Merkezi Sinir Sisteminin Diğer Otoimmün ve İnflamatuar Demiyelinizan Hastalıkları
Yazanlar: Tuncay Gündüz, Gülşen Akman-Demir
Son güncelleştirme tarihi: 03.12.2020
AKUT DİSSEMİNE ENSEFALOMİYELİT
Akut dissemine ensefalomiyelit (ADEM), merkezi sinir sisteminin (MSS) birçok bölgesinin eş zamanlı olarak tutulduğu demiyelinizan bir hastalıktır. En sık çocukluk çağında görülür ve olguların büyük bölümü infeksiyon veya aşılamadan belli bir süre sonra ortaya çıkar. Yıllık insidansının 0,1 – 0,6 /100,000 kişi olduğu bilinmektedir. Bazen sekelsiz, bazen ağır sekeller ile iyileşebilen bu hastalığın ilk tanımlamalardan beri monofazik olduğu kabul edilse de, son yıllardaki bilgiler, ADEM’in alt formlarının olduğu ve başka bazı hastalıklarla iç içe geçebildiğini göstermiştir.
Etyoloji
Bazı çocukluk çağı infeksiyonlarının ardından ADEM gelişebilir. Bu nedenle post-infeksiyöz ensefalomiyelit olarak da isimlendirilir. Bu infeksiyonlardan boğmaca, kızıl, kızamık, kızamıkçık ve suçiçeği başı çeker. Özellikle suçiçeği sonrası gelişen ensefalomiyelit tablosu olguların yarısından fazlasında izole bir serebellar sendrom şeklinde karşımıza çıkar ve çok iyi prognozludur. Bunların dışında çocuklarda ADEM tablosuna yol açabilen başka ajanlar da tanımlanmıştır: enteroviruslar (ECHO, Coxackie), herpes simpleks, mikoplazma ve HIV gibi. Erişkinlerde de non-spesifik üst solunum yolu infeksiyonları, adenoviruslar, mikoplazma ve HIV gibi ajanlar sorumlu tutulmaktadır. Aşılanma sonrasında da ADEM tablosu (post-vaksinal ensefalomiyelit) ortaya çıkabilir. Örneğin çiçek aşısı sonrası ortaya çıkan ADEM, aşının yapıldığı dönemde büyük bir sorun iken, aşının uygulamadan kalkması ile çok nadir görülen bir durum haline gelmiştir. Bir diğer sorun da nöral doku içeren kuduz aşılarından sonra gelişen ensefalomiyelit tablosudur. Bu da tipik ADEM özellikleri gösterir. Yeni tip insan diploid hücre aşısının (HDCV) kullanılmaya başlanmasıyla oldukça seyrekleşmiştir. Diğer pek çok aşılanmadan sonra da ADEM gelişebilir. Bunlar arasında hepatit B, kızamık, boğmaca, difteri aşıları sayılabilir. Fakat ADEM olgularının bir kısmında herhangi bir öncü infeksiyon veya aşılanma saptanamayabileceği akılda tutulmalıdır.
Patoloji
ADEM hastalığında başlıca ak maddeyi tutan, ancak gri maddeyi de etkileyen yaygın perivenöz inflamasyon söz konusudur. Tablonun ortaya çıkışında moleküler taklit mekanizmasının rol oynadığı düşünülmektedir. Örneğin MSS’de bulunan yapılardan özelikle miyelin “basic” protein (MBP), proteolipid protein (PLP), ve miyelin oligodendrosit glikoproteinin bazı viral antijenler ile benzerlik gösterdiği bilinmektedir. Sonuçta oluşan geniş inflamasyon alanlarında demiyelinizasyon, lipid içeren makrofajlar, T ve B lenfositleri ve ara ara plazma hücreleri ve granülositler bulunur. Lezyonlar histolojik olarak aynı yaştadır ve bazı bölgelerde aksonal hasar görülebilir.
Klinik özellikler
Belirtiler infeksiyon veya aşılanmadan sonraki birkaç gün ile birkaç hafta içinde ortaya çıkar. Klinik olarak ADEM ile MS ayrımında en önemli bulgunun ensefalopati varlığı olduğu düşünülmektedir. Başağrısı ve uyanıklık kusuru sıktır; bazen komaya kadar gidebilir. Ense sertliği ve meningeal iritasyon bulguları nadirdir. Davranış-kişilik değişiklikleri, konvülziyonlar ve etkilenen bölgeye göre fokal bulgular (paraparezi, hemiparezi, beyinsapı bulguları) görülebilir. Optik nöropati de sıktır. Nadiren ADEM tablosuna periferik sinir sistemi tutulumu (akut inflamatuar demiyelinizan polinöropati) de eşlik edebilir.
İlk olarak 2007 yılında Uluslararası Pediatrik MS Çalışma Grubu (UPMSÇG) pediatrik ADEM tanı kriterlerini yayınlamış, bu kriterler 2013 yılında revize edilmiştir (Tablo 1).
Tablo 1. UPMSÇG pediatrik ADEM tanı kriterleri 2013 revizyonu (Krupp ve ark, 2013’den uyarlanmıştır.)
* Aşağıdaki bütün kriterler karşılanmalıdır
|
MSS’yi polifokal olarak tutan, inflamatuar demiyelinizan karakterde ilk atak |
|
Ateş, sistemik hastalık ve postiktal dönem ile açıklanamayan ensefalopati bulguları |
|
Akut fazda (İlk üç ayda) anormal beyin MR |
|
Hastalık başlangıcından >3 ay sonra yeni MR veya klinik bulgu olmaması |
|
Akut faz (ilk üç ay) beyin MR lezyon özellikleri: Başlıca serebral ak maddede , yaygın, sınırları belirsiz ve büyük (> 1-2 cm) lezyonlar ortaya çıkar. Lezyonlar T1 sekansında nadiren hipointens olarak izlenir. Talamus veya bazal ganglia gibi derin gri maddede lezyonlar olabilir. |
Seyir çoğu zaman monofaziktir. Bununla beraber değişen literatür bilgileri ile alt tiplerin var olduğu öne sürülmüştür (Tablo 2). Olguların büyük bölümünü monofazik ADEM ve multifazik ADEM (en az 3 ay ara ile iki atak) oluşturur. Daha önce başka bir alt tip olarak kabul edilen ADEM-ON ‘nin ise artık MOG ilişkili hastalığın bir komponenti olduğu düşünülmektedir.
Tablo 2. ADEM’in klinik sınıflaması (Cole ve ark, 2019’dan alıntılanmıştır).
|
Monofazik ADEM |
Tek bir klasik ADEM atağı |
|
Multifazik ADEM |
En az 3 ay ara ile 2 ADEM atağı (ikinci atağın semptomları, bulguları ve MR özellikleri ilki ile aynı ya da farklı olabilir. (3 veya daha fazla atak olmuşsa başka tanı düşünülmeli) |
|
ADEM-ON |
Monofazik ya da multifazik ADEM + ≥1 optik nevrit atağı |
|
Akut hemorajik lökoensefalit |
Hızlı kötüleşme ve yüksek mortaliteli, nekroz ve hemoraji alanları ile karakterize fulminan ADEM prezentasyonu (Bu antitenin durumu tartışmalıdır) |
Bazen MS’in ilk atağı da ADEM’e benzer klinik ve radyolojik bulgularla ortaya çıkabilir. Bu nedenle ADEM ile ilk atak MS ayrımı dikkatle yapılmalıdır. Bulguların yaygın olması, hastada ensefalopatik bir tablo bulunması daha çok ADEM lehine ise de, MS de nadiren bu şekilde karşımıza çıkabilir. Genel olarak ADEM ve MS’i ayırmaya yönelik ipuçları Tablo 3’te verilmiştir.
Tablo 3. ADEM tanısını telkin eden klinik ve görüntüleme bulguları (Santoro ve ark, Neuropediatrics, 2019 ‘dan uyarlanmıştır).
|
Klinik bulgu |
Görüntüleme |
|
Ateş ve meningismus |
Ak madde boyunca yaygın, çok sayıda, birleşme eğilimi gösteren lezyonlar |
|
Prodromal viral hastalık ve yakın zamanda aşılanma |
MS’e göre lezyon sınırları daha zor seçilir. |
|
Ensefalopati veya çok odaklı nörolojik bulgular |
Aynı yaşta çok sayıda lezyon ADEM’i telkin eder. Farklı yaştaki lezyonlar MS ile uyumludur. |
|
Beyin omurilik sıvısında oligoklonal band olmaması |
Talamik ve derin gri madde lezyonları ADEM de çok daha sık görülür |
Son yıllarda tanımlanan anti-MOG antikoru ilişkili hastalık hakkında bilgiler biriktikçe ADEM, NMO ve MS hakkındaki bazı bilgiler de değişmiştir. Şu anda özellikle çocukluk çağında ADEM ile başvuran hastaların önemli bir bölümünde anti-MOG antikorunun pozitif olduğu ve bunların bir kısmının (%34) demiyelinizan ataklar geçirmeye devam edeceği bilinmektedir. Monofazik ve multifazik ADEM tabloları yatıştıktan sonra iyileşme dönemi oldukça uzun sürebilir. Literatürde değişik süreler ve değişik iyileşme skalaları kullanıldığından iyileşme verileri çok net değildir. Ancak hastaların %56-94 ‘ünün tam, %6-56’sının hafif-orta defisitler ile ve %0-18 ‘inin orta-ağır defisitler ile iyileştiği bildirilmiştir.
Radyolojik bulgular
ADEM’de görülen ak madde lezyonları daha yaygın, geniş ve simetrik olma eğilimindedir. Görülen bütün lezyonların benzer kontrast tutma paterni göstermesi, lezyonların aynı yaşta olduğunu gösterir (Şekil 1). Yine de bazı lezyonlar kontrast tutarken bazılarının kontrast tutmaması da söz konusu olabilir. MR’da bilateral simetrik paternin görünmemesi, T1 ağırlıklı kesitlerde kara deliklerin görülmesi ve 2 veya daha fazla periventriküler lezyon bulunmasının MS lehine kabul edilebileceği belirtilmektedir. Daha kesin bir ayrım için izleme döneminde ataktan en az üç ay sonra MR kontrolü yapılmalı ve ortaya yeni lezyonların çıkıp çıkmadığı araştırılmalıdır. Yeni lezyon gelişmesi veya üç ay sonraki MR’da kontrast tutan lezyon saptanması tanıyı MS lehine çevirecektir. Klinik olarak kesin bir ayrım yapılabilmesi için hastanın en az 6 ay takibinin gerektiğini ileri süren görüşler vardır. Aslında klinik olarak bu ayrımın yapılmasının güç olması kadar, histopatolojik ayrımı da güç olabilir.
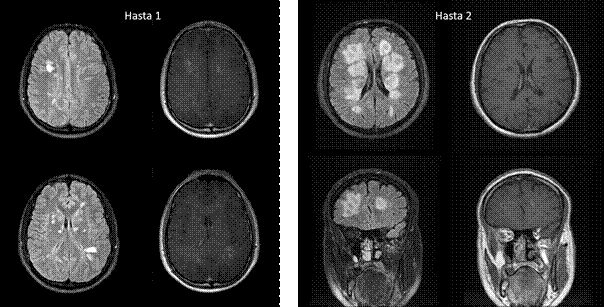
Şekil 1. ADEM tanısı alan iki farklı hastanın T2-FLAIR ve kontrastlı T1-ağırlıklı MR kesitleri (Görüntüler İstanbul Tıp Fakültesi, Nöroloji Anabilim Dalı Multipl Skleroz Birimi materyalinden sağlanmıştır).
Laboratuvar İncelemeleri
Hastaların beyin-omurilik sıvısında (BOS) ılımlı lenfositik pleositoz bulunabilir, BOS nadiren hücresiz de olabilir. Protein artmıştır, şeker normal veya hafifçe azalmış olabilir. Oligoklonal bandlar (OKB) sık olmamakla birlikte görülebilir. Ancak MS’de görülenin aksine, zaman içinde kaybolması beklenir. ADEM hastalarında kanda saptanabilen spesifik bir bulgu yoktur. Ancak olası öncü infeksiyonlar ve karışabilecek diğer hastalıklar açısından bazı incelemeler yapılmalıdır. Bunlar arasında tam kan sayımı, saatlik sedimantasyon hızı, sistemik lupus eritematozus (SLE) ve diğer sistemik hastalıkların belirteçleri ve viral serolojik incelemeler sayılabilir. Mikoplazma antikorları ve titrede olası yükselmeler de araştırılmalıdır. Eğer BOS hücre düzeyi doğrudan infeksiyöz bir süreci düşündürürse mutlaka gerekli kültürler alınmalıdır.
Tedavi
Bugün ADEM tedavisinde ilk yaklaşım yüksek doz intravenöz (IV) metilprednizolon verilmesidir. Genellikle 3-10 gün arka arkaya 1000 mg/gün (çocuk ise 20 mg/kg/gün) IV metilprednizolon verildikten sonra oral metilprednizolona (genellikle 32 mg/gün) başlanır ve doz yavaş yavaş azaltılır. İdame oral steroid süresi hakkında bir konsensus yoktur ancak 3 ay içinde dereceli olarak kesmek doğru bir yaklaşım olabilir. Diğer bir steroid seçeneği 16-32 mg/gün deksametazon verilmesidir. Yakın zamanda yürütülen randomize ve kontrollü bir çalışmada steroide cevapsız olguların plazmafereze iyi cevap verdiği bildirilmiştir. Eğer doğrudan infeksiyöz bir süreç dışlanamamışsa mutlaka uygun antibakteriyel ve antiviral tedaviye de başlanmalıdır.
AKUT HEMORAJİK LÖKOENSEFALİT (Hurst hastalığı)
Akut hemorajik lökoensefalit (AHLE), diğer bilinen adı ile Hurst hastalığı, ADEM’in en şiddetli formu olarak tanımlanmaktadır. Çok nadir olan bu hastalık hem erişkin hem de çocuklarda görülür ve ortaya çıkan nörolojik bulgular hızla ilerler. Kısa zamanda kafa içi basınç artışı sendromu, epileptik nöbetler, ensefalopati, koma tablosu gelişebilir ve sıklıkla ölümcüldür. Hastalığın çocukluk çağı ADEM’lerinin %2’sini oluşturduğu bildirilmektedir. İlk dönem radyolojik bulguları ADEM ile benzerlik gösterir fakat hızlıca hemoraji ve nekroz geliştiği görülür.
Patolojik incelemede ADEM gibi perivasküler inflamasyon ve demiyelinizasyon (bazen demiyelinizasyon olmayabilir) ile birlikte iltihap alanlarında granülositlerden baskın bir iltihabi hücre infiltrasyonunun olduğu, fibrinoid vasküler nekroz ve perivenöz mikrohemorajilerin geliştiği dikkati çeker. Zaman içinde bu bölgelerde aksonal hasar, kanama ve ödemin ortaya çıktığı görülür. Bütün bu bulgular adeta tablonun ne kadar katastrofik olacağının habercisi gibidir.
BOS’ta bu patolojik süreç ile uyumlu olarak nötrofil hakimiyetli pleositoz ve protein yüksekliği saptanır. Bu nedenlerle bakteriyel infeksiyonlarla karıştırılabilir. Tedavi yaklaşımı klasik ADEM’de olduğu gibidir. Tedavinin erken başlanması hayat kurtarıcı olabilir.
Yakın zamanda yayınlanan AHLE tanısı alan iki çocuğun, 16 ADEM ve 20 noninflamatuar nörolojik birey ile karşılaştırıldığı bir çalışmada, AHLE hastalarının BOS’unda yüksek oranda IL-6, IL-17A ve IL-8 olduğu görülmüştür. Buna ilave olarak CCL2, CCL3 ve CCL4 gibi kemoatraktanların daha yüksek olduğu bulunmuştur. Yazarlar bu bulgular ile AHLE hastalarında tosilizumab gibi sitokin fırtınasını önleyebilecek ajanlar kullanılmasının işe yarayabileceğini öne sürmüşlerdir.
AHLE olgularının yaklaşık %70’i ölümle sonuçlanmaktadır. Sağ kalan hastalar ise değişen düzeylerde ağır sekeller ile iyileşmektedir. Bu nedenle klinik tablonun tanısının hemen konarak tedaviye başlanması çok önemlidir.
MULTIPL SKLEROZ VARYANTLARI
Balo’nun Konsantrik Sklerozu
Balo’nun konsantrik sklerozu (BKS), MSS’de tek veya çok sayıda lezyon ile ortaya çıkar. Lezyonlarda demiyelinizasyon olan ve olmayan, iç içe geçmiş halkaların tipik görüntüsü saptanır. Hastalık ilk olarak Marburg tarafından 1906 yılında tarif edilmiş ve 1928 yılında Macar patolog Joseph Balo tarafından bir otopsi olgusunda “encephalitis periaxialis concentrica” adıyla tanımlanmıştır (Şekil 2).
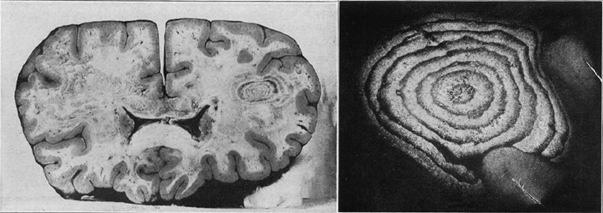
Şekil 2. Balo’nun orijinal olgusu. Soğan zarı gibi içi içe geçmiş demiyelinize olan ve olmayan alanların oluşturduğu lezyon (Arch Neurol Psychiatr 1928;19:242–264)
Lezyon alanlarının histopatolojik incelemesinde miyelin ve oligodendrositlerin korunduğu ve hasarlandığı iç içe geçmiş halkalar görülür. Ancak miyelinin korunduğu bölgeler incelendiğinde normal miyelin değil, ya yeni başlayan ya da kısmi miyelin hasarı olduğu dikkati çeker. Lezyon alanlarında T lenfositleri, makrofajlar ve CD-20(+) B lenfositleri görülebilir. İmmünfenotip olarak MS immünopatolojik patern-3 saptanır. Bununla beraber MS’ten farklı olarak kortikal gri madde etkilenmesi olmaz. Genellikle perivenüler bölgelerde oluşmaya başlayan lezyonların neden halkasal formda geliştiğine yönelik çeşitli hipotezler vardır. Bunlardan birinde en dıştaki inflamasyon sınırında hipoksi geliştiği, bunun hipoksi ile indüklenen faktörleri sentezlettiği ve takiben olası bir oligodendrosit nöroprotektif etki ile demiyelinizasyonun engellendiği öne sürülür. BKS lezyonlarının tümefaktif demiyelinizasyon ve MS lezyonları ile ciddi benzerlik ve ilişkisi olduğu ve BKS lezyonlarının bu ikisi arasında bir ara form olabileceği öne sürülmüştür.
Oldukça nadir bir hastalık olan BKS daha çok genç erişkinlerde görülür ve klinik olarak MS’e benzer belirti ve bulgular ile ortaya çıkabilse de daha çok beyin tümörü gibi davranır. Bazı olgularda ancak biyopsi ile tanı konabilir. Hastalık monofazik olabildiği gibi nüks ve iyileşmeler (%10-20) ile giden ve hızlı progresif olgular bildirilmiştir. Ayrıca bazı MS hastalarında zaman zaman bu tipte lezyonların çıktığı, ayrıca BKS lezyonları ile başvuran bazı hastaların MS’e dönüştüğü bilinmektedir. Hastalığın ayırıcı tanısında glioblastoma multiforme, primer merkezi sinir sistemi lenfoması, iskemik infarkt, beyin apsesi, tümefaktif demiyelinizasyon, sarkoidoz ve serebral tüberküloma yer alır.
Hastalığın akut faz tedavisi, MS tedavisine benzer. Öncelikle kortikosteroid, plazmaferez ve IV immünglobulin (IVIG) denenebilir. Eğer yanıt alınamaz ise siklofosfamid önerilmektedir. Uzun dönem tedavisinde azatioprin ve mitoksantron önerenler vardır. Hastaların bir kısmı sekelsiz iyileşmekle beraber, ağır sekelli iyileşen ve kaybedilen hastalar bildirilmektedir.
Schilder Hastalığı
Miyelinoklastik diffüz skleroz (ya da Schilder hastalığı), sentrum semiovalede daha çok bilateral, az veya çok simetrik büyük demiyelinizan lezyonlar ile karakterize bir tablodur. En sık çocukluk döneminde görülür.
Miyelinoklastik diffüz sklerozun patolojik bulgularının MS’e çok benzediği bilinmektedir. Bununla beraber hastalığını immünolojik BOS profilinin MS’ten belirgin derecede farklı olduğu, bu nedenle MS ile farklı hastalıklar oldukları düşünülmektedir.
Klinik olarak lezyonun yerine göre değişmekle beraber çeşitli fokal bulgular, ensefalopati ve nöbetler görülebilir. Hastalık monofazik olarak kabul edilmekle beraber nüks gösteren olgu bildirimleri de yapılmıştır.
Poser ve arkadaşları tarafından 1986 yılında önerilen tanı kriterlerinde, bu lezyonların en az 3x2 cm boyutlarında olması, fokal bulgu veya ensefalopatinin olması, MSS dışında tutulum olmaması, adrenal fonksiyonların ve uzun zincirli yağ asitlerinin normal olması gerekliliği belirlenmiştir. Ayrıca ADEM kliniğinden önce olan ateş ya da prodromal belirtilerin olması ve BOS’ta OKB bulunmasının tanıdan uzaklaştırdığı öne sürülmüştür. MR lezyonları genelde hafif kontrast tutulumu gösterir ve kronik dönemde lökodistrofik MR paternine yaklaşır. Klasik adrenolökodistrofi lezyonları, tümefaktif demiyelinizan lezyonlar ve ADEM lezyonları ile karışabilir.
Tanının konması büyük oranda biyopsi gerektirmekle beraber MR ve diğer laboratuvar yöntemlerin gelişmesi ile biyopsi yapılmadan tanı konması da mümkün olmuştur. Bu şekilde bir klinik ve lezyon ile başvuran bir hastada, KİBAS bulgusu olmaması, bir-iki subkortikal kistik lezyon (açık halka kontrast tutan) olması, iskemi, apse ve metastazın ekarte edilmesi, perfüzyon MR ve MRS’te glutamat/glutamin oranının primer beyin neoplazmını ekarte etmesi sonrası biyopsi yapılmadan Schilder hastalığı tanısı konabileceği öne sürülmüştür.
Tedavide pulse ve uzun dönem oral steroid, IVIG ve steroide yanıtsız olgularda sitotoksik ajanlar kullanılmaktadır ve iyileşmenin genel olarak iyi olduğu bilinmektedir.
Marburg MS Variantı
Bu hastalık MS’in akut fulminan bir formu olarak 1906’da Otto Marburg tarafından tanımlanmıştır. Hastalık tipik olarak başağrısı, kusma, nöbetler, bilateral optik nevrit, kuadriparezi ile başlar. Bununla birlikte çeşitli multifokal kognitif bulgular dikkati çeker. Tipik olarak akut başlayan hastalık progresif olarak ilerler ve yüksek oranda aylar içinde ölüme yol açar.
Lezyonlar klasik MS ile aynı lokalizasyonlarda görülür. Kontrast tutulumu görülebilir ve lezyonların etrafında şiddetli ödem mevcuttur.
Patolojik incelemede lezyonlarda MS ile benzer bulgular görülmesine rağmen, yoğun makrofaj, nötrofil ve eozinofil infiltrasyonu, ciddi doku hasarı ve nekroz da gelişmiştir. Bu katastrofik tablonun bazı hastalarda görülmesinin sebebinin, daha az katyonik ve daha az stabil bir MBP varyantı olabileceği ileri sürülmüştür.
BOS’ta OKB genellikle negatif bulunur. Tedavide kortikosteroidler, plazmaferez ve sitotoksikler kullanılmaktadır Ancak hastalık tedaviye cevap vermeyebilir. Beyinsapı etkilenmesi ve herniasyon sendromları hastaların kaybına neden olur.
NÖROMİYELİTİS OPTİKA SPEKTRUM HASTALIKLARI
İlk olarak 19. yüzyılda Eugène Devic tarafından transvers miyelit ve optik nevrit sonrası ölen bir hastanın otopsi bulguları yayınlanmış ve sonrasında nöromiyelitis optika hastalığı tanımlanmıştır. Uzun yıllar boyunca benzer yerleri tuttuğu için MS ile ilişkili bir hastalık olduğu düşünülmüştür. Ancak 2004 yılında, bu hastalarda astrosit ayaklarında bulunan akuaporin-4 su kanalına karşı antikorlar (anti akuaporin-4) saptandıktan sonra hastalık anlayışı oldukça değişmiştir. Artık Nöromiyelitis Optika Spektrum Hastalıkları’nın (NMOSH) patogenez, klinik özellikler, radyolojik ve laboratuvar bulguları ile ayrı bir hastalık spektrumu olduğu, medulla spinalis ve optik sinir dışında da merkezi sinir sistemini tutabildiği görülmüştür. Bununla beraber antikorun tanımlanmasından sonra, antikorun negatif olduğu (seronegatif) ancak klinik olarak aynı bulgular görülen NMOSH olguları hakkında da bilgiler birikmeye başlamıştır.
Patofizyoloji
Hastalığın merkezinde olan akuaporin-4-IgG (NMO-IgG kan-beyin bariyerini geçerek, endoteli saran astrosit ayaklarındaki hedefine bağlanır. Başlıca IgG1 alt tipinde olduğundan özellikle kompleman sistemini harekete geçirir. Bunu takiben, ortama NK hücreleri, eozinofiller, nötrofiller ve makrofajlar çağrılır. Oluşan inflamatuar ortamda salınan sitokinler ile beraber kan-beyin bariyerinin geçirgenliği daha da artar ve daha fazla akuaporin-4-IgG etkileşime girer. Bu şelale reaksiyonu sonucunda etraftaki oligodendrositler, nöronlar, akson ve miyelin ve diğer ve destek yapılarda topyekün hasar gelişir. Bununla beraber, antikorun beyin parenkimine geçişinin ise otoreaktif (bu durumda akuaporin-4‘ e karşı) B ve T hücreleri sayesinde olduğu öne sürülmektedir. Periferde aktive olan bu lenfositler bir kez astrosit ayaklarındaki antijenlerini tanıdıktan sonra olayların başladığı düşünülmektedir.
Yukarıdaki patofizyolojik olaylardan anlaşılacağı gibi, bu süreç MS ve diğer demiyelinizan hastalıklardan oldukça farklıdır. En önemlisi NMOSH’nin son zamanlarda bir astrositopati olarak sınıflandırılma eğilimi ortaya çıkmıştır. Hastalarda demiyelinizasyonun sekonder olarak geliştiği bilinmektedir.
Patolojik tanımlamalar oldukça değişkendir, çünkü bu hastalar karakteristik olarak heterojen bir gruptur. Klasik olarak, akut medulla spinalis lezyonları, yaygın şişme ve yumuşama gösterir, birkaç seviyede uzanır, ya da tüm medulla spinaliste devamlı veya yama tarzında dağılır. Akut olarak ak ve gri maddeyi tutan yoğun makrofaj infiltrasyonu, miyelin ve akson kaybı ile damarların etrafında lenfosit kümelerinin oluşmasıyla giden bir yıkım söz konusudur. Kronik lezyonlarda, medulla spinalis nekrotik dejenerasyon ve gliozis nedeni ile nekrotik ve atrofiktir. Bu geniş lezyon infarkta benzeyebilir. Medulla spinalisteki ani şişme, sınırlayıcı pia ile karşılaşınca intramedüller basıncı arttırabilir ve küçük parenkimal damarların kapanmasına yol açarak daha ileri doku yıkımına yol açabilir. Kalın ve hiyalinize duvarlı damarların proliferasyonu infarkttan sonra ya da diğer yaygın yıkımlarda görülenlere benzerdir. Optik sinirler, kiazma, ve bazen serebral hemisferler benzer tarzda hem demiyelinizan lezyonlar, hem nekrotizan lezyonlarla ya da ikisi bir arada olarak etkilenebilir.
Epidemiyoloji
Görülme yaşı ortalama 40 olarak bildirilmektedir (3-88 yaş). Kadınlar olguların %70-90‘ınını oluşturur. Tüm dünyadan bildirilen prevalans oranları incelendiğinde, NMOSH’nin 100,000 kişide 0,7-10 oranında bildirildiği görülmektedir. NMOSH’nin ortalama prevalansı 1/100,000 olarak kabul edilebilir. Genel olarak Doğu Asya ve siyah ırkta daha yüksek oranda görülür. Beyaz ırktaki insidans ise 0,5-0,8 / 100,000 olarak kabul edilmektedir. Bununla beraber aynı popülasyondaki ırklar arasında da farklar olduğu göz önüne alınmalıdır.
Klinik spektrum ve tanı kriterleri
NMOSH’da en sık tutulan bölgeler optik sinir, medulla spinalis ve area postremadır. Yukarıda bahsedildiği gibi, nöromiyelitis optika’nın tanımlanmasından sonraki süreçte hastalığın klinik spektrumu ve tanı kriterleri değişikliğe uğramıştır. Şu anda kabul edilen kriterler ise temelde anti-akuaporin-4 (anti-Aqp-4) antikorunun pozitif olup olmamasına göre şekillendirilmiştir. Bu kriterler ile ana klinik tutulum bulguları ve NMOSH’ye has MR özellikleri de tanımlanmıştır (Tablo 4 ve 5).
NMOSH’de nüks paternleri incelendiğinde en sık transvers optik nevrit (%37-54), sonra transvers miyelit (%30-47), sonra area postrema (%3) ve serebral sendrom (%2) gelir (Şekil 3). Bunların kombinasyonları da daha az sıklıkla görülebilir.
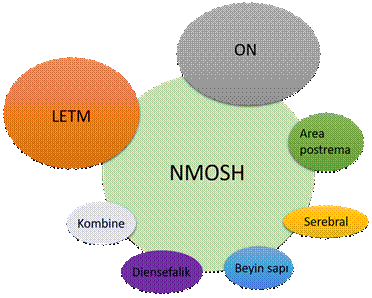
Şekil 3. NMO spektrum hastalıklarında ana klinik tutulum bulguları. LETM, uzun transvers miyelit; ON, optik nevrit; NMOSH, nöromiyelitis optika spektrum hastalığı
Tablo 4. NMOSH’da görülen ana klinik ve MR özellikleri.
|
Ana klinik tutulum bulguları (AKTB) |
NMOSH’ye has MR özellikleri |
|
1- Optik nevrit 2- Akut miyelit 3- Area postrema sendromu (hıçkırık, bulantı ve kusma) 4- Akut beyinsapı sendromu 5- Semptomatik narkolepsi veya akut diensefalik klinik sendrom (NMOSH’ye özel MR görüntüsü ile beraber) 6- Semptomatik serebral sendrom (NMOSH’ye özel MR görüntüsü ile beraber) |
1- Akut optik nevrit: Beyin parenkiminde lezyon olmamalı ya da nonspesifik olmalı. Optik sinir MR’ında optik sinirin ½ sinden fazlasında T2 ya da kontrast tutan lezyon olması ya da optik kiazma lezyonu olması 2- Akut miyelit: ≥3 vertebra segmentinden uzun intramedüller MR lezyonu olması ya da akut miyelit hikayesi olan bir hastada ≥3 vertebra segmentinden uzun omurilik atrofisi 3- Area postrema sendromu: Dorsal medulla/area postrema sendromu 4- Akut beyinsapı sendromu: Periependimal beyinsapı lezyonu |
Tablo 5. NMOSH 2015 tanı kriterleri (Weinshenker ve ark, 2017’den alınmıştır).
|
Akuaporin-4-IgG (+) |
Akuaporin-4-IgG (-) ya da Bilinmiyor |
|
1- ≥1 AKTB* 2- Akuaporin-4-IgG pozitif bulunması (hücre tabanlı testler önerilir) 3- Diğer tanıların dışlanması |
1- ≥1 klinik atakta, ≥2 AKTB* a) En az bir AKTB optik nevrit, akut miyelit ya da area postrema sendromu olmalı b) En az iki farklı AKTB olmalı c) MR bulguları uyumlu olmalı 2- En iyi metod ile yapılan antikor testi negatif olmalı 3- Diğer tanılar dışlanmalı |
· AKTB: Ana klinik tutulum bulguları (Tablo 4’e bakınız)
Halihazırda NMO spektrum hastalığı ile başvuran hastaların %70-80 ‘ininde anti-Aqp-4 antikoru saptanmaktadır. Bununla beraber bu oran testin optimum teknikle yapılıp yapılmadığına göre değişir (bu antikor için canlı hücre temelli teknikler önerilmektedir). Bu antikor saptanmayan grubun bir bölümünde ise son yıllarda ortaya çıkan testlerle anti-MOG (anti-miyelin oligodendrosit glikoprotein) antikorları saptanmıştır. Bu oran %25-40 olarak bildirilmektedir. Bu olguların artık tipik anti-Aqp-4 antikoru pozitif NMO olgularından klinik ve radyolojik olarak farklılık gösteren yeni bir hastalık antitesi olduğu kabul edilmektedir (Şekil 4) (MOGAD- miyelin oligodendrosit glikoprotein antikoru hastalığı).
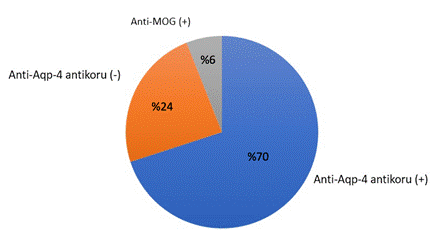
Şekil 4. NMOSH’de anti-Aqp-4 antikor serostatusu ve daha önce seronegatif NMOSH’de olduğu düşünülen MOG ilişkili hastalıkta yaklaşık anti-MOG seropozitifliği.
NMOSH’de klinik bulgular ve eşlik eden belirtiler saatler ya da günler içerisinde yerleşir. Bu bulguların öncesinde başağrısı, bulantı, uyuklama, ateş ve miyalji tabloya eşlik edebilir. Hastaların büyük çoğunluğu iki yanlı optik nöropati geliştirir. Görme kaybına genellikle göz çevresinde ağrı, miyelite ise belli bölgede bel ağrısı ya da radiküler ağrı eşlik edebilir. Çeşitli beyinsapı sendromları (örneğin, okülomotor disfonksiyonlar, kranial sinir felçleri gibi), Lhermitte bulgusu tabloya eşlik edebilir. Medulla spinalis hasarına bağlı olarak tonik spazmlar ve nöropatik alt ekstremite ağrıları sık karşılaşılan sekellerdir. NMOSH olan hastalar genel olarak MS’e göre daha ağır klinik tablolar ile başvururlar ve ciddi sekel görülme oranı daha fazladır. Hastaların önemli bir bölümü akut relaps iyileştikten sonra yıllar süren ciddi nöropatik ağrılardan yakınabilir. Narkolepsi, uygunsuz ADH sendromu, CK yüksekliği ile beraber miyopati, miyeloradikülit, ensefalopati, kognitif bozulma (dikkat eksikliği, yürütücü işlev bozukluğu) az bir oranda hastada ortaya çıkabilir.
Paraneoplastik NMOSH
Kanser dokularında eksprese edilen aqp-4 ‘e karşı ortaya çıkan antikor ile gelişen paraneoplastik MNOSH oldukça nadir görülür. Büyük vaka serilerinde %2-3 oranında bildirilmektedir. Bu hastaların daha çok uzun transvers miyelit ile ortaya çıktığı ve immünoterapiye az yanıt verdiği düşünülmektedir. Paraneoplastik olması olası ve mutlaka kanser taranması gereken hastalar şu şekilde önerilmiştir: 1- beyinsapı tutulumu ile başvuran hastalar (başlıca bulantı ve kusma), 2- uzun transvers miyelit ile başvuran >45 yaş erkek hastalar.
Bazı izole uzun miyelit olgularında aqp-4 antikoru saptanmaz ancak kanser saptanır. Bu olgular izole paraneoplastik miyelopatiler olarak isimlendirilir. Bu tablonun da yine daha çok yaşlı bireylerde ortaya çıktığı, tedaviye iyi yanıt vermediği ve çeşitli paraneoplastik antikorların saptandığı bilinmektedir.
Komorbid otoimmün hastalıklar
SLE, Sjögren sendromu ve antifosfolipid sendromu gibi bazı sistemik otoimmün hastalıklarda NMOSH daha sık oranda görülür. Bu klinik hastalıkları ya da antikorları olan bir hastada NMOSH ortaya çıktığında anti-aqp-4 antikoru saptanabilir. Bu durumda hastada sistemik hastalığın MSS tutulumundan ziyade, beraber olan NMOSH’den söz edilir. Myasthenia gravisin de artmış bir sıklıkla NMOSH ile beraber görüldüğü bilinmektedir. Bu artmış birlikteliklerin, hastada otoimmüniteye yatkınlığı artıracak genetik altyapıdan kaynaklandığı düşünülmektedir.
MR ve laboratuvar bulguları
Klasik MR bulguları optik sinirin yarısından fazla ve/veya optik kiazmada T2 veya kontrast tutan lezyon, daha çok üst servikal medulla spinaliste ve ≥3 vertebra segmentinden büyük, santral yerleşimli, omuriliği şişirebilen, ve kontrast tutan lezyonlardır (Şekil 5). Bununla beraber bazı medulla spinalis lezyonlarının daha kısa olduğu bildirilmektedir (~%15). Bu uzun omurilik lezyonları birleşme eğiliminde olan ancak aslında ayrı lokalizasyonlu MS lezyonları ile karıştırılmamalıdır. Ayrıca çok erken dönemde karakteristik lezyon oluşmayabileceği gibi, geç dönemde de fragmantasyon ve atrofi geliştiği görülebilir. Literatürde bildirilen beyin lezyonlarının oranı çok değişkenlik göstermekle beraber %40-50‘ye kadar saptanabileceği öne sürülmektedir.
Hastaların 1/3’ünde eritrosit sedimantasyon hızında artma, yarısında pozitif antinükleer antikorlar, nadiren diğer otoantikorlar görülebilir. NMOSN’nin değerlendirilmesinde BOS incelemesi esastır ve bazen yinelenmesi gerekebilir. Akut dönemde, MS’de görülenin tersine NMOSH hastaların önemli bir bölümünde (%83) BOS’ta hücre artışı saptanır. Bazen milimetreküpte 100 hücreyi aşan (ve 1000’e varan) hücre artışı görülebilir. Nötrofil hakimiyetiyle karşılaşılabilir. Bu durum MS’de görülmez. Protein artışı genellikle sıktır ve hastaların %41’inde 100 mg/dl’yi aşar. BOS’daki bu yoğun inflamatuar yanıta karşın pozitif OKB olguların %20’sinden azında görülür ve genellikle sonradan negatifleşir.
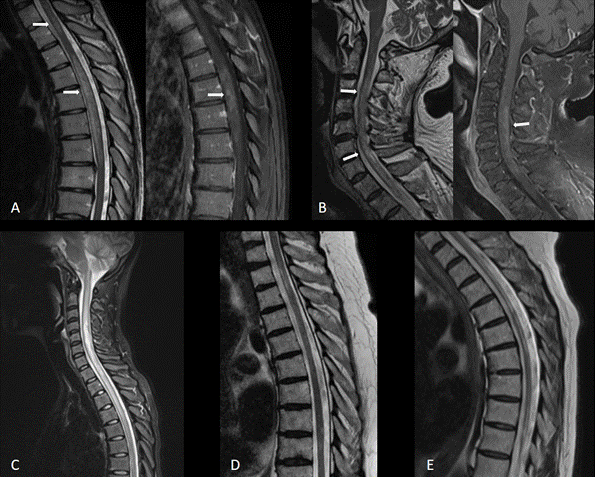
Şekil 5. Anti-aqp-4(+) nöromiyelitis optika spektrum hastalıklarının MR örnekleri. (A), (B), ve (C): Akut dönemde uzun miyelit (A ve B ‘de postkontrast T1 serilerde lezyon bölgesinde kontrast tutulumu görülmektedir) (D) ve (E): Kronik dönemde, farklı seviyelerde sekel lezyonlar ve fokal atrofi bölgeleri (Görüntüler İstanbul Tıp Fakültesi, Nöroloji Anabilim Dalı Multipl Skleroz Birimi materyelinden sağlanmıştır).
Ayırıcı Tanı
Tablo 6. Nöromiyelitis optika spektrum hastalıklarının ayırıcı tanısında düşünülmesi gereken hastalıklar.
|
Multipl skleroz İdyopatik akut transvers miyelit İdyopatik optik nevrit MOGAD (MOG antikoru hastalığı) Nörosarkoidoz Sistemik otoimmün hastalıklar Sjögren hastalığı Sistemik lupus eritematozus MSS lenfoması Nöro-Behçet hastalığı (spinal tutulumlu) Spinal dural AV fistül İnfeksiyon Sifiliz Nörobruselloz Tüberküloz, çeşitli bakteriyel ve viral ajanlar Leber’in herediter optik nöropatisi |
NMOSH’nin ayrılması gereken belki de en önemli hastalık MS’dir (Tablo 6). Bunun nedeni MS tedavisinde kullanılan bazı ilaçların NMO’yu şiddetlendirebilmesidir. Ayırıcı tanıda özetle, relaps şiddetinin (görme kaybı, spinal bulgular) çok ağır olması ve tedaviye iyi yanıt alınamaması; hastalığın ilk ortaya çıkışının nispeten geç yaşta olması; MR’da MS’e tipik periventriküler, küçük, ovoid lezyonlar yerine yukarıda tarif edilen daha dramatik, büyük lezyonların görülmesi; BOS’ta ciddi oranda hücre olması (hatta nötrofil hakimiyetli), proteinin yüksek olabilmesi, OKB’nin negatif olması gibi faktörler tanıyı NMOSH’ye yaklaştırır.
Bir grup hasta tekrarlayan optik nevrit veya miyelit relapsları ile başvururlar ve zaman içinde NMOSH düşündürecek bir bulgu vermezler. Bu hastalar da yine benzer immünsupresif ajanlar ile tedavi edilebilir.
NMOSH ile karışabilecek başka önemli bir hastalık MOGAD’dır ve daha geniş bir klinik spektruma sahiptir. Bu nispeten yeni tanımlanan antiteden ve NMOSH’ile farklılıklarından bir sonraki bölümde ayrıntılı bahsedilecektir.
NMOSH ile başvuran bir hastada mutlaka sistemik bir inflamatuar hastalık olup olmadığı araştırılmalıdır. Bu araştırma süreci benzer klinik ve radyolojik bulgular verebildiğinden, sarkoidoz, Sjögren sendromu, SLE üzerinde yoğunlaşmalıdır. Bu bağlamda uygun vaskülit belirteçleri, toraks ve batın tomografisi, ayrıntılı göz muayenesi, gereğinde PET/CT, lenf bezi/tükrük bezi biyopsisi hastaya göre uygulanabilir. Bu hastalıkların saptanması durumunda tedavi yaklaşım dramatik değişiklikler gösterebilir.
Spinal tutulumlu nöro-Behçet hastalığı özellikle ülkemizde akla gelmeli, oral ve genital aft, üveit, eritema nodozum gibi Behçet hastalığı belirtileri sorgulanmalı ve gereğinde paterji testi yapılmalıdır.
Malignitelerin dışlanması özellikle izole omurilik tutulumu olan olgularda zor olabilir. Bu hastalarda BOS bulguları, BOS sitolojisi, lezyon ve kontrast tutulum paternleri ayrım için yol göstericidir.
Spinal dural AV-fistül tanı konulması gerçekten zor olabilen bir hastalıktır. Klinik gidişi ve radyolojik bulguları büyük oranda NMOSH ile karışabilir. Bununla beraber spinal venöz yapıların MR’da görülmesi, basamaklı bir klinik kötüleşme, BOS’ta inflamasyonun saptanmaması, serumda antikorların yokluğu ile şüphe edilmesi halinde ancak spinal anjiografi ile tanı konabilmektedir. Bununla beraber anjiografinin negatif olabileceği (teknik nedenlerle) akılda tutulmalıdır (Ayrıca bakınız: Omurga ve Omurilik Hastalıkları).
Akut dönemde klinisyeni ayırıcı tanıda en çok zorlayan etyolojik gruplardan birisi de infeksiyonlardır. Çünkü radyolojik bulgular ve BOS bulguları oldukça benzeyebilir ve klinisyeni immünsupresif/antibiyotik-antiviral tedavi seçiminde ikileme sokabilir. Böyle durumlarda bütün infeksiyon belirteçleri, risk faktörleri ve klinik dikkatle gözden geçirilmeli ve “önce zarar verme” prensibi ile hareket edilmelidir.
Tedavi
Hastalığın akut döneminde ilk seçenek IV metilprednizolondur. Değişik merkez ve ülkelerde farklı uygulamalar olmakla birlikte bizim kliniğimizde 5-10 gün, 1 gr/gün IV metilprednizolon uygulaması yapılmaktadır. Tedaviye iyi yanıt vermeyen olgularda ya da daha ilk başta IV steroid verilemeyecek hastalarda plazma değişimi veya immünoadsorbsiyon yöntemleri (5-9 kür) uygulanmaktadır. Bu ilk tedavi sonrasında oral steroid başlanıp (genellikle 1 mg/kg dozunda) aylar içinde azaltarak kesilebilir.
Bu hastalarda uzun dönem koruyucu tedavi başlanması tekrarlayan atakları ve özürlülüğün birikmesini engellemek için çok önemlidir. Çoğu hastada ilk seçenek 2-2,5 mg/kg/gün dozunda azatioprindir (çoğu hastada 150 mg/gün idealdir). İlaç 2x25 mg dozunda başlanır, 15 günde bir hemogram ve karaciğer enzim kontrolü yapılarak 50 mg artırılır. Etkinliğin ortaya çıkması ayları bulabileceğinden oral steroid ile uzun süre devam etmek akıllıca bir yaklaşım olacaktır. Bununla beraber bazı hastalar azatioprini tolere edemez ya da metabolize edemez. Bu hastalarda mikofenolat mofetil oral (2x1000 mg) tedavi iyi bir alternatiftir.
Oral tedaviler altında relaps gelişen hastalarda, hastalığın kötü gitme ihtimali fazla olduğunda üst basamak tedavilere geçilmesi gerekir. Bu anlamda ilk seçenek bir anti-CD20 antikoru olan ve 6 ayda bir uygulanan rituksimabtır. Bu ajan CD20(+) B hücrelerini yıkıma uğratarak hastalık aktivitesini önemli ölçüde baskılar. Rituksimabın NMOSH hastalarında oldukça etkin bir tedavi olduğunu gösteren pek çok retrospektif gözlemsel çalışma mevcuttur. Rituksimab tedavisi altında relaps gelişen olgularda ise IL-6 antagonisti tosilizumab ve kompleman inhibitörü ekulizumab yüksek etkinlikle kullanılabilir.
MOG ANTİKORU İLE İLİŞKİLİ HASTALIK (MOGAD)
Miyelin oligodendrosit glikoprotein memelilerde oligodendrositlerde, bu hücrelerin uzantılarında ve dolayısı ile miyelin yapısında yer alır. Fonksiyonu net olmamak ile birlikte moleküler yapı ve hücre dışı immünoglobulin bölüm göz önüne alındığında, hücre adhezyon molekülü, mikrotübül stabilizatörü olduğu ve miyelin-immün sistem arasındaki iletişimi sağladığı düşünülmektedir. Sitoplazmik kuyruğun hücre içi sinyal iletiminde rol oynayabileceği öne sürülmüştür. Yapılan bir çalışmada dış yüzeye antikor bağlanmasının; hücre içi hayatta kalma sinyallerini, hücre iskeleti yapısını ve hücresel stres yanıtlarını aktive eden kaskadı uyardığı gösterilmiştir.
Anti-MOG antikorunun tespit yöntemlerinin standardizasyonu sonrası MS, ADEM ve NMO ile benzer yakınma ve bulgular ile başvuran bazı hastalarda bu antikorun olduğu görülmüştür. Sonrasında bu hastalığın ayrı bir antite olduğu, ADEM’den farklı olarak relapslar ile gidebildiği, bir astropati olan NMO’dan farklı olarak daha çok demiyelinizasyon ile seyrettiği ve immünopatolojik olarak daha çok MS’e benzediği anlaşılmıştır.
Patofizyoloji
MOG birçok canlı türünde ensefalotijeniktir yani inflamasyon tetikleyicidir. Bununla beraber dolaşımdaki anti-MOG antikorları kan-beyin bariyerini geçmezse hastalık oluşmaz (anti-aqp-4 IgG de olduğu gibi). Anti-MOG antikorları düz peptid zincirinden ziyade, konformasyonel epitop denilen üç boyutlu paternleri tanıyarak MOG’a bağlanır. Sonrasında oluşan inflamasyon ise başlıca T hücreleri ve kompleman bağlayan IgG’ler aracılığı ile ilerler ve başlıca demiyelinizasyona yol açar. Bununla beraber, kişinin MHC allelik varyantı ve T hücre ya da antikor dominansı gibi faktörler klinik tutulum paterni ve patolojik görüntüyü değiştirebilir. Ayrıca insan serumundaki anti-MOG antikorları sıçanlardaki MOG’a bağlanmaz. Bu yüzden in vivo etkilerini değerlendirmek oldukça güçtür. Az sayıda nöropatolojik olgu serisinde, lezyonlarda başlıca antikor ve kompleman birikiminin eşlik ettiği demiyelinizasyon görüntüsü saptanmıştır (MS pattern 2). Ancak bu lezyonlarda MS’teki kronik inflamasyon bulguları yoktur. MOGAD ve aqp-4 IgG (+) NMO lezyonlarındaki histopatolojik farklılıklar ise Tablo 7’de verilmiştir.
Tablo 7. Aqp-4 IgG (+) NMO ve MOGAD lezyonları arasındaki histopatolojik farklılıklar.
|
Anti-Akuaporin 4 (+) |
Anti-MOG (+) |
|
· Astrosit hasarı ve sekonder oligodendrosit hasarı · Demiyelinizasyon
BOS’ta MBP ve GFAP yüksek |
· Astrositler korunmuş · Miyelin içeren makrofajlar · T ve B hücreleri · Demiyelinizasyon
BOS’ta MBP yüksek, GFAP düşük |
MBP:miyelin “basic” protein GFAP:glial fibriler asidik protein
Epidemiyoloji
Henüz insidans ve prevalans verileri yoktur. Daha çok beyaz ırkta görüldüğü düşünülen bu hastalık ile ilgili yayınlanan olgu serilerinde ortalama hastalık başlangıç yaşı 20-36,5 yaş olarak verilmektedir. Benzer şekilde ülkemizden yayınlanan bir olgu serisinde ortalama başlangıç yaşı 30 olarak bulunmuştur. Kadın/erkek oranı diğer MSS inflamatuar hastalıklarından daha düşüktür ve genel olarak eşit olarak kabul edilebilir.
Özellikle çocuk yaşta ADEM ya da demiyelinizan hastalık bulguları ile başvuran hastalarda MOGAD olma ihtimali yetişkinlere oranla oldukça fazladır. Bu nedenle bu hastalarda MOGAD mutlaka araştırılmalıdır.
Klinik spektrum
MOGAD yüksek oranda (%44-83) relapslar ile giden hastalıktır. Hastalığın en sık başlangıç bulgusu ve relapslardaki en sık bulgu optik nevrittir. Bilateral optik nevrit de sık görülür. Buna ilave olarak azalan oranlarda transvers miyelit, ADEM, beyinsapı bulguları ve kombinasyon bulguları saptanır (Şekil 6).
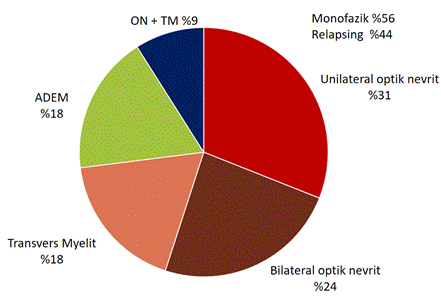
Şekil 6. MOGAD başlangıç bulgusu (bu 252 hastanın verileri Jurynczyk, M ve ark. Clinical presentation and prognosis in MOG-antibody disease: A UK study, BRAIN 2017: 140; 3128–3138’den alınmıştır).
Toplamda 197 hastalık başka büyük bir seride, başlangıç bulguları tek taraflı ON (%35), bilateral ON (%25), miyelit (%22), ON+miyelit (%7), beyinsapı sendromu (%4), ensefalopati (%2) olarak bildirilmiştir.
Relapslar genel itibarı ile NMOSH’den daha az şiddetli ve daha az sekelli iyileşme eğilimindedir. Bu durum patofizyolojik farklılıklar ile açıklanmaktadır. Bununla beraber, olgu serilerinde %50-80 kalıcı kısıtlılık kalabildiği de bildirilmektedir (özellikle transvers miyelit gelişen hastalarda).
MR ve laboratuvar bulguları
Hastaların beyin MR’ları çoğunlukla anormal bulunur. Beyinsapı lezyonları, ADEM benzeri lezyonlar, nadiren periventriküler MS benzeri lezyonlar görülebilir. Ancak lezyonların sınırları çok belirgin değildir. Kontrast tutulumunun ise “bulutsu” tarzda olduğu söylenir. Talamik ve bazal ganglia lezyonları da sıkça görülür. ON olanlarda, optik sinirlerin daha geniş ve ödemli olarak tutulduğu görülebilir. Optik sinir retroorbital bölge ve kiazma genelde etkilenmez. Spinal görüntülemede ise daha çok uzun miyelit (≥3 vertebra segmentinden uzun) ve daha az da kısa miyelit (1/3) ile uyumlu kontrast tutan lezyonlar izlenir. Bununla beraber yamalı tarzda medulla spinalis lezyonları da saptanabilir (Şekil 7). Lezyonların omurilik kalınlığının >%50 tuttuğunu ve şişmeye yol açabileceği bilinmektedir. Özellikle konus medullaris lezyonlarında MOGAD’dan mutlaka şüphelenmek gereklidir.
Beyin-omurilik sıvısında NMOSH’de olduğu gibi daha sık olarak lenfosit ağırlıklı pleositoz görülür. Hücre sayısı ortalama 250/mm3 kadar olabilir ve protein yüksekliği de saptanabilir. Literatürde düşük oranda OKB saptandığı bildirilmektedir. Kliniğimizde ise bazı hastalarda patern 4 OKB olduğu görülmüştür. MOG antikorları yukarıda bahsedildiği gibi MOG antijenin üç boyutlu yapısını tanıyarak bağlanır (konformasyonel antijen tanıma). Bu nedenle serumdaki antikorların saptanması için MOG’un tüm uzunluğu ile eksprese edildiği hücreler kullanılmalıdır. Diğer yöntemlerin duyarlılığı oldukça düşüktür. Antikorun pozitif saptandığı bazı durumlarda ise yeniden test etmek gereklidir. Bunlar; progresif hastalık, semptomların çok ani başlaması ve haftalar boyunca kötüleşmeye devam etmesi, periferik sinir sisteminde de demiyelinizasyon olması, MS benzeri lezyonları olması, relapslar arasında da lezyonların sessizce artması ve anti-aqp-4 ve anti-MOG aynı anda pozitif saptanmasıdır. Bu durumlarda yeniden test etmek yerinde olacaktır.
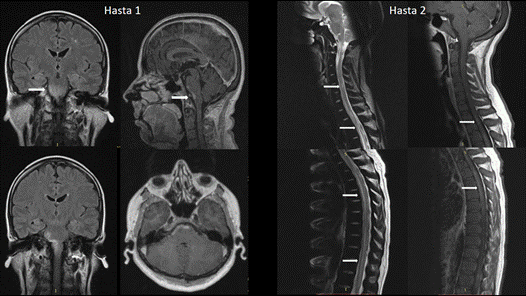
Şekil 7. Solda, FLAIR kesitlerde ponto-bulber ve BOS komşuluğunda yerleşen ve post-kontrast T1 kesitlerde kontrast tutan lezyon. Sağda, üstte servikal ve altta torakal omurilik boyunca yamalı tarzda yerleşmiş, yer yer kontrast tutulumu gösteren lezyonlar (Görüntüler İstanbul Tıp Fakültesi, Nöroloji Anabilim Dalı Multipl Skleroz Birimi materyalinden sağlanmıştır).
Tedavi
Hastalığın tedavisi ile ilgili randomize kontrollü çalışma yoktur. Akut hastalığın tedavisi NMOSH ile aynıdır. Önce pulse IV steroid, cevap yoksa plazma değişimi ya da immünadsorbsiyon uygulanabilir. Erken dönemde oral kortikosteroid başlanıp (48-64 mg/gün metilprednizolon) aylar içerisinde azaltılarak kesilmelidir. Yine erken dönemde NMOSH tedavi dozlarında azatioprin ya da mikofenolat mofetil başlanarak hasta takip edilir. Oral immünsupresifler yerine yükleme dozu sonrası aylık 0,4 mg/kg/gün dozunda IVIG uygulayan merkezler de vardır. Yanıtsızlık (relaps) durumunda rituksimab uygun bir seçenektir.
SİSTEMİK İNFLAMATUAR HASTALIKLARDA MSS TUTULUMLARI
Nöro-Behçet Hastalığı
Behçet hastalığı pek çok sistemi tutan, tekrarlayıcı ve otoinflamatuar bir hastalıktır. Hulusi Behçet bu hastalığı 1937 yılında oral ve genital ülserler ve üveit triadı ile tanımlamıştır. Sonraki yıllarda eklemler, vasküler sistem (başlıca venler), akciğerler, gastrointestinal sistem ve sinir sistemini de tutabildiği görülmüştür (Tablo 8).

Profesör Dr. Hulusi Behcet : 20 Şubat 1889’da, Istanbul’da doğdu. 1910 yılında Gülhane Askeri Tıp Fakültesi’nden mezun oldu. Ardından Dermatoloji ve Sifiloloji Bölümü’nde ihtisas yaptı. I.Dünya Savaşı sırasında çeşitli askeri hastanelerde görev yaptı. Daha sonra bir yıl süre ile Berlin ve Budapeşte’de dermatoloji alanında çalıştı. Türkiye’ye döndüğünde önce Hasköy Zührevi Hastalıklar Hastanesi’nde başhekim, daha sonra da Guraba Hastanesi’nde uzman olarak çalıştı; 1933 Üniversite Reformu ile İstanbul Üniversitesi-İstanbul Tıp Fakültesi’ne bağlanan bu kurumda profesörlüğe ve kürsü direktörlüğüne atandı.
1924 yılında oral aftları, genital ülserleri, eritema nodosum, ve görme kaybı bulunan ilk olgusunu görmüştür. Önceden tüberküloz veya sifiliz tanıları almış olan bu olguyu yıllarca izler, ancak onun tanısı bir viral infeksiyondur. Benzer bir klinik tablo gösteren diğer iki olguyu da 1930 ve 1936 yıllarında görmüştür. Bu gözlemlerini ilk kez 1937 yılında yayınlamıştır.
1936 yılında Dermatologische Wochenschrift ve Medizinische Welt gibi dergilerin yayın kuruluna davet edilmiştir. Fransız, Alman, Avusturya, Macaristan ve Yunanistan Dermatoloji Topluluklarının üyesi olmuştur. 8 Mart 1948 tarihinde kalp krizi nedeniyle 59 yaşında yaşama veda etmiştir.
Tablo 8. Behçet hastalığı’nda klinik tutulumlar.
|
Mukokutanöz
o Rekürren oral ve genital ağrılı ülserasyon o Akne benzeri veya papulo-pustuler lezyonlar o Eritema nodosum o Paterji reaksiyonu
Göz
o Ön üveit (en sık), arka üveit o Retinal vaskülit, retinal kanama o Retinal ven dal oklüzyonu o Vitrit, kistoid maküler ödem o Konjonktival ülserler o Sekonder glokom, katarakt o Akut optik nöropati
|
Vasküler
o Derin veya yüzeyel venlerde tromboflebit o Arteryel anevrizma ve tromboz (vasa vasorumda olan nötrofilik vaskülite sekonder arteryel duvar dejenerasyonu sonucu) o Pulmoner arter ve aort anevrizmalarında yüksek mortalite ve morbidite
Gastrointestinal
o Anoreksi, kusma, dispepsi o Diyare ve abdominal ağrı o Budd-Chiari sendromu
Nörolojik
|
Etyopatogenez
Behçet hastalığı uzun zaman önce tanımlanmış olmasına rağmen, halen hangi patofizyolojik kategoride olduğu bile tartışmalıdır. Literatürde bu hastalığı yanlış olarak vaskülit olarak sınıflayan yazarlara da rastlamak mümkündür. Şu anda hastalığın temel özelliğinin doğal bağışıklık hücrelerinin non-spesifik stres uyaranlarına karşı artmış yanıtı olduğu bilinmektedir. Bunun en iyi göstergelerinden biri ise Behçet hastalığında sık saptanan paterji reaksiyonudur.
Histopatolojik incelemelerde nötrofil, makrofaj ve mononükleer hücre infiltrasyonları saptanmaktadır. Kronolojik sıralamayı inceleyen 8 hastalık bir çalışmada 4. saatte iğnenin batırıldığı yerde vaskülit olmadan nötrofil infiltrasyonu olduğu, 48. saatte dermiste başlıca T lenfosit, monosit ve makrofaj birikimi olduğu saptanmıştır. Bu bulgular travma sonrası önce doğal bağışıklık hücrelerinin uyarıldığını, adaptif bağışıklık hücrelerinin ise sonradan sürece katıldığını işaret etmektedir. Behçet hastalığının immünopatogenezinde hem otoinflamatuar hastalıklar hem de otoimmün hastalıklardan özellikler bulmak mümkündür (Şekil 8). Bu kendine has özellikler nedeni ile şu anda bu iki kategori arasında yer aldığı düşünülmektedir. Yapılan genetik ilişkilendirme çalışmalarında sıklıkla bulunan gerek HLA-B51 alleli gerekse bazı SNP’ler (tek gen polimorfizm) ile ilişkilerse yine MHC-1, antijen sunumu ve doğal bağışıklık hücre reaksiyonlarında bozulmaların göstergeleri olarak kabul edilmektedir. Büyük bir GWAS (genome wide association study) Behçet hastalarında IL-10, IL23R-IL12RB2 gen lokuslarında, HLA*B51 ve HLA*B bölgelerinde telomerik bir alanda ortak varyantlar olduğunu göstermiştir. Behçet hastalığının, karşıt görüşler olsa da, MHC1-opati (MHC1 bozuklukları) sınıflandırmasına dahil edilmesi önerilmiştir. Bütün bu hipotezlerin başka bir destekçisi doğal bağışıklık yanıtının önemli bir sitokini olan TNF’nin (tümör nekroz faktörü) süreçteki rolüdür. Bilindiği üzere TNF’nin anti-TNF monoklonal antikorlar ile baskılanması Behçet hastalığı aktivitesini önemli ölçüde azaltmaktadır.
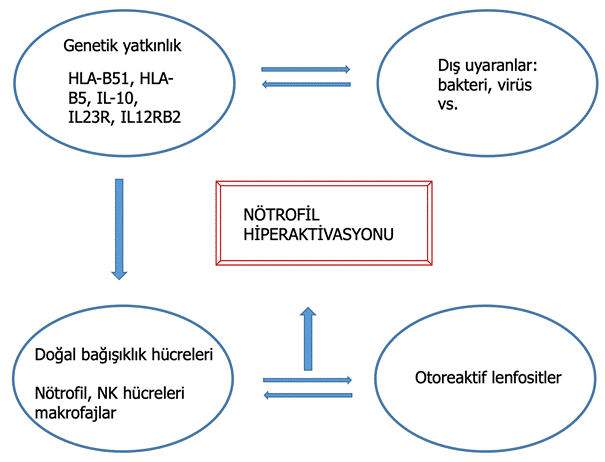
Şekil 8. Behçet hastalığı immünopatogenez özeti.
Behçet hastalığı olanların belli bir bölümünde ortaya çıkan nöro-Behçet hastalığı ise histopatolojik çalışmaların azlığı nedeni ile daha az anlaşılabilmiştir. Bununla beraber görüntüleme ve BOS incelemelerindeki inflamasyon bulguları nedeni ile vücudun diğer bölgelerindeki hastalık süreçleri ile benzer olduğu kabul edilebilir. Akut nöro-Behçet atağı sırasında alınan bir biyopsi örneğinde, küçük damarlar çevresinde çoğunlukla T lenfosit ve monositlerden, çok az sayıda da B lenfositlerden oluşan mononükleer hücre infiltrasyonu olduğu ve çoğu nöronun apoptoz ile ölmekte olduğu görülmüştür. İlginç olarak, damar duvarında fibrinoid nekroz veya inflamatuar hücre infiltrasyonu yoktur. Bu nedenle bu hastalığın özünde vaskülit değil perivaskülit olarak sınıflandırılması daha doğru görünmektedir. Parenkimal tutulumun neden beyinsapı ve diensefalonda yoğunlaştığı ise çok net değildir. Bu konudaki tek yorum bir MR çalışmasında yapılmıştır. Yazarlar bu seçici dağılımın beyinin değişik bölgelerindeki venöz yapıların farklı anatomik özelliklerinden kaynaklanabileceğini öne sürmüştür (veno-venöz anastomozlar daha seyrek ve kan akımı sentrifugal) (Koçer ve ark) Akut nöro-Behçet lezyonlarının, genellikle büyük T2 lezyonları olduğu, ancak çoğunlukla bunların küçük bir bölgesinde kontrast madde ekstravazasyonu olduğu bilinmektedir. Ayrıca steroid sonrası lezyonlar hızla kaybolmakta ve klinik tablo iyileşmektedir. Bu bulgular ise akut lezyon sahasının büyük bölümünün ödem olabileceğini akla getirmektedir. Venöz tromboz gelişen damarlarda endotelyal inflamasyon olduğu ve trombozun buna sekonder geliştiği bilinmektedir. Bu nedenle, venöz tromboz gelişen hastalarda antikoagülasyon yanında kortikosteroid de kullanılmaktadır. Kliniğimizde dural sinüs trombozunda antikoagülan kullanılmamaktadır.
Epidemiyoloji, Tanı ve Klinik Spektrum
Behçet hastalığı en sık olarak, İpek Yolu etrafındaki ülkelerden, Japonya’dan ve Kuzey Afrika’dan köken alan insanlarda görülür. Türkiye’deki prevalansı 20-141 /100,000, İngiltere’de 0,64 / 100,000, İspanya’da 6,4 /100,000 olarak bildirilmiştir. En sık 30-40’lı yaşlarda ortaya çıkmakla beraber daha erken yaşlarda da görülebilir. Sıklık açısından cinsiyet farklılığının olmadığı bilinmekle beraber erkeklerde daha kötü seyrettiği bildirilmektedir.
Behçet hastalığı için ilk tanı kriterleri 1990 yılında Uluslararası Behçet Hastalığı Çalışma Grubu tarafından oluşturulmuştur. Uzun yıllar kullanılan bu kriterlerden sonra 2014 yılında nörolojik ve vasküler tutulumların dahil edildiği yeni kriterler önerilmiştir. Bu iki kriter Tablo 9’ da verilmiştir.
Tablo 9. Behçet hastalığı tanı kriterleri. 1990 ve 2014 tarihli kriterlerin karşılaştırılması.
|
1990 A. Tekrarlayan oral aftöz ülserler (≥ 3/yıl)
Ve
B. Aşağıdakilerden en az ikisi · Genital ülserler · Göz tutulumu (üveit, retinal vaskülit) · Deri tutulumu (eritema nodozum, psödofollikülit, deri ülserleri) · Pozitif paterji testi |
2014 · Oral aft · Genital aft · Oküler lezyon · Deri lezyonu · Nörolojik tutulum · Vasküler bulgular · Pozitif paterji testi
ilk 3 maddeye 2’şer, son 4 maddeye 1 puan verilir. Behçet Hastalığı tanısı için ≥4 puan gereklidir. Sensitivite: %94 Spesifisite: %92 |
Behçet hastalığı yaklaşık %5-10 hastada nörolojik tutulum yapar. Bu hastaların yaklaşık %80’inde parenkimal ve yaklaşık %10-20’sinde vasküler (başlıca dural sinüs trombozu (DST)) etkilenme ortaya çıkar. Nöro-Behçet hastalığı (NBH) erkeklerde daha sıktır (erkek / kadın oranı: 2,8-4). Parenkimal NBH ile başvuran hastalarda genellikle beyinsapı ve diensefalik etkilenme bulguları saptanır (göz hareket kusuru, dizartri, parezi, serebellar bulgular ve kognitif etkilenme gibi). Daha nadir olarak miyelit, hareket bozuklukları (hemikore, hemiballismus, hemidistoni) ve epilepsi görülebilir. İzole medulla spinalis tutulumunun %2,5-30 hastada olabileceği bildirilmektedir. DST gelişen hastalar sıklıkla başağrısı ve papilödem ile başvururlar. Bunlar dışında NBH, nadiren pür nöropsikiyatrik semptomlar, optik nöropati, aseptik menenjit, periferik nöropati ve miyozit gibi tutulumlar ile karşımıza gelebilir.
Parenkimal nöro-Behçet (p-NBH) hastalığı relapslar ve iyileşmeler ile seyredebileceği gibi hastanın giderek kötüleştiği progresif bir seyir de izleyebilir. İstanbul Tıp Fakültesi NBH kohortu incelendiğinde relapsa (ikinci atağa) kadar geçen ortalama sürenin p-NBH grubunda 5,4±0,7 yıl ve DST grubunda anlamlı farklı şekilde 13,7±1,3 yıl olduğu hesaplanmıştır. Ayrıca, yıllık atak riskinde %5 altına düşüşün p-NBH ve DST grubunda sırasıyla beşinci ve ikinci yıldan sonra, %2’nin altına düşüşün ise sırasıyla yedinci ve sekizinci yıllarda gerçekleştiği izlenmiştir.
Tipik bir p-NBH atağı subakut-günler içinde kötüleşen bir nörolojik tablo ile karakterizedir. DST de akut-subakut başağrısı ile ortaya çıkar ve tipik KİBAS bulguları gösterebilir. Bununla beraber bazı hastalarda tam rekanalizasyon olmayabilir ve KİBAS kronik hale gelip cerrahi işlem gerektirecek boyutlara ulaşabilir.
Akut relapsın iyileşmesi ile parenkimal tutuluma ait bulgular haftalar içerisinde azalır. Bazı hastalarda tam klinik düzelme görülürken, bazı hastalarda çeşitli derecelerde sekel kalabilir.
Görüntüleme ve Laboratuvar Bulguları
Parenkimal nöro-Behçet hastalarında tipik olarak, beyinsapından diensefalona uzanan, merkezi küçük bir yerinde kontrast tutulumu olan, ödem etkisi oluşturan T2 hiperintens lezyonlar saptanır (Şekil 9 ve 10). Lezyonlar tedavi ile küçülerek daha sınırlı T2 lezyonlar halini alabilir. Geç dönemde, özellikle tekrarlayan ataklardan sonra, beyinsapı ve diensefalik yapılarda atrofi geliştiği görülebilir.
Dural sinüs trombozu bazı hastalarda birden çok lokalizasyonda çıkabilir. Görüntülemede ise sinüslerin içinde trombüs görünümü, kontrast madde dolmaması ve MR venografide akım yetersizlikleri görülebilir (Şekil 11).
Akut dönemde p-NBH hastalarının kan tahlillerinde genellikle anormallik görülmez. BOS’ta ise nötrofil veya lenfosit hakimiyetli pleositoz (birkaç yüz hücreye kadar olabilir), protein yüksekliği görülebilir. IgG yüksek sıklıkla artmış bulunabilir. OKB varlığı ise yaklaşık %16’dır. Hastaların önemli bir kısmında HLA-B51 alleli saptanır.
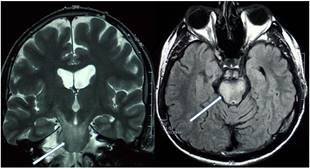
Şekil 9. Akut atak döneminde ponstan mezodiensefalik bölgeye uzanan T2 hiperintens lezyon (Görüntüler İstanbul Tıp Fakültesi, Nöroloji Anabilim Dalı Multipl Skleroz Birimi materyalinden sağlanmıştır).
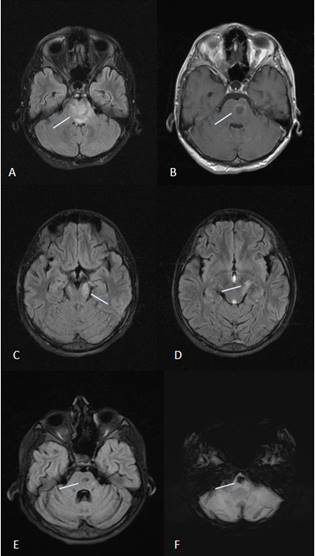
Şekil 10. 22 yaşında bir parenkimal nöro-Behçet hastasının MR görüntüleri (A, B, C ve D akut dönemde; E ve F, 6 ay sonra). A, FLAIR kesitte ponsun neredeyse bütün kalınlığını tutan heterojen hiperintens lezyonlar. B, aynı seviyede daha küçük bir alanda halkasal kontrast tutulumu. C ve D, mezodiensefalik bölgeye kadar çıkan lezyonlar. E, ilk prezentasyonda kontrast tutulumu olan bölge hariç diğer lezyonlar 6 ay sonra kaybolmuş. F, ilk MR’da kontrast tutulumu olan bölgede SWI sekansta hemoraji/demir birikimine bağlı hipointens görünüm (Görüntüler İstanbul Tıp Fakültesi, Nöroloji Anabilim Dalı Multipl Skleroz Birimi materyalinden sağlanmıştır).
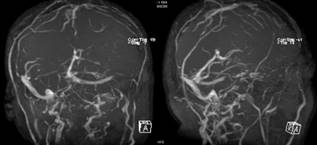
Şekil 11. Non-parenkimal nöro-Behçet Hastalığı (dural sinüs trombozu). MR venografide, superior sagittal sinüs arka yarısında, sol sigmoid sinüs, ve sağ transvers sinüste tromboz ile uyumlu akım yokluğu izleniyor(Görüntüler İstanbul Tıp Fakültesi, Nöroloji Anabilim Dalı Multipl Skleroz Birimi materyalinden sağlanmıştır).
Tablo 10. Nöro-Behçet hastalığında ayırıcı tanı.
|
Nöro-Behçet Hastalığında Ayırıcı Tanı
Multipl skleroz Genç inme Primer MSS vasküliti Primer MSS lenfoması Beyinsapı gliomu Sistemik vaskülitler Nöro-sarkoidoz, MSS tüberkülozu Vogt-Koyanagi-Harada sendromu Reiter sendromu Eales hastalığı Cogan sendromu Susac sendromu Sweet sendromu Otoinflamatuar MSS hastalıkları |
Tedavi
Nöro-Behçet hastalığına hızlıca tanı konup tedaviye başlanmalıdır. Bunun nedeni hastalığın ciddi somatik nörolojik ve kognitif sekellere sebep olabilmesidir. Erken dönemde agresif antiinflamatuar tedavi, ciddi inflamasyonun baskılanması ve temizlenmesi, ardında da rejenerasyonun başlamasına olanak tanıyabilir. Akut ataklarda en etkili ilk tedavi yüksek doz IV metilprednizolondur. Hastalığın şiddetine göre 5-10 gün arasında verilebilir. Sonrasında ise en az 6 ay oral steroid tedavisi kullanılması (32 mg/gün den başlayarak dereceli azaltılır) relapsları büyük ölçüde kontrol altına alacaktır. Akut relaps tedavisinde plazmaferez ve IVIG’in yeri yoktur, ancak gerekirse IV siklofosfamid uygulanabilir. Hastalarda uzun dönemde steroid harici immünsupresif/immünmodülatör ihtiyacı ortaya çıkar. Bu anlamda ilk seçilebilecek tedavi azatioprindir (2-2,5 mg/kg/gün). Azatioprine intolerans durumunda ise mikofenolat mofetil (2000 mg /gün) denenebilir. Bununla beraber bazı hastalarda etkisizlik, kontrendikasyon ya da diğer nedenlerle, başta anti-TNF ajanlar olmak üzere, başka tedavilere geçmek gerekebilir (Tablo 11)
|
|
İlaç |
Etki Mekanizması |
Dozaj |
|
1. Basamak |
Azatioprin |
Purin sentez inhibitörü |
2.5-3 mg/kg/gün po. |
|
Mikofenolat mofetil |
De novo guanosin nükleotid sentez inhibitörü |
2 gr/gün po. |
|
|
Metotreksat |
Dihidrofolat redüktaz inhibitörü |
10-15 mg/hafta po. |
|
|
2. Basamak |
Siklofosfamid |
Alkilleyici ajan. DNA dizilerini çapraz bağlayarak ve DNA sentezini azaltarak hücre bölünmesini engeller. |
6-12 ay boyunca her ay 1000 mg iv. |
|
Infliximab |
Anti-TNF-α |
0, 2 ve 6. haftalarda 5 mg/kg IV yükleme, sonrasında her iki ayda bir 5 mg/kg |
|
|
Etanercept |
Anti-TNF |
Haftada 2 kez 25 mg sc. |
|
|
Adalimumab |
Anti-TNF-α |
160 mg sc., ve 2 hafta sonra 80 mg , sonrasında iki haftada bir 40 mg |
|
|
3. Basamak |
Tocilizumab |
Anti-IL-6 receptör antikoru |
4 haftada bir 8 mg/kg IV |
|
Canakinumab |
Anti-IL-1β |
6 haftada bir 150 mg sc. |
|
|
Anakinra |
Anti-IL-1 receptör antikoru |
Her gün 100 mg sc. |
|
|
Diğer |
Interferon-α |
Immun regülasyon |
1 hafta boyunca 6‒9 MIU/gün, sonra haftada 3 kez 3 MIU |
|
Otolog periferik kök hücre transplantasyonu |
|||
|
Lumbo-peritoneal şant |
|||
Tablo 11. Nöro-Behçet Hastalığı tedavisinde kullanılan ajanlar.
Sistemik Lupus Eritematozus
Sistemik lupus eritematozus (SLE) oto-antikor üretimi ve pek çok organda immünkompleks birikimleri ile karakterize kronik otoimmün bir hastalıktır. Bu sistemik hastalık MSS ve periferik sinir sistemini tutabilir. MSS tutulumu (Şekil 12); başağrısı, duygudurum değişiklikleri, psikoz (%1,5), inme (%5-18), epilepsi (%4,6), PRES (posterior reversible encephalopathy syndrome, %1,5), küçük damar hastalığı, miyelit (%1,2), optik nevrit, kognitif bozulma ve diğer inflamatuar MSS hastalıkları ile birliktelik gibi tablolar ile ortaya çıkabilir. Yukarıda bahsedilen klinik tablolar nöropsikiyatrik lupus başlığı altında toplanmaktadır ve yaklaşık %15-20 sıklıkla görüldüğü bildirilmektedir. SLE hastalarında ortaya çıkan inme ise dikkatle değerlendirilmelidir. Çünkü bu hastalarda sekonder antifosfolipid antikorlar sendromu olabilir ve inmeye yol açabilir. Periferik sinir sistemi tutulumu kitabın başka bölümlerinde anlatılacaktır.
SLE’de MSS tutulumunun patofizyolojisi net bilinmemektedir. Ancak, mikrovaskülopati, lokal inflamatuar mediatörler ve antikorların doğrudan ve dolaylı etkileri kan beyin bariyerinin bozulduğu düşünülmektedir. SLE’de pek çok ana sitokin yolağında disregülasyon olduğu bilinmektedir. Bunlarda tip 1 interferon, IL-6’nın, tip 2 interferon ve TNF yolaklarının aşırı aktivasyonunun nörolojik etkilenmeye yok açabileceği öne sürülmüştür. İlginç olarak nöropsikiyatrik lupus hastalarında yapılan patoloji çalışmalarında sinir sistemi immün hücre infiltrasyonu çok geri plandadır. Bu bulgunun, sistemik sitokin disregülasyonunun MSS hücreleri üzerindeki etkileri ile hastalığa yol açtığı görüşünü desteklediği öne sürülmektedir.
SLE hastalarında ortaya çıkan optik nevrit ve miyelit olgularının bir bölümünde anti-akuaporin-4 IgG veya anti-MOG antikorları saptanmaktadır. Böyle bir durumda optik nevrit ya da miyelitin direkt olarak SLE’ye bağlı olmadığı düşünülmelidir. Bu hastalarda SLE’ye ilave olarak NMO spektrum hastalığı/MOG antikor hastalığı (MOGAD) bulunduğu kabul edilmektedir. Yine de antikor saptanamayan hastalarda, bu tabloların direkt SLE’ye bağlı olabileceği de akla gelmelidir. Lupus hastalarında görme kaybının optik nöropatiye bağlı olmayabileceği akılda tutulmalıdır. Bu hastalarda lupus retinopatisi, retinal vasküler oklüzyon ve retinal vaskülit de görme kaybına neden olabilir.
SLE’de görülen PRES’in endotelyal disfonksiyon ve/veya kullanılan immünsupresif ilaçlara bağlı olabileceği düşünülmektedir. SLE hastalarının önemli bir bölümünde, yapılan beyin MRG de ak maddede yaygın lezyonlar görülebilir. Bu görüntü MS lezyonları ile karışabileceğinden dikkatle değerlendirilmelidir (Şekil 13).
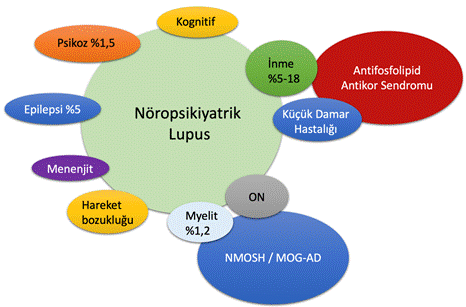
Şekil 12. SLE ‘de merkezi sinir sistemi tutulum paternleri. İnme ve küçük damar hastalığının antifosfolipid antikor sendromu ile miyelit ve optik nöropatinin NMO spektrum hastalığı / MOG antikor hastalıkları ile üst üste bindiği görülmektedir.
Nöropsikiyatrik lupus tedavisi ortaya çıkan klinik tabloya göre değişiklik gösterir. İnme, epilepsi ve hareket bozuklukları için antikoagulan tedavi, antiepileptikler ve psikotroplar kullanılırken, inflamatuar özellikli tablolarda immünsupresifler tercih edilir. Bu anlamda siklofosfamid, oral immünsupresan tedaviler (azatioprin, mikofenolat vd.) ve rituksimab ile başarılı tedavi sonuçları bildirilmektedir.
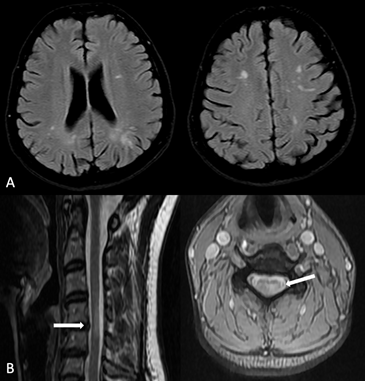
Şekil 13. İki farklı sistemik lupus eritematozus hastasının MR görüntüleri. A, Subkortikal çok sayıda küçük damar hastalığı ile uyumlu lezyonlar. B, ikinci hastada C4 seviyesinde bir vertebra korpus boyunu geçmeyen periferik yerleşimli lezyon (Görüntüler İstanbul Tıp Fakültesi, Nöroloji Anabilim Dalı Multipl Skleroz Birimi materyalinden sağlanmıştır).
Sjögren Sendromu
Primer Sjögren sendromu (pSS), ekzokrin bezlerin (tükrük, lakrimal bez vd.) mononükleer lenfositik infiltrasyonu ile seyreden otoimmün bir hastalıktır. Hastalarda antinükleer antikorlar (özellikle anti-SSa ve anti-SSb) saptanır. Bu bulgular başka bir otoimmün hastalığı bulunan kişilerde saptandığında sekonder Sjögren sendromundan söz edilir. Primer Sjögren sendromunda ekstraglandüler tutulumlar da görülebilir (eklem, kas, akciğer, böbrek ve deri tutulumları gibi). Toplum prevalansı %0,1- 0,6 ve kadın / erkek oranı 9:1 olarak bildirilmektedir.
Primer Sjögren sendromunda yaklaşık %20 civarında nörolojik tutulum bildirilmektedir ve hastalarda nörolojik tutulum ilk bulgu olabilir. Hastalarda %10 periferik sinir sistemi, %2-5 MSS tutulumu olduğunu bildiren yayınlar da vardır. Periferik ve otonom sinir sistemi tutulumundan kitabın başka bölümlerinde bahsedilecektir (Bkz. Periferik Sinirlerin Yaygın ve Çok Odaklı Hastalıkları).
MSS bütün aksı boyunca fokal ya da multifokal olarak tutulabilir ve çok farklı yakınma ve bulgulara neden olabilir (Tablo 12). Akut ya da subakut klinik bulgular vaskülit ya da demiyelinizasyon sonucu oluşur.
Tablo 12. Sjögren sendromunda merkezi sinir sistemi tutulum bulguları.
|
Sık tutulumlar
Optik nevrit (bilateral/ unilateral) Transvers miyelit Beyinsapı sendromları İnternükleer oftalmopleji
|
Nadir tutulumlar
Psikiyatrik/kognitif etkilenme Ensefalopati Parkinsonizm, hareket bozuklukları Aseptik menenjit Serebellar bulgular Fokal/jeneralize nöbetler Motor nöron benzeri hastalık |
Primer Sjögren sendromu olan hastalarda yüksek oranda uzun longitudinal miyelit ve aqp-4 IgG pozitif NMO hastalığı da saptanır. Örnek olarak uzun miyelit ile başvuran pSS hastalarında %50 NMO-IgG saptandığı bildirilmiştir. Böyle hastalarda, nörolojik tablonun pSS’ye bağlı değil, komorbid olarak bulunan NMO’ya bağlı olduğu düşünülmelidir. Ancak aqp-4 IgG negatif olan NMOSH li olgularda, klinik tablodan aksi kanıtlanıncaya dek pSS sorumlu tutulmalıdır (Şekil 14). Neyse ki, iki hastalığın tedavisi de büyük oranda benzer olduğundan tedavi kararı çok etkilenmez.
![]() Şekil
14.
Sjögren sendromu ve sekonder MSS tutulumu olan bir hastanın spinal ve beyin
MR görüntüleri. Solda T9 vertebra düzeyinden başlayarak lomber segmentlere
kadar uzanan, kısa bir alanda kontrast tutulumu gösteren uzun longitüdinal
miyelit görünümü. Sağda, beyinde iki farklı bölgede kontrast tutulumu da
gösteren FLAIR lezyonları (Görüntüler
İstanbul Tıp Fakültesi, Nöroloji Anabilim Dalı Multipl Skleroz Birimi materyalinden
sağlanmıştır).
Şekil
14.
Sjögren sendromu ve sekonder MSS tutulumu olan bir hastanın spinal ve beyin
MR görüntüleri. Solda T9 vertebra düzeyinden başlayarak lomber segmentlere
kadar uzanan, kısa bir alanda kontrast tutulumu gösteren uzun longitüdinal
miyelit görünümü. Sağda, beyinde iki farklı bölgede kontrast tutulumu da
gösteren FLAIR lezyonları (Görüntüler
İstanbul Tıp Fakültesi, Nöroloji Anabilim Dalı Multipl Skleroz Birimi materyalinden
sağlanmıştır).
pSS tanısı alan bir hastada demiyelinizan MR lezyonları saptandığında dışlanması gereken diğer bir hastalık MS’tir. Hastada MS’te görmeye alıştığımız küçük, iyi sınırlı, periventriküler yerleşme eğiliminde kontrast tutulumu gösterebilen lezyonlar saptanmış ise (özellikle OKB da saptanmışsa) yüksek ihtimalle MS beraber bulunmaktadır. Dikkat edilmesi gereken nokta, pSS hastalarının önemli bir kısmında ak maddede nonspesifik denebilecek, iskemik mikroanjiopatiye benzeyen, ama oldukça yüklü T2 lezyonları saptanabilmesidir (%52-80) (Şekil 15). Herhangi bir nörolojik defisit ile ilişkili olmayan bu lezyonlar MS lezyonlarından dikkatle ayırt edilmelidir.
Hastaların bir kısmında nispeten kötüleşmeyen kognitif bir etkilenme olduğu bilinmektedir. Genellikle bellek, dikkat ve yürütücü işlevlerde etkilenme olmaktadır.
Tedavide randomize kontrollü çalışma yoktur, ancak akut dönemde pulse steroid ve plazmaferez, uzun dönemde oral kortikosteroidler, rituksimab, siklofosfamid, azatioprin, mikofenolat kullanılabilir. Bunlardan ülkemizde öncelikle azatioprin ve siklofosfamid tercih edilmekte, eğer yanıt alınamaz ise rituksimab tedavisine geçilmektedir. Bazı hastalar, özellikle miyelit ön planda olanlar, agresif tedaviye yanıt vermeyebilir ve progresif olarak kötüleşebilirler. Uzun dönemde mobilite anlamında dışa bağımlı hale gelen ve bilateral optik nevrit nedeni ile ciddi vizyon kaybı yaşayan hasta sayısı az değildir.
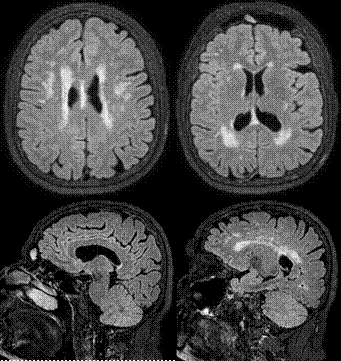
Şekil 15. Sjögren sendromu tanısı olan ancak nörolojik defisiti olmayan bir hastanın beyin MR FLAIR sekans görüntüleri. Sınırları net olmayan, nispeten simetrik, subkortikal alanlarda ve ventrikül etrafında yoğunlaşmış lezyonlar görülmektedir. Korpus kallozum nispeten korunmuştur (Görüntüler İstanbul Tıp Fakültesi, Nöroloji Anabilim Dalı Multipl Skleroz Birimi materyalinden sağlanmıştır).
Nörosarkoidoz
Sarkoidoz etkilediği bölgelerde granülomatöz iltihaba yol açan oto-inflamatuar bir hastalıktır. Vücuttaki bütün dokular tutulabilir ancak en sık akciğer, göz ve deri etkilenir. Hastaların %95’inde başlangıçta akciğer tutulumu saptanır. Tüm dünyada insidans, ırklara göre farklılık göstermekle beraber, 100,000’de 0,2-80 olarak bildirilmektedir. Hastaların tanı yaşı yoğunlukla 25-45 yaş (%70) aralığındadır. Sarkoidoz tanısı alan hastaların ise %5-%10‘unda nörolojik tutulum ortaya çıkmakta, nörolojik tutulumlu hastaların yaklaşık %15’inde sistemik sarkoidoz saptanamamaktadır.
Patofizyoloji
Sarkoidozdaki granülomlar muhtemelen ortamdan temizlenemeyen bir antijenin izolasyon çabası ile oluşmaktadır. Süre içinde makrofajlar epiteloid hücrelere dönüşmekte, T lenfositleri ve fibroblastların da katılımı ile non-kazeifiye granülomlar oluşmaktadır. MSS’de en sık bazal meninkslerde oluşan ve buradan kranial sinirleri infiltre eden granülomlar, perivasküler yapılarda oluşma eğilimindedir. Zaman zaman granülomların beyin parenkimi içine de geçtiği ve büyük inflamasyon alanları oluşturduğu görülür.
Epidemiyoloji, klinik ve radyolojik bulgular ve tanı
Sistemik sarkoidoz kendisini ateş, yorgunluk, gece terlemeleri, kronik öksürük, nefes darlığı, göğüs ağrısı, artrit, eritema nodozum, hiler lenfadenopati, parotis iltihabı, granülomatöz üveit ile gösterebilir. Bunun haricinde deri, kardiyak, hepatik ve renal tutulum olabilir.
Tablo 13. Nörosarkoidoz tutulum bölgeleri.
|
Nörosarkoidoz tutulum bölgeleri
Kranial sinirler Hipofiz ve hipotalamus Meninksler (leptomeninksler / dura) Beyin parenkimi Omurilik ve kauda ekuina Periferik nöropati ve miyopati |
Nörosarkoidozda en sık nörolojik tutulum (Tablo 13) izole kranial nöropatidir (%50-60). Kranial nöropatilerin; (i)-Epinöral inflamasyon, (ii)-Perinöral inflamasyon, ve (iii)-Leptomeninkslerdeki granülomatöz kitlenin basısına bağlı olarak ortaya çıktığı düşünülmektedir. Kranial nöropatiler içinde en sık fasial nöropati görülür (%70). Daha çok tek taraflı olsa da, tekrarlayan ve bilateral tutulumlar da görülebilir. Diğer kranial nöropatilere göre daha iyi seyirli oldukları ve tedaviye daha iyi cevap verdikleri bilinmektedir. İkinci en sık olarak subakut progresif optik nöropati görülür. Bu durum saptandığında kalıcı görme kaybını engellemek için vakit kaybetmeden tedavi uygulanması büyük önem taşır. Optik kiazma tutulumu da görme kayıplarına yol açabileceğinden, lezyon lokalizasyonunda dikkatli olunmalıdır. Bu iki kranial sinir haricinde, diğer kranial sinirler izole ve çeşitli kombinasyonlar halinde tutulabilirler. Örneğin vestibülokoklear sinirin tutulumuna bağlı kalıcı sensorinöral duyma kaybı ve vestibüler disfonksiyon %3-17 oranında bildirilmektedir. Kranial nöropatili hastalarda MR’da uyan bölgelerde leptomeningeal kontrast tutulumu, ya da nodüler oluşumlar görülebilir.
Etkilenen bir diğer alan, bazal meninkslerle yakın ilişkili olmasından dolayı hipofiz ve hipotalamustur. Progresif diyabetes insipitus, uygunsuz ADH salınımı, hiperprolaktinemi, panhipopituitarizm ya da izole gonadotropin ya da tirotropin yetmezlikleri görülebilir. Beyin MR’ında tutulan bölgelerde meningeal ya da parenkimal kontrast tutan lezyonlar görülebilir (Şekil 16)
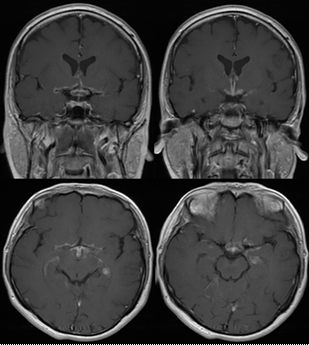
Şekil 16. Nörosarkoidoz tanısı alan bir hastada optik kiazma, hipofiz ve 3. ventrikül etrafında kontrast tutulumu ile karakterize inflamasyon görüntüsü (Görüntüler İstanbul Tıp Fakültesi, Nöroloji Anabilim Dalı Multipl Skleroz Birimi materyalinden sağlanmıştır).
Beyin parenkimi içinde oluşan granülomatöz inflamasyon (nörosarkoidozların %15’inde ortaya çıkar), lezyon yerine göre çeşitli fokal bulgular, ensefalopati ve epileptik nöbetler ile kendini gösterebilir. Beyin MR’ında değişen oranda kontrast tutulumu olan nodüler T1 izointens lezyonlar saptanır. Bazı olgularda granülomatöz vaskülit oluşabilir ve buna sekonder vasküler sendromlar, iskemik inme, intrakranial kanama ve dural sinüs trombozları ortaya çıkabilir. Ayrıca damara dışardan baskı yapan bir granülom da iskemik hasara neden olabilir.
Meningeal tutulum oldukça sık görülür ve leptomenenjit (pia-araknoid) ya da pakimenenjit (dural) formunda olabilir. Menenjit başta bazal beyin olmak üzere bütün bölgelerde görülebilir. Yaygın olabileceği gibi nodüler/kitle lezyonları ile de karşımıza çıkabilir. Bu hastalarda klinik tablo genellikle subakut başlar, başağrısı-ense sertliği, ateş görülebilir ve kronik hale gelebilir. Seyir sırasında komunikan ya da obstrüktif hidrosefali ortaya çıkabilir.
Medulla spinalis ve kauda ekuina tutulumu genellikle serebral tutulum ile beraber görülür ancak izole de olabilir. Klinik olarak subakut spinal sendrom / kauda ekuina sendromu ile prezente olur ve MR’da servikal / torakal omurilikte 3 vertebra boyunu geçebilen uzun miyelit ve eşlik eden meningeal kontrastlanma saptanabilir. Bu hastalık NMOSH ayrıcı tanısında düşünülmelidir. Bunların haricinde nörosarkoidozda daha nadir olarak periferik nöropati ve miyopatik tutulumlar da olabilir.
Hastaların büyük kısmında BOS anormaldir. Protein yüksektir (ortalama 80 mg/dl) ve lenfosit ağırlıklı bir pleositoz bulunur. OKB genellikle negatiftir. Hastalık tanısını ve aktivitesini öngörecek biyobelirteç araştırmaları olumlu sonuçlanmamıştır. Araştırılan biyobelirteçler yeterli özgüllük ve duyarlılığa ulaşamamıştır. Nörosarkoidoz tanısı için 2018 yılında bir uzlaşma raporu yayınlanmıştır (Tablo 14).
Tablo 14. Nörosarkoidoz tanı kriterleri 2018 (Nörosarkoidoz konsorsiyumu konsensus grubu, Stern ve ark, 2018 ‘den uyarlanmıştır)
|
Olası nörosarkoidoz
1. Klinik ve laboratuvar testler nörosarkoidozu destekler (Sinir sisteminin granülomatöz inflamasyonu ile uyumlu klinik bulgular, MR, BOS, EMG. Aynı zamanda diğer nedenler dışlanmıştır) 2. Patolojik doğrulama yok
Muhtemel nörosarkoidoz
1. Klinik ve laboratuvar testler nörosarkoidozu destekler (Sinir sisteminin granülomatöz inflamasyonu ile uyumlu klinik bulgular, MR, BOS, EMG. Aynı zamanda diğer nedenler dışlanmıştır) 2. Sistemik granülomatöz hastalığın patolojik doğrulaması vardır
Kesin nörosarkoidoz
1. Klinik ve laboratuvar testler nörosarkoidozu destekler (Sinir sisteminin granülomatöz inflamasyonu ile uyumlu klinik bulgular, MR, BOS, EMG. Aynı zamanda diğer nedenler dışlanmıştır)
2. Sinir sisteminden alınan biyopsi sarkoidoz ile uyumludur Tip a. Sinir sistemi dışı sarkoidoz da vardır Tip b. Sinir sistemi dışı sarkoidoz yoktur (izole nörosarkoidoz) |
Nörosarkoidoz erkenden etkili tedavi edildiğinde iyi bir prognoza sahiptir. Akut dönemde tedavide pulse ve oral steroidler kullanılır ancak uzun dönemde immünsupresif ajanlara gereksinim olmaktadır. Başlangıç olarak mikofenolat (2000 mg/gün), azatioprin (çoğunlukla 150 mg/gün) veya metotreksat (haftalık 15-20 mg) uygulanabilir. Ancak şiddetli vakalarda önce IV siklofosfamid ile indüksiyon yapıldıktan sonra oral immünsupresif ajanlarla devam etmek iyi bir seçenek olabilir. İlk basamak oral immünsupresiflere yanıt alınamadığı durumlarda anti-TNF ajanlar (örneğin, infliksimab) kullanılmaktadır. Bunlardan başka IL-6 antagonistleri (tosilizumab) ve anti-CD20 ajanlar (rituksimab) ile iyi yanıt alındığı bildirilmektedir.
Igg4 İlişkili Hastalık
Bilindiği gibi insan immünoglobulin G molekülünün IgG 1,2,3, ve 4 olmak üzere 4 alt tipi vardır. Nispeten yeni tanınmaya başlanan bir hastalık olan IgG4 ilişkili hastalık pek çok organ sistemini, merkezi ve periferik sinir sistemlerini tutabilir. IgG4 antikorlarının patogenezde doğrudan rol almadığı ama bir epifenomen olarak oluştuğu düşünülmektedir. Hastalığın, nispeten az tanındığı için, gerçek prevalansı bilinmemektedir.
Hastalığın bilinmeyen bir antijen ile tetiklendiği, lenf bezleri ve hastalığın oluştuğu organdaki CD4 T ve B lenfositlerinin etkileşmesi ile sürecin oluştuğu, B lenfositleri ve plazma hücrelerinin sürekli antijen sunumu yapması ile de sürekli hale geldiği düşünülmektedir. Hastalığın B hücre deplesyon tedavisine yanıt vermesi de bu görüşü desteklemektedir.
IgG4 ilişkili hastalık çoğunlukla relaps ve remisyonlar ile giden bir seyir izler. Sistemik olarak, lakrimal inflamasyon, yaygın lenfadenopati, otoimmün pankreatit ve buna sekonder bulgular, sklerozan kolanjit (intra- ve ekstrahepatik safra kanalları), karaciğerde kitle lezyonları, retroperitoneal fibroz, aortit ve buna bağlı anevrizma ve diseksiyon, pulmoner nodüller ve interstisyel akciğer hastalığı, tübülointerstisyel nefrit görülür.
Hastalık sinir sisteminde ya doğrudan invazyon ya da kitle etkisi ile kendini gösterir. Başlıca hipofiz ve dural etkilenme görülür. Hastalarda daha az oranda beyin parenkimi ve periferik sinirler de etkilenebilir. MSS dışında olsa da nörolojik bulgular ile ortaya çıkabilen orbital tutulum belirgindir. Orbitayı etkileyen inflamatuar ve malign durumların %20’sinden IgG4 ilişkili hastalık sorumlu tutulabilir. Orbital çukurda bulunan bütün yapılar (kaslar, kranial sinir 2,3,4 ve 5 ve yağlı doku) etkilenebilir ve buna bağlı bulgular saptanır. Bu semptomlar ile başvuran bir hastada saptanan infraorbital sinir kalınlaşması yüksek oranda IgG4 ilişkili hastalığı düşündürmelidir.
Pakimeningeal (dural) etkilenme daha çok izole (başka sistemik tutulum olmadan) seyretme eğilimindedir. Bir vaka serisinde spinal dural tutulumlu hastaların %73’ü ve serebral dural tutulumlu hastaların %60 ‘ının MSS’ye izole olarak ortaya çıktığı bildirilmiştir. Dural etkilenme sonucunda (i)- traksiyona bağlı ağrı; (ii)- infratentoryal bölgede servikal üst köklerde basıya bağlı boyun, kulak ve oksiputta ağrı; (iii)- kranial sinirlerin tuzaklanmasına bağlı kranial nöropatiler; ve (iv)- BOS akım engellenmesine bağlı hidrosefali ortaya çıkabilir. MRG de yaygın ya da nodüler dural kontrast tutulumu yapan lezyonlar görülebilir. Tanımlanamayan dural inflamasyon görüldüğünde mutlaka IgG4 ilişkili hastalık akla gelmelidir.
Hipofiz ya da hipofiz sapı etkilendiğinde çeşitli derecelerde hipopituitarizm bulguları ortaya çıkabilir. Bu duruma IgG4 ilişkili hipofizit de denmektedir. MRG’de hipofiz ve çevresinde homojen kontrast tutan lezyonlar görülür. Aynı bulgu, sarkoidoz, granülomatöz polianjiit, tüberküloz, Langerhans hücreli histiositoz, lenfositik hipofizitte de görülebileceğinden dikkatli olunmalıdır.
Beyin parenkim tutulumu oldukça nadir olup sadece bazı vaka raporları bildirilmiştir. Bu nedenle izole beyin parenkimi tutulumu klinik ve radyolojik bulguları ile başvuran hastalarda IgG4 ilişkili hastalık ilk planda akla gelmemelidir. Bu hastalıkta uzun süredir omurilik parenkim tutulumu olmadığı düşünülse de yakın zamanda spinal parenkimal tutulumu olan ve steroide yanıt veren iki olgu bildirilmiştir.
Bu hastalığa tanı koymak oldukça zor olabilir. Hastaların serumunda IgG4 yüksek olması tanıyı destekler ama %30 normal olabileceği akılda tutulmalıdır. Bundan başka sistemik hastalıkta periferde plazmablast, eozinofil ve IgE düzeylerinde artış olabileceği bildirilmektedir. Dural etkilenme olan hastaların BOS’unda lenfositik pleositoz, IgG4 yüksekliği ve OKB bulunabilir. Hipofizer ve orbital etkilenme olan hastalarda BOS genellikle normal bulunur. FDG PET-CT sistemik tutulumu olan pek çok hastada tanıyı destekleyebilir. Histopatolojik incelemede, plazma hücreleri içinde IgG4 artışı (genellikle hücrelerin %40’ından fazlasında) tanıyı büyük oranda destekler. Bununla beraber tanı klinik, radyolojik ve laboratuvar bulgular beraber değerlendirilerek konulmalıdır.
Tedavide ilk seçenek kortikosteroidlerdir. Erken başlandığı takdirde iyi yanıt alındığı bilinen bu hastalıkta pek çok merkezde steroidlerin yanında rituksimab uygulanmaktadır.
Primer ve Sekonder Merkezi Sinir Sistemi Vaskülitleri
Primer merkezi sinir sistemi vaskülitleri (pMSSV) sistemik tutulum olmadan, serebral damarların iltihabı ile giden hastalıklardır. Eğer bu iltihaplanma sistemik vaskülit zemininde gelişirse sekonder olarak adlandırılır. Bu son tablo, granülomatöz polianjiitis (Wegener granülomatozisi), eozinofilik granülomatozis ve polianjiitis (Churg-Strauss sendromu), poliarteritis nodoza, dev hücreli arterit zemininde gelişebilir.
pMSSV insidansı milyonda 1-2 civarında olup son derece nadir bir hastalıktır. Ortalama başlangıç yaşı 50 yaş civarındadır. En sık başlangıç semptomu başağrısıdır. Yeni başlayan günlük başağrılarına kognitif bozulmalar, davranış ve kişilik değişiklikleri eşlik edebilir. Vasküler etkilenme ön planda olduğu için, çeşitli lokalizasyonlarda iskemik ve hemorajik inmeler, subaraknoid kanamalar oldukça sık görülebilir (%30-40). Kilo kaybı, ateş, konstitüsyonel semptomlar ve başka organ tutulumları varsa, sekonder MSS vasküliti daha ön planda düşünülmelidir.
Hastalık küçük ve orta boy leptomeningeal ve kortikal arterleri tutar. Tetikleyen etken bilinmemektedir. Histopatolojik anlamda (i) granülomatöz, (ii) lenfositik, (iii) akut nekrotizan ve (iv) amiloid-beta ilişkili olarak sınıflandırılır. Tanı koymak için arterde transmural inflamasyon ve damar duvarı hasarı olması gereklidir.
Hastaların beyin MR’larında atipik lokalizasyonlarda, genellikle kontrast tutulumu gösteren, subkortikal ve kortikal bölgelerde yoğunlaşma eğiliminde lezyonlar görülebilir (Şekil 17). Bundan başka vasküler komplikasyonlara bağlı, iskemik ve hemorajik infarkt bulguları, hemosiderin birikimleri görülebilir.
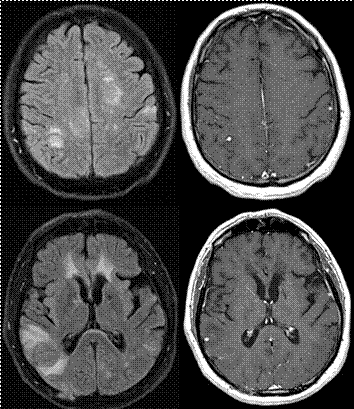
Şekil 17. Biyopsi ile granülomatöz primer merkezi sinir sistemi vasküliti tanısı konan bir hastanın MR görüntüleri. FLAIR hiperintens lezyonların merkezlerinde küçük bir alanda kontrast tutulumu görülüyor (Görüntüler İstanbul Tıp Fakültesi, Nöroloji Anabilim Dalı Multipl Skleroz Birimi materyalinden sağlanmıştır).
MSS vasküliti şüphesi olan hastada, rutin kan tahlillerinin yanında, ESR, kompleman seviyeleri, romatoid faktör, protein elektroforezi ve immünfiksasyon, ANA, Anti- SSa, SSb, Sm, c-ANCA, p-ANCA, HIV ve hepatit serolojileri, BOS’ta IgG sentezi, gram boyama, kültür ve viral polimeraz zincir reaksiyonu incelemeleri yapılmalıdır. Hastalardan BOS alınmasındaki en önemli neden infeksiyöz ve malign süreçlerin dışlanmasıdır. Hastaların BOS’u büyük oranda anormal bulunur (%80-90). Lenfosit ağırlıklı pleositoz, protein yüksekliği en sık bulgulardır.
Orta büyüklükte arterlerin tutulduğu durumlarda konvansiyonel anjiografi (DSA)’de multifokal ve bilateral bir görünüm saptanabilir. Bununla beraber reversibl vazokonstriksiyon sendromunda benzer anjiografik bulgular olabileceği akılda tutulmalıdır. Küçük damarları etkileyen bir MSS vaskülitinde DSA yüksek oranda normal bulunur. Patolojik olarak pMSSV tanısı alan hastaların %30 ‘unda DSA’nın anormal olduğu bildirilmektedir. Bu nedenle DSA negatif vakalarda tanıyı koymanın tek yolu beyin biyopsisi olabilir.
Bu hastalardan yapılacak biyopsinin açık “wedge” yöntemle yapılması, alınan materyalin 1 cm3 lük hem ak hem gri maddeyi, leptomeninks ve en az bir kortikal damarı içermesi önerilmektedir. Beyin biyopsisinin, hem tanı koymak hem de diğer tanıları dışlamak için yapılması önerilmektedir. Stereotaktik biyopsinin morbiditesinin az olmasına rağmen yeterli doku alınamaması olasılığı yüksektir.
PMSSV hastaların ayırıcı tanısında infeksiyöz vaskülitler de bulunmaktadır. Viral, bakteryel ve fungal ajanlar ile bu durum oluşabildiğinden, bu infeksiyonlar dışlandıktan sonra immünsupresif tedavi verilmesi çok önemlidir.
Tedavide ilk seçenekler IV ve oral steroidlerdir. Akut atak döneminde yapılan 5-10 gün 1 gr/gün metilprednizolon sonrasında oral metilprednizolon (en fazla 80 mg/gün) uygulanır ve aylar içinde doz azaltılır. Yine atak döneminde aylık IV siklofosfamid (ortalama 1 gr/ ay) başlanarak hastalık remisyonu sağlanmaya çalışılır. Yanıta göre bir-iki yıl sonra oral immünsupresif (azatioprin, mikofenolat mofetil, metotreksat) tedaviye geçilmesi önerilmektedir. Bununla beraber siklofosfamid ile remisyon sağlanamaz ise anti-TNF ajanlar ve rituksimab kullanılabilir. Hastalık, özellikle geç kalınan olgularda, tedaviye dirençli olabilir ve ciddi kognitif ve somatik sekeller kalabilir.
CLIPPERS (Steroide yanıtlı pontin perivasküler kontrastlanma ile giden kronik lenfositik inflamasyon)
İlk olarak 2010 yılında Pittock ve arkadaşları, 8 hastada başlıca beyinsapını tutan, MR’da noktasal çok sayıda kontrast tutulumu gösteren lezyonları olan ve steroide çok iyi yanıt veren inflamatuar bir hastalığı tanımlayarak, bu hastalığa CLIPPERS ismini vermişlerdir. Başlıca subakut pontoserebellar bulgular (dizartri, diplopi, trigeminal alanda his kaybı, ataksi gibi) ile kendini gösteren hastalıkta görülen kontrast tutan lezyonlar perivasküler lenfohistiositik infiltrasyon (başlıca T lenfositleri) alanlarıdır. Önceleri ponsa lokalize olduğu düşünülen lezyonların yukarıda jukstakortikal alanlara, aşağıda ise konusa kadar uzanabileceği bilinmektedir. Ak ve gri madde etkilenirken, korteks korunur. CLIPPERS için 2017 yılında tanı kriterleri önerilmiştir. Aynı yazıda, CLIPPERS tanısı konan bazı hastalarda aynı zamanda B veya T hücreli lenfoma olduğu (3/23), bir hastada da lenfoma öncesinde CLIPPERS bulgularının çıktığı belirtilmiştir. Bu bulgular ile CLIPPERS ile lenfoma arasında ilişki olduğu ve prelenfomatöz bir durum olabileceği öne sürülmekle beraber bu durum halen tartışmalıdır. Bunlardan başka, MS ve MOGAD’ın CLIPPERS benzeri MR ve klinik bulgular verdiği olgular yayınlanmıştır. Bütün bu bulgular ile CLIPPERS’ın tek bir hastalık olmayıp, farklı klinik durumların ortak sonucu olabileceği ifade edilmektedir.
Hastaların beyin-omurilik sıvısında hafif protein artışı, 5-50 /mm3 kadar hücre (lenfomonositik ağırlıklı), geçici OKB saptanabilir.
Tedavide önerilen ilk seçenek olarak pulse steroid vermek, arkasından oral steroid (1 mg/kg/gün) başlamak ve yanıt alındıktan sonra dereceli azaltmaktır. Sonrasında metotreksat, mikofenolat ya da azatioprin başlanmalıdır. Bununla beraber yetersiz yanıtlı olgularda uzun dönemde rituksimab ve tosilizumaba iyi yanıt veren hastalar bildirilmiştir.
Kaynaklar
1) AbdelRazek, M.A., N. Venna, and J.H. Stone, IgG4-related disease of the central and peripheral nervous systems. The Lancet Neurology, 2018. 17(2): 183-192.
2) Baptista, B., ve ark., Neurological manifestations of IgG4-related disease. Curr Treat Options Neurol, 2017. 19(4): 14.
3) Bevan, C.J. and B.A. Cree, Fulminant Demyelinating Diseases of the Central Nervous System. Seminars in Neurology, 2015. 35(6): 656-666.
4) Bhattacharyya, S., Helfgott S.M. Neurologic complications of systemic lupus erythematosus, sjögren syndrome, and rheumatoid arthritis. Semin Neurol, 2014. 34(4): 425-36.
5) Byram, K., R.A. Hajj-Ali, L. Calabrese, CNS vasculitis: an approach to differential diagnosis and management. Curr Rheumatol Rep, 2018. 20(7): p. 37.
6) Cole, J., ve ark., Acute disseminated encephalomyelitis in children: An updated review based on current diagnostic criteria. Pediatr Neurol, 2019. 100: 26-34.
7) Flanagan, E.P., Neuromyelitis Optica Spectrum Disorder and Other Non-Multiple Sclerosis Central Nervous System Inflammatory Diseases. Continuum (Minneap Minn), 2019. 25(3): 815-844.
8) Fujihara, K., Neuromyelitis optica spectrum disorders: still evolving and broadening. Curr Opin Neurol, 2019. 32(3): 385-394.
9) Gül, A., Pathogenesis of Behçet’s disease: Autoinflammatory features and beyond. Seminars in Immünopathology, 2015. 37(4): 413-418.
10) Gündüz, T., ve ark., Laboratory and clinical correlates of brain atrophy in Neuro-Behçet's disease. J Neurol Sci, 2020. 414: 116831.
11) Hardy, T.A., ve ark., Atypical inflammatory demyelinating syndromes of the CNS. Lancet Neurol, 2016. 15(9): 967-981.
12) Hoftberger, R., H. Lassmann, Immune-mediated disorders. Handb Clin Neurol, 2017. 145: 285-299.
13) Kidd, D.P., Neurosarcoidosis: clinical manifestations, investigation and treatment. Pract Neurol, 2020. 20(3): 199-212.
14) Koçer N, ve ark., CNS involvement in neuro-Behçet syndrome: an MR study. AJNR Am J Neuroradiol. 1999 Jun-Jul;20(6):1015-24.
15) Krupp, L., ve ark., International Pediatric Multiple Sclerosis Study Group criteria for pediatric multiple sclerosis and immune-mediated central nervous system demyelinating disorders: revisions to the 2007 definitions. Mult Scler, 2013.19:1261-7.
16) Limaye, K., E.A. Samaniego, and H.P. Adams, Jr., Diagnosis and treatment of primary central nervous system angiitis. Curr Treat Options Neurol, 2018. 20(9): 38.
17) Mandal, J. and S.A. Chung, Primary angiitis of the central nervous system. Rheum Dis Clin North Am, 2017. 43(4): 503-518.
18) Pawate, S., Sarcoidosis and the nervous system. Continuum (Minneap Minn), 2020. 26(3): 695-715.
19) Pittock, S.J., ve ark., Chronic lymphocytic inflammation with pontine perivascular enhancement responsive to steroids (CLIPPERS). Brain, 2010. 133(9): 2626-34.
20) Reindl, M, P. Waters, Myelin oligodendrocyte glycoprotein antibodies in neurological disease. Nat Rev Neurol, 2019. 15(2): 89-102.
21) Santaro, D.S., ve ark., Diagnostic Considerations in Acute Disseminated Encephalomyelitis and the Interface with MOG Antibody. Neuropediatrics, 2019. 50:273-279.
22) Stern, B,J., ve ark., Definition and Consensus Diagnostic Criteria for Neurosarcoidosis: From the Neurosarcoidosis Consortium Consensus Group. JAMA Neurol, 2018. 75:1546-1553.
23) Tobin, W.O., ve ark., Diagnostic criteria for chronic lymphocytic inflammation with pontine perivascular enhancement responsive to steroids (CLIPPERS). Brain, 2017. 140(9): 2415-2425.
24) Waak, M., ve ark., Acute hemorrhagic leukoencephalopathy: Pathological features and cerebrospinal fluid cytokine profiles. Pediatr Neurol, 2019. 100: 92-96.
25) Weinshenker, B.G. and D.M. Wingerchuk, Neuromyelitis Spectrum Disorders. Mayo Clinic Proceedings, 2017. 92(4): 663-679.
26) Wynford-Thomas, R., A. Jacob, V. Tomassini, Neurological update: MOG antibody disease. J Neurol, 2019. 266(5): 1280-1286.
27) Yilmaz, Y., ve ark., Is the brain biopsy obligatory or not for the diagnosis of Schilder's disease? Review of the literature. Childs Nerv Syst, 2008. 24(1): 3-6.