerde dizigot ikizlere oranla AH birlikteliği anlamlı oranda fazladır.
Down sendromu aile öyküsü olması AH riskini 2 ila 3 misli arttırmaktadır. Ayrıca Down sendromlu çocuk doğuran annelerin AH riski, diğer tiplerde mental retarde çocuklar doğuran annelere göre 5 misli fazla bulunmuştur.
APOE-ε4 aleli taşımak AH için risk iken ε2 aleli bazı çalışmalarda koruyucu gibi görünmektedir. ε2’nin β-amiloid fragmanının temizlenmesinde etkin bir alternatifi temsil ettiği düşünülmektedir.
Diyabetes mellitus, sigara, depresyon, mental inaktivite, fiziksel inaktivite, beslenme yetersizliği veya kötü beslenme, hipertansiyon, obezite ve düşük eğitim hem AH için hem de kognitif bozulma için değiştirilebilir risk faktörleri olarak bildirilmiştir.
Yorumu tartışmalı olsa da düşük eğitimin başlı başına bir risk faktörü olduğu artık yerleşmiş bir bilgidir. Eğitim deneyimindeki artışın ve mental aktivitenin bireyin kognitif rezervinin genişlemesine yol açarak hastalığın ortaya çıkış eşiğini yükseltmesi çekici bir açıklama gibi durmaktadır. Rezerv kavramı AH patolojik değişikliklerinin başlamasıyla AH kognitif bozulmasının başlaması arasında geçen süre ve otopside AH patolojik değişiklikleri saptandığı halde klinik olarak asemptomatik kalan bireyleri açıklama çabası nedeniyle ortaya çıkmıştır. “Beyin rezervi” beynin maruz kaldığı patolojik değişikliklere dayanma ve normal fonksiyonunu sürdürme kapasitesini ifade ederken, “kognitif rezerv” ise patolojinin ortaya çıkardığı etkilerle başa çıkabilmek için alternatif beyin ağları ya da kognitif stratejiler kullanabilme yeteneğini ifade etmektedir. TAPS çalışmasında da düşük eğitim demans için bir risk faktörü olarak belirlenmiştir.
Kardiyovasküler ve serebrovasküler risk faktörleri ve inme öyküsünün AH riskini anlamlı biçimde arttırdığı birçok çalışmada gösterilmiştir. Yüksek kan basıncının AH ve demans riski artışıyla ilişkili olduğu, AH patolojisini arttırdığı bildirilmiştir.
Yüksek kan basıncı kan beyin bariyeri bütünlüğünü bozarak beyin dokusuna enflamatuvar protein sızmasına ve bunun sonucu olarak da hücre hasarı, apoptoz ve Aβ birikiminin artışına yol açarak AH riskini arttırıyor olabilir. Ancak hipertansiyonun Aβ birikimine yanıt olarak serebral hipoperfüzyona karşı protektif bir mekanizmayla ortaya çıktığı da öne sürülmektedir. Bu da hipertansiyon ve AH arasındaki nedensel ilişkinin henüz tartışmaya açık olduğunu göstermektedir.
Diyabet artmış AH riskiyle ilişkilendirilmiştir (RR = 1.39). Hiperinsülineminin beyinde bir amiloid-beta peptidi şaperon molekülü olarak işlev gören insülin-degrade edici enzim (IDE) için yarışarak (MSS’den insülin katabolizması için sistemik kullanıma çekilerek) beyinde Aβ klirensini bozduğu öne sürülmektedir. Bazı çalışmalar ise diyabetin doğrudan AH patolojisini arttırmadığı, bunu serebrovasküler riski arttırarak dolaylı olarak yaptığını öne sürmüştür.
Yüksek kolesterol düzeyleri artmış Aβ seviyesi, kognitif bozulma ve AH progresyonuyla ilişkilendirilmiştir. Buna paralel olarak, kolesterol düşürücü ajanları (statinler) kullananlarda AH prevalansın daha düşük olması yerleşmiş epidemiyolojik bilgilerdendir. Kolesterolün Aβ yıkımını azalttığı ve üretimini arttırdığı öne sürülmüştür.
İnme öyküsü yaşlı hastalarda AH riskini 2 kat arttırmaktadır. İnmeyle birlikte aterosklerotik risk faktörlerinin varlığında risk artar. İnme öyküsü daha kötü kognitif performansla ve klinik olarak daha ağır bir demansla ilişkilidir. Serebral hipoperfüzyonun hipoksiyle sonuçlandığı, hipoksinin ise BACE1 geninin transkripsiyon ve ekspresyonunu arttırarak Aβ üretimini arttırdığı gösterilmiştir. Aynı zamanda bir inme risk faktörü olan yüksek plazma homosistein düzeyinin AH için de bağımsız bir risk faktörü olduğu bildirilmiştir. Atrial fibrilasyon, aritmiler, kardiyak arrest gibi kalp hastalıkları beyin perfüzyonunu bozarak hücre hasarı dolayısıyla kognitif bozukluk ve Aβ üretim artışına yol açarak giderek demansa neden olabilirler.
Sigara içenlerde, kolinerjik nikotinik reseptörlerin yukarı-regülasyonu ve bunun sonucu olarak Aβ düzeylerinin düşmesi ile açıklanan biçimde AH’nin daha az görüldüğü bulgusu, başlangıçta sigara içenlerin ileri yaşlara gelmeden diğer hastalıklar nedeniyle ölmeleri ve vasküler demansa daha yatkın olmaları şeklinde metodolojik olarak eleştirilmişken, Rotterdam ve New York çalışmalarında tam tersine AH riskini birkaç kez arttırdığı yönünde bulgular elde edilmiştir. Sigaranın AH için rölatif riski düşük olsa da (RR = 1.20–1.60), oksidatif gerilim ve enflamatuvar mekanizmalar üzerinden AH riskini arttırabileceği düşünülmektedir.
Çalışmalarda hem düşük hem de yüksek vücut ağırlığının kognitif bozulma ve AH riskinde artışla ilişkili olduğu gösterilmiştir. Özellikle orta yaşta obezitenin AH riskini %60’a kadar arttırdığı gösterilmiştir. Bunun insülin direnci ve diyabet ile komorbiditesinden kaynaklanıyor olabileceği de ileri sürülmüştür. Günlük kalorinin protein ve karbonhidrat yerine büyük oranda yağdan alındığı beslenme biçimi obezite riskini arttırarak dolaylı yoldan kognitif bozulmaya ve AH gelişimine neden olabilir. Yağdan zengin beslenmenin Aβ ya da tau üzerinden etki göstermediği BDNF düzeylerini azaltarak oksidatif gerilimi arttırdığı ve sinaptik plastisitede kayba neden olduğu bildirilmiştir. Yüksek glisemik bir diyetle beslenme insülin direnci ve diyabet gibi önemli AH risk faktörlerinin ortaya çıkmasına neden olmaktadır. Yüksek glisemik bir diyetin Aβ birikiminde artışla korele olduğu gösterilmiştir.
Buna karşılık Akdeniz diyeti bir koruyucu faktör olarak en popüler çevresel faktör araştırma konularından biridir. Akdeniz diyeti (MeDi) ile tahıllar, sebzeler, meyveler, peynir, süt, özellikle balık, zeytinyağı ve kırmızı şaraptan zengin bir diyet kastedilmektedir. Columbia Üniversitesi MeDi Çalışması, New York’ta MeDi uyumuna göre düşük, orta ve yüksek olarak sınıflandırdığı 1393 normal yaşlının MCI ve 482 MCI’lının demans geliştirme sonlanma noktalarında yüksek uyum grubunun düşük uyum grubuna göre avantajlı olduğunu göstermiştir. Rotterdam ve Chicago Sağlık ve Yaşlanma Projesi gibi 2 büyük çalışmada balık tüketiminin daha yavaş kognitif bozulmayla ilişkisi gösterilmiştir. MeDi’nin hücre içi reaktif oksijen türevlerini, apoptozu ve telomer kısalması gösteren hücreleri azaltarak oksidatif gerilimi azalttığı gösterilmiştir. AH’den korunmada özellikle zeytinyağında bulunan polifenollerin etkin olduğu, natürel sızma zeytinyağında bulunan oleuropein aglikonun otofajiyi tetiklediği, protein birikintilerini ve enflamasyonu azalttığı, AH’de kognitif fonksiyonu geliştirdiği gösterilmiştir. Başka bir zeytinyağı ürünü olan hidroksitirosolün de antioksidan ve anti-enflamatuvar özellikleri bulunduğu ve fare çalışmalarında mitokondrial oksidatif gerilim, enflamasyonu ve apoptozu azalttığı gösterilmiştir.
Kafa travması öyküsü ile AH arasında ilişki bildiren önemli çalışmalar olmakla birlikte (örneğin, EURODEM) bunu red edenler de vardır (örneğin, Rotterdam Çalışması ve Kanada Sağlık ve Yaşlanma Çalışması). Bilinç kaybına neden olmuş şiddetteki kafa travmaları (BKKT) ilk yıldan itibaren demans için bir risk faktörü olarak bildirilmiştir. BKKT öyküsü olan bireylerde olmayanlara göre demans riskinin %24 daha fazla olduğu, ağır bir BKKT (başlangıçta Glasgow koma skalası 3-8 arası ve ardından uzamış bilinç kaybı) öyküsü AH riskini %35 arttırırken, tek ılımlı bir BKKT öyküsünün (30 dakikadan kısa süren bilinç kaybı) AH riskini %17 arttırdığı bildirilmiştir. Demans riski BKKT sayısıyla da artmaktadır. 2-3 BKKT öyküsü riski %33 arttırırken, 4 BKKT öyküsü %61, 5 ve daha fazla BKKT öyküsü %183 oranında arttırmaktadır. APOE-ε4 yükü ile kafa travmasının AH’nin başlangıç yaşı üzerine birlikte etkisi olduğunu ileri süren çalışmalar mevcuttur. Bir çalışmada kafa travması tek başına etkisiz görünürken, normalde iki misli artmış olan APOE-ε4 heterozigotlarının riskini 10 misline arttırdığı ortaya konmuştur. Kafa travmasının ilk etkisi fokal ya da difüz aksonal hasardır, ardından oksidatif gerilim, enflamasyon, nöronal eksitotoksisite gibi mekanizmalarla nöron kaybına ve bunun sonucunda kognitif bozulmaya yol açar. Ayrıca BKKT’nin ardından saatler içerisinde Aβ birikimi olduğu gösterilmiştir. BKKT hastalarının post-mortem incelemesinde Aβ plakları ve NFY’ler yaklaşık üçte bir oranında görülmüştür. Ayrıca BKKT hastalarında BOS’ta tau düzeylerinin arttığı da gösterilmiştir. Dementia pugilistica geliştiren boksörlerin otopsilerinde saptanan gevşek neokortikal amiloid plaklar da bu varsayımı destekler niteliktedir.
Depresyonun AH için bir risk faktörü mü yoksa AH’nin erken bir semptomu mu olduğu konusu tartışmalıdır. Depresyon başlangıç yaşına göre erken-yaş-depresyonu (EYD) (60 yaşından önce başlayan) ve geç-yaş-depresyonu (GYD) (60 yaşından daha sonra başlayan) olarak sınıflandırıldığında GYD’nin AH riskini 1,65 kat kadar arttırdığı gösterilmiştir. Hayatı boyunca majör depresyon öyküsü olan bireylerde Aβ birikiminin arttığını gösteren bir çalışma mevcuttur. Komorbid depresif semptomları bulunan MCI hastalarının da AH’ye dönüşme hızlarının depresif semptomları bulunmayanlara göre daha yüksek olduğu ve Aβ yüklerinin de daha fazla olduğu gösterilmiştir.
Uyku bozukluğunun kognitif bozukluk, MCI ve demans için risk faktörü olduğu öne sürülmüştür. Uykunun Aβ klirensini arttırdığı, bu nedenle uyanık kişilerde interstisyel Aβ konsantrasyonunun uyuyanlara oranla daha yüksek olduğu gösterilmiştir. Sadece bir gece akut uyku yoksunluğunun beyinde Aβ düzeylerini arttırdığı gösterilmiştir. Ayrıca uyku interstisyel sıvı tau düzeylerini de düşürmektedir. Uyku yoksunluğu olan hastaların BOS tau düzeylerinde de %50 artış gözlenmiştir. Uyku-uyanıklık siklusunun bozulması AH’nin en erken semptomlarından biri de olabilir. Yani uyku bozukluğu ile AH arasındaki nedensellik karşılıklı gibi görünmektedir. Uyku-uyanıklık siklusu bozukluğunun tedavisi AH patolojik sürecini yavaşlatabilir.
Alkol ve AH patolojisi ilişkisi tartışmalı bir konudur. Birçok çalışmada hafif-orta alkol tüketiminin koruyuculuğu bildirilmiş, giderek bu etkinin bir U eğrisi sergilediği, yüksek tüketimde kaybolduğu ileri sürülmüştür. Ilımlı düzeyde alkol alımının AH riskini azalttığı fakat ağır içiciliğin [erkekler için günde 4’ten ya da haftada 14’ten fazla, kadınlar için ise günde 3’ten ve haftada 7’den fazla sayıda alkollü içecek tüketiminin] artmış riskle ilişkili olduğu bildirilmiştir. Koruyuculuk açısından alkollü içkiler içinde özellikle kırmızı şarap üzerinde durulmaktadır. Kırmızı şarabın içerdiği polifenoller ve özellikle de bir uzun ömür faktörü olarak sirtuin sinyallemesinde rol oynayan resveratrol Aβ agregasyonunu inhibe etmekte, oksidatif gerilim ve enflamasyonu azaltmakta ve protein homeostazını dengeliyor gibi görünmektedir. Bu açıdan demans riskini azaltmada düşük dozlarda şarap tüketimi önerilebilir.
Ayrıca, zihinsel ve fiziksel aktivitenin koruyuculuğundan söz edilebilir. Normal koşullarda demansı olmayan yaşlı bireylerin hippokampuslarında her yıl %1-2 hacim kaybı görülebilir. Orta yoğunlukta 1 yıl süren bir egzersiz programının hippokampus hacim kaybını durdurmakla kalmayıp, hacmi %2 oranında arttırdığı gösterilmiştir. Fiziksel aktivite plastisiteyi aktive ederek, beyin vaskülarizasyonunu ve nörogenezisi arttırarak, enflamasyonu ve amiloid plak formasyonunu azaltarak kognisyon üzerinde olumlu etkiye sahiptir. Ayrıca bir yandan vasküler riskin kontrolü aracılığıyla, diğer yandan da eğitim için olduğu gibi zengin uyarana maruz kalmanın kognitif rezervi arttırması yoluyla etki ediyor olabilir. Osteoporotik Fraktürler Çalışması’nda uzunlamasına izlenen 6000 65 yaş üzeri kadın arasında en fazla yürüyen üst çeyrek, en az yürüyen alt çeyrekle karşılaştırıldığında, 6-8 yıl sonra daha az kognitif kayıp sergilemekteydiler. Colcombe ve Kramer’in meta-analizinde (2003) egzersize tabi tutulan sedanter yaşlıların tutulmayanlarla karşılaştırıldığında kısa süreli kognitif düzelme sergiledikleri ortaya konmuştur. Yüksek düzeyde fiziksel aktivite gösteren bireylerde AH riski sedanter yaşama sahip olanlara kıyasla yarıya düşmektedir (RR = 0,72 tüm demans türleri için ; RR = 0,55 AH için). Fiziksel aktivite AH’yi önlemekle kalmayıp AH hastalarında günlük yaşam aktivitelerine katılımla değerlendirilen klinik kötüleşmeyi yavaşlattığı gösterilmiştir.
Çok sayıda çalışma, okumak, sanatsal faaliyette bulunmak, oyun oynamak gibi zihinsel aktivite gösteren yaşlıların AH geliştirme risklerinin daha düşük olduğunu ileri sürmektedir. Wilson ve arkadaşları Chicago’da ortalama 5,3 yıl izledikleri 4000 yaşlı arasında, kognitif olarak stimüle edici faaliyetlere daha sık katılan yaşlıların daha düşük ihtimalle kognitif bozulma sergilediklerini gösterdiler. Zihinsel aktivite de daha zengin kognitif rezerv oluşturarak etki ediyor olabilir. Kısıtlı sosyal ilişkiler de Kungsholmen, İsveç ve Kuzey Manhattan, ABD gibi önemli toplum temelli kesitsel ve uzunlamasına gözlem çalışmalarında bağımsız bir risk faktörü olarak ortaya konmuştur. Sosyal ilişkiler akraba ve arkadaş ilişkilerinin zenginliği, sinema, lokanta, klüp, dernek faaliyetleri amaçlı sokağa çıkmalar olarak özetlenebilir. Honolulu-Asya Yaşlanma Çalışması’nda bu risk orta yaşlarından itibaren sosyal aktiviteleri düşen bireylerle sınırlı bulunmuştur.
Küçük kafa çevresi ile AH arasında bazı çalışmalarda gösterilen ilişki, eğitim ile olan ilişkide olduğu gibi kognitif rezerv teorisiyle açıklanmaya çalışılmaktadır.
Hipotiroidinin gerek genel olarak demans ve gerekse de AH ile ilişkisi ortaya konmuştur. AH ile ilişkisinin mekanizması aşikar olmasa da potansiyel olarak düzeltilebilir bir durum olması bakımından önemlidir.
TAPS çalışmasında elektro-manyetik alan (EMF) maruziyeti de AH için bir risk faktörü olarak ortaya konmuştur. Elektrikçiler, tamirciler, santral operatörleri, teknisyenleri, kaynakçılar, marangozlar, terziler gibi elektrikli aletlerle çalışanlar aşırı düşük frekanslı (ELF) EMF maruziyeti için risk altında meslek gruplarıdır. Bizim çalışmamızı da içeren ELF-EMF maruziyeti ve AH ilişkisi ile ilgili 12 çalışmanın meta-analizi olan bir çalışmada bu ilişkinin bağımsız bir risk olduğu kabullenilmekle birlikte mevcut çalışmalarda maruziyet dozu ile risk artışı ve maruziyet ile cinsiyetlerin ilişkisinin belirlenememiş olması dolayısıyla daha fazla çalışmanın gerektiğinin altı çizilmiştir. Maruz kalınan diğer toksik ve zararlı durumlar arasında organik çözücüler, alüminyum ve genel anestezi ön plana çıkar gibi görünmektedir. Ancak bunların yerleşmiş tutarlılıkta bulgular olduğu söylenemez.
AH ile açıkça ilişkili bulunan çeşitli risk faktörleri ve koruyucu faktörler hastalıktan primer korunma çalışmalarının ilgi odağı olmuştur. Özellikle dejeneratif hastalıklarda oksidatif gerilim ve eksitotoksisite mekanizmalarının açıklayıcı olarak devreye girmesinden sonra, bu mekanizmaları tersine çevirdiği iddia edilen terapötik ajanların popülaritesi de artmıştır. Antioksidan olarak selenyum ve E vitaminiyle yapılan çalışmalarda etki gösterilememiştir. Anti-enflamatuvar ajan kullananlar arasında AH sıklığının daha az olduğu birçok gözlemsel çalışmada gösterilmiştir. Bu bulgu, söz konusu ajanların AH’deki enflamatuvar süreci tersine çevirmeleri şeklinde yorumlanmıştır. Ancak, özgül olarak sağlıklı yaşlılarda demans gelişimini engellemek veya AH’lilerde hastalık seyrini yavaşlatmak amaçlı tasarlanan anti-enflamatuvar klinik çalışmaları başarılı olamamıştır. Balık yağı, omega-3, B vitamini, Acetyl-l- carnitine ve folik asit tüketiminin de hastalığı önleyebileceği üzerinde durulmuş fakat çalışmalar başarılı olamamıştır.
Östrojen replasmanı kullanan post-menopozal kadınlarda AH’nin daha az görülmesi, östrojenin koruyuculuğu üzerine yoğun bir tartışma başlatmıştır. Ancak, Kadın Sağlığı İnisiyatifi çalışmasında yaşlı kadınlarda östrojen replasmanının jinekolojik kanserlerde artmanın yanısıra, demans riskini düşürmek bir yana tam tersine arttırdığı bulunmuştur. Kolesterol düşürücü ilaçlar, özellikle de statin grubu kullananlarda AH prevalansı kullanmayanlara göre daha düşüktür. Son zamanlarda bu etkinin kolesterol düşürücü etkinin ötesinde, statinlerin doğrudan α-sekretaz proteolitik yolunu uyararak, sAPP oluşumunu arttırmalarıyla ilintili olduğuna ilişkin deneysel deliller bulunmuştur. Yine de, AH’lilerde klinik çalışmalarda sınanan statin tedavisinin faydası gösterilememiştir.
Anti-hipertansif ve anti-diyabetiklerin vasküler risk faktörlerinin kontrolü aracılığıyla demansa karşı koruyucu olabileceği sağduyuyla da çıkarımlanabilir. Nitekim, bu öngörü anti-hipertansifler açısından birkaç prospektif çalışmada doğrulanmıştır. Yukarda değinildiği gibi IDE Aβ klirensinde bir şaperon olarak işlev gördüğü bilinmektedir. İnsüline dirençli tip II diyabetiklerde, IDE beyinde işlev görmek yerine periferde artan insülini parçalamak için kullanılmaktadır. Dolayısıyla, özgül olarak insülin direncini düşürerek işlev gören peroksizom proliferatörü ile aktive reseptör gama (PPAR-γ) agonistleri sınıfı anti-diyabetikler vasküler risk kontrolü yanısıra potansiyel olarak özgül anti-Alzheimer etki de gösterebilirler. Fakat klinik çalışmalarda başarılı olamamıştır.
Genetik
AH’de genetik faktörler büyük oranda hastalığın gelişimi için çevresel faktörlere bir yatkınlık zemini yaratacak şekilde birer risk faktörü niteliğindedirler. Tüm AH olgularının sadece %1-2’si basit Mendelyen OD geçişle hastalığa yakalanırlar. OD-AH’liler tipik olarak erken başlangıçlı (60 yaş öncesi) hastalığa sahiptirler.
Genetik açıdan genel olarak kompleks nörolojik bir hastalık olarak nitelenmesinin yanısıra, AH Mendelyen geçiş açısından da heterojenite gösteren polijenik/multialelik bir hastalıktır: birden fazla kromozomdaki gen lokuslarının çok sayıda farklı mutasyonları aynı hastalığa yol açar. OD geçişten sorumlu olan şimdiye kadar 3 ayrı gen bulunmuştur: amiloid prekürsör protein (APP) geni (APP, 21. kromozom), presenilin 1 geni (PSEN1, 14. kromozom) ve presenilin 2 geni (PSEN2, 1. kromozom). PSEN1 mutasyonları tüm OD vakaların %80’ini oluştururken, PSEN2 %5’ini ve APP mutasyonları %15’ini oluşturmaktadır. PSEN1 için 200’den fazla, PSEN2 için yaklaşık 16 tane ve APP için 50’den fazla mutasyon tanımlanmıştır Her 2 gendeki mutasyonlar da γ-sekretaz aktivitesini etkileyerek Aβ’nın daha toksik formu olan Aβ42 üretiminin artmasına neden olmaktadırlar. OD-AH aileleri genç yaşta ortaya çıkmaları (PSEN1, 25-60 yaş; APP, 40-65 yaş; PSEN2, 45-84 yaş) yanısıra, bellek dışı bir çekirdek sendromla ve/veya erken davranışsal özellikler, miyoklonus ve nöbetler şeklinde atipik klinik tablolarla da çıkabilirler. Genel olarak PSEN1 aileleri APP ailelerine göre daha atipik ve daha kısa süreli ve ağır demans sergilerlerken, PSEN2 aileleri daha fazla sporadik AH’ye benzeyen ve daha yavaş seyirli hastalık tablolarına sahiptirler.
ApoE proteinini kodlayan gen (APOE, 19. kromozom), mutasyonlarıyla doğrudan belirleyici değil fakat polimorfizmiyle sporadik AH’de risk faktörü olarak ortaya konmuştur. ApoE, üç ayrı alelik forma sahiptir: ε2, ε3 ve ε4. Kuzey Amerika ve Kuzey Avrupa’dan normal popülasyonlarda en sık ε3 (%70-80) görülürken, ε4’ün sıklığı %10-20, ε2’ninki ise %5-10’dur. AH’de ε4 sıklığı ikiye katlanarak %40-50’ye ulaşır. ε4 AH riskini doza bağlı bir şekilde arttırır ve hastalık başlangıç yaşını azaltır. TREM2 (triggering receptor expressed on myeloid cells 2 protein) genindeki heterozigot nadir varyantlarının da (R47H) AH’ye güçlü bir şekilde yatkınlık yarattığı gösterilmiştir.
Şimdiye kadar incelenen diğer poliformizlerin hiçbiri APOE-ε4 taşımanın yarattığı risk artışı düzeyinde etkili bulunmamıştır. Diğer yatkınlık genleri içinde özellikle anjiyo-tensin konverting enzim (ACE), GRB2 asosiye bağlayıcı protein 2 (GAB2), transferrin (TF), sortilin ilişkili reseptör (SORL1), clusterin (CLU) ve phosphatidylinositol bağlayan clathrin asamble proteini (PICALM) genlerindeki polimorfizmler öne çıkmaktadır.
Öyle görünüyor ki, her şeye rağmen yeni yatkınlık genlerinin araştırılması ve bulunanların doğrulanmasına yönelik yoğun çabalar sürecektir. Bu konunun bir özeti için Tablo 4’e bakınız.
Tablo 4. Alzheimer hastalığının genetiği (Robinson ve ark., 2017; Karch ve ark., 2015, Karch ve ark., 2014; Guerreiro ve ark., 2013)
Gen ve Proteini
Kromozom
Risk
Mekanizma
APP –
Amiloid prekürsör protein
21q21.3
Tam penetrans
Hasar yanıtı/Amiloid kaskadı
Aβ40 ve Aβ42 üretiminde artış
PSEN1 –
Presenilin 1
14q24.3
Tam penetrans
Amiloid kaskadı
Aβ42 üretiminde artış
PSEN2 –
Presenilin 2
1q31-q42
Tam penetrans
Amiloid kaskadı
Aβ42 üretiminde artış
APOE –
Apolipoprotein E
19q13.2
ε4: 3,68 (3,30-4,11)
ε3: 0,62 (0,45-0,85)
Kolesterol/lipid metabolizması
Aβ klirensinde bozulma
ADAM10
α-secretase A Disintegrin And Metalloprotease 10
15q21.3
Bulunamadı
APP işlenmesi
DSG2
Desmoglein 2
18
0,73
Hücre-hücre bağlantılarının düzenlenmesi
CD2AP-
CD2-assosiye protein
6p12
1,1
Reseptör endositozu ve sitokinezi
SORL1 –
Sortilin ilişkili reseptör
11q23.2-q24.2
1,10 (1,03-1,17)
Endositoz ve kargo, lipid taşınması
Aβ üretiminde artış
TREM2-
Triggering receptor expressed on myeloid cells 2
6p21.1
2,55
İmmün yanıt; kronik enflamasyon
PLD3
Fosfolipaz D3
19q13.2
2,75
Klatrin aracılı endositoz
Patofizyolojideki tam etkisi bilinmiyor.
CASS4 –
Cas scaffolding protein family member 4
20q13.31
0,88
APP ve Tau patolojisi; Hücre iskeleti fonksiyonu ve aksonal taşınma
FERMT2-
Fermitin family member 2
14q22.1
1,14
Anjiogenez, Tau patolojisi
ABCA7
ATP-binding cassette, subfamily A (ABC1), member 7
19p13.3
1,15
Kolesterol/lipid metabolizması; immün ve kompleman sistemleri/enflamatuvar yanıt
RIN3
Ras and rab interactor 3
0,91
Nöral gelişim, sinaps fonksiyonu ve endositoz
SLC24A4-
Solute carrier family 24), member 4
14q32.12
0,91
Nöral gelişim, sinaps fonksiyonu ve endositoz
CR1 –
Kompleman bileşeni (3b/4b) reseptörü 1
1q32
1,18 (1,09-1,28)
İmmun ve kompleman sistemleri
Aβ klirensinde bozulma
HLA-DRB5-DRB1-
Major histocompatibility complex, class II, DR B5-B1
6p21.3
1,11
İmmün yanıt ve enflamasyon
ZCWPW1-
Zinc finger, CW type with PWWP domain 1
7q22.1
0,91
Epigenetik düzenleme
CD33
sialic acid binding immunoglobulin (Ig) like lectin
19q13.3
0,94
İmmün ve kompleman sistemleri
CELF1-
CUGBP, Elav-like family member 1
11p11
1,08
Hücre iskeleti fonksiyonu ve aksonal taşınma
EPHA1-
EDH reseptör A1
7q34
0,9
İmmün ve kompleman sistemleri
PICALM –
Phosphatydinilinositol bağlayıcı klatrin asamble proteini
11q14
0,87 (0,83-0,91)
Klatrin aracılı endositoz
Aβ klirensinde bozulma
CLU –
Clusterin
8p21-p12
0,86 (0,82-0,89)
İmmun ve kompleman sistemleri
Aβ klirensinde bozulma
PTK2B-
Protein tirozin kinaz 2 beta
8p21.1
1,1
Hippokampal sinaptik fonksiyon; hücre göçü
NME8-
NME/NM23 family member 8
7p14.1
0,93
Hücre iskeleti fonksiyonu ve aksonal taşınma
MS4A4A6A
membrane-spanning 4A gene cluster
11q12.1
0,9
İmmün ve kompleman sistemleri
MS4A4A4E
11q12.2
0,9
İmmün ve kompleman sistemleri
BIN1-
Bridging integrator 1
2q14
1.22
Sinaptik vezikül endositozu
MEF2C-
Myocyte enhancer factor 2C
5q14.3
0,93
İmmün yanıt ve enflamasyon; Hippokampal sinaptik fonksiyon
INPP5D-
Inositol polyphosphate-5-phosphatase, 145kDa
2q37.1
1,08
İmmün yanıt ve enflamasyon; APP metabolizması
Koyu Kırmızı: Tam penetrans
Kırmızı: Yüksek risk
Turuncu: Orta risk
Yeşil: Düşük risk
Yukarıdan aşağı yaklaşık olarak toplumdaki sıklığına göre artan şekilde sıralanmıştır.
NÖROPATOLOJİ
Nörodejeneratif hastalıklar (proteinopatiler), genetik ve/veya çevresel faktörlerin etkisiyle normal işlev gören bir hücresel proteinin fiziksel konformasyonunu değiştirerek kendi üstüne katlanması (β-katlı tabaka oluşumu) ve böylece birikerek oligomerleşme ve klirense dirençli hale gelmesi şeklinde bir ortak mekanizmaya sahiptir. β-katlanma sonucu birbirlerine lateral olarak en az 2-3 hidrojen bağlarıyla bağlı β-iplikçikleri oluşur. Bir β-iplikçiği tipik olarak 3 ila 10 amino asitlik bir polipeptid zinciridir. Hidrojen bağları iplikçikler arasında bir çeşit omurga oluşturur.
Tüm nörodejeneratif hastalıklar ya da biyokimyasal perspektiften proteinopatiler, geleneksel isimlerinin yanısıra biriken proteinin adıyla anılırlar (örn., LCD bir α-sinükleopatidir). Bu açıdan AH benzersiz bir şekilde bir çifte proteinopatidir. Dolayısıyla, AH’yi bir amiloidopati (zamansal olarak önce biriken protein) olarak da taupati (klinik bulguların iticisi olan protein) olarak da sınıflamak doğru olmayacaktır. Pick hastalığı, KBD, PSP gibi nörodejeneratif hastalıklar tekil taupatilerdir ama AH bir amiloidozlu taupatidir.
Primer patolojiler olan AP ve NFY’ler yanısıra gliozis-enflamasyon, nöron ve sinaps kayıpları, aksonal ve dendritik morfolojik değişiklikler, kortikal kolinerjik innervasyon ve diğer nörotransmitter sistemlerinde kayıplar AH nöropatolojisinin eşlikçi bileşenlerini oluştururlar.
![]() Amiloid
Plaklar ve Amiloid-β
Fizyopatolojisi
Amiloid
Plaklar ve Amiloid-β
Fizyopatolojisi
NFY’lerin tersine yükleri hastalık şiddetiyle korele etmeyen senil plakların (Şekil 3) ana bileşenlerinin amiloid beta peptidi (Aβ) olduğu 1984’ten beri bilinmektedir (yani, senil plak = amiloid plak). Aβ, 21. kromozomda kodlanan bir transmembran protein olan amiloid prekürsör proteininin (APP) katabolizma ürünlerindendir. APP geninin yok edildiği transjenik farelerde (APP nakavt) nöral plastisitede rolü olabileceğini düşündüren ılımlı bulgular mevcuttur. APP yokluğu onun gibi davranan ve “amiloid prekürsör benzeri proteinler” adı verilen APLP1 ve APLP2 tarafından kompanse ediliyor olabilir. Nitekim, APP/APLP2 çifte nakavtlar sağ kalamamaktadır.
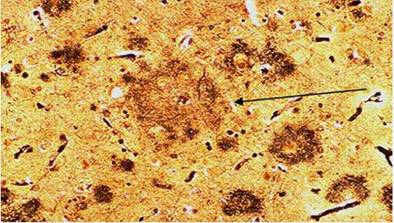
Şekil 3. Amiloid Plak (Kaynak: http://www-edlib.med.utah.edu/WebPath/CNSHTML/CNS090.html)
Bütün transmembran proteinlerde olduğu gibi APP’nin de bir hücre içi karboksi ucu (CT), bir membran kateden parçası, bir de hücre dışı amino ucu (NT) vardır (Şekil 4). 770, 751 ve 695 rezidülük üç izoformu olan APP’nin intramembranöz parçası 700-723. rezidülere rastlar. β-amiloid, 671 ila 711-713. rezidüler arasında yer alan 40 veya 42 rezidülük, distal kısmı ekstraselüler, proksimal kısmı intramembranöz yerleşmiş bir parçasıdır. APP bir dizi proteolitik enzimle kesilerek metabolize edilir. Bu enzimlere α, β ve γ-sekretaz adları verilir. α-sekretaz APP’yi tam da Aβ’nın ortalarına rastlayan 687. ekstraselüler bölgede keser. Bu kesim sonunda distalde parenkime atılan çözülebilir APP ya da sAPPα adı verilen ekstraselüler parça oluşurken geride hala membrana bağlı 83 rezidülük C-terminal fragmanı-α (CTF-α) kalır. sAPPα’nın hücre kültürlerinde nörotrofik etkileri bulunmuş ve APP nakavt transjeniklerdeki plastisite defisitlerini geri çevirdiği gösterilmiştir. Oysaki, alternatif distal kesimi yapan β-sekretaz enzimi APP’yi Aβ’yı salim bırakacak şekilde, ekstraselüler bölgede α-sekretazın daha distalinden 671. pozisyondan keserek akson budanması ve dejenerasyonuna neden olduğu düşünülen distal parça sAPPβ’yı parenkime atar ve geride salim Aβ fragmanını da içeren 99 rezidülük membrana bağlı CTF-β’yı bırakır.
Proksimal kesme ise intramembranöz bölgede γ-sekretaz enzimi aracılığıyla sekansiyel olarak yapılır. γ-sekretaz CTF’yi ilk olarak 720/721. pozisyondan epsilon (ε) kesimine uğratır ve nükleer sinyallemede rol oynadığı bulunan amiloid intraselüler domeni (AICD) isimli fragman sitoplazmaya salınır. İkinci olarak 717. pozisyondan zeta (ζ) kesimi ve nihayet üçüncü olarak ağırlıkla 711, daha az olarak 713. pozisyondan gamma (γ) kesimi yapar. γ-sekretaz CTF-α’yı γ-kestiğinde distalde bu kez parenkime Aβ fragmanının budanmış ve zararsız p3 adı verilen parçacığı atılır. Buna karşılık, γ-sekretaz CTF-β’yı γ-kestiğinde 711 veya 713. pozisyonlardan kesilmeye göre daha ağırlıkla daha kolay klirensi sağlanan Aβ40, daha az oranda da, daha toksik formu olan Aβ42 fragmanı, yani klirensi başarıyla sağlanamazsa oligomerleşip, sonra fibrilize olup nihayet amiloid plaklarda birikecek materyal ortaya çıkar (Şekil 4).
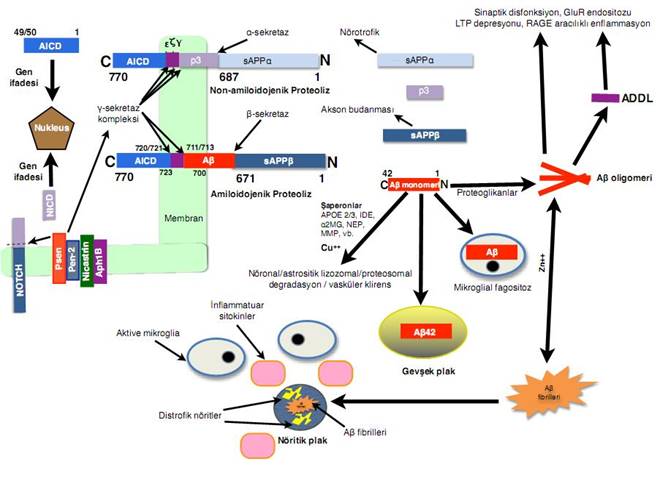
Şekil 4. APP proteolizi ve Aβ birikimi.
Amiloid prekürsör protein (APP) 770 aminoasidlik (aa) bir transmembran (TM) proteindir. 700 ve 723. aa’lar membranı kateden parçasına rasgelir. Aβ karboksi ucuna (CT) yakın 40-42 aa’lık bir fragmanıdır. Aβ’nın 700 ila 711-713. aa’lara karşılık gelen kısmı membran içinde (transmembran domen – TMD), 671 ila 699. aa’lar arasında kalan kısmı ise ekstraselüler (jukstamembran domen – JMD) yerleşimlidir. APP proteolizi distalde ekstraselüler parçada amiloidojenik veya non-amiloidojenik olarak yapılır. Amiloidojenik proteoliz Aβ fragmanını salim bırakacak şekilde 671. pozisyondan β-sekretaz kesimiyle başlatılır. Bu kesim amino ucunda (NT) akson budanmasında rol oynadığı bulunan sAPPβ ve CT’de membrana bağlı 99 aa’lık (C99) fragmanı oluşturur. Non-amiloidojenik proteoliz ise APP’yi Aβ-JMD içinden, 687. pozisyondan kesen α-sekretaz aracılığıyla başlar. Bu kesim de NT’de nörotrofik etkileri bilinen sAPPα ve CT’de yine membrana bağlı 83 aa’lık (C83) fragmanı oluşturur. C99 ve C83 intramembranöz parçada γ-sekretaz kompleksi (GS) ile bir dizi ardışık kesimden geçer. İlk kesim ε kesimi adı verilen 720 veya 721. pozisyonda meydana gelir. Oluşan 49-50 aa’lık APP intraselüler domeni (AICD) isimli fragmanın hücre çekirdeğine transloke olarak protein sentezini desteklediği bilinmektedir. İkinci kesim ζ kesimidir ve 717 veya 718. pozisyonda ve nihayet 3. kesim olan γ kesimi 711 veya 713. pozisyondadır. Bu son kesim non-amiloidojenik C83’te ise ortama 24-26 aa’lık inert bir budanmış Aβ parçacığı olan p3, amiloidojenik C99’da ise 40-42 aa’lık salim bir Aβ fragmanı üretir. GS APP’nin yanısıra bir dizi TM proteinin daha proteolizinden sorumlu olan bir aspartil proteaz kompleksidir. Aktif katalitik bölgesi presenilin 1 veya 2 (PSEN) olmak üzere 4 ayrı proteinin bileşimidir (Pen-2, Nicastrin, Aph1B). GS’nin diğer önemli proteolitik hedefi nöro-gelişimde önemli rol oynayan NOTCH proteinidir. GS kesimi sonrası NOTCH intraselüler domeni (NICD) adı verilen fragman, gen ifadesini ve hücresel farklılaşmayı başlatmak üzere nukleusa transloke olur. Ortama salınan Aβ monomerleri mikroglial fagositozla temizlenebilecekleri gibi şaperon moleküller tarafından vasküler endotelden klirensi sağlanabilir veya yine şaperon moleküller tarafından lizozomal veya proteozomal degradasyona maruz kalacakları nöronlar ve astrositler içine sokulur. Bu yöntemlerin herhangi biriyle ortamdan uzaklaştırılamayan, özellikle de potansiyel olarak daha toksik form olan Aβ42 gevşek plaklarda birikir. Bu son savunma mekanizması da başarısız kaldığı takdirde Aβ monomeri çözülebilir oligomerler halinde bir araya gelir. Çözülebilir oligomer bir dizi sinaptik disfonksiyon mekanizmasını tetikleyerek AH’nin en erken fizyopatolojik özelliği olan LTP depresyonuna neden olur. Oligomerler daha toksik ADDL’lere budanabilecekleri gibi çözülemeyen Aβ fibrillerine doğru da evrilebilirler. Aβ fibrilleri de hücre yıkıntı ürünleri olan distrofik nöritlerle birlikte nöritik plaklarda hapsedilirler. Fibriler Aβ gibi nöritik plaklar da kararlı yapılar olmaktan çok toksik oligomerler için bir tür rezervuar işlevi görmektedirler. Nöritik plaklar çevrelerinde oksidatif gerilim yanısıra aktive mikroglia aracılığıyla enflamasyona yol açarlar.
Normal yaşlanmada α-sekretaz aktivitesi baskınken, AH’de denge β-sekretaz alternatifine kaymaktadır. OD-AH özellikle Aβ42 üretimini arttırırken, başta APOE olmak üzere bütün yatkınlık genleri Aβ klirensini etkileyerek risk oluşturmakta veya koruyucu olmaktadırlar. α-sekretaz aktivitesini çok sayıda transmembran proteinin proteolizinde görevli olan bir disintegrin ve metalloproteinaz (ADAM) ailesinden bir dizi metalloproteinazın yerine getirdiği bilinmektedir. Bunlar arasında en aktiflerinden biri ADAM17 olarak da bilinen tümör nekrotizan faktör-α çevirici enzim (TACE), diğeri ADAM10’dur. β-sekretazın 11. kromozomdaki geni bulunmuş ve BACE (beta-site APP cleaving enzyme) adı verilmiştir. BACE nakavt farelerde kayda değer bir değişiklik olmamakla birlikte APP/PS1 gibi son derece virulan bir AH transjeniği ile çaprazlaştırılan BACE nakavt farelerin soyunda, plak birikimi için BACE’in temel önemde olduğunu gösterecek şekilde birikim olmamaktadır. Bu da β-sekretaz inhibisyonunu çekici bir tedavi hedefi kılmaktadır. γ-sekretazın ise aktif katalitik bölgesinde presenilini (PS1 veya PS2) de içeren ayrıca nikastrin, APH-1 ve PEN-2 isimli proteinlerden oluşan bir proteaz kompleksi olduğu bilinmektedir. Daha önce de söz edildiği gibi γ-sekretaz aynı zamanda nöro-gelişimde temel olan NOTCH sinyalizasyonunda da rol oynamaktadır ve PS1 nakavt fareler ağır gelişimsel kusurlar sergileyerek sağ kalamamaktadırlar.
Parenkimdeki yaklaşık 4 kilo Daltonluk (kD) Aβ monomerleri kendi başına zararsızdır ve mikroglia tarafından doğrudan fagositoza maruz bırakıldıkları gibi APOE alelik formları, insülin degrade edici enzim (IDE), α2-makroglobulin (α2-MG), neprilizin ve matriks metalloproteinaz (MMP) gibi proteinleri içeren bir dizi “şaperon moleküle” bağlanıp, düşük dansiteli lipoprotein reseptörü ilişkili protein (LRP-1) aracılığıyla astrosit ve nöronlara internalize edilerek otofaji ve proteozomal degradasyona maruz bırakılırlar. LRP1 farklı hücre tiplerinde Aβ’nın internalizasyonu için bir kapıcı olarak düşünülebilir ve nitekim vasküler endotelinden sirkülasyona aktarılarak temizlenmeye de aracılık eder. Degradasyon ve klirens aracılığıyla başa çıkılamayan miktarlardaki monomerler bir araya gelerek dimer ve tetramerler şeklinde moleküler ağırlıklarını arttırarak (8-20 kD) çözülebilir Aβ oligomerlerini oluşturacaklardır. Bu çözülebilir oligomerlere “düşük moleküler ağırlıklı Aβ (LMW Aβ)” adı da verilir. Oligomerler parçalanarak daha toksik olan “amiloid betadan türeyen difüze olabilen ligandlara” (ADDL’lar) değişebilirler veya toplanmayı sürdürerek bir sonraki aşamada artık çözülemeyen protofibrilleri ve giderek toksik fibrilleri oluştururlar. ADDL’a değişme, glutamik asit siklaz (QC) enziminin oligomerleri glutamik asit konumlarında piroglutamatlaması ile olur. Oligomerin N-terminusundan itibaren 3. ve 11. pozisyondaki glutamik asitin piroglutamatlanması sırasıyla [Aβ (3(pE)-42)] ve [Aβ (11(pE)-42)] şeklinde gösterilen ADDL formlarını oluşturur. Toksik potansiyeli en fazla biçim olduğunun gösterilmesinden sonra QC inhibisyonu çekici bir tedavi hedefine dönüşmüştür. Aβ fragmanının önce oligomerizasyonu, sonra da fibrilizasyonuna amino ucuna yakın bir konumda bulunan ağır metal bağlayıcı bölgelerinin aracılık ettiği düşünülmektedir. Buna göre, Cu++ bağlanması oligomerizasyon, Zn++ bağlanması fibrilizasyon için gereklidir. Zn++ yanısıra metal bağlayıcı bölgenin yakınlarındaki proteoglikan polisakkarid makromolekülleri bağlayan başka bir bölgenin de fibrilojenezde etkili olduğu bildirilmiştir. Fibriler Aβ plaklara “hapsedilecektir”.
İlk birikim NFY’lerin tersine limbik sistemde değil, neokortekste ve gevşek (difüz) plaklar şeklindedir. Bu plaklardaki amiloidin büyük çoğunlukla fibriler değil granüler Aβ oligomerlerini içerdikleri bulunmuştur. Gevşek plaklar kognitif bozukluk geliştirmeden ölmüş olan yaşlı beyinlerinde de büyük miktarlarda görülebilir. Geçen yüzyılda bu bulgular AH nöropatolojik değişikliklerinin normal yaşlanmada da görülebileceği şeklinde yorumlanırken, bugün artık bu bireylerin hastalığın pre-klinik evresinde ölmüş subklinik AH-P hastaları olduğunu biliyoruz. Ayrıca, çok sayıda immünositokimyasal çalışma ile gevşek plak içeriğinin daha toksik ve birikime yatkın başlıca Aβ42 formu olduğunu görülmüştür. Dolayısıyla, plak oluşumu Aβ monomerinden birikime doğru giderek daha toksik formlara giden bir patogenetik mekanizmanın daha nihai evresinden çok bir savunma mekanizması olarak düşünülebilir: degradasyon ve klirens mekanizmalarıyla Aβ40 ile başa çıkılmakta, bu mekanizmalar Aβ42 için yetersiz kaldığında plak formasyonu yoluna gidilmektedir.
Buna karşılık diğer plak formu olan katı (kompakt) plaklar çekirdeğini fibriler Aβ’nın oluşturduğu plaklardır. Bu aşamadan sonra dejenere distrofik nöritler içeren katı plaklara nöritik plaklar (NP) adı verilir. NP’ler immün cevabı tetikleyerek mikroglia ve astrosit aktivasyonu ile TNF-α ve IL-β gibi sitokinlerin salınımı, kompleman aktivasyonu ve serbest radikal oluşumuna neden olurlar. Bu immün aktivasyon nörotoksisitenin başlıca etmenlerindendir gibi durmaktadır. NP’ler yalnızca demanslı beyinlerde görülür. Nöritik bileşen sinaptik artık ve nörofilamanlardan oluşur ve çoğu aynı zamanda tau-çift sarmallı filamanları (τ-PHF) için de pozitif immünoreaksiyon gösterir. PHF(+) plaklarda nöritik bileşenin NFY içeren akson kalıntıları olduğu söylenebilir. Böylelikle bölgesel olarak farklı başlangıç yatkınlıklarına sahip olan NFY ve AP’ler hastalığın seyri içinde PHF(+) plaklarla birlikte yan yana gelmiş olurlar. Nöritik plaklar tam boy Aβ40 ve Aβ42’nin her ikisini de içerdikleri gibi ADDL’leri de içerirler. Bunlar arasında yukarda en toksik olduğu söylenilen amino ucundan budanmış Aβ (3(pE)-42 formunun demans şiddetine korele edecek şekilde Braak ve Braak evre IV-VI boyunca nöritik plak kompozisyonundaki oranı giderek artar. Bu bulgu da yalnızca demanslı beyinlerde görülen bu daha olgun plak formunun da bizzat kendisinin bir tetikçi olmasından çok alternatif savunma mekanizmalarının çok yetersiz kaldığı nörodejenerasyonun bu ileri aşamalarına özgü bir ek savunma girişimi olduğunu düşündürmektedir. Son zamanlarda Aβ’nın özellikle Aβ (3(pE)-42 formu ile yalnızca ekstraselüler parenkimal plaklarda değil, agrezom adı verilen intranöronal birikintilerde de biriktiğine dair çalışmalar artmaktadır. Bu çalışmalar sonucunda intranöronal birikimin sinaps ve nöron kaybına neden olan, ekstraselüler plak birikimini önceleyen erken bir fenomen olduğu ileri sürülmüştür. Bu yeni bulgular ışığında Aβ monomerinden nöritik plağa doğru evrim eski perspektifle olduğu gibi giderek daha zararlı bir forma doğru çizgisel bir evrilme şeklinde değil de toksik Aβ oligomeri ile farklı aşamalarda başa çıkma stratejileri olarak görülmelidir. Degrade edilemeyen toksik Aβ’nın fibrilizasyonu ve sonrasında da plaklara hapsolunması toksisitesinin giderilmesine yönelik önlemler olmakla birlikte, bu çözülemeyen birikintilerle çözünür oligomerik formlar arasında sürekli bir dinamik dengenin mevcudiyeti aslında bu formların toksik biçimler için sürekli bir rezervuar işlevi görmeleri anlamına gelmektedir. Aβ oligomerinin yol açtığı sinaptik disfonksiyon ve giderek nöron kaybını önceleyen ve aşan sinaps kaybı mekanizmalarına aşağıda değinilecektir.
AP yükünün AH’de demans şiddetiyle korele etmediğini gösteren nöropatolojik çalışmaların ötesinde, PIB’den başlayarak PET amiloid ligandlarının geliştirilmesinden sonra AP yükünün bir kısım kognitif yakınması olmayan yaşlılar ve bir kısım MCI’lılarda AH tanısı almış demanslılar düzeyine yaklaştığı, hatta AP yükü açısından AH’ye benzeyen MCI’lıların uzunlamasına izlemlerinde demansa dönüştükleri, bir kez demans geliştikten sonra ise AP yükünün fazlaca değişmediği ortaya konmuştur. PET ile ölçülen amiloid yükü esas olarak MCI’dan da önce pre-klinik aşamada ilerleyici olarak artıyor, pre-demans MCI ve demans aşamalarında ise büyük ölçüde değişmeyecek denge durumuna ulaşıyor gibi durmaktadır. Amiloid yükü kognitif bozukluk şiddetiyle korele etmese de AH-P’den AH-K’ya dönüşme olasılığıyla korele etmektedir. Amiloid PET görüntülemenin bu yüzyılın ilk onyılında erişilebilir olması nöropatolojinin o tarihe kadar ortaya koyamadığı bir hakikati de berrak biçimde ortaya koymuştur: amiloid birikimi o zamana kadar sanıldığı gibi belli bir coğrafi yatkınlık göstermeden yaygın neokortikal özellikte değil fakat öncelikle beynin orta hat yapılarını tutmakta ve bu yapılarda ağırlığını giderek arttırmaktadır. Bu yatkınlık olağan durum ağı DMN ile büyük ölçüde örtüşmektedir.
Nörofibriler Yumaklar
NFY’lerin (Şekil 5) temel bileşeni hiperfosforile tau (τ) proteinidir. τ 17. kromozom tarafından kodlanan mikrotübül asosiye proteinler (MAP) ailesinden bir proteindir (MAP-τ). Kargo veziküllerinin hücre gövdesinden akson terminaline anterograd ve bunların degradasyon ürünlerinin akson terminalinden geriye hücre gövdesine retrograd nakliyesinde önemli roller oynayan mikrotübüller başlıca MAP-τ ile stabilize edilir. MAP-τ görevi bir demiryolunda rayları paralel stabilize eden traverslerin işlevlerine benzetilebilir. Stabilizasyonu bozulmuş bir mikrotübül sistemi ile nakliye işlevi de mümkün olmayacaktır. Kargo vezikülleri eskiyen yapıların replasmanı ve sinaptik aktivite için gerekli protein ve enzimleri içerir. Mikrotübüller tau-tubulin etkileşimi ile stabilize olabilen labil yapılardır. Bu etkileşim tau proteininin izoformuna ve fosforilizasyon durumuna bağlıdır.
Tau proteini karboksi ucunda mikrotübüllere bağlanma bölgesi sayısına göre üçü üç tekrar (3R), üçü ise dört tekrar (4R) taşıyan toplam altı izoforma sahiptir. 4R tau mikrotübül stabilizasyonunda daha etkindir. İkincil mesajcıların uyarımı post-sinaptik nörondaki tüm intraselüler proteinleri olduğu gibi tauyu da protein kinazların aktivitesiyle fosforile eder. Fosforile tau mikrotübülden ayrılır. Yine bütün fosforile intraselüler proteinler gibi tau da işlev bittikten sonra fosfatazlar aracılığıyla defosforile edilmelidir ki yeniden mikrotübüle bağlanabilsin. Fosforile taunun defosforilasyonundan başlıca sorumlu fosfataz protein fosfataz 2A’dır (PP2A). AH patogenezinde hiperaktif kinazlar ve/veya hipoaktif fosfatazlar tau proteininin hiperfosforilizasyonuna yol açarak mikrotübüllere bağlanma yeteneğini bozarlar. Bu kinazlar arasında özellikle glikojen sentetaz kinaz 3β’nın (GSK-3β) yalnız AH’de değil, diğer taupatilerde de patolojik hiperfosforilasyondan sorumlu olan enzim olduğu üzerinde durulmaktadır. Bağlanmamış hiperfosforile tau monomerleri fiziksel konformasyonlarını değiştirerek kendi üzerlerine katlanırlar (β-katlanma) ve toksik oligomerlere dönüşürler. Tau oligomerleri bir sonraki aşamada yine toksik çözülemeyen çift sarmallı filamanlara (PHF) polimerize olurlar. PHF’ler ısı şoku proteinleri ve başlıca ubikuitin proteozom sistemlerinin devreye girmesiyle giderek intranöronal NFY’ler şeklinde kondanse olurlar (Şekil 6). Dendritik PHF birikimlerine nöropil iplikçikleri (neuropil threads – NT) adı verilir. NFY’lerin, AP’lere benzer şekilde, toksik PHF’lerin etkisini sınırlamaya çalışan bir savunma mekanizması olduğu giderek geçerlilik kazanan bir görüştür. Yetersiz bir savunma olan NFY ile sonunda hücre iskeleti bütünlüğü ve aksonal nakliye bozularak hücre ölümü meydana gelir. Hücre ölümüyle ortaya çıkan ekstraselüler NFY’ye “hayalet yumak” adı verilir. NFY’den hayalet yumağa geçişin yılları kapsayan uzun bir süreç olduğu düşünülmektedir.

Şekil 5. İntraselüler Nörofibriler Yumak (Kaynak: http://www-edlib.med.utah.edu/WebPath/CNSHTML/CNS094.html)
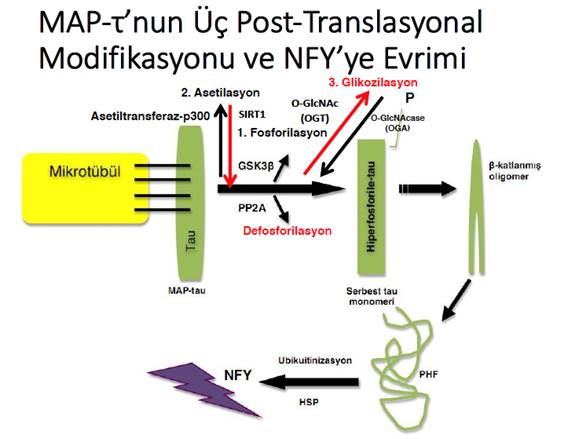
Şekil 6. Mikrotübül asosiye tau proteinin NFY’ye evrimi. Üç veya dört tekrar bölgesinden mikrotübüle bağlı olan tau proteini hiperaktif kinazlar (özellikle GSK3β) ve/veya hipoaktif fosfatazların (özellikle PP2A) varlığında hiperfosforile olarak mikrotübüllerden serbestlenir. Serbest tau patolojik olarak katlanarak oligomerleşir. Oligomerler çift sarmallı filamanlara (PHF) kondanse olurlar. PHF ise ısı şoku proteinleri (özellikle HSP90) ve proteozomal ubikütinizasyon sistemi aracılığıyla NFY’lere hapsolurlar.
GSK3β: glikojen sentetaz kinaz 3β; HSP: ısı şoku proteini; MAP: mikrotübül asosiye protein; NFY: nörofibriler yumak; P: fosfor; PHF: çift sarmallı filaman; PP2A: protein fosfataz 2A
NFY’ler beyinde rasgele değil fakat belli bir bölgesel yatkınlığı yansıtacak şekilde yerleşirler. “Normal yaşlılık” sürecinde limbik (entorhinal ve hippokampal) NFY sayısıyla kronolojik yaş arasında korelasyon gösterilmiştir. Braak ve Braak’ın I ve II. evrelerine karşılık gelen bu aşamanın normal kognisyondan sübjektif bellek yakınmalarına geçişin nöral altyapısı olduğu ileri sürülebilir. En erken değişiklikler entorhinal korteksin II. piramidal tabakasında (ECx-II) oluşmaktadır. ECx limbik sistemin dış dünyayla arayüzü olarak değerlendirilebilir. Lateral sektörü (LECx) infero-temporal girus ve perirhinal cortex (PRC) aracılığıyla gelen nesne ve yüz bilgisini, mediyal sektörü (MECx) posterior parietal korteks ve parahippokampal girus (PHG) aracılığıyla gelen bağlam ve mekan bilgisini paralel olarak hippokampal formasyona sunar. Hatırlanmak üzere kaydedilmesi gereken her türlü yeni bilgi limbik sisteme ECx II aracılığıyla girer ve bu tabakadaki nöronların aksonları perforan traktusu (PT) oluşturarak ECx’i hippokampal formasyonun giriş kapısı denilebilecek olan dentat girusunun (DG) granüler tabakasına bağlarlar. DG ile lokal entrensek hippokampal devreye (EHD) girilmiş olur (Şekil 7A). EHD içinde trisinaptik bir uzun yol ve bu yol üzerindeki bir veya daha fazla durağa uğramadan kestirmeden daha ilerdeki duraklara bağlanan daha kısa yollar bulunur. Trisinaptik uzun yolda DG yosunsu lifler aracılığıyla Ammon boynuzunun 3. sektörünün (CA3) piramidal tabakasına bağlanır. Schaffer kollateralleri bu kez CA3’ü CA1’e bağlarlar. CA3 sahip olduğu Schaffer liflerinden ayrılarak kendi üzerine geri dönen oto-asosiyatif lifleri ile EHD içinde benzersiz bir anatomik yapıdır (Şekil 7B). CA1 ise subikuluma (SUB) bağlanır. SUB, EHD’nin çıkış kapısı konumundadır; ECx alt tabakalarına (IV-VI) bağlanarak EHD kapanacak ve ECx IV-VI işlenmiş limbik bilgiyi multimodal asosiasyon kortekslerine geri bildirebilecektir. Kestirme yollara ilk örnek PT’den ayrılan bir kollateralin DG’yi atlayarak doğrudan CA3’le bağlanmasıdır. ECx-III’ten çıkan bir efferent ise PT’ye katılmadan doğrudan CA1’e hatta bu efferentin bir kollaterali doğrudan son durak olan SUB’a bağlanır. Bu alternatif yolaklar gelmekte olan bilgi ne kadar yeni ise uzun yolu kullanmalı, ne kadar aşina ise EHD’yi kısa yoldan dolaşabilmeli şeklinde yorumlanabilir. SUB aynı zamanda geniş boyutlu Papez devresinin (Şekil 7C) de hem başlangıcı hem de devrenin dönüp kendi üstüne kapandığı çıkış kapısıdır. Papez devresi ise SUB’dan fimbria-fornix aracılığıyla hipotalamusun mamiller cisimciklerine (CM) olan bağlantıyla başlar. CM, mamillo-talamik traktus (MTT) aracığıyla talamusun limbik çekirdekleri de denilen anterior grup çekirdeklerine bağlanır. Talamus anterior grubu posterior singulat girusla (pCG), ve retrosplenial girusla (rSG) talamo-kaudal singulat traktus (TKST) aracılığıyla bağlantılıdır. rSG’den SUB’a ventral singulum (vCING) aracılığıyla olan bağlantılarla Papez devresi kapanır. Papez devresi içinde de LECx ve MECx lokal devresindeki özelleşme daha geniş bir şekilde izlenir: bağlam ve mekan belleğinden sorumlu bir postero-mediyal sistem (PMS) ve nesne ve yüz belleğinden sorumlu bir anterior temporal sistem (ATS). PMS, DMN’nin posterior düğümü precuneus ile bağlantılı DMN anterior düğümü (ventromediyal prefrontal korteks) ve lateral düğümünü (anguler girus) PHG aracılığıyla MECx’e bağlar. ATS ise temporopolar korteks (TPCx), lateral orbitofrontal korteks (lOFCx) ve amigdalayı PRC aracılığıyla LECx’e bağlar. Ranganath ve Ritchey 2012 tarihli derlemelerinde PMS ve ATS’nin göreli özelleşmelerinin EHD içinde DG-CA3 bağlantısallığında entegre edildiğini ileri sürmüşlerdir. Maass ve arkadaşları 2019 yılında serebral amiloid yükünün PMS disfonksiyonu olarak yorumlanabilecek şekilde ön planda sahne belleğinde, tersine serebral tau yükünün ATS disfonksiyonu olarak yorumlanabilecek şekilde ön planda nesne belleğinde bozukluğa neden olduğunu gösterdiler.
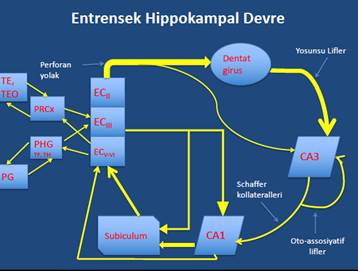
Şekil 7A. Entrensek Hippokampal Devre. Multimodal asosiasyon alanları ECx’in üst tabakaları ile ileri besleme bağlantılarına sahiptir. ECx II moleküler tabaka nöronlarının aksonları perforan yol aracılığıyla DG’nin granüler tabakasına bağlanır. DG yosunsu lifler aracılığıyla CA3 piramidal tabakasına, CA3 ise Schaffer kollateralleri aracılığıyla CA1’e bağlanır. CA1 ise EHD’nin çıkış kapısı konumunda olan subikuluma bağlanır. Subikulum ECx alt tabakalarına bağlanır ve EHD kapanmış olur. Ayrıca perforan yoldan ayrılan bir kollateral DG’yi atlayarak doğrudan CA3’le bağlanırken, ECx-III’ten çıkan bir efferent ise doğrudan CA1’e hatta bu efferentin bir kollaterali doğrudan son durak olan subikuluma bağlanır.

Şekil 7B. Transenthorinal NFY evresinde Perforan Yolak Disfonksiyonu ve CA3 kompansasyonu. CA3’ün %95 girdisi kendi oto-asosiyatif lifleri aracılığıyladır. NFY patolojisi ECxII dejenerasyonuna ve dolayısıyla DG diskonneksiyonuna neden olduğunda multisinaptik uzun yol fonksiyonu bozulur. Bunun sonucunda CA3 oto-asosiyatif liflerinde kompansatuar bir hipertrofiyle epizodik bellekteki eksikliği aşinalık ile kompanse etmeye başlayacaktır.

Şekil 7C. Epizodik Bellek için Papez Devresi. Papez devresi subikulumdan fimbria-fornix aracılığıyla hipotalamusun mamiller cisimciklerine olan bağlantıyla başlar. CM, mamillo-talamik traktus aracığıyla talamusun anterior grup çekirdeklerine bağlanır. Talamus anterior grubu posterior singulat girusla ve retrosplenial girusla talamo-kaudal singulat traktus aracılığıyla bağlantılıdır. rSG’den subikuluma ventral singulum aracılığıyla olan bağlantılarla Papez devresi kapanır.
PRCx: perirhinal kortex, PHG: parahippocampal girus, ECx: entorhinal korteks, Ant. Th: talamus anterior grup çekirdekler; CA1 ve 3: cornu ammonis 1 ve 3; CM: corpus mamillare; DG: dentat girus; rSG: retrosplenial girus, HF: hippokampal formasyon, dHipCom: dorsal hippokampal kommissur, vCING: ventral cingulum, Fx: fornix, TCP: talamosingulat projeksiyon, ACC: anterior singulat korteks, MCC: mediyal singulat korteks.
AH’de nörodejenerasyonun ağırlığı ve süresiyle paralel biçimde NFY’nin önceden görüldüğü bir bölgedeki yoğunluğu artarken o bölgeyle bağlantılı olan komşu bölgeye transsinaptik olarak yayılır. Dolayısıyla, ECxII dejenerasyonuyla DG diskonneksiyonu büyük ölçüde yenilik bilgisinin değerlendirildiği multisinaptik uzun yolu disfonksiyonel kılar. Bunun sonucunda DG girdilerini yitiren CA3 oto-asosiyatif liflerinde kompansatuar bir hipertrofi izlenir. CA3 aşırı aktivitesinin sonucu olarak değişerek reorganize olan entrensek hippokampal devre artık yenilik bilgisini epizodik bellekte eski deneyimler dolayısıyla kaydedilmiş bilgiyle karşılaştırarak yeniliği aşina olandan ayırt etmede yetersiz kalacağından (örüntü ayrıştırma bozukluğu), henüz salim kalmış kestirme yolları sayesinde bu eksikliğini aşinalık ile kompanse edecektir (örüntü tamamlama). SCI’daki bellek bozukluğunun nöropsikolojik karşılığı yenilik işleme bozukluğu ve aşinalık kompansasyonu, nöral temeli ise CA3 reorganizasyonu olabilir. Braak ve Braak 1995 yılında, I’den VI’ya sıraladıkları evreler boyunca NFY’lerin entorhinal kortekste (transentorhinal evreler I ve II) görülmeye başlayıp önce EHD ve akabinde Papez devresine yayıldıkları (limbik ve paralimbik evreler III ve IV), sonunda da heteromodal asosiasyon alanlarına (neokortikal evreler V ve VII) yayılıp sayılarının artışını izleyen bir evreleme sistemi geliştirdiler. Yazarlar, 2011 NIA-AA söylemini öngörmüşçesine Evre I-II’ye “klinik olarak sessiz olgular”, Evre III-IV’e “başlangıç halinde Alzheimer hastalığı, Evre V-VI’ya “tam olarak gelişmiş Alzheimer hastalığı” adı verdiler. Evre I-IV’te amiloidozsuz izole taupati olguları görülebilirken Mesulam, Braak ve Braak sistemine paralel bir klinikopatolojik korelasyon evrelemesi önererek NFY yayılımını normal yaşlanmadan (EvreI/II), MCI (Evre III/IV), hafif ve ağır demans evreleri (Evre V/VI) sürekliliği içinde izler. Bu evreler boyunca entrensek hippokampal devrenin ve Papez devresi yapılarının giderek daha fazla NFY ile yüklendikleri ilerleyici süreç AH’nin KN-SCI-MCI-demans sürekliliği boyunca merkezi ilerleyici bellek bozukluğu tipik fenotipinin de açıklamasıdır.
Nöron kaybı
AH’de nöron kaybı ECx’ten başlar. Limbik sistemi terkettiğinde superior temporal sulkusta tespit edilebilir. Nöron kaybının zaman içinde ilerleme ve anatomik yatkınlık tarzı genel anlamda NFY’nin tarzına benzese ve NFY ile nöron sayılarının arasında anlamlı negatif korelasyonlar saptansa da, nöron ölümünden tek başına NFY’ler sorumlu tutulamaz. Bunun nedenlerinden biri, yukarda değinildiği gibi, taunun sadece fosforillenmesinin nöron ölümü için yeterli olmaması, ayrıca asetillenmesinin de gerekmesi olabilir. Subkortikal çekirdekler gibi NFY’lerin bulunduğu bölgelerde ille de nöron kaybının olması gerekmediği gibi, NFY’lerin az sayıda olduğu ya da hiç bulunmadığı bölgelerde de ağır nöron kaybı görülebilir. Amiloid nörotoksisitesinin neden olduğu sinaps kaybı ve transsinaptik dejenerasyon hücre ölümünde rol oynayan bir diğer etmen olabilir.
Sinaps kaybı
Sinaps kaybı kortikal biyopsi örneklerinde klinik demans ağırlığıyla, nöron kaybından dahi daha kuvvetli korelasyon gösteren yapısal değişikliklerin başında gelir. Sinaptofizin gibi sinaptik proteinlerin miktarlarının da demans ağırlığıyla korele ettiği gösterilmiştir. Sinaps kaybı başlangıçta NFY ve nöron ölümünün anterograd Wallerian dejenerasyon sonucu sekonder etkisi ile açıklansa da, primer hasarın özellikle de intraselüler çözülebilir Aβ oligomeri aracılığıyla sinapslara olması ve bozukluğun retrograd olarak hücre gövdesine taşınarak, NFY oluşumu ve nihayetinde hücre ölümüne yol açması artık daha muhtemel görünmektedir.
Çözülebilir Aβ oligomerinin toksisitesi ve neden olduğu erken sinaptik disfonksiyon üzerine odaklanan çalışmalar çözülemez fibriler formun veya daha olgun plakların parenkimde oynadığı rol yerine toksik çözülebilir oligomerin veya ADDL’ın hücre yüzeyinde uygun bir reseptör bulup bağlandıktan sonra veya membrandan intraselüler mesafeye geçtikten sonra başlattığı bir dizi zincirleme reaksiyonun doğasını aydınlatmaya başlamıştır. Bu mekanizmaların basamaklarının aynı zamanda nedene yönelik tedavi hedefleri de oluşturmaları heyecan vericidir.
Gliozis ve Enflamasyon
Gliozis AH nöropatolojisinin bir başka özelliğidir. Aβ’nın nöritik plaklarda birikiminin mikroglial ve astroglial hücreleri aktive ettiği AH patogenezinde göreli geç bir olay olarak öteden beri iyi bilinmektedir. Mikroglial aktivasyon sitokinlerin salgılanması, akut faz reaktanları ve komplemanın aktive edilmesiyle enflamasyonu harekete geçirir. Son zamanlarda plaklardan bağımsız olarak doğrudan Aβ oligomerinin ileri glikasyon son ürünleri reseptörü (RAGE) ile bağlanarak MSS’deki başlıca pro-enflamatuvar sinyalleme sistemi olan aktive B hücrelerinin kappa hafif zincir güçlendiricisi nükleer faktörünü (NF-κB) tetiklemesinin AH’de erken bir fenomen olarak enflamasyona yol açtığının üzerinde durulmaktadır.
Mesulam ve çalışma arkadaşları asetilkolinesteraz (AChE) histokimyası ve kolin asetil transferaz (ChAT) immünhistokimyası kullanarak bazal önbeyindeki ChAT-immünopozitif nöronları sınıflamak üzere morfoloji ve rostro-kaudal organizasyona dayanan “Ch” sektörel terminolojisini önererek dört sektör ayırt ettiler: Ch1-4. Ch1, mediyal septal nukleus (MSN) ve Ch2, Broca’nın diagonal bandının vertikal bacak nukleusu (nvlDBB) hippokampusun kolinerjik aferentlerini sağlar. Ch3, Broca’nın diagonal bandının horizontal bacak nukleusu (nhlDBB) bulbus olfactorius’un kolinerjik afferentidir. Bütün korteksin ve amigdalanın kolinerjik innervasyonu ise Ch4 veya Meynert çekirdeğinden (Meynert’in bazal çekirdeği - nbM) sağlanır. Bu innervasyon dikkat ve bellek işlevlerinin optimal sürdürülebilmesi açısından büyük önem taşır. Yukarda değinildiği gibi tau hiperfosforilasyonun ilk görüldüğü alanlardan biri de Meynert çekirdeğidir (Braak Evre c). AH’de limbik alanlardaki yaygın NFY formasyonu ve nöron kaybı Meynert çekirdeğini de etkiler. Kolinerjik aksonların kaybı diğer patolojik özellikler gibi bir bölgesel yatkınlık gösterir. Lokal internöronlardan sağlanan striatal kolinerjik innervasyon ve talamusun beyinsapı pedinkülopontin çekirdek kaynaklı kolinerjik innervasyonu etkilenmediği gibi, etkilenen Meynert kaynaklı kolinerjik innervasyon da aynı tarzı yansıtır: kortekste en fazla etkilenen bölgeler limbik ve asosiasyon korteksleri iken, primer sensoryal ve motor korteksler göreli olarak salim kalırlar.
Kolinerjik buton da denilen pre-sinaptik kolinerjik terminalde ChAT bir yandan mitokondrilerden asetil koenzim A aracılığı ile serbestlenen kolinden, diğer yandan da yüksek afiniteli kolin geri alınım sistemi aracılığıyla alınan kolinden asetil kolin (ACh) üretir. ACh molekülleri veziküllere paketlenip depolarizasyonu izleyerek sinaptik aralığa salgılanır. Sinaptik aralıkta difüzyonla ilerleyen ACh post-sinaptik membranda nikotinik reseptörlere bağlanarak doğrudan, muskarinik reseptörlere bağlanarak ise G-proteini ilişkili ikincil mesajcılar üzerinden etkisini gösterir. Nikotinik etkilerin hücrenin uyarılabilirliğini arttırarak dikkat tonusunun sağlanmasında rol oynadığı, muskarinik etkilerin ise kalıcı sinaptik değişikliklerle yeni bilginin depolanması şeklindeki nöroplastisite mekanizmalarının unsuru olduğu bilinmektedir. Beyinde α7 nikotinik reseptörler (α7nAChR) pre-sinaptik yerleşimlidir ve uyarılmaları ACh’nin pre-sinaptik akson terminalinden salınımını arttırırken, yine pre-sinaptik muskarinik 2 ACh reseptörleri (M2AChR) tam tersine ACh salınımı inhibe ederler. AH’nin erken döneminde α7nAChR’de kompansatuar bir artış söz konusuyken pre-sinaptik denge giderek nikotinik aleyhine ve muskarinik lehine dönmeye başlar. Post-sinaptik membranda bulunan ve her ikisi de AH sürecinde erken dönemde kaybolmaya başlayan M1AChR ve hem pre- hem de post-sinaptik yerleşmiş olan α4β2nAChR’lerin uyarılması yukarda değinilen hızlı (nikotinik) ve yavaş (muskarinik) kolinerjik nörotransmisyona aracılık eder. Hem nikotinik, hem de muskarinik stimülasyon kaybının gerek α-sekretaz etkinliğinin azalmasıyla Aβ oluşumunun artması ve gerekse de GSK-3β etkinliğinin artmasıyla taunun patolojik fosforilizasyonunun da artması şeklinde in vitro etkileri gösterilmiş, diğer yandan Aβ’nın α7nAChR blokajı üzerinden ACh’nın sentez, salınım ve post-sinaptik etkinliğini azaltabileceği de ortaya konmuştur. Bu durum Aβ ve kolinerjik kaybın birbirlerini pozitif geri besledikleri bir kısır döngü oluşturmaktadır. Muskarinik kolinerjik nörotransmisyonun tetiklediği fosfoinositol sistemi üzerinden anti-Alzheimer mekanizmalar Şekil 8 ve 9’da gösterilmiştir. Bu mekanizmalar α4β2 ve α7 nikotinik ile M1 muskarinik agonizmanın bir tedavi hedefi olarak önemini göstermektedir. M2 antagonizmasının hayvan modellerinde M1 agonizmasına benzer anti-AH etkileri gösterilmiştir. Asetilkolinesteraz (AChE) enzimi katalitik iyonik kısmının aktivitesiyle ACh’yi asetat ve koline hidrolize ederek ACh’nin post-sinaptik aktivitesini durdurur. AChE’nin diğer yandan periferik aniyonik kısmı aracılığıyla Aβ oligomerleriyle oluşturdukları fibriler kompleksin de tek başına oligomerinkini aşan toksik etkileri gösterilmiştir. AChE inhibitörleri AH tedavisinin ilk yerleşik ajanları olarak terapötik cephanede çoktan yerlerini almışlardır.

Şekil 8. Kolinerjik sinaps. Pre-sinaptik akson terminalinde ChAT, asetil CoA aracılığıyla kolini asetilleyerek ACh oluşturur. ACh veziküller içine paketlenerek sinaps aralığına salınır. Presinaptik nikotinik α7 reseptörlerinin uyarılması ACh salınımını stimüle ederken (yeşil ok), muskarinik M2 reseptörlerinin uyarılması inhibe eder (kırmızı çubuk). Sinaptik aralığa salınan ACh post-sinaptik nikotinik α4β2 reseptörleriyle bağlandığında Na+ kanalı açılır ve içeriye Na+ girerek dikkatten sorumlu hızlı kolinerjik nörotransmisyonu başlatır. Muskarinik M1 reseptörlerine bağlanan ACh, reseptöre bağlı Gq proteinini aktive ederek PLC’yi fosforiller. Fosforile PLC bir yandan ikincil mesajcılardan membrana bağlı diasil gliserolü (DAG) aktive ederek PKC’yi doğrudan fosforillerken, diğer yandan da fosfoinositol difosfattan (PIP2) inositol trifosfat (IP3) üreterek endoplazmik retikulumdaki (ER) kendi reseptörüne bağlanacak diğer ikincil mesajcıyı hücre içine salar. IP3, ER’den Ca++salınımını sağlar ve artan intraselüler Ca++ böylece daha fazla PKC’yi dolaylı yoldan fosforillemiş olur. Fosforile PKC, mitojen aktive protein kinaz (MAPK) aracılığıyla nükleer transkripsiyonu devreye sokar ve öğrenmenin temeli olan yeni sinaps yapımına aracılık eder. Sinaps aralığında işlevi biten ACh, AChE aracılığıyla asetat ve koline parçalanır. Kolin pre-sinaptik terminalden kolin taşıyıcısı aracığıyla geri alınır.

Şekil 9. AH’li sinapsta Aβ’nın anti-glutamaterjik ve anti-plastisite etkileri.
Şekil 9a. Glutamaterjik sinaps, GABA-erjik olanın tersine AH’de erken dönemde saldırı altındadır. APP’nin amiloidojenik proteolizi sonrası ortamdan temizlenemeyen çözünür Aβ oligomeri post-sinaptik membranda bir dizi patolojik zinciri tetikler. Bunların sonucunda, önce NMDA, sonra da AMPA olmak üzere glutamaterjik (GluR) sinapslar endositozla kaybedilir. Sonuçta glutamaterjik aktiviteye dayanan ve öğrenmenin moleküler mekanizması olan uzun süreli potansiasyon (LTP) giderek azalırken, uzun süreli depresyon (LTD) artar. Bu etkinin GluR ile doğrudan etkileşimle değil fakat başka bir transmembran protein (TMP) olan bir aracı üstünden olduğuna dair deliller mevcuttur. Bu aracılardan biri olarak normal TMP prion proteini (PrPc) gösterilmiştir. Diğeri ise yalnızca glutamaterjik sinapslarda bulunan ve GluR stabilitesini sağlayan neuroglin 1’dir. Neuroglin 1-Aβ kompleksinin Wnt sinyalizasyonunu başlatacak ligandın reseptörüne bağlanmasını engellediği, bunun sonucunda da GSK3β-Axin-APC’den oluşan “yıkım kompleksi”nin inhibe edilemeyerek bir nükleer transkripsiyon faktörü olan β-kateninin işlevini göremeden yıkıldığı bilinmektedir. Çözünür Aβ oligomeri ileri glikasyon son ürünleri reseptörü (RAGE) ile de etkleşir. Nöronal membranda etkileşim temel enflamatuvar sinyalizasyon olan NFκB sinyalizasyonunu tetikler ve böylelikle ortama enflamatuvar sitokinler salınır. Aβ fagositozu için aktive olmuş mikroglia da RAGE-Aβ etkileşimi ile salınan enflamatuvar sitokinlerin bir diğer kaynağıdır. Bir TMP olan RAGE, bu tür proteinlerin proteolizinden sorumlu olan bir metalloproteinaz olan ADAM10 aracılığıyla kesilerek parenkime salınan çözünür RAGE (sRAGE) oluşur. sRAGE de Aβ bağlar, ancak artık membrana bağlı olmadığından bir patolojik zinciri başlatmanın tam tersine Aβ’yı nötralize eden bir işlev görür. Aβ düşük yoğunluklu lipoprotein reseptörü 1 (LRP1) aracılığıyla kan-beyin bariyerinden dolaşıma verilir. Vasküler endotel membranındaki RAGE Aβ’yı yeniden beyne alırken, dolaşımdaki sRAGE Aβ bağlayarak buna karşıt çalışır. Dolayısıyla, RAGE antagonistleri ve sRAGE agonistlerinin potansiyel anti-AH farmakolojik stratejiler olduğu söylenebilir. Yerleşik tedavi stratejilerinden biri olan memantinin bilinen etki mekanizması NMDAr’da Mg++ tıkacı işlevi görerek mevcut Ca++ sızıntısı kaynaklarından birini engellemek iken hücre içinde de hiperfosforile tauyu parçalayacak fosfataz agonizması yaptığına dair de deliller vardır.
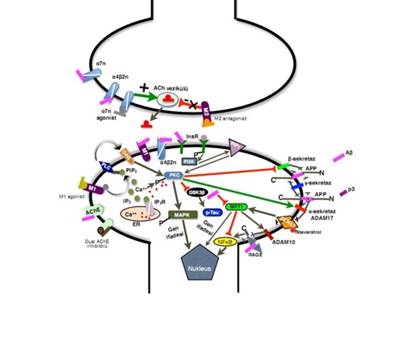
Şekil 9b. Kolinerjik sinaps da AH’de erken tehdit altiındadır. Çözünür Aβ oligomerleri pre-sinaptik membranda asetil kolin (ACh) salınımını inhibe eden M2 muskarinik reseptörlere değil de, tam tersine salınımı arttıran α7 ve α4β2 nikotinik reseptörlere bağlanarak kolinerjik nörotransmisyonu azaltırlar. M2 antagonizması ve/veya α7 ve α4β2 agonizması yapan ajanlar bu dengeyi tersine çevirebilirler. Post-sinaptik M1 kolinerjik aktivite gibi insulin sinyalizasyonu da protein kinaz C (PKC) fosforillenmesi üzerinden anti-AH etkiler (ADAM17 aktivasyonu, β-sekretaz ve GSK3β inhibisyonu) gösterir. Dolayısıyla, M1 agonistleri ve insulin potansiyel anti-AH etkilere sahiptirler. Asetil kolin esterazın (AChE) periferik kısmına bağlanan Aβ ile oluşturduğu kompleksin oligomerin toksisite aracılarından biri olduğu gösterilmiştir. Dolayısıyla, katalitik kısma bağlanan klasik inhibitörlerden farklı olarak ayrıca periferik kısmı da bloke edecek “dual inhibitörler” daha güçlü bir anti-AH etki gösterebilirler. DNA onarımıyla görevli sirtuin sinyalizasyonunun Aβ ile tam tersi etkilere sahip olduğu bilinmektedir. İntraselüler Aβ sirtuin sinyalizasyonunda rol oynayan başlıca moleküllerden biri olan SIRT1’i inhibe ederken, bir TMP olan fraktalcine’nin (CX3CL1), örneğin kırmızı şarapta da bulunan bir polifenol olan resveratrol ile uyarılması CX3CL1’in, aynı zamanda APP’nin non-amiloidojenik proteolizini sağlayan ADAM17’yi aktive etmesini sağlamakta, bunun sonucunda aktive olan SIRT1 ise RAGE’i kesecek ADAM10’u aktive ederek pro-enflamatuvar NFκB sinyalizasyonunu durdururken, diğer yandan da pro-plastisite nükleer transkripsiyon faktörlerini harekete geçirmektedir.
Diğer nörotransmitter kayıpları
Noradrenerjik kayıp
Yukarda değinildiği gibi ön-yumaklar AH ilintili nöropatolojik değişiklik olarak kabul edildiği takdirde beyinsapındaki noradrenerjik (NA-erjik) çekirdek olan LC intranöronal birikintileri AH’deki iki yaygın ön kabulün de değişmesini gerektirecek özelliktedir: 1. AH’de progresif amiloidoz kronolojik olarak AH-P’nin ilk nöropatolojik fenomenidir; 2. AH’de tau patolojisi entorhinal kortekste başlar, patolojik tau fosforilasyonu ilk olarak nöroplastisite potansiyeli en yüksek olan limbik sistemde görülür. Beyinsapında ön-yumaklar gerek en erken amiloidoz delilleri ve gerekse de ilk NFY’lerin transentorhinal kortekste görülmesinden dekadlar önce özellikle de LC’de görülmektedir (Braak Evre a ve b). Ancak LC’de ön-yumakların görülmesi ile ilk nöron kaybının görülmesi arasında da dekadlar süren bir zaman aralığı olduğu anlaşılmaktadır. Bu sırada NFY yayılımı da orta evrelere (Braak Evre III-IV) ulaşmış ve NFY’ler beyinsapında da görülür olmuştur. Bu bulgular ön-yumakların hücre ölümüne ve belki de NA-erjik disfonksiyona neden olmadığı, hücre ölümünden kortekste olduğu gibi beyinsapı NFY’lerinin sorumlu olduğunu düşündürmektedir. LC’deki ön-yumaklar seçici yatkınlığa uygun biçimde LC’nin kortikal projeksiyonları olan anteriyor ve mediyal bölümlerini tutarlarken spinal ve serebellar projeksiyonları içeren kaudal ve lateral bölümleri salim kalır. İyi bilinen NA-erjik işlevler olan dikkat, gerilim cevapları ve uyanıklık yanısıra NA MSS’de glial β-adrenerjik reseptörler aracılığıyla anti-enflamatuvar etkiler de gösterir. Bu etki mikroglial aktivasyonla Aβ klirensinde çok önemli rol oynar. LC nöronları NA yanısıra bir nörotrofik faktör olan BDNF de salgılarlar. BDNF fonksiyonu hiperfosforile tau toksisitesine karşı bir direnç faktörü olarak da düşünülebilir. Nöropatolojik çalışmalarda gösterilen LC’de nöronal kayıp ile kognitif kayıp arasındaki korelasyon, LC için özel geliştirilmiş ve LC nöronal kaybına duyarlı görüntüleme teknikleri ile (LC-I) yaşayan hastalarda da tekrarlanmıştır.
Serotoninerjik kayıp
AH’de serotoninerjik (5-HT-erjik) dorsal raphe çekirdeği ön yumakların, yukarda değinildiği gibi, LC’den sonra en erken görüldüğü beyinsapı yapısıdır (Braak Evre c). LC’ye benzer şekilde dekadlar sonra burda da NFY’ler görülür ve belki LC’de olduğu kadar olmasa da nöron kaybına neden olur. Kortikal 5-HT-erjik akson terminallerinde 5-HT’nin salınımı ve geri alınımı ciddi düzeyde bozulmuştur. AH’de 5-HT-erjik kayıpla depresyon ve saldırgan davranışın korelasyonu bildirilmektedir. 5-HT geri alınım blokerlerinin (SSRI) AH’deki depresyon ve ajitasyonun tedavisindeki rasyoneli bu gözlemlere dayanmaktadır.
Dorsal raphede bildirilen bu kayıp ve pre-sinaptik serotonerjik aktivitedeki bu eksikliklere rağmen AH’de serotonerjik sistemin hiperaktif olabileceğine dair görüşler mevcuttur. Bu sağduyuya karşıt gibi görünen durumun nedeni farklı post-sinaptik 5-HT reseptörlerinin işlevsel farklılıkları ve yerleşimleri ile ilintilidir. Bunlar arasında 5-HT1a ve 5-HT6 reseptörler siklik AMP’nin intraselüler düzeylerini azaltarak ve dolayısıyla plastisite karşıtı bir mekanizmayla işlev görürler. Her iki reseptör tipi de limbik sistemde zengin bulunur. 5-HT6 özellikle glutamaterjik ve kolinerjik nöronları doğrudan inhibe edecek pozisyonda bulunan GABA-erjik nöronlarda zengindir.
Spesifik 5-HT antagonizması aracılığıyla cAMP intraselüler düzeylerinin azaltılmasına karşı durmanın pro-plastisite etkilerinin gösterilmesinden sonra cAMP’yı yıkan enzim olan fosfodiesterazın (PDE) inhibisyonunun terapötik poyansiyeli üzerinde durulmuştur.
Dopaminerjik kayıp
AH ileri evrelerinde parkinsonizm sıradışı bir olgu değildir. Bu olguların patolojik karşılığı büyük sıklıkla substantia nigra pars compacta (SNc) dopaminerjik (DA-erjik) nöronlarında kayıp ve Lewy cisimcikleridir (LC). LCD kendine özgü bir antite olarak ortaya çıkmışken LCD ve AH’nin içiçe geçtiği durumlar olan hem NFY-AP hem de kortikal Lewy cisimciklerinin sonucu olan klinik durumların LCD mi yoksa AH’nin Lewy varyantı (AH-LV) olarak mı adlandırılacağı tartışmalıdır. LC’ler ve NFY-AP birlikteliği için 2 ayrı nöropatolojik örüntü sınıflandırmaya yardımcı olabilir. LC’ler gerek sporadik ve gerekse de ailesel AH’de en sık amigdalada ve %60’a yaklaşan bir oranda görülürler. Diğer limbik alanlar, özellikle de periamigdaloid ve entorhinal korteks amigdaladan sonra en sık görüldükleri yerlerdir. LCD’ye özgü neokortikal ve beyinsapı yerleşimi çok daha seyrektir. Buna karşılık LCD’de yaygın neokortikal ve beyinsapı yerleşimine değişken miktarlarda AH tipi patoloji eşlik edebilir.
Alzheimer Hastalığının Nedenselliği Üzerine Son Değerlendirmeler
AH araştırmacıları plaklar ve yumaklar temelinde, hangisinin birincil rolü oynadığı üzerine nerdeyse uzlaşmaz iki kampa büyük ölçüde ayrılmış durumdadırlar. Bu kamplaşmanın dinsel fanatizme benzerliğine ince bir eleştiriyle de, tau patolojisini (yumakları) ön plana alanlara Tauistler, β-amiloid patolojisini alanlara ise Baptistler dendiği olmuştur.
Gerçekten de, ancak demans evresinde tanınabilen hastalığın klinik evreleri NFY yayılımıyla çok iyi korele ederken, AP ile korelasyonun olmaması Tauistlerin temel tezi olurken, gösterilen bütün genetik bozuklukların Aβ (veya Beta-amiloid protein: Bap) üzerinden etkisini göstermesi, buna karşılık tau mutasyonlarının AH dışı dejenerasyonlara neden olması Baptistlerce tezlerinin kanıtı gibi sunulmuştur.
Günümüzde ilk bakışta Baptizm tartışmasız bir zafer kazanmış görünmekte ve amiloidin hastalığın tetikçisi olduğu, buna karşılık tau patolojisinin bir epifenomen olduğu artık egemen görüş olarak yerleşmiş durumdadır. Tetikçi amiloid olduğuna göre bunun doğal sonucu olarak neredeyse tüm nedene yönelik tedavi çabaları anti-amiloid stratejilere vakfedilmiş durumdadır. Bu çabanın temel rasyoneli, tetikçi ortadan kaldırıldığı takdirde ona sekonder olarak gelişen fizyopatolojik değişiklikler, tau da dahil olmak üzere düzelecektir şeklinde özetlenebilir. Yeni yüzyılın ilk onyılında bu çabaların yarattığı büyük heyecan ve iyimserlik, klinik çalışmalarda nihai gelişim aşamasına kadar gelen hiçbir anti-amiloid aday molekülün ipi göğüsleyememesiyle, büyük maddi ve moral kayıplarla birlikte birbiri ardına hayal kırıklıklarına dönüşmüştür.
Bunlar arasında özellikle de Aβ antijeniyle ilk aktif immunizasyon çalışması olan ve katılımcılar arasında gelişen meningoansefalit olguları nedeniyle durdurulan AN1792 çalışması temel varsayımın sınanması açısından öğretici olmuştur. Katılımcılar arasından ölen olguların otopsilerinde serebral plak yükünde (hem gevşek, hem de sert plaklar) kayda değer bir azalmaya karşın NFY yükünde bir farklılık izlenmemiştir. Dahası, bu çalışmaya katılıp da yeterli antikor geliştiren hastaların uzun süreli izlemleri de dahil olmak üzere, çok sayıda farklı anti-amiloid tedavi çalışmalarına katılan hiçbir hasta grubunda bildirilen yararlanmanın büyüklüğü amiloid hipotezinden yola çıkarak beklenileceği gibi dramatik olmamış, plaseboya karşı farklı sonlanım ölçütleri açısından ancak marjinal yararlar ifade edilmiştir. İlk onyılın sonunda bu durum en militan Baptistler arasında dahi orijinal görüşe yönelik bir skeptisizmle birlikte görüşün revizyonu taleplerinin ileri sürülmesine neden olmuştur.
Daha önce gözden geçirildiği gibi demans AH’nin oldukça ileri bir evresine karşılık gelmektedir ve bu ileri evrede amiloidle başa çıkmak yeterli olmamaktadır. Alzheimerli beyin, orduların sürekli yeniden mevzilendiği bir savaş alanı gibi düşünülebilir. Demans aşaması bu savaşın artık büyük ölçüde kaybedildiği bir evreye karşılık gelmektedir. Öyle görünüyor ki bu savaşın başlangıcında savunmanın henüz başarılı olduğu dönemde kolay degrade edilebilen Aβ40’ın güvenlik kuvvetleri olarak düşünülebilecek şaperon moleküller, NA-erjik mikroglial aktivasyon aracılığıyla klirensini sağlamak ve BDNF gibi nörotrofik faktörlerle nöronların sağkalım dirençlerini arttırmak, kolay başa çıkılamayan daha saldırgan Aβ42’yi ise gevşek plaklarda hapsetmek, ön-plakları onyıllar boyunca fazlaca zarar görmeden hücre içinde tutabilmek temel savunma stratejisidir. Böylesi bir “çatışmanın” henüz hiçbir kognitif yakınması olmayan bir yaşlının (hatta orta yaşlının) beyninde sürmekte olduğu ileri sürülebilir (AH’nin pre-klinik evresi). İlk savunma mevzilerinin düşmeye başlamasıyla saldırganın Aβ (3(pE)-42 gibi daha elit birlikleri hücre içine girip sinapsları yok etmeye başlayacak ve bu kez savunma bunları, yıkılmış sinaps artıklarını da içeren, daha maliyetli ekstraselüler nöritik plaklara, intraselüler agrezomlara hapsetmeye çalışacaktır. Bu aşamada savunma da insülin, sirtuin, WnT, sRAGE gibi kendi elit birliklerini devreye sokarak saldırıya göğüs germeye çalışacaktır. İlk savunmanın böylesi çökmesinin karşılığı AH’nin SCI aşaması olarak düşünülebilir (Braak Evre 1a-1b ve I/II). Bu ikinci savunma mevzisi de çökecek olursa bu kez tau da saldırganın 2. elit birliği olarak devreye girecek ve en başarılı olduğu savaş alanı olan limbik sistemden başlayarak nöronları öldürmeye başlayacaktır. Bu üçüncü aşama AH’nin demans evresidir ve tau yeni mevziler kazanıp yayıldıkça savunma da giderek şiddetlenen demans maliyetini ödemek pahasına, sonuna kadar direnebileceği en tahkim edilmiş bölgeleri olan primer sensori-motor alanlara doğru geri çekilecektir. Bu senaryoya göre bir “tetikçi” tanımlamak gerekiyorsa bu Aβ değil fakat nöronun başlıca katili olan taudur gibi durmaktadır. Aβ’yı ise tetikçiyi tutan sponsor olarak düşünebiliriz. Öyleyse, AH’den korunmanın yolu sponsorun tetikçiyi bulmasına engel olmak gibi durmaktadır. Dolayısıyla, hastalığı demans öncesi aşamasında, hatta pre-klinik döneminde tanımak artık bir zorunluluk olmuştur. Öyle görünüyor ki AH’nin bu demans öncesi evreleri yeterli duyarlılık ve özgüllükte belirlenip tanındıktan sonra çeşitli anti-amiloid stratejileri bu erken aşamalarda denemek, bu ilk savunma kaybedildiğinde veya zaten hastalık gecikmiş demans aşamasında tanındığında bu kez başlıca anti-tau stratejilere yönelmek gerekecektir. Önümüzdeki onyılın genetik ve çevresel olarak AH riski taşıyan kognitif olarak normal yaşlı ve SCI’lı bireylerin ve gerçekte prodromal AH olan MCI’lı bireylerin biyo-işaretleyici kombinezonlarıyla hassasiyetle ayırtedilip, AH-P olanların anti-amiloid, AH-K olanların anti-tau tedavi klinik çalışmalarına dahil edileceği bir dönem olacağını ileri sürmek bir kehanet olmayacaktır.
Mesulam, AH’nin nedenselliği için Baptizm ve Tauizm dışında kalan ve hastalığın yaşlılık riskiyle NT/NFY nöroanatomik yatkınlık tarzını gayet iyi açıklayan “Nöroplastisite Yetmezliği Teorisi”ni ileri sürmüştür. Bu hipoteze göre AH özetle, genetik ve çevresel etkilerle yaşam boyunca artarak biriken plastisite yükünün artık kaldırılamaz olması ile normal yaşlanmadan sapmanın sonuçlarıdır.
Mesulam varsayımını bilinen AH ile ilintili tüm risk faktörleri ve patolojik ürünlerin nöroplastisiteye müdahale ettiğine ilişkin delillerle desteklemektedir. Nöroplastisite potansiyeli, AH’de NFY oluşumunun yayılımı ile örtüşür biçimde en yüksek limbik sistemde iken, azalan sırayla paralimbik yapılar, asosiasyon neokorteksi ve en az da primer sensoryel-motor korteksler şeklinde kendini gösterir. MAP-τ üretimi ve fosforilizasyonunda artış plastik değişiklikler sürecinin temel işlemlerindendir. Özellikle plastisite yükünün yüksek olduğu bölgelerde kinaz-fosfataz döngüsünün dengesinin bozulması plastik talep karşısında fosforillenmiş taunun görevi bittiğinde defosforilizasyonunun sağlanamayarak yukarda söz edildiği gibi patolojik katlanmasına ve birikim eğilimi gösteren oligomerlere dönüşmesine neden olmaktadır. APP, daha önce de söz edildiği gibi, erişkin beyninde başlıca sinaptik bütünlüğün korunması olmak üzere, nöroplastisitedeki rolü deneysel olarak gösterilmiş bir membran proteinidir. APP proteolizi sonucunda α-sekretaz ile kesilen ürün olan sAPP’nin de nörit büyümesi ve LTP’deki rolü bilinmektedir. Buna karşılık, β sekretazla alternatif kesim sonucu oluşan Aβ fragmanı nörotoksiktir, aksonal tomurcuklanma ve LTP’yi inhibe eder. β-sekretazın distal amino terminal ürünü olan sAPPβ’nın da akson budanması tarzı anti-plastisite etkisinden yukarda söz edilmişti. PS1’in, nörogenezde çok önemli işlevi olan NOTCH proteini ile etkileşerek işlev gördüğüne de değinilmişti. Bu etkileşimin erişkin beyninde plastisite yükseltici etkileri gösterilmiştir. PS1 ve PS2 mutasyonları APP proteolizini Aβ42 yoluna kaydırmak yanısıra, NOTCH ile anılan etkileşimi gösteremeyen bozuk ürünlere yol açarak da plastisite potansiyelini baskılıyor olabilir. Kolesterol ve fosfolipidlerin transselüler naklinde rol oynayan ve dolayısıyla aksonal büyüme ve sinaptogeneze katkıda bulunduğu düşünülebilecek ApoE’nin plastisite ile ilişkisine daha önce değinilmişti. Buna göre ApoE-ε4 plastisiteyi baskılarken, ApoE-ε3 ve ε2 yükseltici etki göstermektedir. Yine östrojenin nörotrofik etkilerine değinilmişti. Yaş AH için temel risk faktörüdür. Uzun ömür doğal olarak hem yaşantılama bağımlı plastisite, hem de aşınma, yıpranma ve hasarlanmanın artmasıyla kompansatuar plastisite ihtiyacını arttırmaktadır.
Dolayısıyla da yaşla birlikte artan bu ihtiyacı karşılamak için gerek τ fosforilasyonunda ve gerekse de APP ekspresyonundaki adaptif artışla birlikte denge ne kadar plastisite yükseltici faktörler lehine ise o kadar AH patolojisinden korunulabilmekte, tersine durumda ise bir yandan serbest τ polimerlerinden NFY oluşumu, diğer yandan da Aβ’nın önce gevşek plaklarda birikmesi, sonrasında da bunların katı plaklara dönüşmesi artmaktadır.
Çağdaş uygarlık tek bir bireyin optimal adaptasyonu için yüksek bir kognitif kapasite, özellikle de bellek kapasitesi talep etmektedir. Çağdaş insan genetik yapısını, o dönemin doğal çevresine adaptasyon için seleksiyona uğramış 30.000 yıl önceki avcı-toplayıcı uygarlığının Homo sapiensinden miras almıştır. Avcı-toplayıcı uygarlığına kıyasla gerek bireysel olarak uzayan ömür ve gerekse de sosyal olarak nicel ve nitel açılardan olağanüstü değişen koşullar nöroplastisite yükünü taşınılamaz hale getirerek AH’yi modern insanlık durumunun kaçınılmaz bir eşlikçisi yapmaktadır. Bu perspektifle bakıldığında yukarda resmedilen savaş senaryosunun tarafları da pro-plastisite ve anti-plastisite güçler olmaktadır. Dolayısıyla, nöroplastisite yüküne odaklanılmayan uzun ömür veya AH kesin tedavisi düşleri de anlamsızlaşmaktadır.
TEDAVİ
Kognitif Semptomatik Tedavi
Halen tedavinin çekirdeğini kolinesteraz inhibitörleri (ChEİ’ler) ve memantin ile kognitif semptomatik tedavi oluşturmaktadır. Onaylanan tüm tedaviler AHD içindir. Pre-demans evrelerde onaylanmış tedavi henüz yoktur.
Kolinesteraz İnhibitörleri
Takrin (Cognex®), Amerika Besin ve İlaç İdaresi (FDA) tarafından 1993 yılında Alzheimer hastalığının tedavisi için ruhsat alsa da yan etki profili açısından sorunlu olmasından dolayı günümüzde kullanılmamaktadır. Buna karşın donepezil, rivastigmin ve galantamin sınanan çok sayıda ChEİ’den şimdiye kadar ruhsatlananlardır. Şimdiye kadar piyasaya verilmiş olsun olmasın klinik çalışmalarda düzgün biçimde sınanan tüm ChEİ’ler başarılı olmuştur. Bütün ChEİ’ler için klinik çalışmalarda genel olarak söylenebilecek olan ortak etki plaseboya göre anlamlı farklılığın yanısıra, sonlanım ölçütlerinin zaman içindeki değişim eğrileri izlendiğinde 6. ayda plasebo grubu kötüleşmişken, kötüleşmeksizin hala başlangıç noktasında olma ve 1. yılda başlangıç noktasına göre kötüleşmeye karşın ChEİ öncesi dönemin uzunlamasına izlemlerdeki bozulma hızlarına referansla (”tarihsel plasebo”) daha iyi durumda olmaktır. Plasebo grubundayken Dolayısıyla, ChEİ’lerin AHD’li hastaya yaklaşık 1 yıl kazandırdığı ve tanı konulduktan sonra ilaca ne kadar erken başlanırsa o kadar iyi olduğu söylenebilir.
Donepezil (FDA onayı 1996) günde tek doz (prospektüs bilgisi akşamları önerirken biz birimimizde bazen uykuyu bozması ve fazik fizyolojik kolinerjik aktiviteyle uyumu nedeniyle sabahları tercih ediyoruz), 5 ila 10 mg tabletler şeklinde kullanılan selektif bir ChEİ’dir. Her 2 dozu da etkin olmakla birlikte yüksek dozun etkinliği de daha yüksektir. Başta GİS’e ait olanlar olmak üzere, yan etkileriyle baş edebilmek için 4 haftada titrasyonla yüksek doza çıkmak uygundur. Sonradan piyasaya verilen ağızda eriyen şekli yutma güçlüğü de olan özellikle ileri evre hastalarda yararlı olmaktadır. Rivastigmin (FDA onayı 1997) 1.5, 3, 4.5 ve 6 mg’lık kapsüller şeklinde piyasadadır. Bütiril kolin esteraz (BuChE) inhibisyonu etkisi de olan ve günde iki kez kullanılan rivastigminin en düşük etkili dozu 6 mg/gün olmak üzere, 4’er haftalık titrasyonlarla 12 mg/gün’e yükseltilmesi hedeflenmektedir. Renksiz ve kokusuz sıvı şekli ilaç kullanmayı reddeden hastalarda kullanımı kolaylaştırmıştır. AChEİ’lerin kafa kafaya karşılaştırıldığı en kaliteli çalışma olan, büyük boyutlu ve uzun süreli EXCEED isimli çalışmada ikinci yılın sonunda rivastigmin 2 ikincil sonlanım ölçütünde (demans evrelendirmesi ve GYA’lar) donepezile üstün bulunmuşsa da donepezil de ilk 4 aylık titrasyon döneminde rivastigmine kıyasla anlamlı ölçüde daha iyi tolere edilmiştir. Birincil sonlanım ölçütü (kognitif) açısından her 2 ilacı kullananların da yaklaşık %35’i 2. yılın sonunda başlangıç noktalarından daha iyi durumdaydılar. Rivastigmin deriden emilen bant formunun oral formuna eşdeğer etkinliği ve plaseboya yakın tolerabilitesini gösterdiği IDEAL çalışmasıyla kayda değer bir yenilik getirmiş oldu. İlki 6mg/gün, ikincisi 12mg/gün klasik dozlarına eşit 5 ve 10 ve 15 cm 2 dozlarıyla piyasada olan bant şekli de 4 haftada titre edilmelidir. Gostrointestinal toleransı mümkün olmadığı için oral eşdeğeri olmayan 15 cm 2 ile %50 doz arttırmak mümkün olmuştur. Galantamin (FDA onayı 2001) 8, 16 ve 24 mg/gün aralığında günde iki doz şeklinde kullanılan ve pre-sinaptik allosterik nikotinik reseptör modülasyonu etkisi de olan bir ChEİ’dir. Önce sıvı şekli, daha sonra da yavaş emilen kapsül formu piyasaya verilmiştir. Sıvı form hala günde 2 doz şeklinde 4’er haftalık titrasyonla önce etkisiz 8mg/gün ile başlanıp, 4 hafta sonra etkin olduğu ilk doz düzeyi olan 16mg/gün, son olarak da kompliyans sağlanabildiği takdirde 24mg/gün düzeyine çıkılmalıdır. Kapsül formu günde tek doz benzer titrasyon şemasıyla arttırılmalıdır. Birkaç yıldır ülkemizde piyasadan kalkmıştır. En düşük dozdan başlayıp 4 haftalık aralıklarla yükseltildiğinde üçü için de ortak problem olan bulantı-kusma ile başa çıkmak daha kolay olmaktadır. ChEİ’ler hafif ve orta evre AH’de kullanım için ruhsatlanmış olsa da ağır evrede yararlarına ilişkin gerek klinik çalışma ve gerekse de kendi deneyimizden anekdotal deliller bulunmaktadır. İlaçların etkisizlik ve kesilme kararlarına ilişkin çok aşikar göstergeler olmasa da, düzenli kullanılan ilaca karşın yıl içinde sürekli kötüleşme gözlemleri sonucunda kesme kararı verilebilir. Yine gerek bizim deneyimimiz ve gerekse birinden diğerine geçiş verileri (”switch data”), ilaçlardan biri kesildiğinde diğerinin kullanılmaya başlanmasının hastaların %50’sinde yararlı olabildiğini göstermektedir.
ChEİ’lerin non-kognitif ya da davranışsal belirtiler üzerindeki olumlu etkileri de bildirilmektedir. Buna bir sonraki bölümde değinilecektir. ChEİ’ler büyük ölçüde hafif-orta evre hastalarda çalışılmış ve bu evrelerde kullanım için endikasyon almışlardır. Bununla birlikte, orta-ağır evre çalışmaları da genellikle pozitiftir. Günümüze kadar biriken verilerin kapsamlı bir gözden geçirmesi için Cochrane veritabanına bakınız.
Memantin
ChEİ’lerden sonra bu kez farklı bir etki mekanizmasına sahip olan bir ajan daha onay almış, böylece kolinerjik nörotransmisyonun yanısıra glutamaterjik nörotransmisyonun da NMDA reseptör modülasyonu aracılığıyla düzenlenmesinin AH’de bir tedavi stratejisi olabileceği kanıtlanmıştır. Memantin (FDA onayı 2003) düşük afiniteli bir NMDA reseptör antagonistidir. Memantinin bozuk Alzheimer sinapsında post-sinaptik NMDA reseptöründe magnezyum tıkacı görevi yaparak patolojik intraselüler kalsiyum birikimini engellediği bilinmektedir. Buna karşılık, NMDA antagonizmini geri dönüşümsüz değil fakat düşük afiniteyle yaptığından, NMDA reseptörleri aracılıklı normal glutamaterjik nörotransmisyon için gerekli olan depolarizan bir uyaranla magnezyumun yapacağı gibi reseptörü terketmekte ve böylelikle de LTP mekanizmasına engel olmamaktadır.
Memantin 10 mg’lık tablet şeklinde piyasaya verilmiştir ve günde 2 doz kullanılacak şekilde, haftada 5 mg’lık artışlarla 4. haftada 20 mg/gün dozuna ulaşılır. Sonradan bu günlük dozun gece tek doz verilmesinin de tolerabiliteyi bozmadığı, buna karşılık bir kısım hastada görülebilen gündüz sedasyonuyla başa çıkmayı kolaylaştırdığı görülmüştür. İzleyerek, memantinin de sıvı formu 10 damla = 5mg olacak şekilde geliştirilmiş ve ChEİ’lerde olduğu gibi yutma güçlüğü ve ilaç reddi olan hastalarda kullanım kolaylığı sağlamıştır. Yine de günlük toplam doza 40 damlayla ulaşılması bir güçlük nedeniyken bu sorun da daha sonradan piyasaya verilen pompalı sıvı formuyla aşılmış gibi görünmektedir. Bu yeni formda bir bardak su içine akacak şekilde pompa düzeneğine bir kez basış 5 mg eşdeğeri memantin sağlamaktadır.
AH’de mevcut tedaviler olan ChEİ’ler ve memantinin etkileri genel olarak mütevazi görünse de bu tedavilerin erişilebilir olmasından itibaren hasta ve yakınlarının yaşam kalitelerine katkıları olduğu kuşkusuzdur. Bu yönde özellikle de endüstri sponsorluğunda olmayan, ABD’nin 2 ayrı mükemmeliyet merkezi diyebileceğimiz Alzheimer merkezinden gelen 2 bağımsız çalışmanın verileri oldukça önemlidir. Bunların ilkinde Boston, Massachusetts General Hospital Alzheimer Merkezi’nin 1990-2005 yılları arasındaki verileri analiz edilmiştir. Bu 15 yılın başları ilaçsız dönem, ortaları sadece ChEİ’lere erişilebilen dönem ve sonları ise ChEİ memantin kombinasyonunun mümkün olduğu dönemdir. Bu çalışmanın sonuçları, ortalama 30 ay izlenen 382 hastada, kognitif ve fonksiyonel sonlanım ölçütleri açısından tek başına ChEİ kullanan grubun ilaçsız gruptan daha iyi, ChEİ+memantin kombinasyon tedavisi kullanan grubun ise her ikisinden de daha iyi olduğu yönündedir. İkincisinde Pittsburgh Üniversitesi Alzheimer Merkezi’nin 1983-2006 arası verileri analiz edilmiştir. Bu 23 yıllık dönem de benzer şekilde ilaçsız, tek başına ChEİ’li ve kombinasyon tedavili dönemler olarak ayrılabiliyor. Bu kez bakımevine yatırılma bir sonlanım ölçütü olarak alınmış ve ortalama 60 ay izlenen hastalarda tek başına ChEİ’nin ilaçsız gruba göre bu süreyi geciktirdiği, kombinasyon tedavisinin ise yine her ikisine göre daha da geciktirdiği gösterilmiştir.
Non-Kognitif Semptomatik Tedavi
ChEİ’lerin özellikle halüsinasyon ve hezeyanlar şeklindeki psikotik belirtilere, ajitasyon ve apatiye iyi gelebildiği gösterilmiştir. Benzer şekilde, memantin de psikotik belirtileri ve özellikle de ajitasyonu iyileştirebilmektedir. Biriken verilerin analizi memantinin mevcut ajitasyonu kontrol altına alabildiği gibi uzunlamasına izlemde ilaç kullanan hastalarda daha az ajitasyon ortaya çıktığı gözlemlenmiştir. Dolayısıyla da, bu anılan belirtilerin şiddetleri anti-psikotiklerle derhal müdahaleyi gerektirmediği durumlarda yeni başlanılan bir ChEİ’nin ve/veya memantinin onların üzerine muhtemel etkisini değerlendirmekle gereksiz ilaç kullanımının da önüne geçilmiş olur.
Depresyon, mani ve anksiyete gibi duygudurum bozuklukları AH’de sıktır. Depresyon tedavisinde öncelikle bu yaş grubunda sık kullanılan ve depresyonu bizzat ortaya çıkarabilecek veya şiddetlendirebilecek olan reserpin, α-metil-dopa, β-blokerler gibi anti-hipertansifler kesilerek kalsiyum kanal blokerleri veya ACE inhibitörleri gibi daha uygun ilaçlara geçilmelidir. Trisiklik anti-depresanlar anti-kolinerjik yan etkileri nedeniyle seçilmemesi gereken gruptur. SSRI grubundan ilaçlar, venlafaksin, mirtazapin, duloksetin gibi yeni kuşak hem serotonin, hem de noradrenalin gerialım blokeri olan (SNRI) anti-depresanlar güvenle kullanılabilir. Tam bir mani oldukça seyrek olsa da emosyonel labilite, iritabilite ve öfori gibi özellikleri bazen tedavi gerektirebilir. Bu durumda lityum, karbamazepin ve valproat gibi duygudurum stabilizatörleri kullanılabilir. Anksiyete tedavisinde çok gerekmedikçe kognitif depresan etkileri nedeniyle benzodiazepinlerden kaçınılmalıdır. SSRI’lar ve trazodon bu amaçla öncelikle seçilebilir. Etkisiz kaldığında lorazepam (Ativan®) veya alprozalam (Xanax®) gibi kısa etkili benzodiazepinler tercih edilmelidir.
Hezeyan ve halüsinasyon gibi psikotik belirtilerin tedavisinde ekstrapiramidal ve anti-kolinerjik yan etkilerden arınmışlık başlıca tercih nedenidir. Bu nedenle atipik nöroleptikler öncelikle tercih edilenlerdir. Risperidon ve olanzapinin ekstrapiramidal yan etki potansiyelleri onları gerçek anlamda birer atipik olarak sınıflamayı engellemektedir. Risperidonun aktif metaboliti olan paliperidol kilo aldırmaması ve belirgin sedasyon yapmaması nedeniyle tercih edilebillir. Klozapin çok etkili bir atipik olsa da kemik iliği baskılama potansiyeli nedeniyle haftalık lökosit sayımlarının getirdiği güçlük kullanımını sınırlamaktadır. Atipikler arasında ketiapin diğerleri için sıralanan kısıtlamalardan arınmış ve geniş bir güvenlik aralığında rahatlıkla kullanılabilmektedir. Diğerleriyle kıyaslanamayacak kadar ucuz olması düşük dozlarda haloperidolün tipik bir nöroleptik olsa da hala bir seçenek olarak korunmasına neden olmaktadır. Demansta psikoz tedavisinde çok düşük dozlarda başlayıp optimal dozu olguya göre uyarlamak esastır. Dozun tümünü ya da büyük bölümünü gündüz sedasyonundan kaçınmak amacıyla geceleri vermek uygun olur. Ketiapin 50 ila 100 mg’lık dozlarda genellikle etkin olurken, 400 mg/gün düzeylerine çıkarmak da gerekebilir. Risperidon 0,5 ila 1 mg, seyrek olarak da 2 mg/gün dozlarında kulanılır. Olanzapin dozu 2,5-5 mg, bazen 10 mg/gün’dür; bir klinik çalışmada 15 mg/gün’den itibaren etkinliğinin paradoksal olarak düştüğü gösterilmiştir. Klozapin 6.25 mg ila 50 mg/gün dozlarında yeterli etkinlik gösterse de bazen 200 mg/gün dozlarına kadar çıkmak gerekebilmektedir. Haloperidol dozu 1 ila 3 mg/gün arasında değişebilir ve damla şeklinde kullanımı bir avantaj olarak düşünülebilir.
Ajitasyon ve saldırganlığın tedavisinde yine atipik nöroleptikler ilk seçimlerdir. Ajitasyon için bazen tek başına trazodonla başlayıp cevap alınmazsa nöroleptikleri tercih etmek bir seçenek olabilir. Karbamazepin, valproat ve lityum da tek başına ya da ilave tedavi olarak denenebilir.
İmpulsivite ve dürtü kontrol kusurlarının tedavisinde öncelikle SSRI’lar denenir. Hiperseksüalite için ayrıca anti-androjen ajanlar kullanılabilir.
Uykusuzluk tedavisinde ilaçtan önce güneş batımı sonrası kahve ve çaydan kaçınmak ve yatmadan bir süre önce içilecek ılık süt önerilmelidir. Gerçekte bir anti-depresan olan trazodon 50-200 mg dozları arasında yatmadan 1 saat önce verildiğinde oldukça etkin bir sedatif etki gösterir. Mirtazapin yatmadan önce ½ tablet (15mg) dozunda benzer etkiye sahiptir. Sirkadiyen ritmin düzenlenmesi amacıyla Melatonin 3 mg dozunda başlanıp 12 mg’a kadar yükseltilebilir. Ayrıca bir melatonin reseptör agonisti Ramelteon 8 mg melatoninle benzer etkiye sahiptir.
Non-Farmakolojik Tedavi
Tedaviye non-farmakolojik yaklaşım hasta ve hasta yakını ile kurulan ve hastalıkla başa çıkmaya yönelik ilaç dışı etkileşimin bütününü içermektedir.
Bunlar farklı başlıklarda sıralanabileceği gibi hastalık evrelerine göre de farklılık gösterir. Evresel perspektif şöyle özetlenebilir:
Erken evrede hastalık hasta ve hasta yakınına kültüre uyumlu bir şekilde anlaşılabilir bir şekilde aktarılır ve hastalığın mümkün olduğunca ayrıntılı bir şekilde kavranacağı internet, Alzheimer dernek ve vakıflarının ve benzeri kaynakların dergi, broşür, kitap gibi bilgilendirici yayınlarının okunması teşvik edilir. Hastayla vesayet meselelerini de içerecek şekilde, geleceğin gerçekçi bir biçimde tasarlanmasına yönelik tartışılmaya gayret edilir. Hobiler ve sosyal ilişkileri sürdürmenin önemi aktarılır. Araba kullanmayı sürdürmenin sakıncalarından sözedilir. Hasta yakınına destek grupları hakkında bilgi verilir ve dernek aktivitelerinde yer alması doğrultusunda cesaretlendirilir.
Orta evrede hasta ve hasta yakınına yalnız sokağa çıkmanın sakıncaları anlatılır. Mali ve bunun gibi hukuksal işlerde artık vesayetin zorunluluğu aktarılır. Ev aygıtlarını kullanmadaki güçlükler ve kaza ihtimalleri gözden geçirilir. Giyinmek, yıkanmak ve sofra davranışı gibi güçlükler ele alınır. Artık yalnız yaşamaması yönünde teşvik edilir ve alternatifler ele alınır. Psikotik belirtiler mevcutsa çevresel müdahalelerle başa çıkılmasına gayret edilir. Sfinkter kontrolünü arttırıcı yöntemler üzerinde durulur. Bu tür kuruluşlar o çevrede mevcut olduğunda gündüz bakım evlerinin değerlendirilmesi teşvik edilir. Hasta yakını yükü gözden geçirilir ve muhtemel bir depresyona karşı önlem alınır.
İleri evrede hasta yakınıyla suçluluk duyguları, hastalık yükü ve kayıp üzerine tartışılır. Evde profesyonel yardım ve bir ötesi olarak kalıcı bir bakımevine nakil üzerine tartışılır ve teşvik edilir. Yatak yaraları, kontraktürler, üriner ve solunum sistemi enfeksiyonları riskleri konusunda uyarılır. Sonda ile beslenme gerekliliği ele alınır. Nihayet terminal bakım ve ölüm meseleleri üzerinde durulur.
Spesifik müdahale alanları ise aşağıdaki şekilde sıralanabilir:
İşlevsel performans açısından sözel ipuçlarından fiziksel olarak göstermeye kadar değişen kademeli yardımın demanslı hastanın günlük performansını arttırdığı gösterilmiştir. Davranış modifikasyonu ile tuvaletin uyarılması veya saatlerinin önceden belirlenmesi kuru kalmayı teşvik eder. Uğraşı rehabilitasyonu kognitif performans, psikososyal işlevsellik ve emosyonel dengeyi düzeltir. Işığın azaltılması ve seslerin sükünet verici bir duruma getirilmesi gibi çevresel manipülasyonların yeme davranışını arttırdığı gösterilmiştir.
Davranışsal problemler açısından banyo ve sofra sırasında müziğin ajitasyon ve saldırganlığı azalttığı gösterilmiştir. Mevcudiyetin simülasyonu tedavisi adı verilen aile üyelerinin ses ve görüntü kayıtlarının dinletilmesi veya izletilmesinin de benzer etkisi olduğuna ilişkin bildirimler vardır. Yürüyüş ve benzeri hafif egzersizler gezinme-adımlama davranışının önüne geçebilir. Psikotik belirtiler saldırganlık, kaçma veya yemeyi reddetme gibi tehlikeli veya aşırı sıkıntı verici şiddette değilse yeniden yönlendirme (konuyu değiştirme) en iyi başa çıkma yoludur. Ajitasyon ve saldırganlık için de yeniden yönlendirme, dikkatini çelme ve aktivite tedavisi etkili olabilir. Öncelikle eğer ayırt edilebiliyorsa ajitasyonu tetikleyen olayın önüne geçilmeye çalışılmalıdır (örneğin, aynadaki “yabancı” görüntüden ürküp kavga eden hasta için aynaların kaldırılması).
Hasta yakınları için hastalık hakkında kısa ve uzun süreli eğitim programlarının sıkıntıyı azaltan ve nihai evreyi geciktiren etkileri kesin bir biçimde gösterilmiştir. Hasta yakını destek gruplarının emosyonel denge açısından büyük önemi vardır. Bilgisayar ağları, telefon desteği eğitim için kullanılacak seçeneklerdir. Gündüz bakım evlerinin hasta yakını yükü açısından olumlu etkisi kuşkusuzdur.
İLERİ YAŞIN DİĞER NÖRODEJENERATİF HASTALIKLARI
Arjirofilik Tahıl Hastalığı (AGD)
Arjirofilik tahıl hastalığı, (argyrophilic grain disease -AGD) ilk olarak 1987’de Braak ve arkadaşları tarafından progresif, geç başlangıçlı (85+) bir nörodejeneratif hastalık olarak tanımlanmıştır. Yazarlar nöronal uzantılarda saptadıkları, gümüş boyama ile pozitif küçük iğcik ya da virgül şekilli lezyonlara “tahıl” adını vermiş, gümüşle boyanmaları dolayısıyla da “arjirofilik” sıfatını eklemişlerdir. AGD tipik bir “yaşlanma-ilintili” nörodejeneratif hastalıktır; prevalansı yüzlü yaşlarda %30’lara ulaşır. Bununla birlikte sadece ileri yaşa sınırlı değildir; literatürde ellili yaşlarda olgular da vardır. Dört tekrarlı (4R) tau patolojisi tahılların karakteristik biyokimyasal bulgusudur.
AGD için klinik tanı kriterleri bulunmasa da hastalık kabaca 2 klinik sunumla ortaya çıkabilir:
1- Genellikle çok yavaş seyirli amnestik tipte hafif kognitif bozukluk (MCI), daha seyrek olarak Alzheimer hastalığı demansı (AHD) benzeri klinik;
2- Frontotemporal demansın davranışsal varyantını (dvFTD) andıran kişilik değişikliği ve psikotik ve diğer psikiyatrik özellikler.
Bu sunumlar izole kalabilecekleri gibi sıklıkla birlikte görülürler.
Çok yavaş ilerler. Çok uzun süre MCI kliniğinde kalabilir. Saito ve arkadaşlarının MCI ve demans klinik tanılı 545 olguluk otopsi serilerinde nörodejeneratif MCI olgularının %33’ü Alzheimer hastalığı (AH) iken %31,5’i AGD idi. Demans olgularında ise AH sıklığı %45 iken AGD sıklığı bu kez %18 bulundu.
Tipik olarak, ileri yaşlarda başlayan ilerleyici bellek bozukluğu ve psikoz birlikteliğinde akla getirilmelidir. Tipik laboratuvar bulgusu yoktur. Nörogörüntülemede medial temporal lob (MTL) atrofisi saptanabileceği gibi normal de bulunabilir. Genellikle A-T+(N)+ veya A-T+(N)- şeklinde bir biyoişaretleyici profiline sahiptir.
AGD büyük ölçüde sporadik bir hastalıktır. Şimdiye kadar iki ayrı MAPT mutasyonu ile genetik geçişli olgular bildirilmiştir (S305I ve S305S). Bunlardan ikincisi daha önce frontotemporal demans (FTD) + progresif supranükleer paralizi (PSP) fenotipiyle bildirilmişti.
Kesin tanısı ancak nöropatolojik olarak konulmaktadır.
Makroskopik olarak AGD beyinleri normal veya genellikle MTL’lere sınırlı bir şekilde hafifçe atrofiktir. Mikroskopik olarak üç ana özellik saptanır:
1. Arjirofilik tahıllar;
2. Oligodendrositik inklüzyonlar (sarmallı cisimcikler) ve
3. Nöronal intrasitoplazmik tau-pozitif granüler inklüzyonlar (pre-yumaklar).
Ayrıca balonlaşmış nöronlar ve çalımsı astrositler de ek özelliklerdir.
Patolojik yayılım tarzına göre dört evreye ayrılır:
1. Temporal lobun anterior ve medialinde yer alan ambient girus, amigdala (kortikal ve bazolateral çekirdekler), anterior entorinal korteks, lateral tuberal hipotalamik nükleus,
2. Transentorinal korteks, anterior CA1, presubikulum, dentat girus,
3. CA2, CA3, subikulum, hipotalamusun diğer çekirdekleri, anterior temporal korteks, insular korteks, anterior singulat girus, orbitofrontal korteks, nükleus accumbens, septal çekirdekler
4. Neokorteks ve beyinsapı.
Tolnay ve arkadaşları 19 demanslı AGD olgusu ile 16 demanssız AGD olgusunu karşılaştırdıklarında CA1 kaudalindeki atrofinin AGD’de demansı öngördüğünü bulmuşlardır.
AGD saf bir nöropatolojik antite olabileceği gibi başta AH olmak üzere kortikobazal dejenerasyon (KBD) ve diğer non-AH nörodejenerasyonlara eşlik de edebilir. Hastalığın çok yavaş seyri de göz önüne alındığında, diğer taupatilerin tersine AGD’de sarmallı cisimciklerin asetile tau olmaması ve tek başına hiperfosforile taunun tau birikimi için yeterli olmaması, AGD’nin zararlı bir patolojik antite olmaktan çok diğer taupatilerde hastalığın yayılımına karşı koruyucu bir mekanizma olduğunu ileri süren görüşler de vardır.
AGD’ye özgü bir tedavi yoktur. Kognitif bozukluk için kolinesteraz inhibitörleri, psikiyatrik bulguların özelliklerine göre atipik nöroleptikler, anti-depresan ve anksiyeteye yönelik tedaviler düşünülebilir.
Primer Yaş-İlintili Taupati (PART)
Primer Yaş-İlintili Taupati (primary age-related tauopathy- PART) nöropatolojik olarak amiloid beta (Aβ) plakları olmadan AH-tipi nörofibriler değişiklikleri tanımlamak için öne sürülmüş bir terimdir. PART terimi, bir yanda kognitif olarak normal ileri yaş bireylerde fokal yayılmış nörofibriler yumaklar, diğer yanda demanslı bireylerde gözlenen ve bugüne kadar en bilineni “sadece yumak demansı” (tangle-only dementia – TOD) olmak üzere çeşitli terimlerle adlandırılmış nöropatolojik değişikliklerin dahil olduğu bir sürekliliği tarif eden bir terim olarak öne sürülmüştür. Yazarlar, kognitif değişikliklerin büyük sıklıkla demans evresine evrilmeyen MCI ile sınırlanması nedeniyle TOD yerine PART teriminin daha uygun olduğunu ileri sürmüşlerdir. PART amiloid plak negatif, nörofibriler yumak (NFY) pozitif (NFY+/Aβ−) bir 3R/4R taupatidir.
PART AGD gibi temel olarak ileri yaşın nörodejeneratif hastalığıdır. Büyük çoğunlukla AH’ye göre daha geç yaşta (85+) ortaya çıkan bellek kaybı, diğer kognitif fonksiyonlar ve kişiliğin görece korunması, çok yavaş progresyon sonucunda seyrek olarak diğer kognitif işlevlerin de bozulması klinik özelliklerindendir. Ortalama ölüm yaşı AH patolojisine sahip bireylere göre daha geçtir.
Klinik tanı genellikle mümkün olmaz. İleri yaşta görülen çok yavaş ilerleyici bireylerde, özellikle de görüntüleme veya beyin-omurilik sıvısında (BOS) amiloid negatifken fosfotau düzeyi yüksekse düşünülebilir. Yapısal görüntüleme normaldir veya MTL’ye sınırlı atrofi saptanabilir. Dolayısıyla biyoişaretleyici profili A-T+(N)+/- olmalıdır.
Kesin tanı nöropatolojik olarak konulur. Makroskopik olarak beyin normaldir veya MTL’lere sınırlı atrofi saptanır. Mikroskopik olarak amiloidoz görülmez. NFY’ler MTL’ler yanısıra bazal önbeyin, olfaktor alanlar (bulbus ve korteks) ve beyinsapına sınırlıdır. Dolayısıyla, Braak NFY evresi: >0 ve ≤IV (transentorinal [I-II] ve limbik [III-IV]) ve Thal amiloid faz (AP) evresi: 0 (Aβ yok) olarak özetlenebilir.
Spesifik tedavisi yoktur. Bellek bozukluğu için kolinesteraz inhibitörlerinin kullanımı düşünülebilir.
Limbik-Baskın Yaşla-İlintili TDP-43 Ensefalopati (LATE)
1994 yılında Dickson ve arkadaşları ileri yaşta, demans ile birlikte hippokampal sklerozun (HS) görüldüğü ve AH patolojisinin bulunmadığı 13 hastayı bildirmişlerdir. Bunu demans ile birlikte HS’li hastaların dahil edildiği geniş otopsi çalışmaları izlemiştir. 2006 yılında Frontotemporal lober dejenerasyon (FTLD) ve amiyotrofik lateral skleroz (ALS) hastalarında keşfedilen TDP-43 proteininin HS olan demans hastalarında da gösterilmesiyle kognitif bozuklukla ilişkili olabilecek hippokampal sklerozla birlikte TDP-43 proteinopati, yaşlanmanın hippokampal sklerozu, hippokampal skleroz demansı, sklerozla birlikte serebral yaş ilintili TDP-43, yaşlılıkta TDP-43 patolojisi gibi terimler kullanılmaya başlanmıştır. Limbik-baskın yaşla-ilintili TDP-43 ensefalopati (limbic-predominant age-related TDP-43 encephalopathy-LATE) tüm bu klinikopatolojik tanımları kapsayan bir terim olarak önerilmiştir.
Klinik olarak aMCI kriterlerini dolduran amnestik bulgularla başlar, daha sonra seyrek olarak çoğul kognitif alanları tutarak demans kliniğine ilerleyebilir. AGD ve PART’a benzer şekilde kognitif bozulma AH patolojisinin neden olduğu bozulmaya göre daha yavaştır; sıklıkla stabil bir MCI gibi davranır. AH patolojisiyle birlikte ko-morbid seyrettiğinde saf AH ya da saf LATE patolojisine oranla oldukça hızlı ve ciddi bir kognitif yıkım görülür.
Görüntüleme çalışmalarında LATE hastalarında AH’ye göre daha belirgin hippokampal atrofi gösterilmiştir. Bu atrofi çoğunlukla asimetrik ve hippokampusun rostro-kaudal gradyanında ilerleme eğilimi gösterir. Yine her iki patolojinin birlikte bulunduğu durumlarda hippokampal atrofi saf AH ya da saf LATE patolojisine oranla daha belirgindir. MTL’lerin yanısıra frontal lober atrofi de saptanabilir.
Genetik çalışmalarda beş gende LATE için risk alelleri saptanmıştır: GRN, TMEM106B, ABCC9, KCNMB2 ve APOE. Bu bulgular LATE’in hem FTLD hem de AH ile paylaştığı patogenetik mekanizmalar olduğunu göstermektedir.
Kesin tanı nöropatolojik olarak konulur. Nelson ve arkadaşları 2019’da üç evrelik bir patolojik evreleme sistemi önermişlerdir:
I. Sadece amigdala tutulumu;
II. Evre I + hippokampus;
III. Evre II + orta frontal girus.
LATE patolojisine sahip bireylerin %5-40 oranında bir kısmı hastalık sürekliliğinin erken bir evresi olarak yorumlanabilecek şekilde TDP43 (+), HS (-) iken kalan kısmını TDP43 (+), HS (+) bireyler oluşturur.
Ulusal Yaşlanma Enstitüsü- Alzheimer Derneği (NIA-AA) kriterlerine göre A-T-(N)+ bireylerde, N pozitifliği hippokampal atrofi ya da MTL hipometabolizmasıyla saptandıysa altta LATE patolojisinin bulunabileceği düşünülmelidir.
Spesifik tedavisi yoktur. PART için önerildiği gibi bellek bozukluğu için kolinesteraz inhibitörleri kullanımı düşünülebilir.
Bu bölüm, sayfa sayısı kısıtlaması nedeni ile kitabın basılı halinde kısaltılmıştır. Daha ayrıntılı metin için online edisyona başvurulabilir (bölümün uzun web versiyonu için tıklayınız).
Kaynaklar
1. Albert, M. ve ark. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011; 7: 270-279.
2. Blennow, K.; Hampel, H. CSF markers for incipient Alzheimer’s disease. Lancet Neurol, 2003; 2: 605–613.
3. Crary JF, ve ark. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol. 2014;128, 755-766.
4. Crous-Bou, ve ark. Alzheimer’s disease prevention: from risk factors to early intervention. Alzheimer's Research & Therapy. 2017; 9: 71.
5. Dubois, B., ve ark. Research criteria for the diagnosis of Alzheimer's disease: revising the NINCDS-ADRDA criteria. Lancet Neurol. 2007; 6, 734-746.
6. Dubois, ve ark. Advancing research diagnostic criteria for Alzheimer's disease: the IWG-2 criteria. Lancet Neurol. 2014; 13, 614-629.
7. Fann, J. R., ve ark. Modifiable risk Factors for Alzheimer’s Disease. Front. Aging Neurosci. 2019; 11:146.
8. Gurvit H, ve ark. The prevalence of dementia in an urban Turkish population. Am J Alzheimers Dis Other Demen. 2008; 23: 67-76.
9. Hickman RA, ve ark. Tauopathy (PART): Addressing the Spectrum of Neuronal Tauopathic Changes in the Aging Brain. Curr Neurol Neurosci Rep. 2020;20,39.
10. Jack, C. R. ve ark. NIA-AA Research Framework: Toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018; 14, 535-562.
11. Jorm, A.F., Jolley, D. The incidence of dementia: a meta-analysis. Neurology. 1998 ;51: 728-33.
12. McKhann, ve ark. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011; 7: 263-269.
13. Nelson PT, ve ark. Limbic-predominant age-related TDP-43 encephalopathy (LATE): Consensus working group report. Brain. 2019;142,1503-1527.
14. Rodriguez RD, Grinberg LT. Argyrophilic grain disease: An underestimated tauopathy. Dement Neuropsychol. 2015;9,2-8.
15. Smailagic N, ve ark. 18F-FDG PET for the early diagnosis of Alzheimer’s disease dementia and other dementias in people with mild cognitive impairment (MCI). Vol. 2017, Cochrane Database of Systematic Reviews. Wiley; 2015.
16. Sperling, R. A., ve ark. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011; 7: 280-292.
17. Tijms, B. M., ve ark. Pre-amyloid stage of Alzheimer's disease in cognitively normal individuals. Ann Clin Transl Neurol. 2018; 5:1037-1047.
18. Vos SJB, et al. Modifiable risk factors for prevention of dementia in midlife, late life and the oldest-old: validation of the LIBRA Index. J Alzheimers Dis. 2017;58:537–47.
19. Young, P. N. E., ve ark. Imaging biomarkers in neurodegeneration: current and future practices. Alzheimer's Research & Therapy. 2020; 12: 49.
20. Braak, H., & Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991; 82, 239-259.