Sinir Sisteminin Doğumsal Hasarları ve Gelişimsel
Hastalıkları
Yazan: Zuhal Yapıcı
Son güncelleştirme
tarihi: 01. 06. 2020
EMBRİYOLOJİ
Gestasyonun 2. haftasında ektodermden nöronal tabaka ve
nöral tüp oluşmaya başlar. Yirmibeş ila yirmisekizinci günlerde nöral tüpün
rostral kısmında 3 beyin vezikülü oluşur:prosensefalon
(forebrain),mezensefalon (midbrain) ve
rombensefalon (hindbrain). Altıncı haftadan itibaren bu veziküller farklılaşarak
prosensefalondan telensefalon ve diensefalon oluşur. Bunlardan da korteks,
hemisferler, talamus ve hipofiz gibi yapılar meydana gelir. Mezansefalondan
mezansefalonun içyapısı gelişir, rombensefalondan pons, serebellum ve medulla
oblongata farklılaşırlar.
Dorsal indüksiyonun gerçekleştiği 2-5. haftalar arasında
olan sorunlara bağlı anensefali, miyelosel, Arnold-Chiari I-II gibi
malformasyonlar oluşurken, ventral indüksiyonun olduğu 5 ile 10. haftalar
arasındaki sorunlar Dandy-Walker, holoprosenfaliler, serebellar hipoplaziler,
Arnold-Chiari IV gibi malformasyonlara neden olur.
Gestasyonun 7.
haftasında lateral ventrikülün subepandimal tabakasında proliferasyona uğrayan
kök hücrelerden nöron ve glialar meydana gelir. Bu bölgeye germinal matriks ya
da germinal zon denir. Gestasyonun 8. haftasında ilk genç nöronlar germinal
matriksten serebral korteksi oluşturmak üzere radial olarak göçe başlarlar
(nöronal migrasyon). Bu programlanmış süreç gestasyonun 7-24. haftaları
arasında gerçekleşir. Bu nedenle 7-24. haftalar arasındaki zararlar
(intrauterin enfeksiyonlar, toksinler, kromozomal mutasyonlar, iskemi) serebral
kortikal gelişim malformasyonlarını meydana getirir.
Gestasyonun 8.
haftasında nöronal proliferasyon ve farklılaşmayı etkileyen nedenler nörokütane
hastalıkların ortaya çıkmasına da neden olurlar (tuberoz skleroz,
nörofibromatoz).
Tüm bu gelişim sürecini
etkileyen ajanların etki şiddeti ve süresine göre çoğu zaman tek bir anomali değil
birkaç anomali birlikte meydana gelir. Örneğin aynı olguda agiri-pakigiri hem
polimikrogiri hem değişik derecelerde korpus kallozum anomalisi ya da
nörokütane hastalıklar görülebilir.
NÖRAL TÜP KAPANMA DEFEKTLERİ (Nörilasyon Defektleri)
İntrauterin
(İU) 3. ve 4. haftalarda nöral tüpte kapanma sorunu olursa disrafik tablolar
ortaya çıkar. Anterior nöropor denilen bölge rostral parçanın en geniş kısmını
oluşturur. Anensefali anterior nöroporun, miyelomeningosel ise posterior
nöroporun kapanamaması nedeni ile meydana gelir.
Anensefalilerin yarısından daha
azı canlı doğar ve bir ay içinde kaybedilir. Kafa derisi yoktur ve kafatası da
verteksten foramen magnuma kadar saptanamaz. Hemorajik ve fibrotik görünen
beyinde ön beyin yapıları hiç oluşmazken arka beynin daha küçük bir kısmı da
diensefalondan gelişir. Orbitalar sığ ve gözler ise öne doğru çıkıktır. Boyun
retrofleksiyon postüründe ve kolların proksimal kısımları bacaklara göre daha
uzundur. Anensefalinin ikinci doğumda görülme şansı ilk doğumdan sonra 2-5 kat
artmaktadır. İki anensefalik doğumdan sonra ise bu olasılık ikiye katlanır. Ensefalosel, üzeri cilt dokusu ile
örtülü olarak, korteks ve meninkslerin kafatasındaki defektten dışarı doğru
protrüzyonudur. En çok oksipital bölgede orta hatta rastlanır. Çapı çok küçük
ya da kafatası büyüklüğünde olabilir. Büyük olması içeriğinin belirleyici bir
özelliği değildir. Sapsız olan ensefalosellerde sıklıkla beyin dokusu
bulunmaktadır. Genellikle serebral hemisferler, serebellum ve orta beyin
malformasyonları ile birliktedirler. Manyetik rezonans (MR) incelemesi doku
içeriğini belirlemek amacıyla yapılır. Cerrahi girişim nöral dokunun
bulunmadığı oksipital meningosellerde iyi sonuçlar verir. Beyin dokusu
protrüzyonu ve ek malformasyonların olduğu çocuklar süt çocukluğu (SÇ)
döneminde kaybedilmektedirler.
Arnold-Chiari Malformasyonları
Chiari I malformasyonu posterior
serebellar vermis ve tonsillaların foramen magnumdan aşağıya doğru yer alması
ve spinomedüller bileşkeye bası yapması olarak tanımlanır. Klinik olarak
asemptomatik olabildiği gibi oksipital başağrısı veya boyun ağrısı, alt kranial
sinir parazileri veya sirengomiyeliye sekonder ekstremitelerde disosiye duyu
kusurları görülür. Çocuklarda orofarengeal fonksiyon bozukluğu, uyku-apne
sendromu, aşağı vurumlu nistagmus, boyunda hiperekstansiyon postürü gibi
tablolar da bildirilmiştir. Bu olgularda posterior fossa küçük ve klivus
kısadır. Posterior fossa boyutuyla serebellar ektopi derecesi birbiriyle
ilişkilidir. Bu hastalarda mutlaka spinal görüntüleme yaparak
sirengohidromiyeli araştırılmalıdır (%20-65). Eğer servikomedüller açıya bası
yoksa erişkinlerde çoğunlukla sagital MR kesitlerinde bazisten geçen çizgiyi 5
mm’ye dek geçen serebellar ektopilerin klinik semptom vermediği düşünülür. Milhorat
ve arkadaşlarının serisinde ve başka araştırmalarda da foramen magnumu 5 mm‘den
fazla geçiş ve subaraknoid mesafede kapalılık varsa klinik semptomlar büyük
oranda (%91) beklenir. Ancak 5-15 yaş arası çocuklarda asemptomatik 6 mm
ektopiler söz konusu olabilir. Rutin bir MR incelemesi Chiari I tanısı için
yeterlidir, fakat semptomatik olguların tamamına yakınında foramen magnum
seviyesinde yapılan BOS akımı çalışmalarının patolojik sonuç verdiği de akılda
tutulmalıdır.
Chiari I malformasyonları
genellikle MR’de tesadüfi olarak saptanır. Semptomatik olanlar adölesan veya
erişkin yaşlarda başlayan baş-boyun-omuz ağrıları, başta eğilme (tilt), ataksi,
alt kranial sinir tutulumları, nistagmus, kollarda güçsüzlük, alt
ekstremitelerde refleks canlılığı gibi bulgular sergileyebilirler.
Chiari II malformasyonu, Chiari I’in
özelliklerine displastik alt medullanın kaudale doğru yer değiştirmesi ve
lumbosakral meningomiyelosel eşlik etmesidir. Dördüncü ventrikül sıklıkla
medullanın altında, servikal spinal kordun posteriorunda kistik bir yapı
şeklinde bulunur. Beyinsapı elongasyon ve distorsiyona uğramıştır. Servikal
spinal kord aşağı doğru yer değiştirir ve üst servikal sinir kökleri kendi
nöral foraminalarında asılı kalırlar. Hastaların %70’inde servikomedüller
bileşke karakteristik bir katlanma oluşturur. Miyelomeningosel, hidrosefali ve
posterior fossanın hipoplastik olması gibi anomaliler hemen daima eşlik eder. Miyelomeningoselli
bebekler İU dönemde ultrason görüntüleme (USG) ile ya da amniosentezde
alfa-fetoproteinin (AFP) yükselmesi ile tanınır. Supratentoriel olarak korpus
kallozum hipoplazileri de %70-90 oranında eşlik edebilir.
Chiari III’de serebellum ve bazen
de beyinsapı infratentoriel meningoensefalosele doğru uzanmıştır. Oldukça nadir
görülür.
Chiari IV malformasyonunda
serebellum ve beyinsapı hipoplaziktir. Dandy-Walker malformasyonun bir varyantı
olarak kabul edenler de vardır.
Chiari
malformasyonlarının alt tiplerinde ek olarak kemik anomalileri de (baziler
impresyon, atlasın oksipitalizasyonu, C1 spina bifida) bulunabilir. Bu
malformasyonlar en iyi olarak beyin ve spinal MR incelemelerinin sagital kesitlerinde görünür. Bu incelemelerde
sirenks ve beyinsapı basıları net olarak görülebilmektedir. Semptomlara yönelik
olarak beyinsapı dekompresyonu ya da sirenks drenajına yönelik cerrahi tedavi
yapılır.
PROSENSEFALİZASYON
DEFEKTLERİ
Holoprosensefaliler telensefalondan
iki ayrı hemisfer oluşumu sırasında ayrılma gerçekleşmezse meydana gelir. En
ağır şekli olan alobar holoprosensefali pek çok dismorfik özellikle birlikte
olup klinik olarak da ağırdır. Lobar ve semilobar olanlar göreceli olarak daha
hafif klinik gösterirler.
Tam
bir holoprosensefalide limbik korteks ile örtülmüş orta hat vezikülü ile
birlikte minimal bir beyin dokusu vardır. Hafif olgularda 3. ventrikül ve
diensefalon ayrışabilmiş ve hemisferler oksipital lobda parsiyel olarak
oluşmuşlardır. Korpus kallozum yoktur ya da hipoplaziktir. Bu spektrumun en
hafif olguları (arinensefali) tek ya
da iki yanlı koku yollarının yokluğu ve koku alma bölgesinin aplazisi ile
birliktedir. Bunlarda hemisferler arası yarık tamdır, korpus kallozum tam veya
parsiyel olarak oluşmuştur. Holoprosensefali ile doğan çocuklarda sıklıkla 13.
(Patau sendromu) ve 18. (Edward sendromu) kromozom trizomisi
saptanabilmektedir. Hemen daima kraniofasial displazilerle birliktedirler ve
bunlar da orta hat yerleşimli olurlar (hipotelorizm, yassı burun, yarık dudak
ve damak). Sık rastlanılan organ malformasyonları ise konjenital kalp
defektleri, el ve ayaklarda çomak parmak, polidaktili ve sindaktili,
genitoüriner sistem hipoplazileri, aksesuar dalak ve karaciğer, ve barsaklarda
malrotasyon olarak sayılabilir. Hipotoni, mikrosefali, apne epizodları ve
nöbetler neonatal dönemdeki belirtilerdir. Yaşayan çocuklarda ciddi zeka, motor
ve duysal defektler vardır. Sadece arinensefalisi olan çocuklar fiziksel olarak
sağlıklıdır, ancak öğrenme güçlüğü ve nöbetler gibi minör nörolojik defektler
gösterebilirler. Orta hat fasial deformiteleri olan her çocukta özellikle de
diğer organ malformasyonları varsa mutlaka holoprosensefalilerden şüphe etmek
ve araştırmak gerekir.
Korpus kallozum oluşumu,
7. haftada telensefalondan kommisural tabaka gelişimini takip ederek 8-20
haftalar arasında devam eder. Önden arkaya doğru rostrum, genu, gövde, isthmus
ve spleniumdan oluşur. Embriyolojik gelişim sırasında splenium diğerlerine göre
daha erken gelişir. Bu bilgiye göre eğer genu ve gövdede defekt varsa ve buna
karşın splenium ve rostrum normal görünüyorsa bu durum hipogenetik değil
sekonder hasarlanmadır. Bu kural bir tek holoprosensefalide geçerli değildir ve
bu grupta atipik kallozal anomaliler görülür. Korpus kallozum, anterior kommisur ve hipokampal kommisur anomalileri
prosensefalizasyon defektlerinde hemen daima birlikte bulunmaktadırlar. Aynı
zamanda çeşitli başka malformasyonlarla (Dandy-Walker malformasyonu,
ensefalosel, migrasyon anomalileri), metabolik defektlerle (glisin ensefalopatisi
vb.), ve tek gen defekti olarak da rastlanmaktadır. Kallozal agenezi, Aicardi
sendromu (infantil spazmlar, korpus kallozum agenezisi vd. orta hat
anomalileri, retinal malformasyonlar), Andermann sendromu (otozomal resesif
korpus kallozum agenezisi, mental gerilik ve periferik nöropati), 8, 11 ve 13.
kromozom trizomilerinde de görülür. İzole kallozal agenezi (Şekil 1a ve 1b) klinik olarak belirti
vermeyebilir, ancak interhemisferik transformasyonu gösteren özel testlerle
anlaşılabilir. Çoğu hastada mental retardasyon ve öğrenme güçlüğüne
rastlanmaktadır. Agenezide lateral ventriküller düz ve birbirine paralel olup
frontalde daha belirgin görülürler. Hipogenezide ventriküllerin arka parçası
genişler, oksipital boynuzlar dilate olur. Buna kolposefali denir.
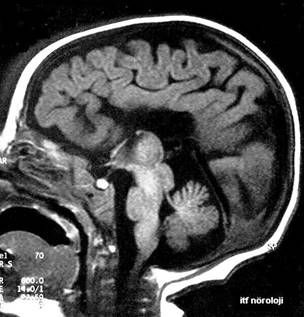
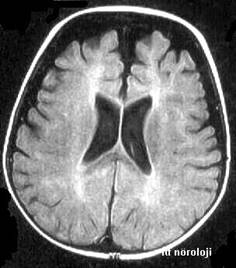
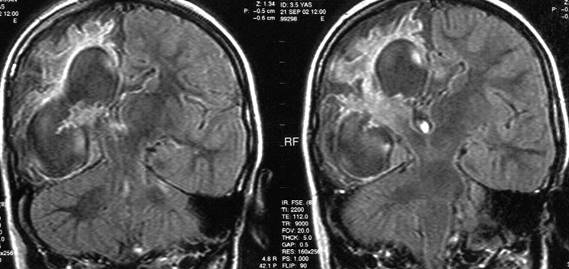
Şekil 1a.
T1 ağırlıklı sagital MR kesitinde korpus kallozum agenezisi (2,5 yaşında,
dirençli epileptik nöbetler, mental gerilik)
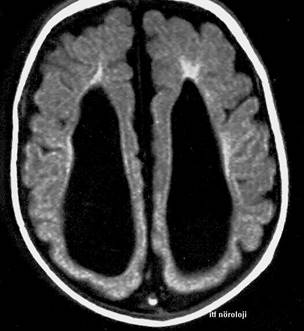
Şekil 1b. T1 ağırlıklı aksiyel kesitte kolposefalik görünüm
SEREBRAL KORTİKAL
GELİŞİM MALFORMASYONLARI
Fetüs beyninde postmitotik
nöroblastların belirli bir hücre pozisyonunun dışına çıkmadan yaptıkları göçe
nöronal göç denir. Gelişimin bir göstergesi olarak merkezi sinir sisteminin (MSS)
her yerinde 7-8. haftada bu göç başlar ve bu sırada nöronlar değişim ve
gelişime uğrar. Nöronal fonksiyonların mükemmelliği, bağlantıların iyi
kurulmasına ve hedefe varılmasına bağlıdır. Bu durum hem presinaptik hem de
postsinaptik iletim için çok önemlidir. Nöronlar serebral korteksi oluşturmak
için ventriküler zondan pial yüzeye kadar belli bir mesafeyi aşmak zorundadırlar.
Tüm bu programlanmış süreç gestasyonun 7-24. haftaları arasında gerçekleşir. Bu
migrasyon sırasındaki zararlar serebral kortikal gelişim malformasyon (SKGM)
spektrumundaki hasarları meydana getirir. SKGM grubu tablolara daha önce
kortikal displazi, kortikal disgenezis veya nöronal migrasyon anomalisi adları
verilmiştir. Gerçek insidans bilinmemektedir, ancak epilepsi cerrahisi yapılmış
hasta gruplarında %12-40 arasında oranlar bildirilmektedir. Genetik veriler
ışığında SKGM’lerin gen, lokus ve ilgili fonksiyonları Tablo 1’de
belirtilmiştir.
Görüntüleme tekniklerinin
gelişmesi ile kortikal malformasyonlar giderek artan sıklıkta keşfedilmiş ve
moleküler genetik çalışmalar da radyolojik bu verilerin sınıflandırılmasına
büyük katkıda bulunmuştur. Klinik olarak nöropsikiyatrik defisit ve epilepsi
görülen çocuk ve erişkinlerde kortikal malformasyon taramak temel bir yaklaşım
olmuştur.
Tablo 1. SKGM’lerde gen, lokus ve ilgili
fonksiyonu.
|
Gen |
Lokus |
Protein-fonksiyon |
|
|
Lizensefali Tip I |
|||
|
Lizensefali (O.D.) |
LIS1 |
17p13.3 |
Mikrotübülle ilişkili protein |
|
İzole lizensefali sekansı (ILS) veya subkortikal band heterotopi (SBH) |
TUBA1A |
12q13.12 |
Mikrotübül oluşumu |
|
Miller-Dieker sendromu |
LIS1 YWHAE |
17p13.3 |
Mikrotübülle ilişkili protein |
|
Lizensefali (X-geçişli) |
|||
|
ILS veya SBH |
DCX |
Xq22.3-q23 |
Mikrotübülle ilişkili protein |
|
X-geçişli lizensefali ile birlikte genital anomali (X-LAG) |
ARX |
Xp21.3 |
Transkripsiyon faktörü |
|
Lizensefali (O.R.) |
|||
|
LCH grup b |
RELN |
7q22 |
Ekstrasellüler matriks serin proteaz |
|
VLDLR |
9p24 |
VLDL’yi bağlar ve endositoz ile hücreye transportunu sağlar |
|
|
Lizensefali tip II: Kaldırım taşı kompleksi (OR) |
|||
|
Fukuyama konjenital kas distrofisi veya WWS |
FKTN |
9q31.2 |
Glikozilasyonda görevli |
|
KGB veya WWS |
POMT1 |
9q34.13 |
Protein-O-mannozil transferaz 1 |
|
POMT2 |
14q24.3 |
Protein-O-mannozil transferaz 2 |
|
|
POMGnT2 |
3p22.1 |
O-bağlantılı mannoz asetil glikozaminil transferaz |
|
|
FKRP |
19q13.32 |
Glikozilasyonda görevli |
|
|
KGB hastalığı |
LARGE |
22q12.3 |
Glikozil transferaz |
|
POMGnT1 |
1p34.1 |
O-mannozil glikozilasyonda görevli |
|
|
Bilateral fronto-parietal polimikrogiri |
GPR56 |
16q21 |
G proteinine bağlı reseptör 56 |
|
CEDNIK sendromu |
SNAP29 |
22q11.21 |
Sinaptozomla ilişkili protein |
|
Kas distrofileri |
ISPD |
7q21.2 |
O-bağlantılı mannozilasyonda gerekli protein |
|
GTDC2 |
3p22.1 |
O-bağlantılı mannoz asetil glukozaminil transferaz |
|
|
TMEM5 |
12q14.2 |
Glikozil transferaz fonksiyonu |
|
|
B3GALNT2 |
1q42.3 |
Beta-1,3-N-asetil galaktozaminil transferaz |
|
|
SGK196 |
8q11.21 |
O-mannoz kinaz proteini |
|
|
B3GNT1 |
11q13.2 |
Lineer poli-N-asetil laktoz aminoglikanların sentezi |
|
|
GMPPB |
3p21.31 |
GDP-mannoz pirofosforilaz |
|
|
Polimikrogiri |
TUBB2B (ILS,PMG-like) |
6p25 |
Mikrotübüllerin yapısında majör rol |
|
GPR56 |
16q21 |
G proteine bağlı reseptör 56 |
|
|
SRPX2 |
Xq22.1 |
Anjiyogeneziste rol |
|
|
TBR2 |
3p24.1 |
Transkripsiyon aktivatörü |
|
|
PAX6 |
11p13 |
Transkripsiyon faktör |
|
|
KIAA1279 |
10q22.1 |
Aksonal mikrotübüllerin organizasyonu |
|
|
RAB3GAP1 |
2q21.3 |
RAB3 GTPaz aktifleyen proteinin subuniti |
|
|
Heterotopi |
|||
|
Heterotopi (X-geçişli, O.D.) |
|||
|
Klasik PVH – difüz |
FLNA |
Xq28 |
Aktin-bağlayan protein |
|
PVH ve frajil-X |
FMR1 |
Xq27.3 |
Translasyon baskılayıcı |
|
PVH - anterior ve Williams sendromu |
WBSCR16 del |
7q11.23 |
Guanin nükleotid değişim faktörü |
|
PVH – anterior |
PVNH3 dup |
5p15.1 |
Periventriküler nodüler heterotopi 3 |
|
PVH – posterior |
PVNH5 del |
5q14.3-q15 |
Periventriküler nodüler heterotopi 5 |
|
Heterotopi (O.R.) |
|||
|
PVH - difuz ve mikrosefali |
ARFGEF2 |
20p13.13 |
Hücre içi vezikül trafiği |
|
PVH ve Donnai–Barrow sendromu |
LRP2 |
2q31.1 |
LDL ile ilişkili protein 2 |
|
FKD |
|||
|
|
TSC1 |
9q34.13 |
mTORC sinyallerinin negatif regülasyonu |
|
|
TSC2 |
16p13.3 |
mTORC sinyallerinin negatif regülasyonu |
|
HME |
|||
|
|
PIK3CA |
3q26.32 |
PI3K/AKT komponenti serin/treonin kinaz
sinyalizasyonu |
|
|
AKT3 |
1q44 |
PI3K/AKT komponenti serin/treonin kinaz sinyalizasyonu |
|
|
MTOR |
1p36.22 |
PI3K/AKT komponenti serin/treonin kinaz sinyalizasyonu |
O.D.: otozomal dominant, O.R.:
otozomal resesif, WWS: ILS: İzole lizensefali sekansı, Walker-Warburg sendromu, LCH: Serebellar
hipoplazi ile birlikte lizensefali, KGB: Kas-göz-beyin hastalığı, CEDNIK: Serebral disgenesiz, nöropati,
iktiyozis ve keratoderma, PVH: periventriküler
heterotopi, FKD: Fokal kortikal displazi,
HME: Hemimegalensefali
KÖK HÜCRELERİN ANORMAL
PROLİFERASYONU VE APOPTOZUNA BAĞLI MALFORMASYONLAR
Mikrosefaliler
Primer mikrosefaliler proliferasyonun bozuk olması ya
da apoptoza yol açan genetik bir nedenle, sekonder mikrosefaliler ise
pre-peri-postnatal nedenlerle ortaya çıkarlar. MR'de görülen morfolojik
karakterlerine göre alt kategoriler incelenir: giral patern, kortikal kalınlık,
heterotopi varlığı, kallozal anomali varlığı. Bu morfolojik özelliklerle
birlikte etyoloji, klinik gösteriler ve diğer MR özelliklerine göre
mikrosefalilerin alt tipleri vardır.
Hemimegalensefali
Bir hemisferin tamamı
veya bir kısmının hamartamatöz büyümesi ile bazı olgularda nöronal
proliferasyon, migrasyon, organizasyon anomalilerinin de birlikte görüldüğü
patolojilerdir. İzole olanlarda hemisfer hipertrofisinin yanı sıra aynı taraf
vücut yarısında da hipertrofi olabilir. Çocuklar genellikle makrosefalik
doğarlar. İlk yaşta hemiparezi saptanır. Hastalar çoğunlukla epileptik
nöbetlerle başvururlar. Nörokütane hastalıklarla birlikte olanlarda epidermal
nevüs sendromu, Proteus sendromu, hipomelanozis Ito, tuberoz skleroz gibi
hastalıklar söz konusudur. Etkilenen hemisferde pakigiri, polimikrogiri,
heterotopi bulunabilir, nöron sayısı azalmış, glia hücreleri artmıştır (Şekil 2).
Şekil 2. T1 ağırlıklı aksiyel MR kesitinde sağ hemimegalensefali,
sağda agiri-pakigiri, subepandimal heterotopi, sağ lateral ventrikül ektazisi
(12 yaşında, ağır mental gerilik, sol spastik hemiparezi, epilepsi).
Fokal Kortikal
Displazi
Kortikal laminasyonda
anomali bulunmaktadır. Histolojik olarak gri-beyaz madde bileşkesindeki
düzensizlik, beyaz maddeye doğru uzanan gri madde parçası olarak fark edilir (Şekil 3). MR’de çok küçük olanların
saptanabilmesi için yüzey koilleri ve üç boyutlu görüntüler kullanılır. FKD
tanısı için yüksek çözünürlüklü MR teknikleri kullanılmalıdır. Anormal giral
yapılar, sulkusların yüzeyelleşmesi, artmış kortikal kalınlık, gri-beyaz madde
ayrımının yapılamaması ve T2 kesitlerinde subkortikal intensite artışı en çok saptanan
bulgulardır. Nöbeti olan bir hastada odağı teyit etmek için PET ve SPECT’ten de
yararlanılabilir. Bu hastaların nörolojik muayeneleri normal olabilmekte ve
genellikle fokal ve/veya sekonder jeneralize nöbetleri görülmektedir.
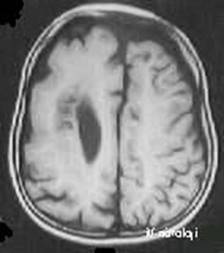
Şekil 3.
T1 ağırlıklı aksiyel MR kesitinde sol frontal fokal kortikal displazi (7
yaşında, perseverasyon, agresyon, anlamsız davranışlar ve epilepsi).
ANORMAL NÖRONAL
MİGRASYONA BAĞLI MALFORMASYONLAR
Anormal nöronal migrasyona bağlı
en önemli malformasyonlar “az sulkus veya sığ sulkus malformasyonları”,
“anormal pial membran yapılanmasına bağlı “kaldırım taşı malformasyonu”
ve nöronal migrasyonun difüz duraksaması ile olan lizensefalik
(agiri-pakigiri) malformasyonlardır.
Lizensefali
Düz beyin anlamına
gelen lizensefali hiç girus olmaması veya az sayıda girus-sulkus bulunmasıdır.
Girus yokluğuna agiri, birkaç kaba,
düz girus varlığına da pakigiri
denmektedir. Lizensefali genel anlamda agiri-pakigiri kompleksi olarak da kullanılmaktadır.
Fakat bazı yazılarda komplet lizensefali agiri ile eş anlamlı olarak yer
alırken, inkomplet lizensefali agiri-pakigiri tablosunu ifade etmek için
kullanılabilmektedir. Nöron migrasyonu durduğunda tip I (klasik) lizensefali,
aşırı migrasyon olduğunda tip II (cobblestone-parke taşı) lizensefali meydana
gelmektedir. Her iki tip lizensefalide de hastalar mental ve fiziksel olarak
başkalarına bağımlıdırlar.
Tip
I (Klasik) lizensefali ve band heterotopi:
Makroskopik olarak beyin yüzeyi düz, transvers kesitlerde
“8” şeklinde (Şekil 4a), silvian
fissürler vertikal yerleşimli, lateral ventrikül oksipital boynuzları embriyodakine
benzer şekilde ektazik, korpus kallozum hipo/agenetik, beyinsapı ve serebellum
değişik derecelerde hipogenetik görülür. Klasik
lizensefalilerin
%65-76’sında LIS1 ya da DCX
mutasyonu saptanmaktadır.
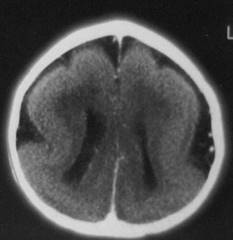
Şekil 4a. Bilgisayarlı tomografi kesitinde Tip I lizensefali (5
yaşında, ağır motor-mental gerilik, epileptik nöbetler, mikrosefali).
a. 17p 13.3 delesyonlu
(LIS1 gen defekti) çoğu hastada agiri parietooksipital, pakigiri de
frontotemporal bölgelerde görülür. Miller-Dieker sendromunda (bitemporal
çöküklük, belirgin alın, küçük çene, düşük kulak, kısa burun, genital ve kardiyak
anomaliler) bu delesyon %90-92 oranında saptanmıştır. Bazılarında ise halka
şeklinde 17. kromozom bulunmuştur. İzole lizensefali sekansında ise dismorfik
özellikler daha hafif olup 17p delesyonu %44 oranında saptanmıştır.
b. X-geçişli
lizensefalide lokus Xq 22.3-q23 olarak belirlenmiştir ve erkek çocuklar
hastadır. Bu grupta agiri-pakigiri frontal bölgede daha belirgin olarak
görülmektedir. Bu çocukların annelerinde band heterotopi saptanabilir.
Klasik
lizensefalilerin bu sayılan sendromlarında MR görüntüleri genellikle komplet
lizensefali şeklindedir. Komplet lizensefali olguları hipotonik doğar ve
hızlıca orofarengeal ve apendiküler spastisite geliştirirler. İnkomplet
lizensefalilerde klinik daha hafiftir. Ciddi olgularda infantil spazm ve
dirençli nöbetlerle kalp, böbrek ve göz anomalileri gibi sistemik problemler de
görülür. Bu olgularda pakigiri ile birlikte normal veya agirik korteks de eşlik
eder.
Band heterotopi (double cortex): Değişik derecelerde
mental gerilik ve çok çeşitli nöbetler ile ortaya çıkar. Herhangi bir yaşta da
başlayabilir. Hafif nöbetler dışında patolojik bulgu saptanmayabilir.
Xq22.3-q23 üzerinde XLIS geni (double cortin) sorumludur ve olguların %90’ından
fazlası kız çocuklarıdır. MR’de lateral ventrikül ile korteks arasında homojen
bir band şeklindedir ve korteks ile band arasında beyaz madde vardır (Şekil 4b). Band heterotopi parsiyel
veya total olabilir. Band ne kadar kalın ise prognoz o kadar ağırdır.
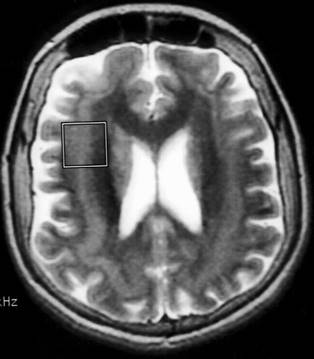
Şekil 4b.
T2 ağırlıklı aksiyel MR kesitlerinde band heterotopi (10 yaşında kız,
Lennox-Gastaut sendromu).
Tip
II (cobblestone) lizensefali: Anormal pial membran yapılanmasına bağlı
malformasyonlardır. Bu tablolarda beyin, göz ve kas tutulumu birlikte
olmaktadır. Konjenital kas distrofisi olan çocukların bir kısmında bu
patolojiler görülmektedir. Fukuyama konjenital kas distrofisi, kas-göz-beyin
hastalığı ve Walker-Warburg sendromu başlıcalarıdır. Bugüne dek pek çok sorumlu
gen keşfedilmiştir (POMT1, POMT2, FCMD, FKRP, LARGE, GPR56, POMGnT1). Klinik
olarak oldukça heterojen olan bu grupta kas biyopsisinde çarpıcı distrofik
bulgular varken MR’de çok çeşitli malformasyonlar saptanabilir. Doğumda
hipotoniktirler ve genel bir kas güçsüzlüğü vardır. Değişik derecelerde eklem
kontraktürleri eşlik eder. Çoğu hastada MSS ve oküler anomaliler de birlikte
bulunur. Bu çocuklarda müsküler disfonksiyonun nedeni bir grup proteininin
eksik olmasıdır. Bu proteinler kas kontraksiyonunun yanı sıra MSS gelişiminde
de etkili oldukları için MSS anomalileri de birlikte görülmektedir. Bu
proteinlerin en iyi bilineni bir laminin formu olan merozindir ve
oligodendrosit prekürsörlerinin migrasyonu için gereklidir. Eksikliğinde
miyelinizasyon da bozulur. Glia ve retina membranında bulunan diğer proteinler
nöronal migrasyonu sonlandırma ve korteks organizasyonunda rol oynarlar.
Bunların eksikliğinde nöronların aşırı migrasyonu olur (cobblestone yapı).
Fukuyama ve kas-göz-beyin hastalığında laminin 1 ve 2
(nöron migrasyonunda uyarıcı ve kılavuz olarak rol oynarlar), Walker-Warburg
sendromunda laminin b-2
eksiktir. Bu tip lizensefalik MR’lerde disorganize kortikal nöronların tipik
olarak bandlar halinde beyaz madde içine doğru uzandığı görülür. Bu bandlar
kimi zaman beyaz maddeden subaraknoid alana kadar uzanırlar.
Fukuyama tip konjenital müsküler distrofi: Pakigiri ve polimikrogiri gibi giral
anomaliler sık görülür. Japon halkında görülen bir hastalıktır.
Walker-Warburg Sendromu (WWS): Tip
II lizensefali bulguları ile birlikte korteks kalınlığı, birkaç sığ sulkus,
serebellar hipoplazi, kallozal hipogenezi ve hidrosefali saptanan belli başlı görüntüleme
özellikleridir. Doğumdan itibaren ciddi nörolojik tutulum vardır. Retinal
displazi, mikroftalmi, katarakt, optik sinir hipoplazisi ve konjenital glokom
olabilmektedir. Kötü prognozludur; hastalar aylar, yıllar içinde kaybedilirler.
Kas-göz-beyin hastalığı (Santavuori): Hastalık
özellikleri WWS’ye benzer ancak daha iyi seyirlidir. Tip II lizensefali
bulgularının yanı sıra korteks kalınlığında artma, sulkus sayısında azlık ve
sığlık, miyelinizasyon geriliği, ventriküler ektazi, silvian fissürlerde
genişlik, septum pellusidum yokluğu, kallozal, pons ve serebellum hipoplazileri
ve serebellar kortikal kistler görülür. Çocuk doğuştan itibaren hipotoniktir ve
vizyon bozukluğu vardır. Epilepsi ve mental gerilik sıktır, spastisite 5
yaşından sonra başlar. Bu gruplar dışında kas-göz bulguları olmayan fakat tip
II lizensefali bulguları olan hastalar da tanımlanmıştır.
Heterotopi
Radial migrasyonun
durmasıyla sinir hücrelerinin anormal lokalizasyonlarda yerleşmesine gri madde
heterotopileri denir. Genellikle diğer anomalilerle birliktedir. Bu tabloda
hemen daima epileptik nöbet vardır. Klinik ve prognostik değerlendirme amacıyla
3 bölümde incelenir: (a) Periventriküler (subepandimal) (b) fokal subkortikal
(c) leptomeningeal heterotopi olmak üzere sınıflandırılmaktadır. Band
heterotopi, önceleri bu bölümde (heterotopi başlığı altında) tartışılırken
artık lizensefalinin hafif bir formu olması nedeni ile "klasik
lizensefali/band heterotopi spektrumu"nun kapsadığı bölümde
sınıflandırılması uygun görülmektedir (yukarda anlatılmıştır).
Subepandimal heterotopi: Çoğunlukla lateral ventrikül trigonu
ile temporooksipital bölgelerde ve asimetrik yerleşimlidirler. İzole olanlar
sporadik veya ailesel, genellikle hafif klinik semptom verirler. Bunların motor
gelişimleri normal olup 2. dekadda nöbetler başlar. Ailesel olanlarda Xq28
mutasyonu saptanmıştır. Bu kız çocuklarda genellikle büyük sisterna magna da
saptanır. MR’de düz, ovoid kitleler şeklinde ve tüm sekanslarda gri madde ile
izointens izlenir. Ovoid olanların uzun aksı komşu ventrikül duvarına
paraleldir. Ayırıcı tanıda tuberoz sklerozun subepandimal hamartomları yer
alır. Bunlarda sınırlar düzensiz, aksları ventrikül duvarına dik ve beyaz madde
ile izo/hipointens görülür.
Fokal subkortikal heterotopi: MR’de tüm sekanslarda
gri madde ile izointens ve heterojen olarak izlenir. Bazen multinodüler gri
madde kitleleri olarak bazen de bandlar şeklinde saptanır. O taraf hemisferde
küçüklük, ince korteks, sığ sulkus görülebilir. Kitle etkisi görülürse tümörler
ile ayırıcı tanı gerekir. Olguların %70’inde kallozal hipogenezi/agenezi
bulunur. Değişik derecelerde motor ve mental gerilik saptanır.
Polimikrogiri (PMG)
Nöronlar migrasyona
uğradıktan sonra kortekste oluşan organizasyon bozukluğu polimikrogirik yapıyı
oluşturur. Ultrastrüktürel olarak hepsinde korteksin normal 6 tabakalı düzeni
bozulmuştur. Tanım kortikal displaziye benzemekle birlikte, bu son durum daha
küçük bir bölgede veya fokal transmental displazi varlığında (anormal kök
hücrelerin oluşturduğu serebral yapı) söz konusudur. PMG ise daha büyük bir
bölgede olur ve çok sayıda minik giruslardan oluşur. İU iskemik olaylar,
kromozomal mutasyonlar, sitomegalovirus enfeksiyonu gibi pek çok nedenler
sorumlu tutulabilir. Son zamanlarda çok çeşitli mutasyonlarla PMG görüldüğü saptanmıştır.
16 O.D., 16 O.R. ve 3 adet de X-geçişli özellikte PMG görülmektedir. Klinik
özelikler tutulan beyin bölgesine bağlı olarak fokal nörolojik bulgular,
epileptik nöbetler, değişik derecelerde motor-mental gerilik olarak
saptanmaktadır. Herhangi bir yaşta klinik belirtiler ortaya çıkabilir. En sık
görülen lokalizasyon silvian fissür çevresidir. Konjenital bilateral
perisilvian sendrom (bilateral operküler PMG), psödobulber felç, epilepsi,
mental gerilik ve bazı dismorfik özelliklerin olduğu özel bir sendromdur (Şekil 5a ve b)
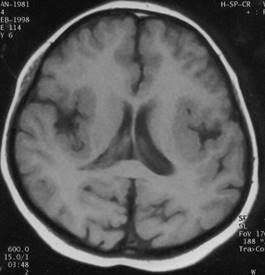
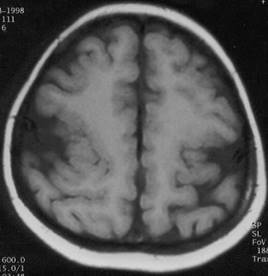
Şekil 5a,b. T1 ağırlıklı aksiyel MR kesitlerinde bilateral perisilvian polimikrogiri
(13 yaşında, mental gerilik, epileptik nöbetler, operküler sendrom).
Şizensefali
Agenetik porensefali olarak da isimlendirilir. Hemisferde
ventrikülün epandimal yüzeyinden korteksin pia örtüsüne kadar uzanan, gri madde
ile çevrili bir yarıktır. Bu gri madde normal olabileceği gibi pakigirik veya
polimikrogirik de olabilir. Bazıları İU 2. trimesterde transmental hasara
uğrayarak, bazıları da EMX2 (homeobox) geninin mutasyonu ile meydana gelir. Yüzde
60’ı tek taraflı, %40’ı iki taraflı (Şekil
6) görülür. Kapalı tipte olanlarda BOS yolu tıkalı iken açık dudaklı
olanlarda BOS ile dolu olan şizensefalik yarık subaraknoid mesafe ile
ilişkilidir. Kliniğin ağırlığı tutulan beyin bölümü ile ilgilidir. Tipik olarak
nöbet, hemiparezi, tetraparezi ve değişik derecelerde gelişme geriliği
saptanır. Bilateral tutulumlarda motor ve
mental gelişme problemleri ve erken başlayan nöbetler ile daha ağır bir seyir
vardır. Bu olguların 1/3'ünde optik sinir hipoplazisine bağlı görme problemleri
de karşımıza çıkmaktadır.
Şekil 6. T1 ağırlıklı aksiyel MR kesitinde sağ frontal, lateral ventrikül trigonuna
uzanım gösteren displastik korteksin çevrelediği kapalı dudak şizensefali, sol
frontoparietal açık dudak şizensefali (6 yaşında, mikrosefali, mental gerilik,
solda belirgin spastik tetraparezi).
KROMOZOM ANOMALİLERİNDE GÖRÜLEN BEYİN MALFORMASYONLARI
En sık görülenleri
arasında Down, Edward, Patau sendromları ile frajil-X sendromu sayılabilir.
Down sendromu (21. kromozom trizomisi) en sık görülen kromozom anomalisi olup
MR’de kranial bölgede brakisefali, mikrosefali, frontal lobların ön-arka
çapında küçüklük, serebellum ve pons hipoplazisi saptanır. Servikal spinal
bölgede ligaman laksitesine bağlı olarak atlanto-aksiyel subluksasyon sık
görülebilmektedir. Mental geriliğin sık rastlanan bir nedeni olan frajil-X’de
sorumlu kromozom anomalisi Xq27.3 olarak belirlenmiştir. Belirgin alın, büyük
kulak, otistik özellikler gibi klinik bulgular ile birlikte MR’de subepandimal
heterotopi ve serebellar vermis hipoplazileri saptanabilmektedir.
Klinik olarak SKGM’lerde motor-mental
gelişme geriliği ve epileptik nöbetler hemen daima ortak özelliklerdir. Tanı
için MR incelemesinin (3 Tesla, yüzey koil uygulaması, MPR metodu ile saptanma
oranı artmaktadır) yanısıra çoğu olguda genetik analiz yapmak gerekmektedir.
Nöbetler tiplerine göre antiepileptik ilaçlarla tedavi edilir. Ağır
malformasyonların bir kısmı SÇ döneminde kaybedilirken bir kısmı da erişkin ama
sekelli olarak daha uzun bir yaşam sürebilir. Sadece epileptik nöbetlerle ya da
minimal problemlerle normal bir yaşam da mümkün olabilmektedir.
MAKROSEFALİ
Baş
çevresinin normal dağılıma göre 2 standart sapmanın üzerinde olduğu durumlara makrosefali denir. Bu terim mutlaka
patoloji varlığı anlamına gelmez. Normal popülasyonun %2’si makrosefaliktir. Ayrıca
hidrosefali ve megalensefali ile eş anlamlı olarak kullanılamaz. Böyle bir
durumla karşılaşıldığında mutlaka anne ve babanın baş çevresi öğrenilmeli ve
hastanın kliniği ile birlikte değerlendirilmelidir. Ailevi makrosefalilere daha çok erkek çocuklarda rastlanır ve
otozomal dominant kalıtılır, fakat penetrans tam değildir.
Makrosefalinin
başlıca nedenleri:
A. Hidrosefali ya da kafa içi BOS miktarının arttığı durumlar: (Tablo 2)
a.
Komünikan (nonobstrüktif): Subaraknoid aralık ve ventriküller arasında BOS
akışı serbesttir.
b.
Nonkomünikan (obstrüktif): BOS akış yolları üzerinde obstrüksiyon vardır.
B. Megalensefali olan durumlar: (Tablo
3)
a.
Anatomik: Çocuklar doğduklarında makrosefaliktirler fakat intrakranial basınç
normaldir.
b.
Metabolik: Doğumda normosefaliktirler ancak neonatal dönemde megalensefali
gelişmeye başlar.
C. Kafa kemiklerinin
kalınlaşmasına
bağlı gelişen makrosefaliler: Kafa çevresi doğumda normaldir ve genişleme
yenidoğan (YD) döneminde değil SÇ döneminde olur. Başlıca nedenler
hiperfosfatemi, osteopetrozis, osteogenezis imperfekta, raşitism, kleidokranial
dizostoz, orodijitofasial dizostozdur.
D.
Subdural veya epidural hemorajilere bağlı makrosefali
Tablo 2. Hidrosefali nedenleri
|
1. Komünikan |
|
Akondroplazi |
|
Baziler
impresyon |
|
Koroid
pleksus papilloması |
|
Meninks
maliniteleri Menenjitler |
|
Posthemorajik,
postenfeksiyöz Subdural
koleksiyon |
|
2. Nonkomünikan |
|
Akuadukt
stenozu (enfeksiyöz, X’e bağlı geçiş) |
|
Arnold-Chiari
malformasyonu |
|
Dandy-Walker
malformasyonu Galen
ven malformasyonu Kitle
lezyonları (apse, hematom, tümör, Galen ven malformasyonu) |
|
Klippel-Feil
sendromu |
|
X-geçişli
formlar |
|
3. Hidranensefali: |
|
Hidranensefali Holoprosensefali |
|
Porensefali Selim
subaraknoid mesafe artışı |
Tablo 3.
Megalensefali nedenleri
|
1. Anatomik megalensefali |
|
Akondroplazi ile birlikte |
|
Genetik megalensefali |
|
Gigantizm ile birlikte (Sotos sendromu) |
|
Nörokütane hastalıklar |
|
Ito hipomelanozu |
|
Incontinentia pigmenti |
|
Linear nevus sebaceus sendromu |
|
Nörofibromatoz |
|
Tuberoz skleroz |
|
2.Metabolik megalensefali |
|
Alexander hastalığı |
|
Canavan hastalığı |
|
Galaktozemi |
|
Gangliosidoz |
|
Globoid lökodistrofi |
|
Glutarik asidüri- Tip I |
|
L-2 (OH) glutarik asidüri |
|
Lökoensefalopatilerin bazı tipleri |
|
‘Maple syrup urine’ hastalığı |
|
Megalensefalik kistik lökoensefalopati |
|
Metakromatik lökodistrofi |
|
Mukopolisakkaridoz |
HİDROSEFALİLER
Komünikan Hidrosefaliler
Komünikan
hidrosefaliler genellikle menenjit
veya subaraknoid kanama sonrasında araknoidin
etkilenmesine bağlı olarak BOS emiliminin bozulması ile oluşurlar.
Hidrosefaliye neden olabilen enfeksiyonlar peri-postnatal bakteriyel,
parazitik, granülomatöz enfeksiyonların yanı sıra İU TORCH grubu etkenler de
olabilir. Başlıca lösemi ve primer beyin tümörlerine bağlı olarak ortaya çıkan meninks infiltrasyonları ise sık
görülmez. Koroid pleksus papillomalarında
BOS aşırı yapılır ancak BOS emilimindeki potansiyel hız kolayca hidrosefali
oluşmasına izin vermez. Bu tümörler daha çok bir veya daha fazla ventrikülde
tıkanmaya neden olarak obstrüktif tipte hidrosefaliye yol açarlar.
Akondroplazi
Otozomal
dominant geçişli, iskelet deformiteleri ve gelişme geriliğine neden olan bir
tablodur. Başlıca klinik özellikleri makrosefali, ekstremitelerde rizomelik
kısalık (ekstremite proksimal parçasının distal parçasından daha kısa olması)
ve enkondral kemik gelişimindeki bozukluğa bağlı anormal yüz görünümüdür.
YD’da
megalensefali vardır fakat hidrosefali saptanmaz. SÇ döneminde posterior
fossadaki venöz sinüslerde darlığa bağlı olarak venöz akım güçleşir ve venöz
basınç artarak BOS geri alınımı bozulur. Bu durum ventriküllerin genişlemesine
neden olur. Hastalarda ayrıca servikomedüller basıya bağlı olarak dispne,
refleks canlılığı, spastisite ve duyu kusurları görülebilir. Mutasyona sık
olarak rastlanır. Tanı klinik özelliklere dayanır. BT’de posterior fossanın
darlığı, sfenoid sinüsün genişliği, bazen baziler bası dikkati çeker. Ventrikül
genişliği YD’da normalken SÇ döneminde orta ya da ileri genişlik görülmeye
başlar. Fakat başlangıçtaki genişlemeden sonra stabilleşerek nadiren cerrahi
girişime ihtiyaç duyulur. Progresif servikomedüller bası durumunda dekompresyon
gerekebilir.
Subaraknoid aralığın
selim genişlemesi (eksternal hidrosefali, ekstraventriküler hidrosefali, selim
subdural efüzyon, selim ekstraserebral sıvı toplanması)
Makrosefali
nedenleri arasında nispeten sık görülmektedir. Bazı olguların babalarında da
makrosefali olması tablonun genetik olabileceğini düşündürmektedir. Erkek
çocuklarda daha sık rastlanan bu durumda doğumda baş çevresinin 90 persantilin
üzerinde olması, seri ölçümlerde 98 persantil civarında seyretmesi ve anterior
fontanelin geniş fakat yumuşak oluşu başlıca klinik özelliklerdir. Bu
çocuklarda motor ve mental gelişim normal olup nörolojik defisit bulunmaz.
BT’de frontal subaraknoid mesafenin, silvian fissürün ve diğer sulkusların
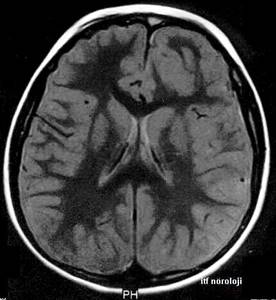
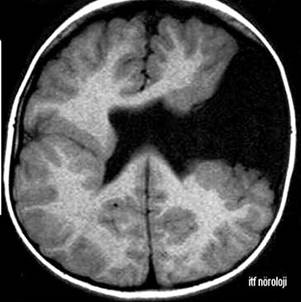
genişlediği, ventriküllerin ise normal veya minimal genişlediği görülür (Şekil 7). SÇ’nda normal subaraknoid
mesafe limiti 5,7 mm, silvian fissür ise 7,6 mm olarak düşünülmektedir. Bu
bebeklerin çoğunda baş çevresi takibi ile tanıdan sonraki 6 ayda baş büyüme hız
eğrisinin normal seyretmeye başladığı saptanır. Büyüme hızı beklenenin
üzerinde, nörolojik muayenede anormal bulgular ve gelişme geriliği söz konusu
olduğunda BT tekrarı, yakın takip ve gerekirse ventriküloperitoneal şant
operasyonu gündeme gelebilir.
Şekil 7. T1 ağırlıklı aksiyel MR kesitinde ön bölgelerde belirgin
subaraknoid mesafe artışı
Subdural kolleksiyon
Başlıca
subdural efüzyon ve subdural kanamalara bağlıdır. Sıklıkla SÇ döneminde ve
genellikle santroparietal bölgede tek veya iki taraflıdır. Doğum ya da başka
nedenli travmalar, menenjit komplikasyonları (H. influenzae, pnömokok), malnutrisyon, kanama diyatezi ve
hipo-hipernatremi başlıca etyolojik faktörlerdir. Klinik olarak kusma, fontanel
kabarıklığı, huzursuzluk, ateş, bilinç değişiklikleri, kafa büyüklüğü, retinal
kanama ve fokal nörolojik bulgular görülebilir. Subdural sıvının ponksiyonla
incelenmesi ve BT/MR tanı koydurucudur. Basit efüzyonlarda prognoz genellikle
iyi seyirlidir. Nörolojik sekelli olan ağır tablolarda cerrahi girişim
gerekebilir ve prognoz iyi değildir.
Meninks maliniteleri
Meninks
ve subaraknoid aralığı infiltre eden tümörler BOS geri alınımını bozarak
komünikan hidrosefalilere neden olurlar. Genellikle agresif bir seyir
göstererek hızla başağrısı, kusma, letarji ve kişilik değişiklikleri eklenir.
Meningismus ve papilödem bakteriyel menenjiti düşündüren özelliklerdir. MR’de
ventrikül sisteminin genişlediği ve subaraknoid mesafenin normal olduğu
saptanır. BOS basıncı ve protein konsantrasyonu artarken glikoz düzeyi normal
veya azalmış olarak bulunur. BOS’ta sitolojik inceleme erken tanı ve tedavi
açısından değerlidir. Pek çok hastada kesin tanı için meninks biyopsisi
gerekir. İntrakranial basıncı ve semptomları hafifletmek için şant, kemoterapi
ve radyoterapi uygulamaları yapılmaktadır. Prognoz genel olarak iyi değildir.
Nonkomünikan Hidrosefaliler
Ventriküllerden
subaraknoid mesafeye geçiş yolu üzerindeki bir tıkanma, proksimalindeki
ventriküllerde dilatasyona ve basıncın artmasına yol açar. Fetal dönemde
görülen hidrosefalilerin en sık rastlanılan mekanizmasıdır. Konjenital
hidrosefalilerin insidansı 1000 canlı doğumda 1’dir. Bunların %40’ında genetik
bir temel düşünülmektedir. Erkek çocuklardaki idyopatik hidrosefalilerin %10
kadarında akuadukt stenozlu X-geçişli hidrosefali (Xq28, L1CAM) bulunmaktadır. SÇ’nda
daha az rastlanırken çocukluk çağında kitle lezyonlarına bağlı olarak stenoz sıklığı
artar. Konjenital hidrosefalilerin yanı sıra epileptik nöbetler de varsa diğer
serebral malformasyonlar gündeme gelir ve bu çocuklarda tabloya mental
retardasyon eşlik eder. Konjenital hidrosefalilerde X-geçişli akuadukt stenozu
dışında, İU dönemde maruz kalınan veya geçirilen radyasyon, alkol veya
enfeksiyonlar da etyolojik nedenler arasındadır.
Konjenital akuadukt
stenozu
Doğumda
12,8 mm boyunda ve en dar çapı 0,5 mm olan bu kanal internal olarak hemoraji ve
enfeksiyonlardan, eksternal olarak da tümör ve anomalilerden kolayca etkilenir.
Tek başına akuadukt stenozu görülebildiği gibi başka anomalilerle birlikte
(X-geçişli hidrosefali, L1 sendromu) de olabilir. Konjenital hidrosefaliler
içinde tek başına akuadukt atrezi veya stenozu %2 oranında görülmekte ve X’e
bağlı iletilmektedir. Akuadukt stenozlu çocuk doğarken hidrosefaliktir (40-50
cm) ve bu durum sefalo-pelvik orantısızlığa neden olur. Muayenede fontaneller
gergin, venler geniş ve sütürler aralanmıştır. Gözlerde batan güneş manzarası
ve abdusens paralizisi bulunabilir. İU dönemde yapılan sonografilerle
genellikle tanı konulur. Makrosefali tespit edildiğinde nöral kanal defektinin
araştırılması için amnios sıvısında alfa-fetoprotein (AFP) bakılması ve
kromozom analizi yapılması gerekir. Postpartum dönemde BT ile lateral
ventriküllerin ve 3. ventrikülün genişlediği, 4. ventrikül ve akuaduktal
kanalın görülemediği tespit edilir. Makrosefali saptandığı zaman amniotik AFP bakılması
nöral tüp defektlerini saptamak için önemlidir. Konjenital akuadukt stenozuna
bağlı konjenital hidrosefaliler BOS’u azaltıcı medikal tedaviye yanıt vermezler
ve ciddi bir seyir gösterirler. BOS’un ventrikül sistemi dışına
ağızlaştırıldığı bir cerrahi yaklaşım gerekir. Konjenital hidrosefalilerin
tedavi ve gidişi, birlikte olduğu diğer malformasyonların varlığına bağlıdır.
%70-80 oranında spina bifida gibi diğer anomalilerle birliktedir.
Ventriküloperitoneal şant, YD ve SÇ’nda ventriküloatrial şanta göre daha iyi
tolere edilmektedir. Hidrosefalinin gerilemesi ile gelişimin daha normal olma
şansı arttırılmış olur. Genellikle diğer anomalilerin varlığı motor kusurlara
yol açmaktadır.
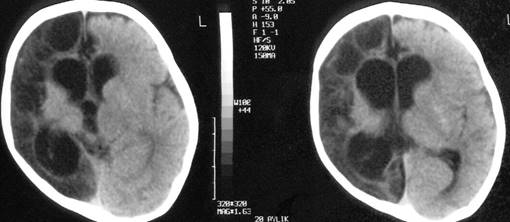
Dandy-Walker
malformasyonu
Bu
malformasyon serebellar vermisin parsiyel veya total agenezisi, posterior
fossada kistik dilatasyon ve hidrosefali olarak tanımlanırdı. Son zamanlardaki
tanımlarda ise 4.ventrikülün posterior yarısında balonlaşma ve sıklıkla eşlik
eden foramen magendi açılmasında yetersizlik, posterior serebellar vermis
aplazisi, inferior oliver nukleusun heterotopisi, serebral kortekste pakigiri
ve diğer serebral ve visseral anomalilerin eşlik edebildiği patolojilerden
oluşabildiği daha çok kabul görmektedir. Hidrosefali doğumda var olmayabilir ve
çocukluk ya da daha ileri dönemde gelişebilir. Lateral ventriküllerin genişliği
ile 4. ventrikülün kistik genişlemesi aynı oranda değildir. Dandy-Walker
varyant terimi ise serebellar vermis hipogenezi ve 4.ventrikülün kistik
genişlemesinin olduğu fakat posterior fossanın genişlemediği durumlarda
kullanılabilmektedir. Mega sisterna magnada ise mega sisterna ile
birlikte posterior fossanın genişlediği görülür, ancak serebellar vermis ve 4.
ventrikül normaldir. Birlikte rastlanılan en sık malformasyon korpus kallozum
agenezisidir. Bunun dışında heterotopi, anormal giral yapı, konjenital tümörler
ve akuadukt stenozu da birlikte bulunabilir.
Hastaların
%25’ine doğum sırasında, %75’ine 1 yaş sonuna kadar tanı konulmaktadır.
Makrosefali başlıca klinik özellik olup oksipital bölge frontal bölgeye göre
daha çıkıntılı durmaktadır. Posterior fossa yapılarındaki basıya bağlı olarak
değişik nörolojik defisitler görülebilir (apne epizodları, nistagmus, kranial
sinir parazileri, gövde ataksisi, piramidal bulgular). BT veya MR tanıyı
sağlamakla birlikte serebral anomalilerin ayrıntılarını seçmek bakımından MR
daha üstün bir incelemedir. Tedavide kist dekompresyonu semptomları azaltsa da
hidrosefalinin yineleme riski nedeniyle ventriküler şant gerekmektedir. Hatta
şant sadece lateral ventrikülden uygulanırsa beyinsapı semptomlarını
düzeltemeyeceğinden, lateral ventrikül ve posterior fossa kistinin dural şant
girişimi birlikte planlanmalıdır. Başarılı operasyonlara rağmen letarji, bu
hastalarda kişilik değişiklikleri ve kusma epizodları görülebilmektedir. Bu
epizodlar şantın işlemediği yönünde şüphe etmeyi gerektirse de çoğu zaman
sebebi bulunamamakta ve fatal seyretmektedir.
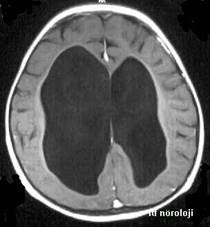
Şekil 8 a ve Şekil 8b. T1
ağırlıklı aksiyel kesitlerde komünikan hidrosefali, geniş posterior fossa ve
kist formasyonu, sağda belirgin hipoplastik serebellum, beyinsapı kompresyonu
(Dandy-Walker malformasyonu). İki buçuk yaşındaki hastanın kliniğinde ağır
motor-mental gerilik, aksiyel hipotoni, alt ekstremitelerde spastisite ve makrosefali
bulunmaktadır.
Klippel-Feil sendromu
Chiari
malformasyonu veya baziler impresyon ile birlikte olabilen ve kranioservikal
bölgede görülen bir malformasyondur. Buradaki hidrosefalinin nedeni BOS’un 4.
ventrikülden subaraknoid aralığa geçişinin engellenmesidir. Üç tipi vardır: I.
Torasik ve üst servikal vertebraların füzyonu, II. Hemivertebra ile birlikte
bir veya iki füzyon ve oksipito-atlantal füzyon (en sık), III. Servikal ve alt
torasik veya lomber füzyon.
Başlıca
klinik özellikler kısa boyun, ense saç çizgisinin aşağı yerleşimi ve boyun
hareketlerinin kısıtlı olmasıdır. Baş ve yüz asimetrileri, skolyoz, ellerde
ayna hareketleri de sıktır. Skapulanın aşağıya doğru migrasyon yetersizliğine
bağlı olarak tek veya iki yanlı skapula yerleşim anomalisi görülebilir
(Sprengel deformitesi). Hidrosefali, posterior fossa yapılarına basarak ataksi,
apne ve kranial sinir disfonksiyonlarına neden olabilir. Tanı için spinal
radyografilerle füzyonların, MR ile de Chiari malformasyonu ve ventrikül
dilatasyonları gibi ilişkili anomalilerin saptanması yeterlidir. Tedavide
miyelopatiyi önleyici servikal füzyon operasyonları, obstrüktif hidrosefali
bulguları için de şant yapılmaktadır.
Konjenital beyin
tümörleri
Erken
embriyogenez döneminde anormal hücre proliferasyonunun bir ajan tarafından
uyarılmasıyla oluştukları düşünülmektedir. Ajanın virulansı, etkili olduğu
dönem, süresi, fetusun sağlığı ve genetik yapı onkojenite ve teratojeniteyi
etkileyen başlıca faktörlerdir. SÇ’da en sık görülen tümörler astrositoma,
medulloblastoma, teratoma ve koroid pleksus papillomasıdır. Konjenital beyin
tümörleri supratentorial ve orta hatta olma eğilimindedir. Hemisferik gliom ya
da teratomu olan YD’larda İU veya postpartum ilk haftalarda genellikle akuadukt
stenozuna bağlı olarak hidrosefali gelişir. Koroid pleksus papilloması ise genellikle
bir lateral ventrikülde lokalize olup perinatal dönemden çok SÇ döneminde
semptomatik olurlar. Bunlardaki hidrosefaliye neden olan mekanizmalar foramen
Monro’nun tıkanıklığı ya da BOS’un aşırı yapımıdır. Medullablastomlar ise
posterior fossada yerleşerek 4. ventrikül ya da akuadukt stenozuna neden
olurlar. Tüm konjenital beyin tümörlerinde klinik semptomlar intrakranial
basınç artışına aittir (makrosefali, sütürlerin ayrılması, letarji, iritabilite,
beslenme güçlüğü, kusma). Posterior fossa yerleşimli olanlarda nistagmus,
gözlerin aşağıya deviasyonu, opistotonus ve apne gibi belirtiler de
görülebilir. Tanı İU dönemde ultrasonografi, postpartum dönemde ise BT ya da MR
incelemeleri ile konur. Koroid pleksus papilloması dışında konjenital beyin
tümörlerinde tam rezeksiyon olanağı azdır.
Galen veni
malformasyonu
Kuadrigeminal
tabakada olan Galen veni genişlediği zaman akuadukta bası yaparak lateral
ventriküllerin dilatasyonuna neden olur. Burada Galen veninin anevrizması söz
konusu değildir. Normal Galen veni gelişmez
ve median prosensefalik Markowski veni ise sabit kalır, dilate olur ve superior
sagital sinüse drene olur. Çok sayıda arteriovenöz fistül eşlik eder. Bu
anomali tüm AVM’lerin %1’i, pediatrik vasküler malformasyonlarının %30’unu
oluşturur. Çoğunlukla erkek çocuklarda görülür (%80). Erken belirtisi ya makrosefali
ya da juguler venin genişlemesi ile yüksek debili kalp yetmezliğidir. Erken
dönemde kanama görülmez, ancak kranial üfürüm hemen hep vardır ve bazı
çocuklarda da açıklanamayan dirençli bir hipoglisemi saptanır. Çocuklar
genellikle kardiyolojik olarak tetkik edilirken kranial patolojisi de açığa
çıkar. Eğer hemodinamik yük sorun oluşturmuyorsa tanı SÇ ve erken çocukluk
dönemine kadar gecikebilir. Bu vakalarda ilk semptom tegmentum ve akuaduktun
basısına bağlı olarak ortaya çıkan obstrüktif hidrosefalidir. Semptomlar
genellikle 5 yaş altında fakat mutlaka 10 yaşından önce ortaya çıkar.
Kontrastlı BT, MR ve kardiyak incelemeler tanıyı koydurur. Mortalite ve
morbiditesi yüksektir.
Walker-Warburg
sendromu (WWS)
O-mannosyltransferase-1 (POMT1)
ve -2 (POMT2) proteinlerini kodlayan gen mutasyonları ile daha az olarak da
fukutin gen (FKTN) mutasyonu ile ortaya çıkar. WWS, lizensefali
tip2 ve oküler anomalilerin (iris hipoplazisi, mikroftalmus, optik disk
kolobomu, optik sinir hipoplazisi, glokom, bustalmus) eşlik ettiği otozomal resesif
bir müsküler distrofidir. Hidrosefali genellikle akuadukt stenozuna ya da
Dandy-Walker malformasyonuna bağlı olarak doğumda saptanır. Ayrıca parsiyel ya
da total agiri (lizensefali) ciddi nörolojik defisite yol açar (bakınız:
serebral kortikal gelişim malformasyonları). Sadece hidrosefaliye yönelik şant
ameliyatı yapılabilir.
Hidranensefaliler
Normal
beyin gelişimi sırasında İU dönemde beyin parenkimini bozan bir olayla ortaya
çıkan, beyin dokusu yerine aşırı miktarda BOS’un biriktiği pek çok tablo bu
başlık altında incelenir. Eğer hidranensefali obstrüktif hidrosefaliye bağlı
ise bu çocuklarda kafa çevresi doğum sırasında büyüktür. Porensefali, İU gelişimin prosensefalizasyonu sırasındaki bir
defekt nedeniyle lateral ventriküller ve subaraknoid aralıkla ilişkili olan
hemisferik kistler için kullanılan bir terim iken, günümüzde İU veya perinatal
infarkt ya da travmaya bağlı gelişen hemisferik kistler için kullanılmaktadır (Şekil 9a ve 9b). Hasarlanan immatür
beyin nöron, glia ve destek dokusunu kaybeder ve yerini sıvı dolu kiste
bırakır. Kist içindeki basınç sıklıkla yüksek olup çevre dokuya bası yapar ve
makrosefaliye neden olur. Konjenital orta
hat porensefalisi konjenital hidrosefali, alopesi, parietal bölgede
ensefalosel ve posterior serebral bölgede orta hat defekti ile şekillenen bir
tablodur. İU dönemde gelişen makrosefaliler doğumda sefalopelvik uyumsuzluğa
neden olurlar. Bu çocuklarda ciddi mental retardasyon ve körlük vardır. Çoğu SÇ
döneminde ya da erken çocuklukta kaybedilirler. BT veya MR’de septum pellusidum
ve korpus kallozumun yapısını bozan büyük dorsal kist görülür. Kist geniş
lateral ventriküllerle ve ensefaloseli oluşturan kemik defekti ile
bağlantıdadır. Makrosefaliye ve ensefalosele yönelik operasyonlar dışında altta
yatan nörolojik hastalığa yönelik tedavi yöntemi yoktur.
Şekil 9a. T1 ağırlıklı aksiyel MR kesitinde sağ hemisfer
parenkiminde sekel yaygın kistik ensefalomalazik alanlar (3,5 yaşında, neonatal
sepsis, sol spastik hemiparezi)
Şekil 9b. Koronal MR kesitinde (FLAIR) sağ hemisferde
ensefalomalazi ve gliotik alanlar
MEGALENSEFALİLER
Anatomik Megalensefaliler
Metabolik
bir hastalık ya da akut bir ensefalopati olmaksızın hücre sayısı ve
büyüklüğündeki artışa bağlı olarak gelişen tablolardır.
Genetik
megalensefaliler
Nörolojik
ve mental fonksiyonları normal fakat kafa çevresi 98. persantilin üzerinde ve
ailesel özellik gösteren tablolardır. Otozomal dominant geçişli olduğu
düşünülmektedir. Doğumda normal olabilirler, fakat SÇ döneminde baş çevresi
büyür. Diğer organlara ait anomali ve iskelet deformiteleri yoktur. BT veya MR’nin
normal olması subaraknoid aralığın selim genişlemesinden ayırt ettirir.
Akondroplazi
Otozomal dominant geçişli genetik bir hastalıktır
(4p16.3). Ekstremitelerde rizomelik kısalık (ekstremite proksimali distalden
daha kısadır), makrosefali, yüzün orta hattında hipoplazi, lomber lordoz,
dirsek ekstansiyon kısıtlılığı başlıca klinik özelliklerdir. Servikomedüller
basıya bağlı olarak dispne, hiperrefleksi, spastisite ve solunum güçlükleri
beklenir. MR’de posterior fossanın küçük olduğu, sfenoid sinüsün geniş olduğu
saptanır. Bazen baziler impresyon görülebilir. Tedavide bazen dekompresyon
cerrahileri gerekebilir.
Gigantizm ile birlikte
megalensefali (serebral gigantizm, Sotos sendromu)
Çocuklar
henüz yenidoğan döneminde iken ağırlık, boy ve baş çevresinin büyüme değerleri
75-90. persantil ve kemik yaşları da 3 yaş ile uyumlu bulunur. Nükleer reseptör
bağlayıcı SET proteini 1’i kodlayan gen (NSD1) mutasyonu (5q35) olguların
%90’ında saptanır. Hemen her olguda makrosefali,
frontal çıkıklık, yüksek damak, hipertelorizm, %80 olguda değişik derecelerde
mental yıkım, otizm, daha az sayıda olguda farklı dismorfik özelliklere
rastlanmaktadır. BT’de ventriküler hafif genişleme dışında özellik saptanmaz.
Endokrin sisteme ait laboratuvar araştırmalarında glikoz intoleransı dışında
belirgin bir patoloji yoktur. İlk yaşta somatomedin seviyesi yüksek bulunabilir
ancak sonra normal seviyeye döner.
Nörokütane hastalıklar
Bu
hastalıklarda başlıca ortak özellikler epileptik nöbetler, mental retardasyon
ve cilt lezyonlarıdır. Cilt lezyonları incontinentia pigmentide (Ito hipomelanozu) olduğu gibi doğumdan itibaren
var olabilir ya da nörofibromatozis ve tuberoz skleroza benzer şekilde sonradan
da gelişebilir. Buradaki makrosefali hidrosefali ya da megalensefaliye bağlı
olabilir. Kinik olarak hemimegalensefali, vücutta hemihipertrofi ve bir
ekstremitede hipertrofi saptandığı zaman mutlaka nörokütane hastalıklar
taranmalıdır. Otozomal resesif geçişli Ito hipomelanoz’unda %25 oranında
megalensefali görülür. Linear nevus sebaceus, epidermal nevus ve Proteus
sendromunda da tam ya da hemimegalensefali görülebilir.
Metabolik Megalensefaliler
Pek
çok doğumsal metabolik hastalıkta madde birikimi ya da serebral ödeme bağlı
olarak megalensefali gelişir. Başlıca motor ve mental gelişme geriliği görülen
ve değişik nörolojik sistemlere ait patolojilerin bir ya da birkaçını birden
sergileyebilen genetik özellikli hastalıklardır. Canavan hastalığı ve ılımlı klinik seyir gösteren varyantları,
Alexander hastalığı, mukopolisakkaridozlar, GM2 gangliosidozlar, L-2 (OH) glutarik asidüri ve megalensefalik
kistik lökoensefalopati (MLC), bilinen
en iyi örneklerdir.
MİKROSEFALİ
Mikrosefali
baş çevresinin normalin 2 standart sapma altında olduğu durumlara denir.
Değişik etnik gruplarda standart değişmektedir. Primer mikrosefalilerde genetik
ve kromozomal nedenlere bağlı olarak geriye dönüşü olmayan bir tablo söz
konusudur. Mikrosefali ile doğanlarda antepartum dönemde beyin gelişiminde bir
hasar olduğu söylenebilir, fakat primer ya da sekonder mikrosefali olduğu ayırt
edilemez. Sekonder olanlarda beyin normal olarak gelişimini sürdürürken dış
faktörler bu süreci bozmaktadır. Çocuk YD döneminde normosefalik olup sonradan
mikrosefali gelişirse sekonder mikrosefali söz konusudur. Perinatal beyin
hasarları postpartum 3-6. aya kadar baş çevresinde bir küçülmeye neden olmazlar.
Beyin gelişimindeki gerilik açık kalan sütürlerin daha erken kapanmasına neden
olabilir. Kraniostenoz gibi kemiğin primer hastalıklarında ise beyin normal
gelişse bile sütürler erken kapanmaktadır. Primer mikrosefalide MR bulguları
normal ya da serebral malformasyonlara ait bulguları gösterir. Sekonder
olanlarda ventriküler genişleme, porensefali ve serebral atrofi gibi patolojik
bulgulardan biri ya da birkaçı bir aradadır. Başlıca mikrosefali nedenleri Tablo
4’te özetlenmiştir.
Tablo
4. Mikrosefali
nedenleri.
|
Primer mikrosefaliler |
Sekonder mikrosefaliler |
|
Mikrosefali
vera |
İU dönemde gelişen
hastalıklar: Enfeksiyon, toksin,
vasküler |
|
Kromozomal
ve genetik bozukluklar |
Perinatal beyin hasarları: Hipoksik-iskemik
ensefalopati Hemoraji vb. vasküler
olaylar Menenjit, ensefalit |
|
Nörilasyon defektleri: Anensefali Ensefalosel |
Postnatal sistemik
hastalıklar: Kronik kardiyopulmoner
hastalık Kronik renal hastalık Malnutrisyon |
|
Prosensefalizasyon
defektleri: Korpus kallozum agenezisi Holoprosensefali |
|
|
Serebral
kortikal gelişim malformasyonları |
|
PRİMER
MİKROSEFALİLER
Mikrosefali vera (genetik)
Otozomal
dominant ve resesif formları olan, beyin gelişme geriliği ile birlikte görülen
genetik defektlerdir. Dominant formu mental gelişimi normal ya da hafif
retarde, sıklıkla öğrenme güçlüğünün görüldüğü, klinik olarak da hafif olan
formdur. Bazı çocuklarda alın yapısının geride, palpebral fissür yapısının
bozuk oluşu ve kulak kepçesinin iriliğine rastlanmasına rağmen diğer dismorfik
özellikler çok sık görülmez. Çocukluk çağında epileptik nöbetler olabilmekle
beraber erişkin yaşlara doğru spontan olarak kaybolma eğilimindedirler. Resesif
formu ise daha ağırdır, yüz ve kafatası arasında karakteristik bir orantısızlık
vardır. Alın arkaya doğru eğiktir, kafa derisi oksipital bölgeye doğru kırışmış
gibi görülür. Alt çene küçük, kulaklar ve burun belirgin olup orta-ileri mental
retardasyonla birlikte değişik nörolojik defisitlere (spastik dipleji,
nöbetler) rastlanabilir. Dominant formda aile anamnezi tanı açısından önemlidir.
MR bulguları ise normaldir. Son yıllarda keşfedilen mikrosefali genleri
arasında MCPH1 29, ASPM30, CDK5RAP2, CNPJ 31, STIL 32, WDR62 2-4, CEP152 33
sayılabilir.
Kromozom bozuklukları
YD
döneminde her zaman mikrosefalik değillerdir. Eğer holoprosensefali gibi
serebral aplazilerle birlikteyse o zaman çocuklar mikrosefalik doğarlar. Bu
çocuklarda genellikle hipotoni ve dismorfik özellikler de vardır. Mikrosefali
genellikle SÇ döneminde belirgindir. Trizomiler (13,18), delesyonlar (4p-s,
5p-s, 13q-s, 18p-s, 18q-s) ve translokasyonlarda mikrosefali görülür. Ayrıca
karyotipi normal bazı dismorfik sendromlarda da mikrosefali vardır: Cornella de
Lange, Seckel, Hallerman-Streiff sendromu.
Nöral tüp kapanma defektleri (örnek: anensefali)
Bakınız:
Nörilasyon defektleri.
Prosensefalizasyon defektleri (örnek: holoprosensefali, korpus kallozum agenezisi, hücre migrasyon defektleri
‘lizensefali, heterotopi’)
Bakınız:
Prosensefalizasyon defektleri ve serebral kortikal gelişim malformasyonları
SEKONDER
MİKROSEFALİLER
İntrauterin dönemde gelişen hastalıklar
İU
dönemde mikrosefaliye yol açan başlıca enfeksiyon sitomegalovirus enfeksiyonudur. Başka sistemik bir hastalık
belirtisi olmadan tek başına mikrosefali yapabileceği bilinmektedir. Maternal
enfeksiyonun asemptomatik olduğu olgularda enfeksiyonun kanıtlanması güç
olmakla birlikte mikrosefalik olgular içinde yüksek seropozitifliğin saptanması
ve normosefaliklere göre anlamlı farklılık göstermesi bunu desteklemektedir.
Sitomegalovirus dışında rubella,
toksoplazma ve HIV de bu grupta sayılabilir. İU dönemde uyuşturucu ve diğer bazı ilaçların kullanımı
(kortizon, nitrojen mustard, valproat), alkolizm, kötü beslenme, radyasyon ve
uygunsuz sağlık koşulları mikrosefaliye neden olabilecek çevre faktörleri
içinde değerlendirilmekte ve beraberinde dismorfik özelliklere, gelişme
geriliğine de yol açabilmektedirler. Yine bu dönemde nadir görülen ve sebebi
bilinmeyen beyin damar malformasyonları
(aplazi, hipoplazi) ve çeşitli nedenlere bağlı infarktlar beslenmeyi bozarak
dolaylı olarak mikrosefaliye neden olabilirler.
Zika Virus
Afrika, Güneydoğu Asya,
Brezilya gibi bazı endemik bölgelerde görülebilmektedir. Etkilenen fetüslerde
ciddi mikrosefali ve hayat boyu nörolojik defisit söz konusudur. Antikor testi
ile tanı konur. Hamile kadınların endemik bölgelere gezilerinin engellenmesi koruyucu
bir yöntemdir.
Perinatal beyin hasarları
Perinatal
beyin hasarları neonatal ve SÇ döneminde ensefalopati tablosu oluşturarak beyin
gelişimini engellemektedir. Bu çocukların kafa çevresi doğumda normaldir.
Perinatal beyin hasarı, mikrosefali ve mental retardasyonu olan bu çocuklar
serebral felç (statik ensefalopati) başlığı altında incelenirler.
Postnatal sistemik hastalıklar
Kronik
sistemik hastalığı ve malnütrisyonu olan SÇ’larında genel bir gelişme geriliği
vardır. Boy ve kilodaki gerilik genellikle baş çevresi geriliğinden daha ön
plandadır. Ancak tedavi edilmezse beyin hasarları, beyin gelişme problemleri ve
mikrosefali gelişebilir.
KAFATASINDAKİ ANORMAL
ŞEKİLLER
Hem intrakranial hem de ekstrakranial nedenler
kafatasında anormal şekil değişikliklerine neden olabilir.
İntrakranial nedenler: Beyin gelişimdeki
anormallikler kemik yapının şekillenmesinde etkili olabilmektedir. Temporal lob
agenezileri kalvaryum şeklinin dar olmasına, serebellar agenezi posterior
fossanın küçük olmasına neden olur. Lateral ventriküllerin geniş olması frontal
kemiğin eğik olmasına, Dandy-Walker malformasyonu oksipital kemiğin eğik
olmasına, SÇ’nda subdural hematom varlığı sagital sütürün ayrılmasına ve
bitemporal geniş görünüme neden olur.
Ekstrakranial nedenler: İU dönemde uterus şekil
bozuklukları ve multipl fetüs gibi kafa kemiklerinin gelişimini fiziksel olarak
kısıtlayan nedenler kafa şeklini bozabilir. Fakat perinatal ve postnatal olarak
böyle bir etkilenme olmaz. Örneğin, uzayan doğum sırasında oluşan kafa şekil
anomalisi kalıcı olmaz. Prematürelerdeki skafosefali (uzun, dar kafa), bebeğin büyümesi
ve farklı yatış teknikleri ile zaman içinde düzelir.
Kraniostenoz
Kraniostenoz ya da kraniosinostoz
bir ya da daha fazla kranial sütürün prematür olarak kapanmasına verilen
isimdir ve bunun sonucunda da anormal kafa şekilleri ortaya çıkar. Bu süreç
SÇ’nun normal beyin gelişimi sırasında erken kapanmanın gerçekleşmesi ile
meydana gelir. Başka nedenlere bağlı mikrosefali gelişen SÇ’larında erken sütür
kapanması klasik kraniosinostozdan farklıdır. Olguların çoğu sporadik olup
diğerleri otozomal resesif ya da dominant
geçişlidir. Kraniosinostoz bir kromozomal bozukluk ya da genetik sendromun
parçası da olabilir. Bu tablolarda genellikle sindaktili, polidaktili ve
değişik ekstremite malformasyonları da eşlik eder. Klinik olarak sabit olan
bulgu anormal şekilli kafa yapısıdır. Skafosefali/dolikosefali sagital sütürün, brakisefali her iki koronal sütürün,
plagiosefali bir koronal veya bir
lambdoid sütürün, trigonosefali metopik
sütürün (İU dönemde iki parçadan oluşan frontal kemikler arasında) ve oksisefali de tüm sütürlerin
prematür kapanmaları ile oluşan kraniosinostozlardır.
Bu prematür kapanmalar intrakranial basıncın artmasına, komünikan ya da
nonkomünikan hidrosefalilerin meydana gelmesine yol açar. Sütürlerin tümü erken
kapandığında kafa normal şeklinde fakat küçüktür. KİBAS’a ait klinik ve
radyolojik bulgular yavaş geliştiğinden gözden kaçabilir, ekzoftalmus, optik
atrofi ve körlük ilk bulgu olabilir. Kraniosinostozlu hastalar altta yatan
başka bir neden yoksa zihinsel yönden normal olabilirler. Tablo ağır ise
zamanla zihinsel duraklama ve gerileme olur. Hidrosefalilerin oluşumunda birden
fazla mekanizma sorumludur. Kraniosinostozların %60’ı sagital sinostozdur ve
erkeklerde sıktır. Koronal sinostoz ise kraniosinostozların %20’sidir ve kız
çocuklarda daha fazla görülür. Tek sütür sinostozlarında gözlem ve palpasyon
tanı için yeterlidir. Konvansiyonel radyografilerde erken kapanan sütürün daha
fazla parladığı görülür. Günümüzde 3 boyutlu tomografi görüntüleri daha fazla
bilgilendirici olmaktadır. Birden fazla sütür kapanması ve hidrosefali şüphesi
varsa BT çekilmesi gerekir. Tedavi, şeklin düzeltilmesi ve hidrosefaliye
yönelik olmak üzere iki farklı amaç içerir.
Akrosefalosindaktili kraniosinostoz ile
birlikte parmakların füzyonunun görüldüğü bazen de mental retardasyonun eşlik
edebileceği bir tablodur. Apert sendromu ise
sindaktili ile birlikte brakisefalinin görüldüğü ve yüz görünümünün Crouzon
hastalığına benzediği bir başka kraniosinostoz tablosudur. Carpenter sendromunda da total kraniosinostoz, sindaktili,
polidaktili, hipogonadizm, obezite ve mental retardasyon vardır ve otozomal
resesif geçişli bir anomalidir. Crouzon
hastalığı (kraniofasial dizostoz) bir ya da tüm sütürlerin kapandığı,
otozomal dominant geçişli ve fasial kemiklerde gelişme anormallikleri ile
seyreden bir tablodur. Fasial deformasyonlar doğum sırasında vardır ve sonra
daha da kötüleşir. Gözler arası mesafe geniş ve gözler çıkık, maksiller
hipoplaziye bağlı olarak alt yüz yarımında çökük görüntü ve gaga şeklindeki
burun başlıca karakteristik yüz özellikleridir.
OMURİLİĞİN KONJENİTAL ANOMALİLERİ (Ayrıca
bakınız: Omurga ve Omurilik Hastalıkları)
Nöral tüpün kapanması nörilasyon olarak bilinir ve muhtemelen
rombensefalondan başlar. Bu kapanma süreci ilk kapanma noktasından kaudale ve kraniale
doğru gerçekleşir. Bazı hayvan deneylerinde nöral tüpün multipl bağımsız
noktalardan kapanabileceği ifade edilmiştir. Nöral tüpün en sefalik ucu
(anterior nöropor) gestasyonun 24. gününde, en kaudal ucu (posterior nöropor)
27. günde kapanır. Nöral tüpün kapanması esnasında ektodermden nöral yapının
ayrılması olayı ayrışma (disjunction)
olarak bilinir. Ayrışmadan sonra ektoderm nöral tüpün dorsalinde orta hatta
birleşir. Ektodermin fokal prematür ayrışması ile spinal lipomalar ve lipomeningoseller,
fokal ayrışma yetersizlikleri ile de dermal sinüslerin oluşum mekanizması
açıklanabilir. Spinal kordun diğer anomalileri, örneğin miyelomeningosellerde
ayrışma yetersizliği daha geniş bölgede meydana gelmektedir.
Spinal disrafizm
bir grup heterojen spinal anomaliyi kapsar. Bu gruptaki tüm lezyonlarda
mezenkimal, osseöz ve nöral dokunun orta hatta kapanma yetersizliğinin
bulguları vardır.
Spina bifida okültada lamina ve spinal çıkıntı
gibi spinal yapının kemik elemanlarının posteriorda kapanma yetersizliği söz
konusudur. L5-S1 bölgesinde sıktır ancak çoğunlukla asemptomatiktirler ve
radyografilerde tesadüfen saptanırlar. Spina bifida apertada
(Spina bifida sistika) spinal yapının tüm elemanları spina
bifidadan protrüde olur. Bu başlık altında basit
meningosel (nöral doku olmaksızın dura ve araknoidin protrüzyonu), miyelosel (nöral doku içeriği cilt ile
ortaya çıkmış fakat ciltten kabarık değil) ve miyelomeningosel (subaraknoid mesafe nöral plağın ventralinden
ekspanse olarak ciltten kabarık miyelosel varlığı) incelenmektedir. Okült
spinal disrafizm dermis ve epidermis altında gelişen bir grup
lezyondur. Sıklıkla subkütan lipom ve basit meningosel gibi cilt altı bir kitle
vardır. Herhangi bir nöral doku içermez. Bu grupta meningosel, diastematomiyeli
ve split notokord sendromu, dorsal dermal sinüs, gergin filum terminale, spinal
lipomalar ve miyelosistoseller sayılabilir.
Ayrışma anomalileri
(nondisjunction)
Miyelosel ve
miyelomeningosel
Nöral tüpün lokalize
kapanma defektleridir. Parapleji, hidrosefali, inkontinans, iskelet
anomalileri, mental problemler bulunabilir. Nörolojik defisitlerin nöral tüpün
açıklığı nedeniyle değil de gestasyon esnasında amniotik sıvının kimyasal iritasyonu
veya mekanik travma sonrası olduğu öne sürülmektedir. İU spina bifidanın
onarılması spinal kord hasarını durdurmaktadır. Bu nedenle meningomiyeloselin
MR veya US ile prenatal tanısı postnatal hastalık görülme oranını azaltacaktır.
Sıklıkla lomber bölgede görülür. Hidrosefali, serebellar tonsil herniasyonu
(Chiari II), siringomiyeli ve polimikrogiri gibi kortikal malformasyonlar eşlik
edebilir. Lomber ve lumbosakral miyelomeningoselli çocuklarda gergin spinal
kord da saptanabilir. Nöral tüp kapanma defektlerinin nedenleri kesin olarak
bilinmemekle birlikte folatın koruyucu rolü üzerinde yoğunlaşılmaktadır. Ayrıca
ailede nöral kapanma defektlerinin varlığı, gebelikte valproat kullanımı da
riskli grupta ele alınır. Gebelik döneminde tarama amacıyla serum AFP seviyesi
ölçülebilir. Miyelosel ve miyelomeningoseli olan yeni doğanlar 48 saat içinde
opere edilmesine rağmen sekel kalmaktadır.
Hemimiyelosel
Diastematomiyelisi olan
miyelomeningoselin özel bir formudur ve miyelomeningoselli hastaların %10’unda
görülür. İki hemikordun birinde küçük miyelomeningosel bulunur, diğer hemikord
sağlamdır. Filum terminale kalın ve gergindir. Etkilenen hastalarda
hemimiyelosel tarafında nörolojik defisit vardır. Miyelomeningoselli hastaların
%46’sında diastometamiyeli veya dermal sinüs görülür. Hidrosefali ve Chiari II
malformasyonları da eşlik edebilir.
Dorsal dermal sinüs
Cilt yüzeyinden içeri
doğru uzanan (%60-70 spinal kanala) epitel kaplı dural kanallardır. Bu anomali
nörilasyon sırasında kütane ektodermin nöral ektodermden ayrışmasındaki
yetersizlikle ortaya çıkar. Spinal kord daha sonra mezenşim ile kaplanınca bu
yapışıklık devam eder. Sıklıkla lumbosakral bölgede görülür. Her iki cinste de
eşit rastlanır ve fizik muayenede orta hatta, nadiren paramedian yerleşimli
hiperpigmente, kıllı nevüs veya kapiller anjiyom ile ilişkili nokta gibi bir
ağız saptanır. Genellikle enfekte olduklarında semptomatik hale gelirler.
Servikal miyelosistosel
Genişlemiş santral
kanalın dorsal subkütanöz yumuşak dokulara doğru spina bifidadan protrüzyonudur.
Kalınlaşmış skuamöz epitelin altında leptomeninkslerle birlikte displastik
nöral doku bulunabilir. Servikal miyelomeningosel olarak tanımlanması
yanlıştır. Çünkü servikal kordun etkilendiği seviyede kapanma tamdır.
Miyelosistosel ile doğan bebeklerde dorsal ya da servikodorsal orta hatta kalın
cilt dokusu ile kaplı kistik bir kitle vardır. Nörolojik muayeneleri genellikle
normaldir.
Kaudal bölge anomalileri
Embriyonal dönemde
kloakadan farklılaşıp gelişmeleri nedeniyle anorektal ve genitoüriner
anomaliler genellikle birlikte görülürler. Erken embriyonal dönemde spinal
kordda her nöral segment kendi seviyesindedir. Her sinir kökü kendi nöral
forameninden direkt olarak çıkar. Kemik yapı spinal kanala göre daha hızlı
büyüme gösterdiğinden zaman içinde spinal kord kanal içinde asılı kalır. Konus
medüllarisin seviyesi kesin olmamakla birlikte hayatın 3. ayında L2 vertebranın
üzerindedir. L2-L3 aralığından aşağıda saptanırsa anomali düşünülmelidir.
Filum terminalenin
fibrolipoması
Normal filum terminale
konus medullarisin ucundan başlar, ekstra ve intradural yerleşimlidir ve
kaudale doğru ilerleyerek koksigeal vertebranın dorsaline yapışır. Bu fibroz
ligamanın fibrolipomları ise miyelomeningosellerin %67’sinde
saptanabilmektedir. Eğer bir hastada gergin kord semptomları varsa fibrolipom
mutlaka araştırılmalıdır.
Gergin Filum Terminale
(Tethered Spinal Cord, Tight Filum Terminale)
Bu sendrom bir grup
nörolojik ve ortopedik deformiteden oluşur. Bulgular kısa kalın filum terminale
ve konus medullarisin aşağıda yerleşimi ile ilişkilidir. Retrogresif
farklılaşma ve filumu oluşturan nöral liflerin uzamasındaki yetersizlik sonucu
oluşur. Hastalar tüm yaşlarda birden ortaya çıkan nörolojik semptomlarla
gelebilirler. Alt ekstremitelerde asimetrik alt motor nöron bulguları, mesane
disfonksiyonları, duyu kusurları, sırt ağrıları, ayaklarda ortopedik anomaliler
eşlik edebilir. Bu sendrom disk dejenerasyonunu da hızlandırır. Yetişkinlerde
skolyoz ve sırt ağrılarına mesane disfonksiyonlarına göre daha sık rastlanır.
Normalde filum terminale çapı L5-S1 düzeyinde 2 mm veya daha altında iken bu
hastalarda 2 mm‘den kalındır. MR incelemelerinde dural kese, filum nedeniyle
posteriora doğru çadır şeklinde genişlemiş olarak görülür ve küçük bir lipoma
ile sonlanır. Cerrahi girişim sonrası yürüme bozuklukları 1/3 oranında
düzelebilmektedir.
Notokord gelişim anomalileri
Split notokord sendromu
Gestasyonun 3. haftasında
dorsal bölgede kalınlaşma, fokal hücre proliferasyonu ile Hensen nodülü
oluşmaktadır. Bu nodülün farklılaşması ve migrasyonu ile ektoderm endodermden
ayrılır ve notokord meydana gelir. Eğer ektoderm endodermden tam olarak
ayrılamazsa notokord ya bölünür (split) ya da sağa/sola doğru yerleşim
gösterir. Split notokord sendromu ventralde endoderm, dorsalde ektoderm ile
persistan bir bağlantının olduğu ayrık ya da yana doğru yerleşim göstermiş
notokord spektrumu için kullanılır. En ciddi formu dorsal enterik fistül olup
intestinal kavite dorsalde cilt yüzeyi ile bağlantılıdır.
Split kord malformasyonu
(diastematomiyeli)
Bu terim spinal kordun
sagital planda simetrik veya asimetrik iki hemikorda ayrılmasını içerir. Her
hemikord araknoid ile çevrilidir. Çoğu olguda alt torasik ya da lomber bölgede
rastlanır. Bölünme tüm kord kalınlığını içerebildiği gibi sadece anterior ya da
posterior yarıyı da içerebilir (parsiyel diastematomiyeli). Semptomlar herhangi
bir yaşta ortaya çıkar. Kızlar erkeklerden daha fazla etkilenir. Çoğu hastada
nevus, hemanjiyom gibi cilt belirtileri, %50 hastada da ortopedik ayak sorunları
görülür.
Sebebi bilinmeyen anomaliler
Dorsal meningosel
Dura ve araknoidin BOS sıvısını da içeren herniasyonu
dorsal meningosel olarak tanımlanır. Basit meningoselde bazen bir sinir kökü
dural keseye girse de genelde sinir dokusu içermez. Konus medullaris spinal
kanalda normal yerindedir. Kese subaraknoid aralık ile ilişkili olduğunda
Valsalva manevrası ya da hastanın pozisyonu ile kesenin çapı etkilenir.
Kompleks meningosele spinal anomaliler de eşlik etmektedir. Bunlar lokalize bir
spina bifidadan multisegmental spina bifida ve genişlemiş spinal kanala kadar
değişik derecelerdedir.
Genişlemiş bir nöral
foramenden BOS içeren dura ve araknoid protrüzyonudur. Kadın ve erkekler eşit
etkilenir ve genellikle asemptomatiktirler. Hastalarda nörofibromatozis veya
Marfan sendromu saptanabilir. Meningosel çapı küçük veya çok geniş olabilir.
Şant operasyonu ile iyi sonuçlar alınabilmektedir.
SEREBRAL FELÇ, (Beyin Felci, Statik Ensefalopati,
“Cerebral Palsy)
“Cerebral palsy” (CP)
hızlı beyin gelişimi döneminde meydana gelen lezyon veya anomalilere bağlı
gelişen, ilerleyici olmayan ve ağırlıklı olarak motor fonksiyonları bozan
tabloları ifade eden ortak bir terimdir. Günümüzde CP yerine statik
ensefalopati (SE) terimini kullanma eğilimi vardır. Tanımı nedeniyle heterojen
bir grup hastalık ve semptom kompleksi bu başlık altında yer almaktadır. Tarihte
ilk kez 1862’de Little ve 1897’de Freud tarafından doğum hipoksisine bağlı
diplejilerin geliştiği bildirilmiştir. Son yıllarda riskli gebeliklerin takibi,
doğum şartlarının düzeltilmesi ve yenidoğan yoğun bakım ünitelerinin gelişmesi
ile birlikte sağkalım oranı yükselmiş, bununla birlikte SE sayısında da artma
olduğu kanısı uyanmıştır. Gelişmiş ülkelerde SE insidansı ortalama olarak %0,2
olarak belirlenmiştir. SE gelişimine neden olan iskemik olaylar gebelik, natal
veya postnatal dönemde meydana gelebilir. Obstetrik ve perinatal bakımı
gelişmiş olan ülkelerde doğum anoksisi ve buna bağlı SE oranı oldukça düşük
olduğundan prenatal faktörler SE etyolojisinde ön plana çıkmaktadır. Buna
karşın perinatal/postnatal bakımı henüz yeterli olmayan gelişmekte olan
ülkelerde doğumda hipoksi, düşük doğum ağırlığı ve prematürelik etyolojide daha
fazla önem kazanmaktadır. Önceki yıllarda pek çok çalışmada kriter olarak
kullanılan APGAR skorlaması, ağlama zamanı gibi veriler şimdilerde çok
güvenilir bulunmamakta, ilk günlerdeki solunum desteği gereksinimi, konvülsiyon
geçirme, kalp atım hızı, kan pH, pO2 ve pCO2 takibi gibi
faktörler daha fazla değer taşımaktadır. Yine de SE gelişme riskini kesin
olarak yansıtan parametreler elde edilememiştir.
Etyopatogenez
Anoksiye bağlı olarak
dokularda laktik asit, eksitatör amino asitler, serbest oksijen radikalleri
artmakta, hücrelere kalsiyum girişi ile doku hasarı oluşmaktadır. Düşük doğum
tartılı bebeklerde normal ağırlıktaki bebeklere göre SE riski 3-10 kez artmakta,
bu grubun bir kısmını da prematüreler oluşturmaktadır. Prematürelerde beyin
damar yapısındaki immatürite (bazal lamina yetersizliği, lif azlığı vb.)
nedeniyle ani sıvı volüm değişiklikleri ve hipoksi intraventriküler ve
subepandimal kanamaların daha kolay oluşmasına yol açmaktadır. Annede
enfeksiyon, bebekte sepsis gibi durumlarda periventriküler lökomalazi görülmesi
enfeksiyon sırasında salınan sitokinlerin (TNF-alfa, IL-6) de SE’deki rolü
olduğunu düşündürmektedir. Son zamanlarda yapılan araştırmalar term
bebeklerdeki CP’nin az oranda perinatal olaylara (hipoksi-iskemi), daha çok da
prenatal problemlere bağlı olduğunu göstermektedir. Normal bebekte olmayan
ancak CP’li bebeklerde saptanan inflamasyon (IL-1, TNF-alfa) ve koagülasyon
anomalilerinin (faktör V Leiden mutasyonu, protein C ve S) etyolojide rol
oynadığı ileri sürülmektedir. Özellikle in utero vasküler olaylar ve hemiplejik
CP ile faktör V Leiden mutasyonu bağlantısı 1997’de gösterilmiştir. Kan-beyin
bariyerinde bozulma ve trombosit agregasyonuna yol açması nedeniyle tümör
nekroz faktör de (TNF) etyopatogenezde yer almaktadır. Tüm bu değişikliklere
rağmen akut beyin hipoksisi beyin tarafından çeşitli derecelerde dengelenmekte,
sinir sisteminin temel görevi olan adaptasyon kuralına göre ayrıntılarını tam
olarak açıklayamadığımız plastisite süreciyle beyin hiç ya da az zarar
görebilmektedir. Bu açıklanmaya çalışılan mekanizmaların yanı sıra bazı CP ailelerinin
tanımlanması ile CP’nin genetik yönü gündeme gelmiş ancak yeterli kanıtlar elde
edilememişken; 2000’li yılların ilk dekadında baş döndürücü şekilde gelişen yeni
genomik teknolojiler nörogelişimsel hastalıkları da aydınlatmaya başlamıştır. Bu
gelişmeler doğrultusunda 2016 yılında “The genetic basis of cerebral palsy”
başlığı ile yayınlanan makalede, “National Institute of Health Workshop on
Basic and Translational Research in CP” konusunda CP’nin 1/3’ünün genetik bazlı
olabileceğine dair giderek olgunlaşan ve kabul gören ortak fikirler olduğu
açıklanmıştır. Şimdiye kadar olan bilgilerimizden çıkardığımız sonuca göre CP
patogenezinde 4 temel genetik mekanizma tartışılmaktadır: Mitokondriyal
kalıtım, kopya sayısı değişiklikleri (copy number variation-CNV), epigenetik
adaptasyon ve genomik varyasyon/mutasyonlar. Şimdiye dek spastik para-kuadriplejik
CP’de adaptor protein 4 kompleks subunitleri (AP4M1, AP4B1, AP4S1, and AP4E1);
hemiplejik CP’de DIP2C, GRIK2, LAMA1, DMD, PTPRM; diplejik CP’de ADD3
mutasyonları ve muhtelif CP formları için TUBA1A, SCN8A, KDM5C, AGAP1, JHDM1D, MAST1,
NAA35, RFX2, ve WIPI2 gibi varyantlar üzerinde çalışılmaktadır.
SE Oluşumunda Risk Faktörleri
Prenatal dönemde annenin
metabolik, endokrin, kardiyak, hematolojik hastalıkları, mental geriliği,
alkol/kokain bağımlılığı, epilepsi, polihidramnios, koriyoamnionit, İU enfeksiyonlar,
plasental anomaliler, proteinüri, düşük ve ölü doğum öyküsü, iki gebelik
arsında kısa (3 ay altı) ya da uzun zaman aralığı (3 yıldan fazla), eklampsi,
preeklampsi, fiziksel ve kimyasal travmalar SE riskini arttırmaktadır. Yine
prenatal dönemde konjenital malformasyonlar, kromozom anomalileri, enfeksiyon
(TORCH), çoğul gebelik, ikiz eşinin ölümü, hidrops fetalis, kafa çevresinin
küçük gelişimi fetusa ait risk faktörleri olarak sayılabilir. Başka bir bakış açısıyla
kronik prenatal hipoksinin konjenital malformasyonlara yol açarak SE
tablolarını oluşturmasının yanısıra, bazı ağır malformasyonların güç ve
hipoksik doğuma neden olması, bu faktörlerin hem sebep hem sonuç olduğunu
düşündürmektedir. Bu durumda prenatal faktöre bağlı gelişecek SE’lerde doğum
şartlarının düzeltilmesi de SE gelişimini engelleyemeyecektir.
Natal dönemde makat
gelişi başta olmak üzere diğer geliş anomalileri, fetal kalp hızının düşüklüğü
(60/dk altı), uzamış doğum, müdahaleli doğum, plasentanın erken ayrılması,
amniotik sıvının mekonyumla boyanması, mekonyum aspirasyonu, fetusta metabolik
asidoz gelişimi, prematürite ve postmatürite SE için risk faktörleri grubunda
yer almaktadır.
Yenidoğan döneminde
asidoz, sepsis, apne, anormal nörolojik bulgular (hipotoni, zayıf emme,
konvülsiyon vb.), hipoglisemi, düşük APGAR değerleri (3’ün altı), ilk 5
dakikada ağlamama, yüksek hiperbilirubin değerleri de halen SE risk faktörleri
listesi içinde sayılmaktadır.
Patoloji
Otopsi çalışmalarında
saptanan başlıca bulgular intraventriküler ve periventriküler kanama, damar
tıkanmaları ve gelişim malformasyonlarıdır. İntraventriküler ve periventriküler
kanamaya sıklıkla prematüre bebeklerde, nadiren de term bebeklerde rastlanır.
Doğum ağırlığı 1500 gr’ın altında olanlarda %20, 500-700 gr arasında %50 gibi
bir oranda görülebilmektedir. Prematürelerde oluşan kanamalar %90 oranında kaudat
çekirdek başındaki germinal matrikste ve %50 oranında bilateral (sol taraf daha
fazla etkilenmek üzere) olur. Hayatın ilk 48-72. saatinde ve sıklıkla respiratuvar
distres sendromu gibi hipoksik durumlarda ortaya çıkar. Görüntüleme
yöntemlerinde saptanan bulgulara göre kanama evreleri:
Evre I: İzole
subepandimal kanama,
Evre II: Ventriküler dilatasyon
olmaksızın intraventriküler kanama,
Evre III: Ventriküler
dilatasyon ile birlikte intraventriküler kanama,
Evre IV: Evre III’e ek
olarak parenkim içi kanama olarak belirlenmiştir.
Kanamaların görüldüğü bu
olgularda sekonder gelişen BOS dolaşım bozuklukları ile oluşan hidrosefaliler
morbiditeyi etkileyen önemli bir faktördür. Kanamanın yanı sıra periventriküler
venöz infarktlar da zaman içinde yerini nekrotik dokuya bırakır.
Hipoksik iskemik
ensefalopati gestasyon yaşına ve hipoksinin şiddetine göre başlıca 5 tipte
serebral lezyona neden olur:
1. Status marmoratus, bazal ganglionların (özellikle putamen, kaudat
çekirdek) bilateral, simetrik, mermer görünümüne verilen isimdir. Bu görünümden
sorumlu olan patoloji nöron kaybı, gliozis ve hipermiyelinizasyondur. Perinatal
dönemde ve term bebeklerde meydana gelir. Başlıca ekstrapiramidal sistem
belirtilerine yol açar.
2. Simetrik talamik lezyon, nöron kaybı ve astrogliozisin neden olduğu,
bazen de kalsifikasyonların eşlik ettiği bir tablodur. Perinatal asfiksi kanıtı
olmayan term bebeklerde görülmekte ve kötü prognoz göstermektedir. Doğumdan 2-4
hafta önce meydana gelen olası hipoksik-iskemik bir olayı düşündürür. Doğumda
ya da hemen ilk günlerde spastisite, emme-yutma güçlüğü, primitif refleks
yokluğu saptanır.
3. Watershed infarktlar, tipik olarak parasagital bölgelerde, ana
serebral arter beslenme alanları arasındaki sistemik kan basıncı
değişikliklerine hassas sınır bölgelerinde, bilateral, bazen asimetrik görülen
infarktlardır. Sıklıkla term nadiren preterm bebeklerde görülür. Spastik CP,
görme ve işitme problemleri saptanabilir.
4. Periventriküler lökomalazi immatür beyinde periventriküler beyaz
maddedeki nekrotik alanlardır ve özellikle de lateral ventriküllerin posterior
ve yan komşuluklarında saptanır. Bu lezyonların ventriküllerin arka
boynuzlarında genişleme ile belirlenen görüntüsü tipiktir (Şekil 10a ve 10b). Lokalizasyonu nedeniyle motor korteksten inen
liflerin geçtiği alanlar söz konusu olduğundan alt ekstremiteleri ilgilendiren
spastik dipleji sıktır. Lezyonun daha laterale uzanması ile kollara giden
lifler de etkilenir ve terapleji saptanabilir. Postnatal olaylarla
periventriküler lökomalazi arasında zayıf bir birliktelik bulunması bu
patolojiye yol açan kaskadın doğumdan önce başladığını düşündürmektedir.
Gerçekten de sıklıkla preterm ve 1500 gr altı bebeklerde görülür.
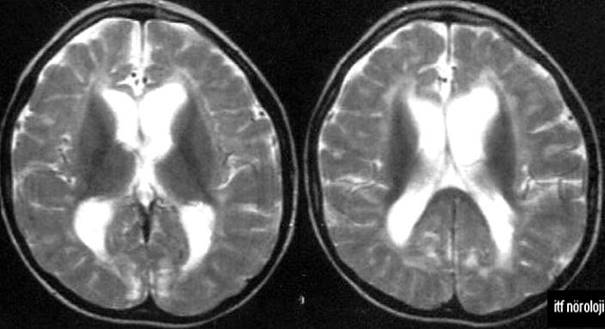
Şekil 10. T2 ağırlıklı aksiyel MR
kesitlerinde periventriküler lökomalazi
5. Selektif nöronal nekroz ise term bebeklerde serebral ve serebellar
korteksin değişik derecelerde etkilendiği, genellikle bilateral, bazen de
asimetrik olan etkilenmelerdir. En çok parietooksipital bölgelerde yer
aldıklarından parezi üst ekstremitelerde hakimdir. Kranial MR’de bulgu
vermeyebilir. Yaşayan bebeklerde spastik tetraplejik, hemiplejik, ataksik CP,
epilepsi, mental gerilik gibi bulgular saptanır.
6. Fokal ve multifokal beyin nekrozları
ana serebral damarların dağılım alanlarına uygun bölgelerdeki nekroz alanlarıdır.
1/3 olguda orta serebral arter beslenme alanında (Şekil 11 a, 11b) ve genellikle 32 haftalık gestasyon yaşından büyük
bebeklerde meydana gelir. Çoğunlukla hemiplejik ya da tetraplejik CP olarak
karşımıza çıkarlar. Nekroz ve kavite oluşumundan kaynaklanan büyük lezyonlar
porensefali, hidranensefali ve multikistik ensefalomalazi olarak
adlandırılmaktadır (Şekil 12).
Gelişimsel
malformasyonlar ise embriyonal süreçte kök hücre proliferasyonu, farklılaşması,
nöron migrasyonu ve organizasyonu dönemlerinde meydana gelen olaylara bağlı
olarak bir ya da daha fazla anomalinin (lizensefali, heterotopi, şizensefali,
polimikrogiri vb.) görüldüğü SE tablolarıdır (Bakınız: Serebral kortikal
gelişim malformasyonları).
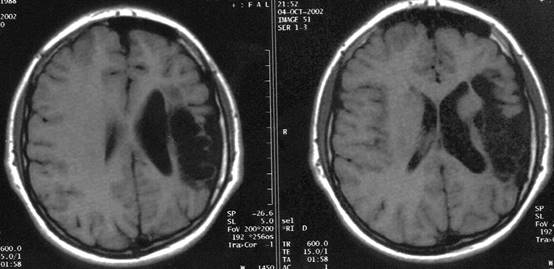
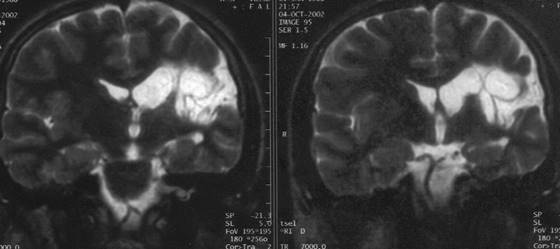
Şekil 11. T1 ağırlıklı aksiyel (üst) ve T2 ağırlıklı koronal (alt) MR kesitlerinde sol orta
serebral arter alanında sekel kistik ensefalomalazi, lateral ventrikülde
çekilme (4 yaşında, siyanotik doğum, sol spastik hemiparezi, hafif mental
gerilik).
Şekil 12. T1 ağırlıklı aksiyel MR kesitinde yenidoğan döneminde
ağır hipoksi sekeli olarak yaygın multikistik ensefalomalazi, volüm kaybı,
sekonder ventriküler dilatasyon (16 aylık, aksiyel hipotoni, altta belirgin spastik
tetraparezi, ağır mental gerilik)
CP’nin
çeşitli klinik formları:
1. Hemiplejik CP
2. Tetraplejik CP
3. Diplejik CP
4. Diskinetik CP
5. Ataksik CP
6. Mikst tip CP
1. Hemiplejik CP: Yaşamın ilk 3 ayında fark
edilmesi güçtür. Sonraki aylarda çocuğun bir taraf el ve kolunu daha az
kullandığı, zamanla klasik spastik postürün yerleştiği fark edilir. Hemiparezi,
çocuk objeleri yakalamaya başlayıncaya ve elini kullanmada yetersizlik
gösterdiği zamana kadar fark edilemeyebilir. Bu da lezyonun postnatal geliştiği
izlenimine neden olabilmektedir. Bu çocuklar yürüyebilseler de normalden daha
geç zamanda ve etkilenmiş ayağın parmak ucuna basarak yürürler. Lezyon parietal
lobu etkilemişse ilgili ekstremiteler ince ve kısadır. Etkilenen bölgelere göre
kortikal duyu bozuklukları, hemianopsi ve ihmal tabloları da saptanır.
Spastisite ilk iki yılda artış gösterir. Korteks etkilenmişse epileptik
nöbetler ve davranış bozuklukları görülebilir. Konuşma gecikebilir, fakat zeka
genellikle normaldir.
2. Tetraplejik CP: En şiddetli CP formudur.
Çeşitli derecelerde mental yetersizlik de eşlik eder. Çocuklarda çoğunlukla
yürüme fonksiyonu gelişemez, anormal baş ve ekstremite postürleri görülebilir.
Genel olarak başkalarına bağımlı yaşarlar. Psödobulber tablo ağır ise konuşma
yetersiz olabilir ve beslenme için de gastrostomiye ihtiyaç duyabilirler.
Epileptik nöbetler sık ve tedaviye dirençli tiptedir. Mikrosefali sıklıkla eşlik
eder. İlkel refleksler de geç dönemlere kadar saptanabilir.
3. Diplejik CP (Little hastalığı): CP’nin
en sık görülen formudur (%40-50). Prematüre CP grubunda diplejik CP daha yüksek
bir orandadır. Genellikle dört ekstremite de etkilenmiş olmakla birlikte
spastisite bacaklarda ön plandadır. Spastisitenin belirginleşmesi ilk yıllarda
yavaş ve sinsidir. Emekleme döneminde bacaklarını kullanamadıkları için
karınları üzerinde emekleme çabası gözlenebilir. Aksiller asma muayenesi
sırasında tipik makaslama ya da parmak ucuna basma postürü belirginleşir. Şiddetli
olgularda kalça subluksasyonları ve dislokasyonları gelişebilir. Sfinkter
bozukluğu genellikle görülmez. Zeka ve konuşma sıklıkla korunmuştur, fakat elin
ince motor hareketlerinde beceriksizlik saptanır. Epileptik nöbetlere 1/3
olguda rastlanabilir.
4. Diskinetik CP: Bazal ganglion
lezyonları sonucu koreatetoz, distoni gibi ekstrapiramidal semptomların
görüldüğü formdur. Çocukluk çağı distonilerinin en önemli bölümünü oluşturular.
Bilirubin yükselmeleri günümüzde daha iyi tedavi edildiğinden bu grupta
kernikterus eskisi kadar önemli bir etyolojik faktör oluşturmamaktadır. Olguların
2/3’ü perinatal-neonatal dönemde hasar görmektedir. Ciddi anoksiye bağlı olarak
gelişen bu tablolarda fasial diskinezi nedeniyle çocuklar konuşamazlar, işitme
ve el kullanımı ile ilgili problemleri vardır. Üst ekstremite tutulumu %85 ve
oromandibular fonksiyon bozuklukları %60-70 gibi bir oranda rastlanır. Kognitif
fonksiyonlar ve alt ekstremite motor fonksiyonları etkilenebilmekle birlikte
diğer CP tiplerine göre şiddetli olmayabilir. İstemsiz hareketler doğumda
mevcut değildir; ilk bir yıldan sonra belirginleşmeye başlar. Erken bebeklik
döneminde hipotoniktirler. Distoni fenomenolojisi %70, koreatetoz ise %30 gibi
bir sıklıkta görülür. İstemsiz hareketlerin sonraki yıllarda hafif progresyon
gösterebilmeleri nedeniyle idyopatik torsiyon distonisi ve diskinezi ile
seyreden metabolik ve dejeneratif hastalıklardan ayırt edilmesi gerekebilir. MR’de
bazal ganglion lezyonları %65-70, ek olarak ak madde hasarı %15-25 oranında
görülürken %5-15 oranında MR normal de bulunabilir.
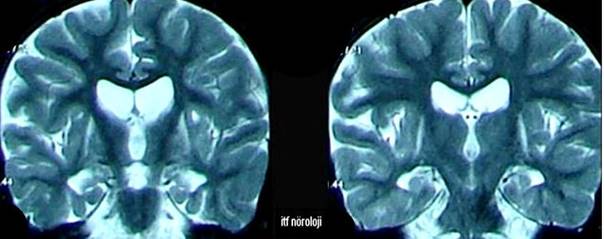
Şekil 13. T2 ağırlıklı koronal kesitlerde bilateral
putamen lateralinde sinyal artışı (7 yaş kız çocuğunda distonik CP tablosu,
evde zor doğum, ağlamada gecikme, siyanotik olma, 15. gün JK, motor-mental
gelişim geriliği).
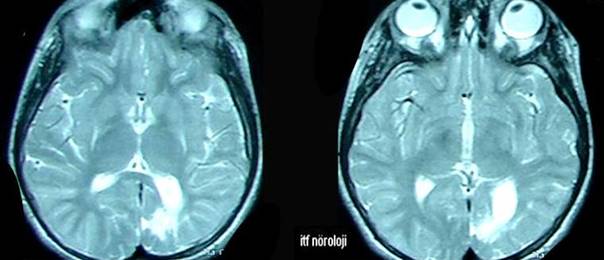 Şekil 14.
T2 ağırlıklı aksiyel kesitlerde sol parietooksipital ak maddede malazik
değişiklikler ve iki taraflı lentiform çekirdeklerin posterior kısmında sinyal
artışı izlenmekte (13 yaş kız çocukta distonik CP, 15 günlükken 1 saat süren
ağır asfiksi, 1 hafta solunum cihazına bağlı kalma, oturma ve yürüme yok).
Şekil 14.
T2 ağırlıklı aksiyel kesitlerde sol parietooksipital ak maddede malazik
değişiklikler ve iki taraflı lentiform çekirdeklerin posterior kısmında sinyal
artışı izlenmekte (13 yaş kız çocukta distonik CP, 15 günlükken 1 saat süren
ağır asfiksi, 1 hafta solunum cihazına bağlı kalma, oturma ve yürüme yok).
5. Ataksik CP: En nadir formdur.
Sıklıkla serebellum ve bağlantılarında hasar ve anomaliler söz konusudur.
Çoğunlukla tabloya hipotoni hakimdir. Gövde ve yürüme ataksisi ekstremite
ataksisine göre daha belirgindir. Yürüme geç dönemlerde gerçekleşebilir, ancak
sık düşmeler gözlenir. Nistagmus sık görülmez ve konuşma dizartriktir.
6. Mikst tip CP: Diskinetik ve spastik CP
ya da ataksik ve diskinetik CP formlarının kombinasyonları olan tablolardır.
Tüm bu klinik formların
yanısıra günümüzde motor defisit olmaksızın dil gelişim problemleri, özel
öğrenme güçlükleri gibi bazı tablolarda doğumda asfiksi anamnezinin alınması
farklı boyutlarda SE’lerin olabileceğini düşündürmektedir.
Tanı
CP klinik olarak
tanınabilen, ilerleyici olmayan, sıklıkla motor semptomların oluşturduğu bir
sendromdur. Tablonun erken tanınması zihinsel ve bedensel özürlülüğün önlenebilmesi
amacıyla tedavi stratejisinin oluşturulması ve prognozun belirlenmesi açısından
önem kazanmaktadır. Spesifik bir laboratuvar metodu olmamakla birlikte
ultrasonografi, BT, MR ve MR-spektroskopi gibi görüntüleme yöntemlerinden ve
görsel/işitsel uyandırılmış potansiyellerden tanı desteği olarak
yararlanılmaktadır. Ön fontanelin açık olduğu zamanlarda ultrasonografi pratik
ve ucuz bir yöntem olarak yardımcı olur. Ancak küçük heterotopiler, fokal
displaziler gibi gelişim malformasyonlarının saptanması ve miyelinizasyonun
takibi MR ile yapılabilir. Bazı displazileri ayırt edebilmek için T1 inversion
recovery ve FLAIR sekansları ile 1,5-3 mm‘lik
ince kesitlere de gereksinim duyulabilmektedir. MR ile elde edilen bu
verilerle etkilenme zamanı tahmin edilebilir. Örneğin orta serebral arter
alanındaki bir ensefalomalazik alan bir term hasarını düşündürürken lizensefali
saptanması gestasyonun 7-24. haftaları arasındaki döneme yönlendirir. Ayrıca CP’nin
diğer yavaş seyirli santral sinir sistemi hastalıklarından ayırt edilmesinde
çoğu zaman MR yardımcı olmaktadır. Son yıllarda İU dönemde fetal anomalileri
saptamak ve cerrahi müdahale şansını değerlendirmek amacıyla fetusa yönelik MR
incelemeleri yapılmaya başlanmıştır.
Tedavi
Bu çocukların değerlendirilmesi
multidisipliner bir yaklaşımla, tüm sorunlarına yönelik olarak nörolog ya da
çocuk nöroloğu, fizyoterapist, ortopedist, sosyal danışman, pedagog ve psikologdan
oluşan profesyonel bir ekibi gerektirmektedir. Görme ve işitmenin değerlendirilmesine,
konuşma ve beslenmeyle ilgili uzman kişilere de ihtiyaç vardır. Gelişmiş
ülkelerde ayrı merkezlerde konuyla ilgili uzmanlar bu çocuklara mental ve
fiziksel rehabilitasyon uygulamaktadırlar.
Motor defisitlerin
derecesine göre fizyoterapi başta olmak üzere spastisiteye yönelik baklofen,
botilinum toksini, dantrolen, diazepam, intratekal baklofen, hipersalivasyon
için skopolamin, distoniye yönelik triheksifenidil, L-dopa gibi medikal
tedaviler ve tenotomi, selektif dorsal rizotomi, stereotaktik talamotomi gibi
cerrahi girişimler uygulanabilmektedir. Kognitif olarak daha az etkilenmiş
olanlar özel sınıflarda eğitilerek topluma kazandırılabilirler. Translasyonel
yaklaşımlar CP’de de kişiye özel tedaviler üzerinde araştırma yapmak gereğini
vurgular.
Güncel bilimsel
konseylerin görüşü, CP için hem tanı, hem tedavi ve hem de araştırma
konularında sadece tıbbi branşların (nöroloji, çocuk hastalıkları, fizik tedavi
ve rehabilitasyon, ortopedi, göz hastalıkları, kulak-burun-boğaz, radyoloji,
gastroenteroloji, diş hekimliği, psikiyatri) bir araya gelmesinin değil, aynı
zamanda translasyonel bir yaklaşımla tıp, moleküler tıp/biyoloji, sosyoloji,
mühendislik, bilişim, etik, felsefe, istatistik gibi pek çok bilim dalının
birlikte çalışmasının gerektiği yönündedir.
KAYNAKLAR
- Barkovich AJ ve Raybaud CA. Congenital Malformations of the Brain and
Skull. Içinde: Barkovich J ve Raybaud
CA editörler. Pediatric Neuroimaging. 6.baskı. Philedelphia:
Wolters-Kluwer; 2019. s.565-859.
- Dobyns WB, Leventer RJ, Guerrini R. Malformation of Cortical
Development. İçinde: Swaiman KF, Aschwal S, Ferriero D, Schor NF, Finkel
RS, Gropman AL, Pearl PL, Shevel MI editörler. Swaiman’s Padiatric
Neurology. 6.Baskı. China: Elseiver; 2017. s.218-225.
3.
Fahey
MC, Maclennan AH, Kretzschmar D, Gecz J, Kruer MC. The genetic basis of cerebral
palsy. Dev Med Child Neurol. 2017; 59 :
462-9.
- Krageloh-Mann I. Cerebral Palsy and Related Movement Disorders. Alexis
Arzimanoglou with Anne O’Hare, Michael Johnston ve Robert Ouvrier editörler.
İçinde: Aicardi’s Diseases of the Nervous System in Childhood. 4.Baskı. London:
Mac Keith Press; 2018. s.347-364.
- Mirzaa MG ve Dobyns WB. Disorders of Brain Size. İçinde: Swaiman KF,
Aschwal S, Ferriero D, Schor NF, Finkel RS, Gropman AL, Pearl PL, Shevel
MI editörler. Swaiman’s Padiatric
Neurology. 6.Baskı. China: Elseiver;
2017. s.208-217.
- Pina-Garza JE ve KC James. Disorders of Cranial Volume and Shape . İçinde.
JE Pina-Garza ve KC James editörler. Fenichel’s Clinical Pediatric
Neurology. 8.Baskı. Philedelphia: Elseiver; 2019. s. 346-364.
- Sanchez P, Graham JM. Congenital Anomalies of the Skull. İçinde:
Swaiman KF, Aschwal S, Ferriero D, Schor NF, Finkel RS, Gropman AL, Pearl
PL, Shevel MI editörler. Swaiman’s Padiatric Neurology. 6.Baskı. China:
Elseiver; 2017. s.233-241.
- Yapıcı Z. Serebral Palsi, Güncel Yaklaşım ve Genetik. İçinde: Uğur Özbek, Nihan Hande Akçakaya
editörler.“Cerebral Palsy’ye Güncel Yaklaşım ve Genetik. İstanbul: Boyut
Yayınları; 2019. S.6-8.
- Yapıcı Z. Serebral Kortikal Gelişimsel Anomaliler ve Epilepsi. İçinde.
İbrahim Bora, Naz Yeni, Candan Gürses editörler. Epilepsi. 2.Baskı.
İstanbul: Nobel Tıp Kitabevleri; 2018. s.129-163.
- Yıldız E. Kraniosinostozlu Olgularda Radyolojik Değerlendirme. Türk
Nöroşir Derg 2017; 27 : 263-277.
11.
Zarrei
M, Fehlings DL, Mawjee K, Switzer L, Thiruvahindrapuram B, Walker S, Merico D,
Casallo G, Uddin M, MacDonald JR, Gazzellone MJ, Higginbotham EJ, Campbell C,
deVeber G, Frid P, Gorter JW, Hunt C, Kawamura A, Kim M, McCormick A, Mesterman
R, Samdup D, Marshall CR, Stavropoulos DJ, Wintle RF, Scherer SW. De novo and rare inherited
copy-number variations in the hemiplegic form of cerebral palsy. Genet Med. 2018; 20
: 172-180.