FRONTOTEMPORAL DEMANS
Yazanlar:
İ. Hakan Gürvit, Bedia Samancı
(kapsamlı web versiyonu)
Son güncelleme tarihi: 14.10.2020
Frontotemporal demans (FTD) tipik olarak ilerleyici davranış değişikliği ve ilerleyici dil bozukluğu tablolarıyla sunulur. Nasıl ki Alois Alzheimer'in Aguste D. isimli kadın hastası Alzheimer hastalığının ilk olgusu olarak değerlendirilmekteyse, Arnold Pick’in 1892 yılındaki “Afazi ve Beynin Senil Atrofisi Arasındaki İlişki Üzerine” isimli makalesinde, bildirdiği bir ilerleyici afazik olan adaşı August H. isimli erkek hasta FTD tarihinin miladı sayılabilir. Bu olguyla aynı zamanda FTD şemsiyesinin 3 temel klinik sunumundan biri olan dilsel varyant da ilk kez tanımlanmış olmaktadır. “Erişkinlerin Primer Progresif Demansı Üzerine” isimli, 1904 tarihli bir sonraki makalesinde ise bu kez de en sık klinik sunum olan davranışsal varyantı ilk kez tarif edecektir. Pick’in bu klinik tablonun nöropatolojik karşılığı olarak ortaya koyduğu intranöronal globüler arjirofilik inklüzyonlu cisimcikler için Pick cisimcikleri (PiC) terimini 1911 yılında ilk kez Alzheimer kullanmıştır. İzleyerek PiC yanı sıra balonlaşmış şiş nöronlar (Pick hücreleri), yüzeyel kortikal spongioz, glioz ve nöron kaybı olarak belirlenen bu antite 1922’de Gans tarafından Pick atrofisi ve nihayet 1926’da Onari ve Spatz tarafından Pick hastalığı (PiH) olarak adlandırıldı. Hastalığın baskın ailevi özelliğinden de ilk kez sözeden Gans olmuştur. Formel tanı kriterleri arasına dahil edilmese de 3. temel klinik sunum olduğuna dair yaygın bir uzlaşım bulunan motor varyantın antiteleri olan progresif supranükleer paralizi (PSP) ve kortiko-bazal dejenerasyon-sendromun (KBD-KBS) tanımlanması için 1960'lara gelinmesi gerekti.
PiH için ilk büyük serinin yayınlanması ise Pick'in 2. olgusundan 70 yıl sonra gerçekleşti. Cenevre Beyin Bankası’ndan bildirilen 40 olguluk Constantinidis serisinde Pick hastaları klinik olarak davranışsal artı dilsel veya yürütücü bozukluk artı parkinsonizm olarak özetlenebilecek 2 tarzda sunulmuşlardı. İlk sunum temporo-orbitofrontal, ikincisi frontal konveksite atrofisi ile ilişkiliydi. İlk atrofi tarzını sergileyen 10 olguda PiC ve Pick hücreleri (PiHc) birlikte görülürken (Tip A), aynı atrofi tarzına sahip 8 olguda iki işaretleyici de görülmemişti ve izole gliozis mevcuttu (Tip C). Frontal konveksite atrofisi şeklindeki ikinci tarz toplam 14 olguda mevcuttu. Bunların 10’unda PiC olmaksızın sadece PiHc (Tip B), dördünde ise yine PiH işaretleyicilerinin ikisi de olmaksızın sadece gliozis mevcuttu (Tip D). Bu bildiri ilk kez PiH klinik sunumunun kayda değer bir oranının altında PiH’e özgü işaretlerin (yani, PiC ve PiHc) olmayabileceğine dikkat çekmekteydi.
Tau proteini ilk kez 1977’de pürifiye edildi ve mikrotübül asamblesinden sorumlu olduğu bulundu.
Bilgisayarlı tomografinin (BT) erken dönemlerinde Cummings ve Duchen tarafından 5 ağır anteriyor temporal atrofili PiH olgusu bildirildi. Tümünde de atrofi PiC ile ilişkiliydi ve amygdalayı içine almaktaydı. Yazarlar davranışsal özellikleri “Kluver-Bucy sendromu” unsurları olarak özetlediler. Dilsel bozuklukların da ön planda olduğu, bellek ve mekansal oryantasyonun göreli korunmuş olduğu vurgulanarak bu klinik tablonun PiH’in bu varyantı için ayırt edici olabileceği ileri sürüldü. Böylelikle, PiH’in nöro-görüntüleme karşılıkları ve klinik sunumunun Alzheimer hastalığı (AH) ile kontrast oluşturan yönlerine ilk kez vurgu yapılmış oldu. Ertesi yıl Mesulam sonradan primer progresif afazi (PPA) adını vereceği ilk ilerleyici afazi serisini yayınladı. Bildirilen 6 hasta uzun izlem süreleri içinde başkaca demans özelliği sergilemeden sadece ilerleyici dilsel bozukluk göstermişlerdi. BT sol perisylvien bölgeyi işaretliyordu ve içlerinden birinin kortikal biyopsi örneğinde ayırt edici histopatolojik bir bulgu saptanmamıştı. Böylelikle Pick’den 90 yıl sonra progresif afazi ilerleyici kognitif yıkımın farklı bir sunumu olarak bir kez daha canlandırılmış oluyordu. Mesulam’ın orijinal adlandırmasıyla “demanssız yavaş progresif afazi” hastalarına dair ilk otopsi bulguları 1987’de Kirshner ve Tanrıdağ tarafından yayınlandı. Yazarlar bildirdikleri 2 olguda yine ayırt edici histopatolojik bir özellik bulmaksızın sol inferiyor frontal giruslarında, başlıca II. tabakada olmak üzere spongiozis ve hafif astrositozis gördüler. Constantinidis serisinden sonra bu olgular da asimetrik fokal serebral dejenerasyonların nöropatolojik karşılığı olarak PiH’den farklı bir “ayırt edici histopatolojik özelliği olmayan demans” (DLDH) kavramının dayanacağı rasyoneli oluşturacaktır. DLDH terimi ilk kez 1990 yılında David Knopman tarafından, 14 olguluk bir otopsi serisi bildirimi sırasında kullanıldı. Yazarlar DLDH patolojisinin PiH’in yanı sıra frontal demansların başlıca nöropatolojik karşılığı olduğunu ileri sürdüler.
Diğer yandan, bir Japon araştırıcı grubunun PiC’lerin AH nörofibriler yumaklarındaki (NFY’ler) çift sarmallı filamanlara benzerliğini vurgulamalarından bir yıl sonra AH NFY’lerinin mikrotübül asosiye tau proteinine (MAP-τ) immunoreaktif olduğu, ondan bir yıl sonra ise NFY’lerin temel bileşenlerinin MAP-τ olduğu bulunmuştu. Aynı yıl MAP-τ’nin 17. kromozomda q21 lokusuna haritalandığı gösterildi. Bu 10 yılın sonlarındaki önemli bir gelişmenin de gerek AH ve gerekse de Parkinson hastalığı (PH), PiH ve progresif supranükleer paralizi (PSP) gibi farklı nörodejeneratif hastalıklardaki nöronal inklüzyonların anormal proteinlerin degradasyonunda rol aldığı bilinen bir proteosomal protein olan ubikuitini içerdiklerinin bulunması olduğu söylenebilir. Nihayet, geçen yüzyılın son 10 yılına girildiğinde PiC’lerin temel bileşeninin de MAP-τ olduğu ortaya çıktı. Bu tarihten sonra tau ve ubikuitin immun-boyaması birlikte kullanılarak FTD’lerin nöropatolojik sınıflamasında kullanılacaktır. Bu yeni nosolojiye göre PiH bir Ub+T+, kortikobazal dejenerasyon (KBD) Ub-T+, FTD-amyotrofik lateral skleroz (ALS) Ub+T- demans olarak adlandırılacak, frontal tipte klinik sunumların kayda değer bir bölümünün PiH tarzı değil de Ub+T- olduğu görülecek ve nihayet DLDH terimi Ub-T- olgularla sınırlanacaktır. İlerleyen yıllarda ubikuitin immun-boyamasındaki gelişmeyle birlikte önceden DLDH olarak sınıflanan olguların yeniden değerlendirildiklerinde büyük bir bölümünün Ub+T- olarak yeniden sınıflandırılabildikleri görülmüştür. Günümüzde DLDH terimi inklüzyon içermeyen FTD (FTLD-ni) olgularını kastedecek şekilde çok küçük bir orana sınırlanmış durumdadır.
1980’lerin sonlarından itibaren, daha sonra yayınlanacak ilk kriterler dizisine de isimlerini verecek olan İsveç’ten Lund, İngiltere’den Manchester gruplarından araştırıcıların bir dizi çalışmasıyla, PiC ve PiHc’nin bu antitenin sık görülen bir bileşeni olmadığı gerekçesiyle, Pick’in ismi kaldırılarak atrofi tarzı (fronto-temporal) antitenin adı haline geldi. Bu çalışmalar özetle frontal lob demansının (FLD) AH’den ayırt edilebilir bir sendrom olduğunu, tüm demansların %15-20’sini kapsadığını, olguların çoğunda PiC bulunmadığını ileri sürmekteydi. Bu 2 grup 1994’te bir araya gelerek “Manchester-Lund Fronto-temporal Demans (FTD) Kriterleri” olarak anılacak ilk tanı kriterleri dizisini yayınladılar. Bu kriterlerde davranışsal özellikler ayrıntılı bir şekilde tanımlanırken, dilsel özelliklerin oldukça yüzeysel bir tanımına rastlanır. Parkinsonizm ve ALS bulgularının görülebileceği vurgulanır. Erken inkontinans, normal EEG ve görüntülemede frontal, anteriyor temporal veya bunların kombinasyonunun bu tanıya özgü olduğunun altı çizilir. Erken başlangıç ve sık aile öyküsü AH ile kontrast oluşturan öykü özellikleri olarak belirlenir.
Aynı yıl içinde familyal FTD’de bağlantı analizinin 17q21.22 lokusuna anlamlı bir bağlantı sergilediği gösterildi. Sonrasında 1996’da o zamana kadar aynı bağlantının saptandığı 13 geniş aileyi tanımlamak üzere gerçekleştirilen bir uzlaşım toplantısında “Kromozom 17’ye bağlı fronto-temporal demans ve parkinsonizm (FTDP-17)” terimi önerildi. Ertesi yıl FTDP-17 ailellerinde ilk MAPT mutasyonları çeşitli gruplar tarafından bildirildi. Bu gelişmeler bir nörodejeneratif hastalığın ilk kez protein biyokimyasına dayanılarak adlandırılması önerisine yol açtı: tauopati.
Aynı yıl içinde Lund ve Manchester gruplarına Fransız, Kanadalı ve ABD, California’dan araştırıcıların katılımıyla isim bir kez daha değiştirilip bugünkü haline getirildi: frontotemporo lobar dejenerasyon (FTLD). Neary ve ark. Tarafından yayımlanan bu yeni kriterler dizisinde davranışsal varyant FTD (dvFTD), ilerleyici tutuk afazi (progressive non-fluent aphasia: PNFA) ve semantik demans (SD) biçimlerinde 3 farklı sunumunun ayrıntılı tanımları sıralandı.
FTLD “Uzlaşım” Kriterleri’nin yazarları arasında bulunan Kertesz, Pick isminin bir klinik tanı olarak ortadan kaldırılıp sadece PiC’lerin göründüğü, klinik karşılığı müphem ve dolayısıyla tanısı çok güç ender bir histopatolojik tanıya indirgenmesine baştan beri itiraz etmekteydi. Kertesz, ilk kez 1994’te PPA, FLD ve KBD gibi fokal kortikal dejenerasyonlarla karakterize nörodejeneratif hastalıkları “Pick Kompleksi (PK)” terimiyle adlandırarak Pick eponimini yeniden canlandırmayı önerdi. Zaman içinde Kertesz için PK dilsel ve davranışsal bileşenlerle birlikte KBS ve PSP’yi içeren bir motor bileşenin de bulunduğu bir klinik spektrum haline geldi. Klinik sunumlar bu bileşenlerden biriyle başlayıp seyir boyunca izole kalabildikleri gibi, daha sıklıkla sendrom transformasyonuna uğramakta ve diğer bileşenlerden özelliklerin de katıldığı karma sendromlar (sekonder ve tersiyer sendromlar) haline dönüşmekteydi. PK spektrumunu oluşturan antitelerin öngörülebilir nöropatolojik ve genetik karşılıkları olduğunu ileri sürdü.
Son olarak 2011 yılı içinde uzlaşım kriterlerinin duyarlılık yetersizliği, görüntüleme ve genetikteki gelişmeler gerekçeleri kalabalık iki ayrı uluslararası uzmanlar topluluğunu biraraya getirerek dvFTD için Rascovsky ve ark. tarafından ve PPA’lar içinde PNFA ve SD için Gorno-Tempini ve ark. tarafından yeni kriterlerin yayınlanmasını tetikledi. Yeni kriterlerde PNFA agramatik-tutuk varyant PPA (atvPPA), SD semantik varyant PPA (svPPA) olarak adlandırıldı.
Yeni yüzyıldan itibaren giderek artan gözlemler 17q21’e bağlantılandığı halde bütün çabalara rağmen MAPT mutasyonu bulunamayan ve aynı zamanda Ub+T- olan ailelerin sayısının FTD-17-tau ailelerini aşmaya başladığını ortaya koydu. Sonunda 2006 içinde 2 ayrı grup tarafından bu Ub+T- FTD-17 ailelerinden sorumlu genin progranulin geni (GRN) olduğu bulundu.
GRN’nin bulunmasından birkaç ay sonra birbiri ardına bildirilen otopsi serilerinde GRN mutasyonlu bireylerin beyinlerindeki bilinmeyen ubikuitinize proteinin transaktif cevaplı-DNA’ya bağlanan protein 43 (TDP-43) olduğu ortaya konmuştur. İzleyerek sporadik Ub+T- FTD’lerin de (FTLD-U) %90’dan fazlasının TDP-43 immünoreaktivitesi gösterdiği bulunmuş ve TDP-43 proteinopatileri sporadik ve familyal FTLD’nin önemli bir bölümünden ve sporadik ALS’den sorumlu olan yeni bir proteinopati olarak nörodejeneratif hastalıklar sınıflamasına dahil edilmiştir: FTLD-TDP.
Nihayet 2009 yılı içinde Ub+T-TDP- %10’a yaklaşan bir grup için de ubikuitinize proteinin “fused-in” sarkoma (FUS) proteini olduğu bulundu ve böylelikle FTLD-U grubu biraz daha küçülerek bir kaç istisnai tek tek olgular dışında esas olarak sadece CHMP2B mutasyonlu FTD-3 olarak adlandırılan olgulara sınırlandı ve yeni antite de FTLD-FUS adını aldı.
Durmaksızın gelişme gösteren FTD araştırmaları tarihinde günümüze yakın bir diğer buluş da 9. kromozomun kısa bacağına bağlantılandırılan, fakat şimdiye kadar geni bir türlü bulunamayan ailevi FTD-ALS'nin geninin bulunması ve 2 ayrı ekip tarafından Neuron dergisinde peşpeşe yayınlanması oldu: c9orf72.
Epidemiyoloji
Az sayıda prevalans çalışması arasından Zuid, Hollanda çalışmasında her 100,000 nüfusta olmak üzere 50-59 yaşları arasında 3,6, 60-69 arasında 9,4 ve 70-79 arasında 3,8 bulunmuştur. İngiltere çalışmasında FTD’nin pre-senil dönemde (45-64 yaş) AH kadar sık olabileceğini gösterecek şekilde, genel olarak erken başlangıçlı demansın prevalansı 81/100,000 iken, her ikisinin de ayrı ayrı prevalansları 100,000 nüfusa 15 olarak saptanmıştır. Daha yakın tarihli bir çalışmada Kuzey İtalya’da FTD’nin sadece pre-senil döneme özgü olmadığını düşündürecek şekilde her 100,000 nüfusta olmak üzere 45-65 yaşları arasında 22, 66-75 arasında 78 ve 75 yaş üzerinde 54 bulunmuştur.
Japonya’dan gelen rakamlar erken başlangıçlı demans prevalansı açısından karşılaştırılabilir iken, bunun içinde FTD’nin yeri açısından oldukça düşük görünmektedir. Lund, İsveç’te 30 yılda biriken toplam 524 otopsi doğrulamalı demans olgusunun %42’sini Alzheimer hastalığı (AH) hastaları oluştururken FTD’lilerin oranı %4 bulunmuştur. Yeni yayınlanmış bir İtalyan çalışmasında ise AH %43,8 ile birinci sırada demans nedeni iken, FTD prevalansı %30,2 bulunmuştur.
İnsidansı gözden geçirildiğinde, ABD’den gelen insidans rakamlarının (100,000 kişilik nüfusta yılda yeni olgu) 40-49 yaş aralığında 2,2, 50-59 aralığında 3,3 ve 60-69 aralığında 8,9 şeklinde olduğu görülmektedir. İngiltere’de ise 45-64 yaş aralığındaki insidans rakamları FTD için 3,5, AH için 4,2’dir.
FTD’nin genellikle cinsiyetleri eşit etkilediği görüşü hakimdir. Zuid-Hollanda ve Cambridge insidans çalışmaları bu düşünceyi desteklerken, Cambridge prevalans çalışmasında %82’lik bir erkek baskınlığı bulunmuştur. Bilindiği gibi düşük eğitim AH için yerleşmiş bir risk faktörüdür. Bir İtalyan çalışmasında FTD için buna ters bulgular saptanmıştır.
Ortalama hastalık süresi AH’ye göre biraz daha kısa, 8 yıldır (2-20 yıl). Amiyotrofik lateral skleroz (ALS) eşliği sağkalımı 2-3 yıla kadar kısaltır. Hollanda’dan bir çalışmada sağkalım ortalama olarak 9,9 yıl bulunmuştur. Hollanda ve ABD’den bir ortak çalışmada FTD-ALS sağkalımı incelenmiş ve kognitif başlayıp motor bulguların sonradan eklendiği hastalarda sağkalımın 67,5 ay ile 28,2 ay olan eşzamanlı başlangıçlı gruba göre anlamlı olarak daha uzun olduğu görülmüştür.
Bir başka çalışmada da davranışsal baskın FTD-ALS ile dilsel baskın FTD-ALS sağkalım açısından karşılaştırılmış ve davranışsal varyantın ortalama 506 gün daha uzun yaşadığı görülmüştür. Dilsel varyantta bulber tutulum daha olasıdır ve spinal tutuluma göre yaşamı kısaltmaktadır. Avustralya’dan bir çalışmada 91 olguluk dvFTD grubunun ortalama sağkalımı genel olarak ortalama 9,0 yıl iken, 24 fenokopi olgusunun dışlanmasından sonra 7,6 yıla kısaldığı görülmüştür.
Hastaların %40’ının 1. derece akrabalarında benzer hastalık öyküsü vardır ve bunların en az yarısı otozomal dominant tarzda bir geçiş sergilerler. Familyal ve sporadik olgular arasında AH için mutad olan başlangıç yaşı farkı yoktur.
Frontotemporal lober dejenerasyon (FTLD)’nin alt gruplarının prevalans ve insidansları spesifik olarak çalışılmamıştır. ABD ve Almanya’dan 353 hastalık ortak bir çalışmada, yüzdeler ve başlangıç yaşları davranışsal varyant FTD (dvFTD) için %56,7 ve 57,5, semantik varyant primer progresif afazi (svPPA) için %18,7 ve 59,3, agramatik tutuk varyant PPA (atvPPA) için %24,6 ve 63 olmuştur. Bu çalışmada dvFTD ve svPPA gruplarında atvPPA grubuna göre daha fazla erkek vardır. Bir derlemede atvPPA’nın tüm FTLD’nin %10 ila 35’ini, svPPA’nın ise %15-25’ini oluşturduğu, başlangıç yaşı ve sağkalım açısından tipik FTD’den farklılık göstermediği ileri sürülmüştür. Bir çalışmada 61 otopsi doğrulamalı olguda ilerleyici tutuk afazi (PNFA) başlangıç yaşının (68,3 ± 2,7 yıl) dvFTD’ye göre (59,9 ± 7,4 yıl) anlamlı olarak daha geç olduğu, fakat sağkalımın farklı olmadığı bulunmuştur. Hodges yakın tarihli başka bir çalışmada 17 yıllık bir zaman dilimini kapsayan dönemde kaydedilmiş birbirini izleyen toplam 100 SD olgusunun analizini bildirmiştir. Sonuçlar beklenenden biraz daha geç tanı yaşı (ortalama 64,2 ± 7,1 yıl, aralık 40-79) ve iyi huylu seyir (ortalama sağkalım 12,8 yıl) göstermektedir. Ailesellik özelliği SD’den beklendiği gibi %2-7 olacak şekilde düşüktür. Kortikobazal sendrom (KBS) ve progresif supranükleer palsi (PSP)'li olgular dahil motor varyantın da dahil edildiği toplam 269 olgudan oluşan bir FTD serisinin aile öyküleri incelendiğinde FTD-ALS'nin en yüksek, svPPA'nın en düşük kalıtılabilirlik alt tipi olduğu görülmüştür.
Belirlenmiş ve yerleşmiş kesin risk faktörleri olduğu söylenemez. Kafa travması öyküsü FTD ile anlamlı ilişkili bulunmuştur. Son zamanlarda, PGRN mutasyonlarının bir pleitropik protein olan progranulinin işlevselliğini azaltan bir haplo-yetmezlik durumu yaratarak etkin olduğu bulunmuştur. Kafa travmasının, sonrasında neden olduğu mikroglia aktivasyonu aracılığıyla, diğer sitokinlerin yanı sıra progranulini pro-inflamatuar granuline kesen elastazların da salgılanması sonucu, potansiyel olarak bir progranulin yetersizliği durumu yaratarak FTD’ye yatkınlığı arttırabileceği şeklinde bir yorum getirilmiştir. Genetik risk faktörlerinden genetik bölümü içinde söz edilecektir.
Klinik Tablo
Bu bölümde FTD'nin üzerinde uzlaşılan klinik sunumları olarak dvFTD ve iki dilsel varyant, SD ve PNFA üzerinde durulacaktır. dvFTD, PNFA ve SD ayrı antiteler olmakla birlikte aralarındaki örtüşmelerin istisna olmaktan çok mutad oldukları vurgulanmalıdır. Bu terimler klinik sunumdaki baskın rengi vurgulamaktadır. Başlangıç tablosu seyrek olarak bütün seyir boyunca izole kalır ve sıklıkla davranışsal, dilsel ve motor varyantlardan biri sunum iken, bir diğeri 2. sendrom olarak ve hiç seyrek olmayarak geri kalan da 3. sendrom olarak zaman içinde eklenir. dvFTD ve PNFA tablolarına ALS de eklenebilir.
Davranışsal Varyant Fronto-temporal Demans (dvFTD)
Geniş deneyime sahip 46 uluslararası uzmandan oluşan grup kendilerine “International Behavioural Variant FTD Criteria Consortium (FTDC)” adını vererek dvFTD için 6 temel erken dönem klinik özelliği sıralamışlardır. dvFTD'li hastalar seyrin erken dönemlerinden itibaren davranışsal disinhibisyon sergilemeye başlarlar. FTDC kriterlerine göre disinhibisyon tanımı sosyal uygunsuz davranış; hal, tavır ve edepli davranışın kaybı; dürtüsel, düşüncesiz ve ihtiyatsız hareketlerden en az birinin varlığında kullanılır. Sosyal uygunsuz davranış yabancılara dokunmak veya öpmek, uygunsuz cinsel eylemler, herkesin gözü önünde işemek, yasadışı davranışlarda bulunmakla örneklenebilir. Hal, tavır ve edepli davranışın kaybı örnekleri arasında başkalarına incitici gelebilecek cinsel içerikli imalar, şakalar; sosyal ipuçlarına dikkatsizlik (örneğin, muhatabın konuşmayı bitirmeye çabalamasına rağmen konuşmayı sürdürmek); gaz çıkarmak, geğirmek, tükürmek, cinsel organlara dokunmak; hijyen veya giyinmede sorunlar (kötü kokan, lekeli, yırtık veya uygunsuz kıyafet); kişilerarası mesafeye saygısızlık gösterilebilir. Dürtüsel, düşüncesiz ve ihtiyatsız eylemler örnekleri ise yeni başlayan kumarbazlık, kendine ait olmayan şeyleri alma, genellikle yiyecek veya görsel olarak uyarıcı nesneleri çalma, sonuçlarını düşünmeksizin mülk alım satımı olabilir. Erken dönemde apati veya atalet diğer bir temel klinik özelliktir. FTDC ikisinden birinin mevcudiyetini şart koşmaktadır. Apati, edilginlik, kendiliğindenliğin kaybı, önceki ödüllendirici aktivitelere ilgi kaybı şeklinde örneklendirilebilecek olan ilgi, dürtü veya motivasyon kaybı olarak tanımlanabilir. Atalet ise rutin aktiviteleri başlatma veya sürdürme için uyarı gerekmesi, konuşmayı başlatma veya sürdürme eğiliminin azalması şeklinde örneklenebilecek şekilde bir davranışı başlatmanın azalmasıdır. Erken dönemde sempati veya empati kaybı 3. temel klinik özelliktir. Bu özelliğin mevcut olarak kaydedilmesi için hastada başkalarının ihtiyaçları ve hislerine tepkinin azalması veya sosyal ilgiler, kişilerarası ilişkiler veya kişisel sıcaklığın azalmasının saptanması gerekir. Başkalarının hislerine tepkisizlik, kırıcı yorumlar, başkalarının acı veya sıkıntılarına umursamazlık, uygunsuz şakalarla örneklenebilecek başkalarının hislerine kayıtsızlık ve anlayışsızlık durumlarıdır. Sosyal ilginin azalması ise kişisel soğukluk, göz kontağının kaybı gibi sosyal angajmanda genel bir azalmadır. Erken dönemde perseveratif, stereotipik veya ritualistik davranışlar 4. temel klinik özelliktir. Mevcut olarak değerlendirilmesi için basit tekrarlayıcı hareketler; kompleks kompulsif veya ritualistik davranışlar veya konuşma stereotipisinden birinin saptanması gerekir. Parmak vurma, alkış tutma, oğuşturma, kaşıma, cildi veya giysileri çimdikleme, dudak bükme, dudak şapırdatma basit tekrarlayıcı hareketler örnekleridir. Sayma ve temizleme ritüelleri, toplama, kontrol etme, tekrar tekrar tuvalete gitme, nesneleri sıralama, sadece belli yollardan yürüme kompleks kompulsif veya ritualistik davranışlar örnekleri arasında sıralanabilir. Konuşma stereotipisi ise iletişimsel değeri olmayan kelime ve cümleciklerin tekrarlanmasıdır. Hiperoralite ve diyet alışkanlıklarında değişiklik 5. temel klinik özelliktir. Mevcut olarak değerlendirilmesi için besin tercihlerinin değişmesi (başlıca şekerlemeye düşkünlük tarzında karbonhidrat açlığı, abur cubur yeme); aşırı yeme, aşırı alkol veya sigara tüketme (doygunluğa rağmen yemeye devam etme, kompulsif sigara veya alkol tüketimi); yenilmeyecek nesnelerin ağza alınması veya yutulması durumlarından en az birinin saptanması gerekir. Nöropsikolojik profil 6. ve son özellik olarak sıralanmıştır. dvFTD nöropsikolojik profili bellek ve görsel-mekansal işlevlerin göreli olarak korunduğu bir yürütücü bozukluktur. Yürütücü işlevlerde bozukluk saptanmalı (İz Sürme B, kelime ve desen akıcılığı görevlerinde bozukluklar, perseverasyonlar ve kural ihlalleri gibi hataların saptanması), epizodik bellek göreli korunmuş olmalı, görsel-mekansal işlevler (basit çizimler yapabilme yeteneği) göreli korunmuş olmalıdır. FTDC kriterlerine göre bu 6 temel özellikten en az üçünün mevcudiyeti mümkün dvFTD tanısı için yeterlidir. Mümkün dvFTD tanısı kriterlerini dolduran hastalar arasında işlevsel bozulmaları uygun bir ölçekle de belgelenen ve dvFTD ile uyumlu yapısal veya fonksiyonel görüntüleme delilleri (frontal ve/veya anterior temporal atrofi, hipometabolizma veya hipoperfüzyon) saptanabilen hastalar muhtemel dvFTD tanısı alırlar. Muhtemel dvFTD tanılı hastalar arasında da histopatolojik deliller veya FTD nedeni Mendelyen geçiş genlerinden birinde mutasyon saptanması kesin dvFTD'ye karşılık gelir.
dvFTD klinik, anatomik, nöropatolojik ve genetik olarak en fazla heterojenite gösteren alttiptir. Yeni tanı kriterleri içinde sıralanan klinik özelliklerin tümü hastalığın seyri boyunca tek bir hastada görülebileceği gibi tek tek hastalar erken evrede apati, disinhibisyon, stereotipi veya beslenme alışkanlığı değişiklerini ön planda sergileyebilirler. Apatik baskın tip dorsolateral ve medial baskın atrofiyle, bunun gibi disinhibisyon baskın tip orbitofrontal, stereotipi baskın tip kaudat çekirdek, hiperoralite baskın tip sağ anterior temporal, amigdala, anterior insular atrofiyle ilişkilendirilmiştir. Yukarıda söz edildiği gibi dvFTD seyri boyunca izole kalabildiği gibi (özellikle FUS inklüzyonları taşıyan bir alttip çok genç yaşta başlayan, psikotik özellikler de gösterebilen bir izole dvFTD sergiler), frontal baskın alttipine PNFA, sağ temporal baskın alttipine SD şeklinde dil bozuklukları da eklenebilir. KBS, PSP ve ALS eklenebilecek motor bozukluklardır. Patolojik olarak %40-45 tau, %40-45 TDP-43, %5-10 FUS birikintileri saptanır. Tek tek vakalarda kimliği belirlenemeyen ubikuitinize protein birikintileri (FTLD-UPS), birikinti saptanamayan non-spesifik atrofi (FTLD-ni) ve hatta atipik sunulmuş AH saptanabilir. Genetik olarak FTDP-17, PGRN ve MAPT mutasyonları ailevi olguların %90'ından fazlasını açıklar. ALS ilişkili ailevi olgulardan 9. kromozomdaki mutasyonlar çok büyük oranda sorumludur. FTLD-UPS nöropatolojisi gösteren dvFTD hemen daima FTD3-CHMP2B mutasyonu taşır. İnklüzyon cisimciği miyopatisi ve/veya Paget hastalığının eşlik ettiği alttip ise hemen daima FTD9-VCP mutasyonlarıyla ilişkilidir. TARDP, FUS ve PSEN1 mutasyonlu olgular da bildirilmiştir.
Cambridge-Sydney grubu Neary kriterlerini dolduran fakat ilerleyici olmayan, yapısal ve fonksiyonel görüntüleme çalışmaları normal olan ve muhtemelen dejeneratif bir etyopatogeneze değil de Asperger sendromu, şizotipal bozukluk gibi bir kişilik bozukluğu, geç yaş kronik manisi gibi psikiyatrik antitelerle daha iyi açıklanabilecek "FTD fenokopisi" adını verdikleri bir duruma dikkat çekmişlerdir. Kognitif, işlevsel ve davranışsal ölçeklerde başlangıçta benzer puanlar alsalar da dejeneratif dvFTD olgularının zaman içinde puanları düşerken fenokopi olguları değişmeden kalırlar. Yeni kriterlerin "muhtemel dvFTD" düzeyi fenokopiyi de dışlayabilmektedir.
Altta yatan nedene bağlı olarak dvFTD’de atrofi genel olarak, anterior singulat korteks (pregenual ve subgenual), anterior insula (ventral ve dorsal), striatum, amigdala, hipotalamus ve talamusu içerir.
Olguların yaklaşık yarısında nöropatolojik verilerin de mevcut olduğu bir çalışmada, voksel temelli morfometri ile analiz edilen MRG verilerine göre toplam 66 dvFTD olgusu, atrofilerinin ağırlıklı bölgelerine göre frontal (F), temporal (T), fronto-temporal (FT) ve fronto-temporo-parietal (FTP) olmak üzere 4 anatomik alt gruba sınıflandırılmışlardır. Nöropsikiyatrik Envanter (NPI) ile belgelenen davranışsal özellikler açısından gruplar arasında farklılık yoktur. Nöropsikolojik olarak F, FT ve FTP gruplar tipik yürütücü bozukluğu sergilerken, T grubu ağırlıkla konfrontasyon adlandırması ve epizodik bellek bozukluğuna sahiptir. Görsel-mekansal bozukluk hafif ve gruplar arasında farklı değildir. FTP grubu daha fazla PGRN mutasyonu taşıma ve daha fazla FTLD-TDP, KBD ve AH nöropatolojisi sergileme eğilimindedir. Enteresan bir bulgu T grubuna ilişkindir. Tüm olgular R>L şeklinde bir anterior temporal atrofiye sahiptirler. Tümünde de aile öyküsü pozitiftir ve genetik analizleri yapılmış olanların tümü de MAPT mutasyonu taşımaktadırlar. Semantik demans bölümünde aşağıda söz edileceği gibi, bu bulgular SD olgularının büyük sıklıkla L>R anterior temporal atrofiye sahip olmaları, pozitif aile öyküsünün çok nadir olması ve büyük sıklıkla tip C FTLD-TDP sergilemeleriyle tam bir kontrast oluşturmaktadır.
Aynı grup bir başka çalışmalarında "sağ temporal varyant" (RTV) olarak adlandırdıkları bu tarz atrofiye sahip 20 olguyu SD olgularıyla karşılaştırdı. RTV'de en belirgin kognitif defisit epizodik bellek bozukluğu ve sokakta kaybolma idi. Prosopagnozi RTV'ye özgüydü. Tüm davranışsal semptomlar genelde RTV'de daha ağır iken, aşırı dindarlık ve görsel hallusinasyonlar yine RTV'ye özgüydü. Yazarlar bu ayırıcı özellikleriyle RTV'nin FTLD'nin 4. alttipi olması gerektiğini savundular. Gonzales-Caballero ve arkadaşları tarafından 2015 yılında yine davranışsal bulguların baskın olduğu, ancak birinde epizodik bellek kusuru ile topografik dezoryantasyonun eşlik ettiği 2 RTV vakası bildirildi ve vakaların birinde PGRN mutasyonunun pozitif olduğu ifade edildi. Sonrasında başka araştırmacılar tarafından da sağ temporal atrofinin baskın olduğu çeşitli vakalar bildirildi ve kimi araştırmacılar buna RTV-SD derken kimisi RTV-FTD demeyi tercih etti. Giderek artan sayıda çalışma yapılan bu antitenin henüz tanı kriterlerinde kendine yer bulamasa da yakın dönemde 4. alttip olarak yer bulacağı düşünülmektedir.
Yukarıda sıralanan ve özellikle de anatomik seçiciliğe özgü birtakım istisnalar dışında, klasik olarak FTD'nin bir yürütücü bozukluk sergilemesi beklenir. Yatak başında yürütücü bozukluk ikili benzerlikler ve atasözü yorumları gibi soyutlama testleri, saat çizimi gibi planlama testi, kelime akıcılığı gibi sürdürülen dikkat testleri, geriye sayı menzili gibi çalışma belleği testleri, Luria desenleri gibi zihinsel esneklik, Vur-Vurma ("Go-No Go") gibi enterferans direnci testlerinde bozulmaların ön planda olduğu, kelime menzili öğrenme gibi epizodik bellek ve geometrik şekil kopyaları gibi görsel mekansal testlerin göreli korunduğu bir profildir. Geleneksel geniş kapsamlı nöropsikolojik test bataryalarında bu profil soyutlama, zihinsel esneklik (örneğin, Wisconsin Kart Eşleme Testi, İz Sürme Testi B), planlama (örneğin, Londra veya Hanoi Kulesi) testlerinde ön planda bir bozukluğa karşılık gelir. Görsel-mekansal testlerden saf algısal testler (örneğin, Benton Çizgi Yönü Belirleme ve Yüz Tanıma, Hooper Görsel Organizasyon) göreli korunmuşken, aynı zamanda bir planlama unsuru da içeren görsel yapılandırma testleri (örneğin, Wechsler Küp Desenleri) bozulmuş olabilir. Nöropsikolojik olarak daha önce de değinilen epizodik bellek bozukluğu, konfrontasyon adlandırma bozukluğu gibi genellikle anatomik seçiciliği yansıtan istisnaların yanı sıra, özellikle erken dönem dvFTD hastaları davranışsal gösterilerinin ağırlığına rağmen geleneksel testlerde normal performans gösterebilirler. Sosyal kognisyon paradigmalarının klinik nöropsikolojiye dahil edilmesi bu tür hastalardaki sorunların daha iyi belgelenebilmesini sağlamaya başlamıştır. Karmaşık sosyal ortamlarda, riskli durumlarda akıl yürütme, karar almayı simüle eden testler (örneğin, Zihin Teorisi [ToM] testleri, Iowa Kumar Testi) geleneksel yürütücü işlevler testlerinin korunmuş olduğu bu erken dönem hastaların sorunlarını duyarlı bir şekilde yansıtırlar. Empati ölçeklerinde bozulma, özellikle öfke, korku, keder, tiksinti gibi negatif fasial ve yine öfke ve keder gibi negatif vokal emosyonların tanınması testlerinde bozulma ToM testlerindeki bozulmayla korele eder. Sarkastik ifadelerin yorumlanamaması, eğlenme, başkasının tavrından sıkıntı duyma gibi kendilik farkındalığı gerektiren emosyonların sergilenememesi gibi bozukluklar yine sosyal kognisyona ilişkin dvFTD'de bozulmaları beklenen ve yeni yöntemlerle ölçülebilecek olan özelliklerdir; sosyal kognisyon testlerindeki bozulma görüntüleme delilleriyle birlikte fenokopi olgularından da ayırt etmeye yarayabilir. Alzheimer hastalığı için geliştirilmiş olan Mini-Mental Durum Değerlendirme testi (MMSE) gibi kognitif, NPI gibi davranışsal, işlevsel aktiviteler anketi (FAQ) gibi işlevsel ve Klinik Demans Ölçeği (CDR) gibi evreleme ölçekleri FTD'de yeterli duyarlılık gösteremeyebilirler. Cambridge-Sydney grubunun FTD'de yüksek duyarlılıklarını gösterdikleri Addenbrook Kognitif Muayenesi-Gözden Geçirilmiş (ACE-R), Cambridge Davranış Envanteri (CBI), Frontotemporal Demans Evrelendirme Ölçeği (FRS) gibi enstrümanlar da mevcuttur.
Fonksiyonel MRG (fMRG) ile beyin istirahat durumu çalışmaları dvFTD'nin teorik olarak anlaşılmasına büyük katkılar yapmıştır. Geleneksel fMRG çalışmaları beynin bir görev veya uyarana cevabına odaklanmışken, beynin açık bir girdi veya çıktı olmaksızın da son derece aktif olduğu bu istirahat durumu çekimlerindeki kan oksijen düzeyine bağımlı (BOLD) sinyalindeki spontan dalgalanmalarla gösterilmiştir. Bu zamansal olarak senkron, mekansal olarak dağınık BOLD dalgalanmaları ile haritalanan geniş boyutlu nöral şebekelere entrensek konnektivite şebekeleri (ICN) adı verilmiştir. ICN'lerin ilk tanımlananı ve yaygın biçimde çalışılanı "kendiliğindenlik şebekesi"dir (default mode network: DMN). DMN aktivitesi bir kognitif görevde ortadan kalkacak şekilde kısmi hippokampus, medyal prefrontal korteks, posterior singulat korteks ve inferior pariyetal korteksi içine almaktadır. DMN aktivitesine hayal kurma, zihinde geçmişe yolculuk yapma, geleceği tasarlama gibi işlevler atfedilmiştir. DMN yapıları en erken döneminde AH tarafından tutulduğu gösterilen yapılardır ve nitekim azalan istirahat DMN aktivitesinin prodromal AH'nin bir işareti olabileceği ileri sürülmüştür. DMN'den sonra bulunan ve dorsal anterior singulat korteks (dACC), frontal insula (FI) ve ventral striatum gibi limbik subkortikal yapıları içeren ICN'nin emosyonel olarak dikkat çekici iç ve dış uyaranlarla tutarlı bir şekilde aktive olduğu gösterilmiş ve "dikkat çekicilik şebekesi" (salience network: SN) adı verilmiştir. DMN ve SN aktiviteleri sağlıklı kişilerde birbirleriyle anti-koreledir. İşlevlerine olan gönderme ile SN'ye "şimdi ve burda", DMN'ye "eskiden, orda" şebekesi de denilmektedir. Seeley SN'yi oluşturan yapıların sadece büyük beyinli memelilerde bulunan, evrimsel olarak yeni bir nöron tipini, von Economo nöronlarını (VEN) içerdiğine dikkat çekmiş ve dvFTD'de en erken dönemde tutulan bu nöronların FTD nörodejenerasyonuna seçici bir yatkınlık gösterebileceğini ileri sürmüştür. Bu varsayım dACC VEN sayıları ve morfolojilerinin FTD, AH ve kontroller arasında karşılaştırıldığı bir çalışmada doğrulanmıştır. FTD'de VEN sayıları normallere kıyasla anlamlı derecede azamış ve arda kalan VEN'ler dismorfik görünümdeyken, AH'de yoğun nörofibriler yumak varlığına rağmen VEN sayı ve morfolojileri normallerden farklı değildir. Aynı grubun farklı bir çalışmasında FTD ve AH'liler normallerle fMRG ile ICN görüntülemesi ile karşılaştırıldılar. Bu enteresan çalışmada AH grubunun normallere kıyasla azalmış DMN, fakat artmış SN aktivitesi gösterirken, dvFTD grubunun tam tersine azalmış SN, artmış DMN aktivitesine sahip olduğu, dahası bu azalma ve artmanın hastalık şiddetiyle korele olduğu ortaya kondu.
FTD Dilsel Varyantları
Dilsel varyantlar, FTDC üyelerinin baskın olduğu bir uluslararası topluluk tarafından gözden geçirilmiş ve 2011 yılında yeni kriterler yayınlanmıştır. Kriterler öncelikle PPA kriterlerinin doldurulmasını şart koşmakta ve sonra da 3 PPA alttipini tanımlamaktadır. Bunlardan ikisi FTD'nin iki dilsel varyantının yeni tanımlarıdır. PPA tanısı için 3 zorunlu kriter belirlenmiştir. İlki önde gelen klinik özelliğin dilsel bir güçlük olması koşuludur. Bu güçlük kelime bulma güçlükleri, parafaziler, efor gerektiren konuşma, gramer ve/veya anlama bozukluklarıyla yansıtılıyor olabilir. İkinci olarak dil güçlükleri, telefonda konuşma, sözel iletişim gerektiren rutin iş sorumlulukları gibi konuşma ve dille ilintili iletişim aktivitesinde problemlerle yansıyacak şekilde günlük yaşam aktivitelerindeki bozulmanın başlıca nedeni olmalıdır. Son olarak da afazi belirtilerin başlangıç özelliği ve hastalığın ilk evresinde önde gelen bozukluk olmalıdır. Erken dönemde kayda değer epizodik bellek bozukluğu, non-verbal bellek bozukluğu, görsel-mekansal bozukluk PPA tanısını ekarte ettirir. Erken dönemde davranışsal belirtiler olabilirse de bunlar işlevsel bozukluğun başlıca nedeni olmamalıdır. Benzer şekilde hafif bir taraf apraksisi veya ince parmak hareketlerinde bozukluk olabilirse de aşikar bir parkinsonyen sendrom bulunmamalıdır.
İlerleyici Tutuk Afazi (PNFA): Yeni kriterlerde bir FTLD alttipi olan PNFA ve pratik olarak onunla aynı şey olarak Mesulam grubunun kullandığı bir PPA alttipi olan gramatik PPA (PPA-G) terimleri yerine PPA'nın tutuk agramatik varyantı (NFAV-PPA) terimi tercih edilmiştir. PPA kriterlerini dolduran hastalar arasında agramatizm veya eforlu konuşma-konuşma apraksisi şeklindeki çekirdek özelliklerden birinin saptanması NFAV-PPA tanısı için zorunludur. "NFAV-PPA klinik tanısı" için ayrıca sentaktik olarak karmaşık cümlelerin anlaşılmasında bozulma ve SD özellikleri olan tek kelime anlamanın ve nesne bilgisinin bozulmamış olması durumlarından en az iki tanesinin bulunması gereklidir. "Görüntüleme destekli NFAV-PPA tanısı" için sol fronto-insuler atrofi, hipometabolizma veya hipoperfüzyon şeklinde yapısal ve/veya fonksiyonel görüntüleme desteği gerekir. "Kesin patolojili NFAV-PPA" için ise klinik tanının bir nörodejeneratif hastalığın histopatolojik delillerinin varlığı veya bilinen patojenik mutasyonlardan birinin saptanmış olmasıyla desteklenmesi gerekir.
Agramatizm tipik olarak kısa, basit cümleciklerden oluşan, gramatik morfemlerin eksik olduğu konuşma ve sıklıkla da yazma biçimidir. Özellikle çok heceli kelimelerde artikülasyon bozukluğu şeklinde ortaya çıkan konuşma apraksisi efor gerektiren, yavaş bir konuşmaya eşlik eder. Hastalar kendilerinin de farkında olduğu şekilde konuşma sırasında hecelerin bozulduğu, yer değiştirdiği hatalar yaparlar. Prozodi bozulmuş ve konuşma hızı azalmıştır. Bu şekilde hastaların konuşması bir yabancının aksanlı konuşmasını andırabilir. Eforlu, duraksamalı konuşma ve hece üretim hataları aşikar konuşma apraksisi veya agramatik hatalardan önce ilk ortaya çıkan semptom olabilir. Böyle olgularda Boston Tanısal Afazi Muayenesi'nin "Kurabiye Hırsızlığı" veya Western Afazi Bataryasının "Piknik" resmi gibi içinde tanımlanabilecek çoğul unsurlar içeren bir resmin yazılı tanımı şeklinde bir dil üretimi testi veya sentaks anlama görevleri çok erken eşlikçi hafif agramatizmi ortaya koyabilir. Mesulam grubu tarafından geliştirilen Northwestern Anagram Test (NAT) agramatizmin belgelenmesinde oldukça duyarlı bir test gibi görünmektedir. Konuşma apraksisi fonemik parafaziler ve dizartriden ayırt edilmelidir. Fonemik parafazilerin de hecelerin yer değiştirmesi veya hatalı hecenin kelimeye sokulması sonucu ("kabak" yerine "kazak" gibi farklı bir kelime veya "kadak" gibi gerçek dışı kelime) olması nedeniyle ilk bakışta konuşma apraksisiyle karışabilirler. Ancak fonolojik temsillerin (kelime seslerinin doğru sırası) aktivasyonu veya korunması sorununa bağlı olan fonemik parafaziler (logopenik varyant PPA'nın temel özelliklerindendir) hastalar tarafından genellikle farkedilmez ve düzeltilmeye çalışılmaz. Artikülasyonun planlanmasının bir bozukluğu sonucu olan konuşma apraksisinde ise yukarda sözedildiği gibi hastalar hatalarının farkındadırlar ve sıklıkla düzeltmek için birden fazla girişim yaparlar. Hatalar bu düzeltme çabaları sırasında tutarlı olarak aynı değildir ve özellikle de çoğul heceli kelimelerde ortaya çıkar. Dizartri ise artikülasyonu gerçekleştirecek kasların zaafına bağlı motor bir bozukluktur. Az heceli kelimelerde de ortaya çıkar; tutarlı, benzer şekilde tekrarlanan hatalar şeklindedir. Karmaşık sentaktik cümlelerin anlaşılması görevleri "Ahmet'i kurtaran kişi Ayşe'dir" gibi bir nesne cümleciğinde kurtarılan kişinin kim olduğunu sormak veya "Bir aslan bir kaplan tarafından parçalanarak öldürüldü" pasif cümlesinde ölen hayvanın hangisi olduğunu sormak şeklinde uygulanır.
Standart bir afazi muayenesi sonrasında NFAV-PPA'lı bir hastanın spontan konuşmasının tutuk, agramatik, apraktik, anlamasının karmaşık sentaktik cümleler dışında göreli olarak korunmuş, tekrarlamasının spontan konuşmaya paralel ve özellikle karmaşık sentaktik cümlelerde bozulduğu, adlandırmasının apraktik olduğu görülür. Erken evrede özellikle bakıldığı takdirde eylem kelimelerinin adlandırılmasında bozukluk saptanabilirse de Boston Adlandırma Testi gibi standart resim adlandırma tarzı konfrontasyon adlandırma testleri normal olabilir. Kelime akıcılığı görevlerinde adlandırma güçlüğü ortaya çıkacaktır. Erken dönemde yazılı dil konuşma diline oranla daha iyi korunmuş olabilir.
Seyir içinde yürütücü bozukluk, epizodik bellek bozukluğu ve giderek jeneralize demans, davranışsal bozukluklar, KBS, PSP ve bazen ALS gibi motor bozukluklar tabloya eklenebilir.
Yapısal görüntülemede sol frontoinsular atrofi erken dönemde aksiyel kesitlerde belli belirsiz bir sol sylvien fissür genişlemesi şeklinde yansıyabilir. Giderek sol frontal operküler ve anterior insular atrofi koronal kesitlerde aşikar hale gelir. Traktografi çalışmalarında sol superior longitudinal fasikulusun öncelikle tutulduğu ortaya konmaktadır. Pittsburgh compound B (PIB)-PET gibi amilod görüntüleme çalışmalarında PNFA olgularında nadiren AH tarzı t utulumsergilenmektedir.
Ailevi olgular sıklıkla PGRN mutasyonu taşırlar ve dolayısıyla FTLD-TDP histopatolojisi sergilerler. Sporadik olgular en sık kortikobazal dejenerasyon(KBD) olmak üzere genellikle bir tauopatiye bağlıdırlar. Bir tutuk afazi tablosunun çok nadiren AH'nin atipik sunumu olduğu genel bir varsayımdır, ancak bir Cambridge çalışmasında şaşırtıcı bir şekilde tüm PNFA olgularının %44'ünün AH patolojisi taşıdığı ortaya konmuştur. AH patolojisi taşıyan grup taşımayanlara göre daha yaşlıdır.
Semantik Demans (SD): Yeni kriterlerde bir FTLD alttipi olan SD ve pratik olarak onunla çok yakın bir durum olarak Mesulam grubunun kullandığı bir PPA alttipi olan semantik PPA (PPA-S) terimleri yerine PPA'nın semantik varyantı (SV-PPA) terimi tercih edilmiştir. "SV-PPA Klinik tanısı" için çekirdek kriterler anomi ve tek kelime anlamada bozulmadır ve her ikisinin birlikte saptanması gerekir. En az üçü bulunması gereken diğer tanısal özellikler nesne bilgisinin kaybı; korunmuş tekrarlama; ve korunmuş motor konuşma ve gramer; ile yüzey disleksisi veya disgrafisidir. "Görüntüleme destekli SV-PPA" yapısal ve/veya fonksiyonel görüntülemenin en az biri ile anterior temporal atrofi, hipometabolizma veya hipoperfüzyonun gösterilmesi ile konulur. "Kesin patolojili SV-PPA" için ise klinik tanının bir nörodejeneratif hastalığın histopatolojik delillerinin varlığı veya bilinen patojenik mutasyonlardan birinin saptanmış olmasıyla desteklenmesi gerekir.
SD'de konfrontasyon adlandırması bozukluğu diğer varyantlara kıyasla çok belirgindir ve göreli salim kalan diğer dilsel işlevlerin yanında çok ön plandadır. Tek kelime anlama ağır bir şekilde bozulmuştur ve bu bozukluk düşük frekanslı kelimeler için çok daha belirgindir (örneğin., "köpek" sık, "gergedan" seyrek). Tek tek olgu bildirimlerine dayanılarak önerilen somut, zihinsel olarak canlandırılabilirlikleri yüksek kelimelerin anlaşılmalarının, soyut ve canlandırılabilirlikleri düşük kelimelere kıyasla (örneğin., "uçurtma" somut, "adalet" soyut) daha bozuk olduğu varsayımı grup çalışmalarında doğrulanmamıştır. SD'li hastalarla yapılan bir grup çalışmasında bunun tam tersinin doğru olduğu, yani düşük sıklıkta, düşük canlandırılabilirlikteki soyut kelimelerin anlaşılmasının çok daha ağır bozuk olduğu gösterildi. Tek kelime anlamada bozulma hastalığın erken evrelerinde en çarpıcı ve tıbbi yardım istemeye neden olan başlıca neden olabilir. Böyle bir senaryo sıklıkla eşler aralarında konuşurlarken hastanın eşini şaşırtacak şekilde bir tek kelimenin anlamını sorması şeklindedir (örneğin, "Bana mutfaktan havuç getirir misin?" "Havuç nedir?"). Konuşmanın motor özelliklerinin korunması NFAV-PPA'dan, tekrarlamanın korunması logopenik varyanttan ayırıcıdır. İrregüler yazılan kelimelerin regülarize edilerek okunması/yazılmasına verilen isim olan yüzey disleksisi ve disgrafisi bizim gibi büyük ölçüde okunduğu gibi yazılan dillerde ortaya konması çok güç olabilir.
Yeni tanı kriterlerinin önce PPA tanısı koşulu, sağ temporal varyant (RTV) olgularında SD kayda değer davranışsal belirtilerle birlikte hatta onlardan sonra başladığı için SV-PPA'yı zorunlu olarak L>R temporal atrofiye sınırlamakta ve RTV'nin 4. alttip olarak önerilmesini haklı kılmaktadır. Davranışsal özellikler başlangıçta yoksa bile hemen daima eklenir.
California grubu 6 L>R SD olgusuyla 6 R>L SD olgusunun doğal seyirlerini karşılaştırdılar. L>R grup tek istisnayla semantik, R>L grup tek istisnayla davranışsal olarak başlarken, her iki grupta da birer olguda başlangıç semantik ve davranışsal olarak eşitti. Semantik özellikler SV-PPA için sıralanan özellikler iken, davranışsal sendrom "emosyonel mesafelilik, iritabilite, fizyolojik dürtülerin (uyku, iştah, libido) bozulması" olarak tanımlanmıştı. İkinci evrede semantik ve davranışsal başlangıçlı gruplar ortalama 3 yıl içinde başlangıçta sahip olmadıkları diğer grubun başlangıç özelliklerini edinerek birbirlerine benzediler. Üçüncü evrede 5-7 yıl içinde disinhibisyon, kompulsiyonlar, prosopagnozi, besin tercihlerinde değişikler ve kilo alma izlendi. Kompulsiyonlar L>R grupta ağırlıkla görsel, non-verbal uyaranlara, R>L grupta ise kelime ve sembollerle oynanan oyunlara yönelikti. L>R gruptaki bu görsel uyaranlarla kompulsif ilgi tek tek olgularda özellikle resim olmak üzere görsel sanatlarda (ama edebiyatta değil) yeni ortaya çıkan artistik yaratıcılık olarak da bildirilmiştir. Bu erken dönem bir SD olgusunda yeni başladığı ressamlığının ürünleriyle girdiği yarışmalarda ödül alacak düzeyde olabilmiştir. SV-PPA'ya KBS, SD veya ALS'nin eşlik etmesi beklenmez.
Mayo grubunun bir çalışmasında RTV ile uyumlu atrofi sergileyen 20 olgu klinik, nöropsikolojik, genetik ve patolojik özellikleri açısından incelendiler. Bu grupta 12 olgunun başlangıç tanısı dvFTD iken, 8 olgunun SD idi. Başlangıçta kişilik değişikliği ve uygunsuz davranışlar dvFTD grupta anlamlı olarak sıkken, anomi, tek kelime anlama bozukluğunun yanı sıra prosopagnozi ve topografagnozi SD grubunda anlamlı olarak daha sıktı. Seyir içinde dvFTD'ye semantik bozulma, SD'ye kişilik değişikliği ve uygunsuz davranışlar eklendi. dvFTD'de sadece tatlıya düşkünlük ve parkinsonizm anlamlı olarak daha sık görülen özellik halini alırken, SD'de prosopagnozi ve topografagnozi bu özelliklerini sürdürdüler. Anatomik olarak dvFTD grubu daha fazla frontal lob kaybı, SD grubu daha fazla fuziform girus kaybı göstermekteydi. Aile öyküsü pozitif olan olguların tümü dvFTD grubundandı. Bunların arasında genetik analiz yapılan 7 olgunun tümü de MAPT mutasyonları taşımaktaydı. Histopatolojik olarak dvFTD grubunda otopsi bulguları mevcut olan 8 olgunun tümü de FTLD-Tau, SD grubunda toplam 3 olgunun tümü de FTLD-TDP tanısı aldılar. Bu bulgular RTV grubunda klinik sunumun histopatolojiyi öngörebileceğini düşündürmektedir.
Yapısal görüntüleme büyük sıklıkla L>R asimetrik anterior temporal atrofi, fonksiyonel görüntüleme ise aynı bölgede hipometabolizma veya hipoperfüzyon gösterir. Voksel temelli morfometri çalışmaları ve kortikal kalınlık çalışmaları superior temporal girusun göreli olarak korunduğu, temporal kutup, amigdala ve hippokampus, anterior entorhinal korteks ve fuziform girusu da içine alan anterior ve inferior temporal atrofi saptanmıştır. Tutulum seyir içinde simetriğine ve orbito-frontal, insular ve anterior singulat kortekse yayılır. Traktografi çalışmalarında her iki unsinat fasikulus ve superior longitudinal fasikulusun temporal parçalarında tutulum görülür. Amiloid görüntüleme çalışmaları bu varyantın AH ile olan çok nadir ilişkisini yansıtacak şekilde negatiftir.
Klasik SV-PPA'da hemen daima aile öyküsü negatiftir ve genetik nedensellik görülmez ve çok büyük sıklıkla FTLD-TDP Tip C patolojisi saptanır. AH ilişkili SD çok nadir bir durumdur. Yukarıda sözedildiği gibi aile öyküsü pozitif, parkinsonizmin de eşlik edebildiği, davranışsal belirtilerin ağırlığından daha çok dvFTD tanısı alan, R>L anterior temporal atrofi tarzı sergileyen nadir bir alttip FTDP-17T'nin sunumu olabilir.
Genetik
Tau Geni (MAPT)
Tau proteini geni MAPT 17. kromozomun uzun bacağında 21.1 lokusunda (17q21.1) yerleşmiştir. Tau mikrotübüllere (MT) karboksi ucundaki (CT) tekrar bölgelerinden bağlanarak MT asamblesinin bütünlüğünden ve dolayısıyla akson boyunca MT’lerin aracılık ettiği organel ve vezikül nakliyesinden sorumludur. Taunun amino ucundaki (NT) aminoasid (AA) eklentileri MT’lerin nöron membranıyla etkileşimini sağlar. Ekzon 0 kodlamayan ve sonraki 14 ekzon kodlayıcı olmak üzere toplam 15 ekzonu vardır. Ekzon 10’daki alternatif düğümlenme 4 tekrarlı (4R) ve 3 tekrarlı (3R) 2 izoform üretirken, ekzon 2 ve 3’teki alternatif düğümlenmelerle 3R ve 4R izoformları NT ucuna 29 AA’lık 0, 1 veya 2 tane eklenti sokarlar ve böylece toplamda 6 izoform oluşur.
GRN mutasyonları kadar hızlı olmasa da sayılar düzenli olarak artmaktadır ve şimdiye kadar 40’tan fazla patojenik mutasyon bildirilmiştir. Bu mutasyonlar kodlayıcı bölgelerdeki hatalı anlamlı mutasyonlar ve ekzon 10 sonrasında AA silinmeleri ve intronik düğüm yeri mutasyonlarıdır. MAPT mutasyonlu olgular tüm FTLD olgularının %5-10’unu, familyal FTD’nin ise %10-20’sini oluşturur. GRN mutasyonlarının da aynı kromozomda bulunması sonrasında bu antite FTDP-17T olarak adlandırılmaktadır ve C9orf72'nin de bulunmasıyla üçüncü sıklıktaki familyal FTD nedeni durumundadır. Otozomal dominant kalıtım paternine sahip olan MAPT mutasyonunun penetransı %95’in üzerindedir.
Farklı MAPT mutasyonlu aileler FTD-Pick Kompleksi'nin tüm varyantlarını ve hatta AH benzeri tabloları içeren oldukça geniş bir fenotipik varyasyon sergilerler. Bu durum bazen aynı mutasyona sahip bir ailenin farklı üyelerinin fenotipik değişkenliği olarak da görülür. Belli bir mutasyonla klinik sunum arasında aşikar bir korelasyon yoktur. Başlangıç yaşı 25-65 yıl arasındadır, 70 yaş sonrası başlangıç çok nadirdir. Sağkalım çoğunlukla 3-10 yıl arasında değişmekle birlikte 1 yıldan 25 yıla kadar değişen hastalık süreleri bildirilmiştir ve cinsiyet farkı yoktur.
En sık görülen mutasyon 1998-2008 arası toplam 32 aile ile ekzon 10 P301L hatalı anlamlı mutasyonudur. Kodlayıcı bölgedeki bir nokta mutasyonu AA yer değişikliğine yol açar. Ortalama başlangıç yaşı 52,6 yıl, ölüm yaşı 59,3 yıldır. Sıklıkla 45-65 arasında başlar. Fenotip başlıca dvFTD tarzındadır. İkinci sırada 1994-2009 arasında toplam 27 aile ile intron 10, IVS10+16C>T mutasyonu vardır. İntronik bölgedeki nokta mutasyonu ekzon 10 düğümlenmesini etkileyerek tau transkriptine ekzon 10 dahil edilmesini arttırır. Bu mutasyonu taşıyan toplam 33 bireyin retrospektif analizlerinde ortalama başlangıç yaşı 50 yıl (aralık: 42-72 yıl) ve ortalama sağkalım 11 yıl (aralık: 3-22 yıl) olarak bildirilmiştir. Fenotip ağırlıkla dvFTD şeklinde olmakla birlikte PSP tarzında sunum veya PSP/KBS şeklinde sekonder sendromlar bildirilmiştir. Üçüncü sırada 1992-2004 arası 7 aile ile yine ekzon 10’daki N279K mutasyonu bulunmaktadır. Bu kodlayıcı bölgedeki nokta mutasyonu yine bir AA yer değişikliğine yol açsa da (asparagin yerine lizin) bir intronik mutasyon gibi ekzon düğümlenmesini etkiler. Başlıca fenotip dvFTD + Parkinsonizm (bazen PSP tarzında) şeklindedir.
En sık görülen klinik sunum zaman içinde sıklıkla parkinsonizm ve/veya afazi sendromlarının da eklendiği dvFTD tablosudur. Manchester serisinde FTLD tanısı almış birbirini izleyen 233 olgunun genetik analizleri de yapılmıştır. MAPT mutasyonu bulunan 16/17 olgu yukarda anılan intronik mutasyonu taşımaktadırlar. Tümü de disinhibisyon baskın bir dvFTD tablosu ve bunların ¾’ü de zaman içinde eklenen “semantik bozukluk” sergilerler. Söz konusu semantik bozukluk ilk yazar tarafından daha önce svPPA’da görülmeye alışılan tek kelime anlamada bozukluk, aşina meşhur kişilerin tanınamaması şeklinde tarif edilmiştir. Zaman içinde dil bozukluğu mutizme evrilir. Daha önce göreli geç başlangıçlı ve selim seyirli atvPPA ile ilişkilendirilmiş olan MAPT ekzon 12 V363I mutasyonu, erken başlangıçlı klasik bir svPPA fenotipi sunan başka bir olguda da bildirilmiştir. MAPT mutasyonu (ekzon 10, S305S) PSP nöropatolojisi sergileyen bir ailede de 2000 yılında bildirilmiştir. Ailenin 2 üyesi dvFTD ile başlayıp vertikal bakış parezisi ve parkinsonizmin seyir içinde eklendiği bir tablo sunarlarken, üçüncüsü asimetrik distoni ve dizartri ile başlayıp sonradan supranükleer paralizi ve düşmelerin eklendiği bir tabloya sahiptir. Aynı mutasyon daha sonra başka bir ailede de bildirildi; bu ailenin 2 üyesinde tablo dvFTD olarak sunulmuştu ve zaman içinde L-dopa dirençli parkinsonizm bulguları eklendi. Kırklı yaşlarda PSP klinik sunumu yukarıda anılan en sık intronik mutasyon olan IVS10+16C>T ile de bildirilmiştir. Yirmili yaşlarda MAPT ekzon 10, P301S mutasyonu sonucu hızlı seyirli demans geliştiren baba dvFTD ile sunulurken oğul KBD sergilemiştir. Ekzon 13 R406W mutasyonu şimdiye kadar 6 farklı ailede göreli geç başlangıçlı AH benzeri amnestik klinik ve temporal atrofiyle giden tablolarla ilişkilendirilmiştir. İspanya, Bask bölgesinden bildirilen ekzon 11, K317M mutasyonuna sahip ve haplotip ortaklığı nedeniyle muhtemelen birbirleriyle akraba 2 aileden 13 olgu ile MAPT mutasyonlu ilk FTD-ALS olguları sunulmuştur. Tablo ortalama 48 yaşında genellikle dizartriyle başlıyor, parkinsonizm ve piramidal bulgular ekleniyor, bir bölümünde amiyotrofi ortaya çıkıyor, ortalama 6 yıllık seyirde demans göreli olarak geç görünürken, davranış değişikliği ön planda olmuyordu. Yine nispeten nadir bir mutasyon olan ekzon 10, P301S mutasyonlu olgularda ise Parkinson hastalığı demansına benzer bir tablo olarak daima parkinsonizmli başlangıç ve demansın sonradan eklenmesi tanımlanmıştır.
Bir çalışmada FTDP-17T grubu içinde başlıca tau proteini yapısını etkileyen hatalı anlamlı ekzon mutasyonlarından (örneğn, P301L) oluşan grup, başlıca tau proteini pro-haberci RNA'sını etkileyen intronik düğüm yeri mutasyonları (örneğin, IVS10+16) grubuyla VBM ile karşılaştırıldığında ilk grubun başlıca lateral temporal, ikinci grubun ise başlıca medial temporal lob hacim kaybıyla birbirlerinden ayrıldıkları görüldü.
MAPT mutasyonları bir dizi farklı tau patolojisine yol açar. Bunların ilki başlıca 4R taudan oluşan, AH’ye benzemeyen nörofibriler inklüzyonlardır ve 4R tau mikrotübül bağlanmasını bozan veya 4R/3R oranını arttıran 2 türde ekzon 10 mutasyonları sonucu görülürler (en sık olan P301L birinci türe neden olur). Bu birikintilerde AH tipi nörofibriler yumakların (NFY) eşleşmiş helikal filamentlerinden (PHF) farklılıklar izlenir. Hem nöronlar hem de glia etkilenir. Buna karşılık, ekzon 9, 11, 12 ve 13 mutasyonları 3R ve 4R tauyu birlikte etkiler veya NFY-PHF’lere veya PiC’lere benzeyen inklüzyonlar yaratırlar. Glial tutulum daha azdır.
Progranulin Geni (GRN)
Şimdiye kadar 70’ten fazla mutasyon bildirilmiştir. Tüm FTLD’lerin %5-10’unu, familyal FTD’lerin ise %15-25’ini oluşturdukları tahmin edilmektedir. Bu rakamlarla, C9orf72 bulunana kadar en sık ailevi FTD nedeni olarak kabul edilirken artık daha yaygın kanı ikinci sıraya düştüğü yönündedir.
Bildirilen mutasyonların tümüne yakını prematür bir stop kodonla sonuçlanan anlamsız (”nonsense”) veya çerçeve kayması (”frameshift”) mutasyonları olan sıfır (”null”) mutasyonlardır. Gen delesyonu ve hatalı anlamlı (”missense”) mutasyonların da sıfır mutasyonlar gibi bir haplo-yetersizliğe yolaçan protein işlev yitimi mutasyonları olduğu ortaya konmuştur.
Progranulin (PGRN), 17q21.31 lokusunda kodlanan 593 AA’lık bir prekürsör proteindir. MSS’de nöronlar ve mikrogliada bulunur. Elastazlar tarafından proteolitik kesilmesi 7 farklı granulin peptidini (grn) oluşturur. Genelde hücre büyümesi ve farklılaşması, yara iyileşmesi yanı sıra tümör gelişiminde de rol oynadığı düşünülen grn’nin MSS’de nükleer transkripsiyon sinyallemesinde görev aldığı ve anti-enflamatuvar etki gösterdiği bulunmuştur. Buna karşılık PGRN’nin tam tersine proenflamatuvar sitokinlerin üretimini teşvik ettiği bilinmektedir. GRN 13 ekzon ve 4, 5, 9 ve 13. ekzon dışında kalan ekzonların kompleks alternatif düğümlenmesinden türetilen 3 ayrı isoformda bulunur.
GRN mutasyonu otozomal dominant bir kalıtım paternine sahiptir ve penetransı 60’lı yaşlarda %60-70 iken, 70’li yaşlarda %90’ın üzerine çıkmaktadır. Bu mutasyona sahip grubun ortalama başlangıç yaşı 60, aralığı 35-83 yıl, sağkalım aralığı 1-15 yıldır (MAPT'a göre biraz daha geç ve biraz daha kısa). Cinsiyetler eşit tutulur. Davranış ve kişilik değişikliği, yürütücü bozukluk ve dil bozukluğu en sık raslanan bulgularken, parkinsonizm bunları izler; apraksi, bellek bozukluğu ve görsel-mekansal bozukluğa da rastlanır. Dil bozukluğu hemen daima atvPPA ile uyumlu tutuk afazi tarzındadır; şimdiye kadar svPPA tarzı tek kelime anlamanın bozulduğu şeklindeki FTLD'nin diğer dil bozukluğu fenomeni bildirilmemiştir. MAPT mutasyonlarında beklenilmeyen bir belirti olan hallusinasyonlar GRN mutasyonlarında %25 sıklığında görülebilir. En sık parkinsonizmin de eşlik edebildiği dvFTD şeklinde sunulur. atvPPA ve KBS de sık sunumlardır. AH demansı benzeri bellek baskın bir sunum da çok ender değildir; LCD, PHD, saf parkinsonizm şeklinde daha ender sunumlar da bildirilmiştir.
Enteresan biçimde, FTD-ALS ile TDP-43 inklüzyonlarını paylaşsa da şimdiye kadar FTD-ALS fenotipi ile ilgili bir çalışmada 49 FTD-ALS olgusu içinde sadece bir olguda sonradan patojenik olmadığı kararlaştırılan bir ekzon 2, Asp 33 (p.S120Y) mutasyonu tespit edilmiş, daha sonra ise aşağıda söz edilecek olan Brescia, İtalya çalışmasında 4 familyal FTD-ALS olgusunun ikisinde ekzon 8, Thr272fs mutasyonu bildirilmiştir. Başka bir çalışmada ise GRN polimorfizmlerinin sporadik ALS hastalarında daha erken başlangıç ve daha kısa sağ kalımla ilişkili bir hastalık modifiye edicisi olarak rol aldığı gösterilmiştir.
En sık bildirilen mutasyon ekzon 12 Arg493X’dir. Kodlama bölgesindeki nokta mutasyonu prematür bir sonlanma kodonuna yol açarak anlamsız bir kod oluşturur. Ortalama başlangıç yaşı 56,7 yıl, ortalama ölüm 63,2 yıldır. Fenotipik olarak en sık dvFTD görülürse de atvPPA ve KBS de bildirilmiştir. İkinci sıklıktaki mutasyon ekzon 8, Thr272fs'dir. Kodlama bölgesindeki tetranukleotid silinmesi çerçeve kayması ve prematür tercüme sonlanmasına neden olur ve sonuç olarak yine anlamsız bir kod oluşur. Ortalama başlangıç yaşı 60,1 yıl, ortalama ölüm 71 yıldır. Fenotipik olarak ilkine benzer. Üçüncü sıklıktaki ise intronik mutasyon IVS7, Ala237fs'dir. İntron 7 düğüm kabullenici bölgedeki nokta mutasyonu ekzon 8'in atlanması, çerçeve kayması ve prematür tercüme sonlanması ile anlamsız bir kodun oluşmasını öngörür. Ortalama başlangıç yaşı 58,3 yıl, ortalama ölüm 66,6 yıldır. Fenotip yine öncekilere benzer.
En sık görülen mutasyonlardan olan ekzon 12, Arg493X bir uluslararası işbirliği çalışmasında görünüşte birbirleriyle ilgisiz 30 aileden 37 olguda analiz edilmiştir. Olguların ortalama başlangıç yaş ve sağkalım süreleri 56,4 yıl ve 6,1 yıldı. Klinik sunumlar dvFTD %75, atvPPA %12, AH demansı %9, KBS %6 şeklinde dağıldı. Seyir içinde dil bozukluğu %79, parkinsonizm %47 olguda gelişti. Haplotip analizi 27 aile için 300 yıl önce İngiltere kökenli bir ortak kurucuyu işaretledi. Enteresan biçimde bu mutasyona Londra, İngiltere’den bildirilen geniş bir seride hiç raslanılmamıştır. Araştırıcılar FTLD, KBD, PSP klinik tanısı alan toplam 212 olguları arasında 25 PGRN mutasyonlu olgu buldular. Bunların yirmisi haplotip analiziyle tek bir kurucuya geri götürülebilen ekzon 2, Cys31fs mutasyonuydu. Olguların %75’i apati baskın bir dvFTD ile sunulurken %25’i atvPPA ile sunuldular. dvFTD’li olgular seyir içinde mutizme kadar giden dil bozukluğu da geliştirdiler. Olguların yarısında zaman içinde KBS de görüldü.
Diğer bir sık mutasyon olan ekzon 8, Thr272fs bir Kuzey İtalya (Brescia) serisinde özellikle incelendi. Aynı grup daha önce söz konusu mutasyonu 2 ayrı aileden toplam 9 hastada göstermişlerdi. Yazarlar bu mutasyonu 82 familyal FTD olgusunun 19'unda ve 69 sporadik olgunun 3'ünde buldular. Familyal olguların 4'ü FTD-ALS idi ve bunların ikisinde yukarda sözedildiği gibi bu mutasyon vardı. KBS grubunda ise 4 familyal olgunun 3'ünde, 9 sporadik olgunun 1'inde mevcuttu. Enteresan biçimde MSA ve PSP'li olguların hiçbirinde görülmezken, 45 LCD'li olgunun 1'inde de bulundu. Yazarlar hareket bozukluğu ile sunulan veya eşlik eden ailevi FTD olgularında öncelikle düşünülmesi gereken mutasyonun bu olduğunu ileri sürdüler.
Fransız serisinde dvFTD, FTD-ALS, PPA, KBS tanısı almış 502 olgu arasında 32 PGRN mutasyonlu olgu belirlendi. Araştırıcılar 24 farklı ailede toplam 18 farklı mutasyon buldular. GRN mutasyonlarının FTLD alt grupları içindeki sıklığına bakıldığında dvFTD’de %5,7, atvPPA’da %4,4, KBS’de %3,3 bulundu. FTD-ALS grubunda PGRN mutasyonuna raslanmadı. 20/32 olgu dvFTD şeklinde sunulmuştu. Bunların ikisine afazi, birine ise hallusinasyon ve hezeyanlı bir psikoz tablosu eşlik etmekteydi. 5/32 olgu Mesulam’ın PPA tanımıyla uyumlu biçimde ilk 2 yıl boyunca izole kalan ilerleyici afazi şeklinde sunuldular. 2/32 olgu KBS şeklinde asimetrik parkinsonizm ve apraksi ile sunuldular, ayrıca davranışsal semptomlar da sergilemekteydiler. Enteresan şekilde 3/32 olgu AH demansı, 2/32 olgu ise LCD şeklinde sunulmuşlardı. Belli bir genotip-fenotip korelasyonu olmadığı gibi aynı aile içinde de geniş bir fenotipik değişkenlik söz konusuydu. Grubun ortalama yaşı 59,1 yıl (49-79) idi ve bunların arasından ölen 20 olgu için ortalama sağkalım 6,5 yıl idi.
ABD, Mayo Klinik’ten tümü de farklı mutasyonlara sahip 8 aile bildirildi. Bu aileler, başlangıç yaşları 64,5 yıl (49-88 yıl), ortalama sağkalımları 7,0 yıl (2-14 yıl), 25’i kadın olan toplam 44 hasta bireye sahipti ve bunların 18’i klinik olarak incelendi. Bunların %55’i kişilik değişikliğiyle sunuldu ve seyir içinde %94’ü tarafından sergilendi. Dil bozukluğu ve parkinsonizmin sunum/erken dönem ve seyir içindeki yüzdeleri sırasıyla %44-%89 ve %17-%61 idi. Sunum/erken dönem özelliği olarak bellek bozukluğu sıklığı %44 idi ve 6 olguda şiddeti AH demansı ya da amnestik MCI tanısı alacakları düzeydeydi. Görsel hallusinasyonları ve parkinsonizmi olan bir olgu LCD tanı kriterlerini doldurmaktaydı.
Yu ve ark. tarafından yapılan bir uluslararası işbirliği çalışması farklı alt tipleri içeren toplam 434 FTLD olgusu içinde 28 farklı aileden 22 farklı mutasyona sahip 31 GRN mutasyonlu olgu buldu. Ortalama başlangıç yaşı 57,7 yıl (39-73 yıl) idi. FTD spektrumu tanısı 26 olguda konulurken (22 dvFTD, 3 atvPPA, 1 KBS) bunların dokuzuna parkinsonizm eşlik ediyordu. Geri kalan olguların dördü AH, biri LCD tanısı almaktaydılar.
GRN mutasyonları, tam penetransa sahip olmamaları dolayısıyla sporadik FTD’de de saptanmıştır. Dolayısıyla klinik (apraksi, tutuk afazi, sıradışı bellek ve/veya görsel-mekansal bozukluk bulguları, hallusinasyonlar) ve görüntüleme (parietal lobu da içine alan belirgin asimetrik atrofi, hipometabolizma) düşündürdüğünde görünüşte sporadik bir olguda da GRN mutasyonu tanı çalışmasına dahil edilebilir. Bir Belçika çalışmasında 103 sporadik FTD olgusu içinde bir olguda GRN genini de içeren bölgede, 267 kontrol bireyinde olmayan bir genomik silinme tarzında bir mutasyon saptandı. Bir Fransız-Belçika işbirliğinde 158 sporadik FTD olgusu içinde GRN mutasyonları için klasik toplam 5 anlamsız veya sıfır (”nonsense” veya ”null”) mutasyonlu olgu mevcuttu.
Nöropsikolojik muayenede FTD fenotipi için tipik yürütücü bozukluk ve tutuk afazinin yanı sıra atipik olarak epizodik bellek bozukluğu ve "parietal" bulgular saptanabilir.
MRG'de FTDP-17T'nin simetrik fronto-temporal atrofisiyle kontrast oluşturacak şekilde asimetrik bir fronto-temporo-parietal atrofi daha olasıdır. Ayrıca ak madde bozukluğunun da eşlik edebileceği bilinmektedir. GRN mutasyonunda parietal atrofi MAPT mutasyonuna göre çok daha belirgindir (Şekil FTD1).
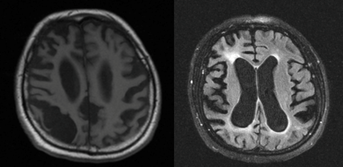
Şekil FTD1. Progranulin gen mutasyonu olan bir hastanın manyetik rezonans görüntülemelerinde dikkati çeken asimetrik parietal baskın atrofi ve ak madde bozukluğu.
GRN mutasyonları nöropatolojik olarak yüzeyel kortikal tabakalarda spongiform değişikliklerle Ub+T- inklüzyonlara yol açar. Bunlar genellikle nöronların sitoplazmalarında çok sayıda olmakla birlikte nöronal sitoplazmik inklüzyonlar (NCI), az sayıda lentiform intranükleer birikintiler de olabilirler (NII). Distrofik nöritler (DN) de görülür. Giriş bölümünde de sözedildiği gibi GRN mutasyonlu bireylerin beyinlerindeki bilinmeyen ubikuitinize proteinin TDP-43 olduğu bulunmuştur. Birtakım varsayımlara rağmen PGRN eksikliğinin tam olarak hangi mekanizmalarla TDP-43 birikimine yolaçtığı henüz aydınlatılabilmiş değildir. GRN mutasyonu ilintili FTDP-17 (FTDP-17U veya FTDP-17P) başlıca II. kortikal tabakada çok sayıda NCI ve kısa DN'leri birlikte gösteren tarzıyla FTLD-TDP alttiplerinden Tip A olarak sınıflanılır.
Kromozom 9 Açık Okuma Çerçevesi 79 Geni (c9orf72)
Uzun süre önce 9. kromozoma bağlantılanan fakat büyük bir rekabete rağmen bir türlü bulunamayan FTD-ALS'den sorumlu gen nihayet 2011 yılı sonlarına doğru bulundu. Türler boyunca yaygın biçimde korunmuş, MSS'de yaygın biçimde ifade edilen, başlıca sitozolik yerleşimli bu proteinin 9p21.2 lokusunda konumlanmış geninin kodlamayan düzenleyici bölgesindeki GGGGCC hekzanukleotid tekrar genişlemesinin gen ifadesini azalttığı veya RNA metabolizmasını bozarak toksik işlev kazanmasına yolaçtığı düşünülmektedir.
Neuron dergisinde yayınlanan ilk çalışma Vancouver, Mayo ve San Francisco gruplarının bir ortak çalışmasıydı. Yazarlar önce VMS-20 ailesi olarak adlandırılan ve daha önce 9. kromozomla bağlantılandırılmış olan geniş bir FTD-ALS ailesinde saptadıkları 4G2C tekrar genişlemesinin sorumlu mutasyon olduğunu gösterdiler. Klasik PCR ile nicelleştirilemeyen tekrar sayısının Southern blot ile 700-1600 gibi dev miktarlarda olduğunu buldular. Sonrasında arşivlerindeki ailevi FTD-ALS, FTD ve ALS fenotipli klinik ve FTD-TDP patolojik serilerinde aynı mutasyonu aradılar ve ailelerin çoğunluğunda buldular. Kombine ailevi FTD-ALS olguları yanı sıra bu mutasyon hem ailevi FTD (%11,7) hem de ailevi ALS (%25,3) olgularında saptanan en sık genetik nedendi. Aynı dergide hemen ardından yayınlanan ikinci çalışma bir Avrupa ve ABD işbirliği çalışmasıydı. Yazarlar daha önce Finli ailevi ALS olgularıyla yapılan bir genom boyu asosiasyon çalışmasında (GWAS) 9p21 lokusuyla bağlantılandırmış ve büyük bir bölümünde MOBKL2b, IFNK ve C9orf72 genlerini kapsayan bir kurucu haplotipini yüksek risk olarak belirlemişlerdi. Yazarlar aynı risk haplotipini taşıyan 4 kuşaklık ve 9 bireyi ALS veya ALS-FTD tanısı almış Galler kökenli bir ailede C9orf72 geninde 4G2C tekrar mutasyonunu gösterdiler. Sonrasında aynı mutasyonu 6 kuşaklık, 8 bireyi ALS veya FTD tanısı almış bir Hollandalı ailede de buldular. İzleyerek önceki GWAS çalışmasında Finli ALS serisi ve yanı sıra yine Finli izole FTD, ayrıca ABD'li, Alman ve İtalyan ALS serilerinde bu mutasyonu aradılar. Finli olgular arasında bu mutasyonun sıklığı ailevi ALS olgularında %46, sporadik ALS olgularında %21, ailevi FTD olgularında %29 olarak bulundu. Mutasyonu taşıyan FTD olgularının 1/3'ünde ALS de vardı, çoğunluğu dvFTD, geri kalanı ise atvPPA klinik fenotipine sahiptiler. Finli olmayan ailevi ALS serisinde ise sıklık %38 olarak bulundu.
Bu öncü çalışmaları izleyerek dünyanın her tarafından çok sayıda yeni çalışma yayınlandı. Bunları gözden geçiren bir derleme Kuzey Amerikan ve Avrupalı popülasyonlarda bu mutasyon sıklığını ortalama olarak ailevi ALS için %37, sporadik ALS için %6, ailevi FTD için %21 ve sporadik FTD için %6 olarak bildirdi. Bu durumda C9orf72 mutasyonu ailevi ALS için SOD1 mutasyonlarından dahi daha sık, ailevi FTD için ise GRN mutasyonları ile karşılaştırılabilir bir sıklıkta idi. Bu çalışmalardan biri büyük bir işbirliği çalışmasıydı ve dünyanın 17 farklı popülasyonundan 4500'e yakın ALS ve 1500'e yakın FTD olgusunda bu mutasyonu aradı. Bu sayıların büyük bölümünü oluşturan beyaz popülasyonda yukarıdaki gözden geçirmenin rakamlarına çok yakın rakamlar buldular. Buna karşılık her biri az sayıda hasta gruplarından oluşan Asyalı, Pasifik Adalarlı, Hintli ve Amerikalı Yerli popülasyonlarında bu mutasyona rastlamadılar. Sporadik ve ailevi hastalığa sahip analiz edilen 262 olgunun tümünde yukarıda sözü edilen Finli risk haplotipini buldular.
C9orf72'nin bulunmasının tüm dünyada yarattığı heyecan ve şevki en iyi yansıtan olgulardan biri de daha önce benzeri görülmemiş bir şekilde Brain dergisinin aynı sayısında birbiri ardına basılan 8 ayrı makaledir. Bunlar arasında dördü klinik seri makaleleriydi. Hollanda, Manchester, Londra ve ABD Mayo'dan gruplar toplamı 1200 sporadik ve ailevi olguya yaklaşan kendi FTD serilerinin C9orf72 için taranmasının sonuçlarını bildirdiler. Genel olarak mutasyon taşıyan olguların sıklığı bu serilerde %7 ila %12 arasında değişmekteydi. Vancouver çalışması yukarıda sözedilen VMS-20 ailesi dahil 16 farklı aileden toplam 30 hastanın klinik ve patolojik bulgularını bildirdi. Mayo'dan bir başka makale C9orf72 alt grubu dahil farklı ailevi FTD alt gruplarını sporadik FTD alt grubuyla yapısal MRG bulguları açısından karşılaştırdı. Kalan 2 makale ise sporadik ve ailevi ALS olgularından oluşan İngiltere ve Avrupa anakarasından iki geniş seride C9orf72'ye ait bulguları bildirdiler. C9orf72 mutasyonu Mayo ve Hollanda serilerinde ailevi olguların %33 kadarını oluştururken bu sayılar Mayo için en sık, Hollanda için MAPT mutasyonlarının çok yakınında ikinci sırada yer almaktaydı. Londra serisinde ise MAPT %34 ile en sıkken, C9orf72 ve GRN %24 ile ikinci sırayı paylaşıyorlardı. Klinik serilerin tümünde de dvFTD en sık sunum idi ve hastalığın ilerlemesiyle genellikle ALS eşlik etmeye başlıyordu. Dilsel varyant sunum ve/veya eşliği konusunda Mayo serisi dışındaki seriler bunun daha sıklıkla atvPPA şeklinde %12-%28 aralığında olduğu konusunda uzlaşmaktadırlar. Manchester serisindeki 32 olgudan üçü atvPPA, biri svPPA, Hollanda serisindeki 42 olgudan ikisi svPPA, altısı sınıflanamayan PPA, Londra serisindeki 19 olgunun biri atvPPA şeklinde sunulmuştu. Buna karşılık Mayo serisinde mutasyonun saptandığı 53 olgunun hiçbirinde PPA yoktu. Olgularda FTD ve ALS’nin yanı sıra daha az sıklıkta bellek kusuru, psikoz, ekstrapiramidal hareket bozuklukları (özellikle akinetik-rijid sendrom), serebellar bulgular da bildirildi. Ayrıca başka bazı çalışmalarda ilginç olarak, bu hekzanükleotid tekrar genişlemesi nörolojik açıdan normal yaşlı bireylerde de bildirildi.
Otozomal dominant kalıtım paternine sahip bu mutasyonda başlangıç yaşı genellikle 18-83 yaş (ortalama 50 yaş), hastalık süresi ise 1-22 yıldır. Yapısal görüntülemede sıklıkla frontotemporal bölgede belirgin, ancak diğer bölgeleri ve hatta nadiren talamus ile serebellumu da içerebilen simetrik bilateral atrofi görülmesi beklenmektedir. Frontal bölgede atrofi çok daha belirginken, temporal atrofi MAPT ve GRN mutasyonlarına göre daha azdır. Nöropsikolojik profil tipik olarak yürütücü işlev bozukluğunu gösterir, ancak nadiren epizodik bellek kusuru ve vizyospasyal bozukluk da bildirilmiştir. TDP-43 birikintileri tüm kortikal tabakalarda orta düzeyde NCI’lar ve az sayıda distrofik nörit şeklindedir ve önboynuzda da görülür. Bu birikinti tarzı Tip B olarak sınıflanmıştır.
Valosin İçeren Protein Geni (VCP)
İnklüzyon cisimciği myoziti (IBM) ve Paget hastalığı ile birlikte herediter FTD sergileyen aileler (IBMPFD) valosin içeren proteini (VCP) kodlayan 9. kromozom kısa bacağında 13.3 lokusuna haritalanırlar. VCP bir AAA-ATPaz süper-ailesi üyesidir ve hücre çevrimi kontrolü, membran füzyonu ve ubikuitin-proteozom degradasyon yolağı gibi bir dizi hücresel aktiviteden sorumludur.
Hastalık klinik olarak erişkin başlangıçlı kavşak tipi miyopati, omurga ve kalça ağrılarıyla karakterize Paget hastalığı ve tüm olguların %35 kadarında görülen 40’lı yaşlarda başlayan dilsel varyant ağırlıklı FTLD ile karakterizedir. Triad bütün hastaların ancak %12’sinde görülür ve en sık izole sunum %30 kadar IBM şeklindedir. Dolayısıyla bu hastalara daha sıklıkla nöromüsküler hastalıklar birimlerinde rastlanılması beklenir. Bununla birlikte FTD’nin daha sıklıkla görüldüğü aileler de bildirilmiştir. Kalp, karaciğer, periferik sinir sistemi gibi pek çok başka organ ve sistem tutulumu da gözlenebildiğinden Benatar ve ark. tarafından “multisistem proteinopati (MSP)” olarak adlandırılan grup içindeki en sık mutasyon olarak gösterilmiştir. Şu ana kadar VCP dışında MSP nedeni olan genler HNRNPA1, HNRNPA2B1 ve IBM ile birlikteliği olmasa da MSP’nin diğer özelliklerini taşıyan, aşağıda kısaca söz edilecek olan SQSTM1 olarak bildirilmiştir.
VCP otozomal dominant kalıtım paternine sahiptir ve penetransı fenotipe göre değişmektedir. Ekzon 5 mutasyonları en sıktır. Bunlar arasında 9 ailede bildirilen R155H nokta mutasyonu birinci sıklıktadır. Hastalık başlangıç yaşı ortalama 55,5 yıl, ölüm yaşı ortalama 63,1 yıldır. Sonrasında 8 ailede bildirilen R155C nokta mutasyonu yer almaktadır. Hastalık başlangıç yaşı ortalama 42 yıl, ölüm yaşı ortalama 57,9 yıldır. Diğer bir sık görülen mutasyon ise R159H mutasyonudur ve bu mutasyonda hastalık başlangıç yaşı ortalama 57,5 yıl, ölüm yaşı ortalama 68,8 yıldır.
Fransa’dan bildirilen 2 ailenin birinde ekzon 3 R93C, diğerinde ikinci sıklıkta görülen mutasyon olan ekzon 5 R155C mutasyonları bulunmuştur. İlk ailede klinik sunum tümüyle FTD şeklindeyken, 2. ailede FTD sıklığı %70’dir. Bir Belçika-Avusturya ortak çalışmasında ekzon 5, R159H yanlış anlamlı mutasyonunu taşıyan 3 aile bildirilmiştir. Haplotip analiziyle bu 3 ailenin birbiriyle akraba olmadıkları ortaya konmuştur. Belçikalı ailelerden biri sadece FTD ile sunulurken, diğerinde IBM olmaksızın izole FTD, izole Paget veya her ikisini birden gösteren bireyler vardı. Avusturya ailesi ise IBM ve Paget bileşiminden oluşmaktaydı. IBM bulunduğunda ALS tanısının çok sayıda farklı miyopati ile birlikte başlıca yanlış tanılardan biri olduğuna dikkat çekilmektedir. FTD-ALS sunumu bu mutasyonla bildirilmemiştir. Bununla birlikte geniş bir İtalyan serisinde ailevi ALS'nin nedenlerinden biri olarak 4 ayrı VCP mutasyonu gösterilmiştir. Bunlarda biri olan ekzon 5, R159H yakınlarda ailesinde çok sayıda FTD tanısı almış birey olan bir ALS olgusunda da saptanmıştır. Böylelikle VCP mutasyonu FALS14 olarak sınıflanmıştır.
Duyarlılıkları çok yüksek olmamakla birlikte IBM tanısı için kas biyopsisinde inklüzyon cisimciklerinin gösterilmesi, Paget tanısı için serumda yüksek alkalen fosfataz ile kemik sintigrafisi yardımcı olabilir.
Nöropatolojik olarak hippokampusun göreli kurtulduğu başlıca tüm kortikal tabakalarda çok sayıda kısa DN, az sayıda NCI ve az sayıda lentiform NII inklüzyonları (Tip D) şeklinde TDP-43 birikintileri görülür. Ubikuitin pozitif inklüzyonlarda VCP’nin değil de TDP-43’ün bulunduğunun gösterilmesi VCP mutasyonunun TDP-43’ün degradasyon bozukluğuna yolaçan bir VCP işlev değişikliğine neden olduğu varsayımını desteklemektedir.
Yüklü Multiveziküler Protein 2B geni (CHMP2B)
Daha 1987 gibi erken bir tarihte tanımlanmış olan ve 45-65 yaşları arasında sunulan, baskın olarak davranışsal değişiklikler ve bir ölçüde dilsel özellikler ve geri planda KBD benzeri motor özellikler sergileyen geniş bir Danimarka ailesinde bağlantı analizi kromozom 3’ü işaretlemişti. Geninin bulunması için bir 10 yıl daha geçmesi gerekti: kromatin modifiye edici protein 2B veya diğer ismiyle yüklü multiveziküler protein 2B geni (CHMP2B).
CHMP2B, ESCRT-III kompleksinin bir bileşenidir. Bu kompleks endositik protein trafiğinde rol oynar ve endositoza uğrayan proteinlerin degradasyonu amacıyla lizozomla füzyona uğrayan bir endozomal yapı olan multiveziküler cisimciğin (MVC) işlevi için elzemdir. Kısa bacakta 11.2 lokusunda yerleşik bu gende gösterilmiş olan mutasyonların CHMP2B’nin mutant formunun MVC’lere bağlanarak, Rab7 gibi endosom-lizozom füzyonunu gerçekleştirecek proteinlerin bağlanmasını engelledikleri ileri sürülmüştür.
Anılan bu Danimarkalı aile dışında aynı yayında akraba olmayan başka bir Danimarkalı olgu, sonradan bir de Belçikalı aile bildirilmiştir. ABD, İngiltere, Finlandiya, Fransa ve Hollanda'dan geniş ailevi FTD serilerinde çalışılıp bulunamayan bu mutasyon İngiltere'den ailevi ve sporadik ALS serilerinde saptanmıştır.
Danimarkalı bir ailenin 22 üyesinde saptanan mutasyon intron 5'in düğüm kabul bölgesindedir ve karboksi ucundan budanmış CHMP2B proteinleri üretmektedir (p.M178VfsX2 ve p.M178LfsX30). Olgular ortalama 57 olmak üzere 46-65 yaş aralığındadırlar. Aile 6 kuşağı kapsayan 450 bilinen üyeye sahiptir. Aynı çalışmada bildirilen aileyle akraba olmayan ve aile öyküsü bulunmayan birey ekzon 5, G442T mutasyonu taşımaktadır. Başlangıç yaşı 69'dur ve klasik svPPA ile sunulmuştur.
Belçika serisinde sporadik ve ailevi FTD, KBS ve PSP olgularında (n=146) CHMP2B mutasyonu tek bir ailevi FTD olgusunda ekzon 5'te bulunmuştur (c.493C>T). Bu anlamsız mutasyon Danimarka mutasyonu benzeri karboksi uçtan budanmış mutant protein (p.Gln165X) üretmektedir. Olgu 58 yaşındadır ve fenotipik olarak yazılı dilin ön planda bozulduğu progresif afazi ve davranışsal özelliklerle sunulmuştur.
İngiliz ALS serilerinde ilk çalışmada bir ailevi ALS olgusu progresif müsküler atrofi (PMA) ile alt motor nöron tutulumu tarzında sunulmuştu ve ekzon 6, Q206H mutasyonu bu gende bulunan 4. mutasyon oldu. Aynı grubun bir ailevi FTD-ALS olgusunda saptadıkları ekzon 2, Ile29Val mutasyonunun patojenisitesi henüz kanıtlanmamıştır. Sonraki çalışmada Kuzey İngiltere'den 433 ALS olgusunun taranması ile 4 PMA olgusunun ikisinde Ile29Val, birinde Q206H mutasyonlarını yeniden buldular. Sonuncuda ise yine patojenitesi halen kanıtlanamamış yeni bir mutasyon olan ekzon 3, Thr104Asn gösterdiler. Hiçbirinde FTD eşliği yoktu ve sadece Q206H olgusu aileviydi.
FTD-3 nöropatolojik olarak bir FTLD-U’dur. Ancak, TDP-43 immünoreaktivitesi göstermediği gibi %10 kadar TDP-43 negatif FTLD-U’larda mevcudiyeti ortaya konan (FUS) immunoreaktivitesi de negatiftir. Böylelikle, artık FTLD nöropatolojisinin %1’inden azına sınırlanmış olan FTLD-U veya yeni nomenklatürdeki şekliyle FTLD-UPS (UPS: ubikuitin-proteozom sistemi) kategorisinin başlıca temsilcisi, buradaki ubikuitinize protein de keşfedilene kadar FTD-3’tür diyebiliriz. FTD-3 beyinleri makroskopik olarak en belirgin frontal kortekste olmak üzere global atrofi sergilerler. Mezial temporal ve subkortikal yapılarda aşikar atrofi görülmez. Kortikal nöron kaybına gliosis ve mikrovakuolizasyon eşlik eder. Ub+T-TDP-FUS- NCI’ler hippokampal dentat granül hücrelerinde ve fronto-temporal neokortekste yüzeyel tabakalarda görülür ve p62 antikoruyla da pozitif olarak boyanırlar. Ekzon 6, Q206H mutasyonlu olgunun otopsisinde spinal önboynuz ve hipoglossal çekirdek hücrelerinin kaybolduğu görüldü. Artakalan hücrelerde klasik ubikuitinize inklüzyonlar, tipik Bunina cisimcikleri olmaksızın mevcuttu. p62/sequestosome1 antikoru ile boyama sonucunda önboynuz inklüzyonları yanı sıra, baskın olarak motor korteks olmak üzere, premotor korteks ve prefrontal Brodmann 9'da da oligodendrogliaya lokalize boyanma saptandı. Boyanma nöritik profiller ve bobinsi cisimcikler tarzı inklüzyonlar ortaya koydu.
Diğer Mendelyen Geçiş Genleri
Kromozom 1’in kısa bacağında 36.22 lokusunda kodlanan TDP-43 genindeki (TARDBP) mutasyonlar ailevi ALS ile ilişkilendirilmiş ve TARDBP mutasyonlu ailevi ALS olguları ALS10 olarak da sınıflanmıştı. Bildirilen patojenik mutasyonların tümü de ekzon 6'dadır. Başlangıçta ALS olmaksızın FTD’ye neden olan 2 ayrı TARDBP mutasyonu bildirildi. Bunlardan ilki daha önce FALS nedeni olarak bildirilmiş ekzon 6, Asn267Ser mutasyonlu dvFTD fenotipi sergileyen bir kadındı. Diğeri ise yeni bir ekzon 6, Lys263Glu mutasyonu sonucu 35 yaşında dvFTD, PSP özellikleri ve kore sergilemekteydi; nöropatolojik olarak başlıca subkortikal çekirdekler ve beyinsapının TDP-43+ inklüzyonlarla tutulmuş olduğu görüldü. Bir Fransız çalışması 71'i ailevi 149 FTD-ALS olgusunda TARDP mutasyonunu spesifik olarak aradı. Gly295Ser mutasyonu iki olguda saptandı. Bunların ilki 52 yaşında dvFTD ile sunulan, ikincisi ise 59 yaşında svPPA ile sunulan ve ALS'nin zaman içinde eklendiği hastalardı. Aile öyküsü ilkinde negatif, ikincisinde pozitifti. Bir İtalyan çalışmasında ise 3 ayrı ailenin etkilenmiş 9 üyesinin hepsinde de en sık ALS10 nedeni olan ekzon 6, Ala382Thr mutasyonu bulundu. Olguların hepsi de ALS ile sunulmuş sonradan dvFTD eklenmişti. FTD spektrumunda FTD-ALS, svPPA-ALS, KBS, svPPA (RTV ve bitemporal dvFTD dahil) sunumları dışında kore, supranükleer bakış parezisi, progresif anartrinin eşlik ettiği atipik vakalar da bildirilmiştir.
Kromozom 16'nın kısa bacağında 11.2 lokusunda kodlanan ve TDP-43-negatif FTLD-U olgularının büyük bölümünde ubkuitinize edilmiş protein olduğu yakınlarda gösterilmiş olan “fused in” sarkoma proteini (FUS) genindeki mutasyonlar da FALS olgularıyla ilişkilendirilmiş ve FUS mutasyonlu FALS olguları ALS6 olarak sınıflandırılmıştı. Bir Belçika çalışmasında aile öyküsü olmayan bir FTD olgusunda saptanan ekzon 6, Met254Val mutasyonunun patojenitesi henüz kesinleşmemiştir. Bu çalışmada taranan 15 FTD-ALS olgusunun hiçbirinde FUS mutasyonu saptanmamıştır. Bir Fransız çalışmasında dvFTD ile sunulup hızla ALS geliştirmiş olan bir FTD-ALS olgusunda FALS ailelerinde sıkça bildirilmiş FUS mutasyonu olan ekzon 6, Arg521His saptanmıştır. Bir Amerikan çalışmasında 76 ailevi FTD-ALS olgusu arasında biri yeni (ekzon 6, G206S), diğeri ise AD&FTD Mutasyon Veritabanı web sayfasında "patojenik değil" olarak sınıflanan (ekzon 5, p.G174-G175del) FUS mutasyonları saptanmıştır. Her 2 olgu da ALS ile sunulmuş, FTD sonradan eklenmişti. Görüldüğü gibi demansla birlikteliği çok nadir olan bu mutasyon, ALS olmadan yalnızca dvFTD kliniği gösteren çok nadir olguda gösterilmiş, ancak gösterilen mutasyonların patojenitesi tartışmalı kalmıştır.
Her ikisi de DNA/RNA bağlayıcı proteinler olan TDP-43 ve FUS arasında çarpıcı fonksiyonel ve yapısal benzerlikler mevcuttur. Bu durum RNA metabolizma bozukluğunun kritik rolünü işaretlese de TDP-43 ve FUS'un birikimi ve sonrasında yolaçtıkları nörodejenerasyonun mekanizmaları halen aydınlatılabilmiş değildir.
Kromozom X'in kısa bacağında 11.21 lokusunda kodlanan ubiquilin 2 (UBQLN2) genindeki mutasyonların da ailevi FTD-ALS'nin nadir nedenlerinden biri olduğu gösterilmiştir. Ubikuitinize proteinlerin degradasyonundan sorumlu ubikuilin ailesi üyesi bir proteini kodlayan bu gendeki mutasyonu taşıyan olguların %20'sinde ALS yanı sıra dvFTD fenotipi mevcuttur, ALS olmadan FTD gelişmesi ise çok nadirdir.
Kromozom 10'un kısa bacağında 13 lokusunda kodlanan optineurin (OPTN) genindeki mutasyonlar ise açık açılı glokoma dışında ALS12 olarak sınıflanmış bir ailevi ALS nedenidir. Otozomal resesif mutasyonlar dominantlara göre daha sıktır. FTD-ALS sunumu da bildirilmiştir. Biz 6 kardeşlik bir ailenin etkilenmiş 3 bireyinde OPTN mutasyonu saptadık. Bu bireylerin ikisi ALS fenotipi sergilerken biri KBS olarak sunulmuştu. Anne-baba 2. derece akrabaydı ve halen sağ olan anne mutasyonu sağlıklı 2 kardeş gibi heterozigot taşımaktaydı. Etkilenmiş 3 birey ve henüz etkilenmemiş 32 yaşındaki en küçük kardeşte ise homozigot bir p.359 AA silinme mutasyonu mevcuttu. Yapılmış olan bir çalışmada OPTN hatalı anlamlı mutasyonunun nükleer faktör kappa B (NF-κB) aktivasyonunun inhibisyonunu bozduğu ortaya kondu ve bir olgunun OPTN-immunoreaktif sitoplazmik inklüzyonlar gösterdiği bulundu.
Kromozom 21'in kısa bacağında 22.11 lokusunda kodlanan süperoksid dismutaz 1 genindeki (SOD1) mutasyonlar ailevi ALS'nin C9orf72 bulunmadan önce bilinen en sık nedeni (tüm ailevi olguların yaklaşık %20'si) iken (ALS1), bu mutasyonla son 10 yıla kadar bildirilmiş izole FTD veya FTD-ALS olgusu yoktu. Son 10 yılda aile öyküsü pozitif, ALS bulguları geliştirmeden 2 yıl önce ilerleyici kognitif yıkım ve davranış değişikliği ile sunulan SOD1 mutasyonlu (I113T) bir FTD-ALS olgusu ilk kez 2012de bildirildi. Daha önceki bir çalışma ise SOD1 mutasyonlu ALS olgularını SOD1 dışı ailevi ALS olgularıyla nöropsikolojik olarak karşılaştırmış ve SOD1 dışı grubun yürütücü bozukluk ve anomi sergilerken, SOD1 mutasyonlu grubun normal kontrollerden farksız bir performansı olduğunu bildirmişti.
Mitokondriyal proteini kodlayan bir gen olan CHCHD10 (Coiled-Coil-Helix-Coiled-Coil-Helix Domain Containing 10) genindeki varyantlar da son dönemde ALS ve FTD-ALS tanılı birkaç ailede bildirildi.
En sık gözlenen fenotipi Paget hastalığı olan SQSTM1 mutasyonlarında sporadik ve ailesel ALS, ailesel dvFTD, ailesel FTD-ALS de tanımlanmıştır. Bunun yanı sıra son dönemde yürüme bozukluğu, ataksi, dizartri, distoni, vertikal bakış parezisi, kognitif bozulma gibi çeşitli fenotiplerle birlikteliği de gösterildi.
2015 yılında keşfedilen ve bugüne kadar 90’dan fazla mutasyonu tanımlanan “Tank-binding kinaz 1 (TBK1)” geninin bulber ALS ve hızlı seyirli dvFTD ile ilişkili olduğu gösterilmiştir. Dynactin-1 (DCTN1) ve Cyclin-F (CCNF) de FTD-ALS ile birlikteliği gösterilmiş genlerdir.
Otozomal dominant familyal AH’nin en sık nedeni olan presenilin 1 geni (PSEN1) mutasyonlarıyla da FTD benzeri fenotip bildirilmiştir. Bildirilen olgu ve aileler çoğunlukla otopsi doğrulaması olmaksızın dvFTD fenotipi sergilemektedirler. Progresif afazi de görülebilmektedir. Otopsileri de bildirilen birkaç olguda AH nöropatolojisi bulunmuş ve FTD tarzı klinik sunuma sahip sporadik olgularla birlikte bu olguları da kapsayacak şekilde bir AH atipik sunumu olarak “frontal varyant AH” terimi önerilmiştir. AH’ye eşlik eden PiC’lerin bildirildiği familyal FTD’ler de mevcuttur. Enteresan biçimde, yeni bir PSEN1 mutasyonu (ekzon 6, G183V) bulunan dvFTD’li bir olguda tipik ağır frontotemporal atrofiye ek olarak amiloid plaklar olmaksızın sadece PiC’ler mevcuttur. Bu bulgu PSEN1 mutasyonlarının genellikle düşünüldüğü gibi sadece Aβ42/Aβ40 oranını arttıran bir toksik işlev kazanma mutasyonları olmayabileceği, bu örnekte olduğu gibi işlev yitimi mutasyonu sonucu, bir yandan γ-sekretaz yetersizliği ile Aβ üretimini azaltırken, diğer yandan da NOTCH veya PS1’in aracılık ettiği başka bir intraselüler sinyalizasyon yetersizliği aracılığıyla taunun hiperfosforilizasyonuna yolaçması şeklinde yorumlanabilir. İnhibe olan bu sinyalizasyon PI3K-Akt olabilir. PS1’in patolojik tau hiperfosforilizasyonuna aracılık eden protein kinaz GSK-3β’yı inaktive eden bu sinyalizasyonu harekete geçirdiği gösterilmiştir. Benzer şekilde daha önce bildirilen PSEN1 ekzon 10 insR352 mutasyonunun da γ-sekretaz aracılıklı APP ve NOTCH proteolizlerini engelleyerek, yani γ-sekretaz işlev yitimi yoluyla FTD fenotipine yolaçtığı bildirilmiştir.
Tüm bunlar dışında klinik olarak dvFTD ile sunulan olguların bir kısmı, otozomal resesif kalıtım paternine sahip olan TREM2 homozigot gen mutasyonu taşıyan Nasu-Hakola hastaları olarak karşımıza çıkabilmektedirler. Bizim kliniğimizde de şimdiye kadar kemik kisti olan ve olmayan toplam 14 Nasu-Hakola hastamız dvFTD kliniği ile sunulmuşlar ve mutasyon tespit edilmiştir. Özellikle akraba evliliğinin yaygın olduğu bölgelerde dvFTD kliniği ile başvuran hastalarda bu mutasyonun da göz önünde bulundurulması önemlidir. DEFALARCA AYNI ŞEY SÖYLENDİ ÖNCESİNDE DE
FTD ile ilişkisi bilinen majör Mendelyen genler Şekil FTD2’de gösterilmişlerdir.
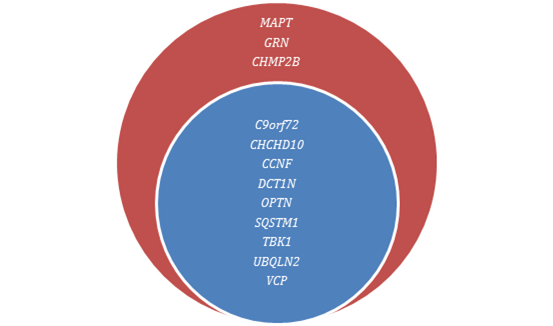
Şekil FTD2. Frontotemporal demans (FTD) ile ilişkisi bilinen Mendelyen genler. Kırmızı alanda majör FTD genleri, mavi alanda ise FTD-amiyotrofik lateral skleroz spektrum genleri görülmekte.
Yatkınlık Genleri
Genetik risk olarak öncelikle MAPT geninin H1 haplotipine sahip olmak ileri sürülmüş ve H1/H1 homozigot genotipi PSP ve KBD için bir risk oluşturduğu doğrulanmıştır.
Daha yakın bir tarihte aynı GRN mutasyonlarının yarattığı PGRN yetmezliğine benzer bir yetmezlik durumuna neden olan GRN polimorfizminin (bir miR-659 bağlanma bölgesi olan 3’-UTR’deki genetik varyant rs5848’de T alleli homozigotluğu) sporadik FTD-TDP riskini 3,2 katı arttırdığı gösterilmiştir. miR-659 yüksek riskli T aleline C aleline kıyasla daha etkin bir şekilde bağlanmakta ve böylelikle GRN'nin translasyonal inhibisyonunu arttırmaktadır. Bu çıkarımları destekler biçimde araştırıcılar homozigot rs5848 T-aleli taşıyıcılarının nöropatolojisinin GRN mutasyonu taşıyanlara benzediğini gösterdiler (Tip A) ve GRN ifadesinin miRNA'lar tarafından düzenlendiğini ve miRNA'ların translasyonal regülasyonunun kompleks nörodejeneratif hastalıklar için ortak bir mekanizma olabileceğini ileri sürdüler.
Bir GWAS çalışmasında 11 ülkeden otopsi doğrulamalı 89’u GRN mutasyonlu toplam 515 FTLD-TDP’li olguda 7. kromozomun kısa bacağında 21. pozisyonla (7p21) 3 SNP için anlamlı ilişki bulunmuştur. Bu lokus işlevi aydınlatılamayan bir transmembran protein olan transmembran protein 106B’yi (TMEM106B) kodlamaktadır. Üç SNP arasında en önde geleni olan rs1990622 için daha sık olan T alleline sahip olmanın OR’si 1,64 bulunmuştur. TMEM106B varyantlarının bu proteinin ifadesini arttırarak FTLD-TDP riskini oluşturdukları düşünülmektedir. Risk GRN mutasyonu taşısın veya taşımasın, aile öyküsü olsun veya olmasın tüm olgular için geçerlidir. Bu açıdan APOE’nin PSEN mutasyonlu olgularda AH başlangıç yaşını değiştirmesine benzetilebilir.
C9orf72 geni lokusunun aynı zamanda sporadik ALS için bir yatkınlık lokusu olduğu Hollanda, Finlandiya ve Birleşik Krallık'tan üç ayrı büyük GWAS çalışmasında ortaya konmuştur. Bunlardan son ikisini Amerikan, İtalyan ve İrlandalı GWAS veri setleriyle birleştiren 2000 olguluk bir meta-analiz Finli kurucu haplotipi olarak anılan bu risk haplotipinin 1500 yıl kadar geriye giden tek bir mutasyonla ilişkilendirilmesinin en makul açıklama olacağını ileri sürmüştür. Yukarıda sözedildiği gibi daha yakın tarihli bir çalışma bu riskin hem sporadik hem de ailevi ALS ve FTD için geçerli olduğunu ortaya koymuştur.
Genetik FTD'lerin Fenotipik Ayırıcı Özellikleri
MAPT olgularının 1. derece akrabalarında demans öyküsü daima mevcuttur. Buna karşılık GRN olgularının soygeçmişleri negatif olabilir. Bir çalışmada MAPT olgularının başlangıç yaşı GRN ve diğer FTD olgularına göre daha erkendi. Ekstremite apraksisi ve atvPPA GRN olgularında diğer FTD olgularına kıyasla daha sıktı. Buna karşılık dvFTD ve semantik bozukluk MAPT için daha kuvvetli bir öngörücü idi. Başka bir çalışmada GRN olguları MAPT olgularına kıyasla daha hızlı tüm beyin atrofisi sergilediler. VBM ile her iki grup da anterior singulat-prefrontal-insüler tarzda bir atrofi sergilerlerken, GRN grubu asimetrik, MAPT grubu ise simetrik bir atrofi tarzına sahipti. VBM kullanılan başka bir çalışmada ise GRN olguları asimetrik inferior frontal, temporal and inferior pariyetal lob gri madde atrofisi ve uzun intrahemisferik asosiasyon beyaz madde yolaklarının tutulumu sergilerlerken, MAPT olguları simetrik anteromedyal temporal lob ve orbitofrontal gri madde atrofisi ve forniks tutulumu gösterdiler. GRN olgularındaki asimetrik atrofiye karşı MAPT olgularındaki simetri atrofi tarzının iki grup arasında ayırıcı bir özellik olabileceği başka bir çalışmada da ileri sürüldü. Yine bir VBM karşılaştırılmasında MAPT olgularının medial temporal lob, insula ve putamende daha fazla gri madde kaybı gösterdiği bulundu.
Yardımcı Tanı Yöntemleri – Biyoişaretleyiciler
Şu anda klinik pratikte FTD'yi teşhis etmek için kullanılacak serum ve beyin-omurilik sıvısı (BOS) biyoişaretleyicisi bulunmamaktadır. Bununla birlikte, son dönemde buna yönelik birtakım gelişmeler olmuştur. Bir aksonal hücre iskeleti proteini olan nörofilaman hafif zincirin (NFL) BOS seviyesinin FTD'de AH'ye göre daha yüksek olduğu ve hastalığın şiddeti ile ilişkili olduğu gösterilmiştir. BOS TDP-43 düzeyinin nonspesifik olduğu, ancak serum ve BOS fosforile TDP-43 düzeyinin C9orf72 tekrar artışı ve GRN mutasyonu olan olgularda diğer FTD alttipleri ve sağlıklı kontrollere göre yüksek olduğu bulunmuştur. PGRN çalışmalarında GRN mutasyonlu olgularda GRN mRNA, serum ve BOS PGRN düzeylerinin düşük olduğu gösterilmiş, ancak serum ve BOS arasındaki korelasyon zayıf bulunmuştur. Şu an için adı anılmakta olan CHIT-1, YKL-40, GFAP gibi diğer işaretleyiciler için yeterli sayıda çalışma henüz yoktur. Tüm bu sayılanlardan rutin klinik uygulamaya girmiş bir biyoişaretleyici maalesef henüz yoktur ve özellikle erken başlangıçlı olgularda FTD-AH ayrımında kullanılabilen işaretleyiciler halen BOS amiloid ve tau düzeyleridir.
Yapısal ve fonksiyonel nörogörüntüleme yöntemleri ise yukarıda pek çok kez sözü edildiği ve detaylandırıldığı gibi FTD tanısını desteklemek, alttipleri ve olası gen mutasyonları hakkında fikir sahibi olmak için kullanılabilmektedir.
Bunların dışında 18F-flortaucipir tau-PET, AH'de nörofibriler yumaklara bağlanıyor gibi göründüğü ve klinik semptomlarla iyi korelasyon gösterdiği için potansiyel olarak AH ve FTD arasında ayrım yapabilir. Bununla birlikte sınırlı duyarlılık ve özgüllüğe sahiptir.
|
|
Nöropatoloji
Nöropatolojik olarak makroskopik açıdan FTLD antitelerinin ayırıcı özelliği daha önce de değinildiği gibi lober atrofidir. En sık olarak asimetrik fronto-temporal kombine atrofi görülürken, bunu izole frontal, anterior temporal ve parietal lob atrofileri izler. Hastalık ilerledikçe atrofi tarzı simetrikleşme eğilimi gösterir. Anterior insula, anterior singulat ve subkallosal giruslar, hippokampus ve amigdala ile subkortikal yapılar bazal ganglia ve talamus tutulan derin yapılardır. AH’nin aksine, tutulum tarzının nasıl ilerleyeceği önceden çok iyi kestirilemez. Yine de dvFTD’nin patolojik ilerleyişine ilişkin bir evreleme sistemi önerilmiştir. Buna göre en erken hafif atrofi orbital ve superior medial frontal korteksler ve hippokampustadır (evre 1), diğer anterior frontal bölgeler, temporopolar ve inferior temporal korteksler ve bazal gangliayı (kaudatta en belirgin olmak üzere azalan tarzda putamen ve globus pallidus) tutacak şekilde ilerler (evre 2). Sonrasında anterior bölgeler beyaz madde de dahil olmak üzere genel olarak tutulur (evre 3). Nihayet anteriorda daha belirgin olan global bir kortikal atrofi ve talamusun tutulumu görülür (evre 4).
Mikroskopik olarak tau+ inklüzyonlar (FTLD-tau), Ub+T- inklüzyonlar (FTLD-U) olarak sınıflanırlar. FTLD-U’nun %90’ına yaklaşan bir kısmında ubikuitinize proteinin TDP-43 olduğu (FTLD-TDP) bulunmuştur. Geri kalan %10 kadar FTLD-U’nun da ubkuitinize proteinlerinin FUS olduğu (FTLD-FUS) bulunmuş ve FTLD-U sınıfı henüz ubikuitinize proteini bulunamayan az sayıdaki CHMP2B mutasyonlu familyal FTLD hastasına sınırlı kalmıştır. Çok az sayıdaki hastada ise geçen yüzyılın “ayırt ettirici histopatolojisi olmayan demans-DLDH” veya non-spesifik gliozis tanımını hakedecek şekilde herhangi bir inklüzyon (FTLD-ni) görülmez. Tümünün de ortak özelliği tutulan alanlarda saptanan nöron kaybı, gliosis ve yüzeyel lineer spongiozistir.
FTLD-Tau
FTLD-taunun Pick hastalığı formunda fronto-temporal yüzeyel kortikal tabakalarda şiş, balonlaşmış görünümlü Pick hücreleri saptanabilir. PiH’in ayırıcı özelliği olan Pick cisimcikleri (PiC) nöronların içinde görülen büyük ve yuvarlak inklüzyonlardır (NCI) (Şekil FTD3). PiC’ler hippokampus ve amigdalanın yanısıra neokortekste II. ve VI. kortikal tabakalarda yoğun olarak bulunurlar. II. tabaka kısa kortiko-kortikal bağlantıların kökeni ve hedefi iken, VI. tabaka talamik difüz projeksiyon sistemini kabul eden ve kortiko-talamik liflerin kökenini oluşturan tabakadır. AH’de NFY’lerin başlıca yerleşim yeri olan ve uzun asosiasyon lifleri ve inen yolların kökeni olan III. ve V. tabakalarda PiC’ler fazlaca görünmeseler de bu tabakalarda ağır nöron kaybı vardır. Tutularak atrofiye katılan ve PiC görülebilen subkortikal yapılar arasında ağırdan hafife doğru kaudat, putamen ve globus pallidus, çok daha hafif düzeyde olmak üzere de substantia nigra bulunur; hipotalamik lateral tuberal çekirdek ağır biçimde, locus coeruleus genellikle tutulmuştur. 3R ve 4R tauyu birlikte içeren AH NFY’lerinden farklı olarak PiC’ler baskın olarak 3R taudan oluşur. Tau antikorlarıyla farklı tarzlarda boyanmaları ve nörofilamanlar, çift sarmallı filamanlar, kısa düz ve uzun kıvrımlı fibriller gibi farklı eleman içerikleriyle, oldukça homojen bir yapı olan NFY’lerden farklı olarak değişkenlik gösterirler. Bunlar PiH’e özgü olarak Bielschowsky boyası ile boyanırken, Gallyas ile boyanmazlar. PiC’ye tutulmamış neokortikal alanlarda raslanılmıyor olsa da sayıları atrofiyle korele etmez ve daima atrofik loba kıyasla hippokampusta daha bol miktarda bulunurlar.
KBD’de başlıca fokal atrofiye uğramış kortikal bölgelerde görülen balon nöronlar ayırıcı özelliklerdendir. NCI’ler PiC’lerden farklı olarak Bielschowsky ile gayet zayıf boyanırlarken, bu kez Gallyas ile iyi boyanırlar. Başlıca 4R taudan oluşan düz, kısa, çok ince fibriler inklüzyonlardır. Yaygın subkortikal tutulum içinde substantia nigra nöronlarında kortikobazal cisimcikler de denilen globoz NFY’ler ayırt edilir. NCI’lerin yanı sıra yaygın astrositik τ inklüzyonları (GCI’lar) tanı koydurucudur. Bunlara amiloid içermeyen glial plaklar adı da verilir. Başlıca astrositlerin distal proçeslerinde birikmeleriyle daha çok perikarya ve proksimal proçesler yerleşimli PSP tipi püsküllü (”tufted”) astrositlerden ayrılırlar. Oligodendrogliada da yaygın biçimde τ ekspresyonu ve arjirofilik helezonsu inklüzyonlar görülür.
PSP’nin klinik sunumları arasında klasik Richardson sendromu, L-Dopa cevaplı parkinsonizm ve yürüyüş bozukluklu saf akinezi (PAGF) gibi klasik motor sunumlarında korteks göreli olarak korunduğu öteden beri bilinirken kortikal tutulum FTD, KBS ve konuşma apraksisi gibi kognitif sunumlarında belirgindir. Bu subkortikal veya kortikal tutulum yatkınlığını yansıtan klinik sunum çeşitliliği dışında histopatolojik ve biyokimyasal özellikler farklı sunumlarda paylaşılmaktadır. NCI’ler 4R taudan oluşan düz filamanlar şeklindeki NFY’lerdir. Arjirofilik (Gallyas +), tau+ astrositik ve oligodendrositik inklüzyonlar görülür. Püsküllü astrositler giderek PSP için patognomonik bir bulgu olarak yerleşmektedir. Oligodendroglial inklüzyonlar perinükleer yerleşimlidir ve helezonsu cisimcikler adı da verilir.
Şekil FTD3. Pick Cisimcikleri. Bielschowsky gümüş boyama ile arjantofilik intraselüler inklüzyonlar (Kaynak: http://www.binderlab.northwestern.edu/pickbodies.html)
FTLD-TDP
FTLD-TDP inklüzyonları daha önce sözedildiği gibi NCI, NII ve DN şekillerinde olur ve bunların biri ya da birkaçının baskınlığı ve tutulum tarzına göre 4 ayrı alt tipe sınıflandırılmıştır.
FTLD-FUS
FTLD-FUS atipik FTLD-U (aFTLD-U), bazofilik inklüzyon cisimciği hastalığı (BIBD) ve nöronal orta boy filaman inklüzyonu hastalığı (NIFID) alt tiplerini kapsar. Kromozom 17 ilintili FTLD ve diğer ailevi FTLD’ler de sporadik FTLD'lerin nöropatolojisini paylaşırlar.
Tedavi
Nedene yönelik bir tedavi doğal olarak FTLD mekanizmasında temel rolü oynayan tau, TDP-43 ve FUS proteinlerinin patolojik katlanmasını engellemeyi hedeflemelidir. AH’nin mekanizma temelli tedavi stratejilerinden tau hedefli stratejilerin ileride FTLD’lerde de kullanılabileceği düşünülebilir. FTLD’de mekanizma temelli tedavilerin yerleşebilmesi için farklı proteinopatilerin de klinik olarak tanınabilmesi tabii ki bir zorunluluk olacaktır. Tau-antikorları, tau fosforilasyon ve asetilasyon inhibitörleri, tau aşıları ve mikrotübül stabilize edici ajanlarla ilgili çalışmalar halen çeşitli aşamalarda devam etmektedir. Bunların yanı sıra, genetik FTD'li olgularda C9orf72 tekrar artışını azaltmak veya PRGN ekspresyonunu artırmak için antisens oligonükleotidler (bir hedef genin mRNA'sını inaktive edebilen sentetik nükleik asitler) kullanan yenilikçi gen düzenleme tedavi yaklaşımları aktif araştırma ve geliştirme aşamasındadır.
Bazı bildirimlerde selektif serotonin geri alım inhibitörlerinin (SSRI) yararı üzerinde durulmaktadır. ChEI’lerin ve memantinin yararı gösterilmemiştir. Çeşitli davranışsal belirtilerin tedavisi için AH tedavisinde önerilenlere paralel bir yol izlenebilir. Parkinsonizm büyük ölçüde L-dopa’ya cevapsızsa da erken dönemde alınabilecek olan sınırlı cevaptan yararlanılabilir.
Kaynaklar
1.
Anonymous. Consensus Statement. Clinical and neuropathological criteria
for frontotemporal dementia. J Neurol Neurosurg Psychiatry. 1994;57(4):416-8.
2. Benatar M, Wuu J, Fernandez C, ve ark. Motor neuron involvement in
multisystem proteinopathy: implications for ALS. Neurology. 2013;80: 1874-1880.
3. Gorno-Tempini ML, Hillis AE, Weintraub S, Kertesz A, Mendez M, Cappa
SF, ve ark. Classification of primary progressive aphasia and its variants.
Neurology. 2011;76(11):1006-14.
4. Johnson JO, Mandrioli J, Benatar M, Abramzon Y, Van Deerlin VM, Trojanowski
JQ, ve ark. Exome sequencing reveals VCP mutations as a cause of familial
ALS. Neuron. 2010;68(5):857-64.
5. Lattante S, Rouleau GA, Kabashi E. TARDBP and FUS mutations associated
with amyotrophic lateral sclerosis: summary and update. Hum Mutat 2013;34:812-826.
6. Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S, et al.
Frequency of the C9orf72 hexanucleotide repeat expansion in patients with
amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional
study. Lancet Neurol. 2012;11(4):323-30.
7. Mesulam MM. Slowly progressive aphasia without generalized dementia.
Ann Neurol. 1982;11(6):592-8.
8. Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, ve ark.
Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria.
Neurology. 1998;51(6):1546-54.
9. Rademakers R, Neumann M, Mackenzie IR. Advances in understanding the
molecular basis of frontotemporal dementia. Nat Rev Neurol. 2012;8(8):423-34.
10. Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J,
ve ark. Sensitivity of revised diagnostic criteria for the behavioural variant
of frontotemporal dementia. Brain. 2011;134(Pt 9):2456-77.
11. Wicks P, Abrahams S, Papps B, Al-Chalabi A, Shaw CE, Leigh PN, et
al. SOD1 and cognitive dysfunction in familial amyotrophic lateral sclerosis.
J Neurol. 2009;256(2):234-41.
12. Yu CE, Bird TD, Bekris LM, Montine TJ, Leverenz JB, Steinbart E, ve
ark. The spectrum of mutations in progranulin: a collaborative study screening
545 cases of neurodegeneration. Arch Neurol. 2010;67(2):161-70.