DEMANS
SENDROMU, ALZHEIMER HASTALIĞI ve ALZHEIMER DIŞI DEMANSLAR
İ. Hakan Gürvit
Son Güncelleştirme Tarihi: 12.5.2010
GİRİŞ
Tıbbi pratikte demans kavramı belli bir
asgari bulgular topluluğu olarak bir sendroma karşılık
gelir. Örneğin, nasıl nefropati sendromunun altında
hipertansiyondan diyabete çok sayıda etyolojik neden sıralanabilirse,
demans sendromu da uzun bir listeye ancak sığdırılabilecek
bir dizi etyolojik etmenin sonucu olarak gelişir. Liste uzun olsa
da, ilerleyen bölümlerde ayrıntıyla görüleceği gibi, tüm
demansların üçte ikisinden fazlası Alzheimer
hastalığına (AH) bağlıdır. Bu yüksek
oran, klinik pratikte belli bir ayırıcı tanı
kolaylığı sağlarken (demans sendromunu tanıyan
klinisyen ileri gitmeksizin AH tanısı koyarsa % 65 doğru
tanı koymuş olacaktır), AH’nin doğasından kaynaklanan
nedenlerle de, kendine özgü güçlükler taşır: diabetes mellitustaki
yüksek kan şekeri düzeyi gibi, AH’nin bir biyolojik işaretleyicisi
olmadığı için kesin tanı ancak nöropatolojik yöntemlerle
mümkündür. Bu durumda klinisyen gördüğü klinik tablo için AH
değil, fakat “Alzheimer Tipi Demans” (bundan böyle İngilizce
karşılığı olan Dementia of Alzheimer Type
kısaltması DAT olarak kullanılacak) sendromu, ya da bu tablonun
kesin olmasa da, yüksek olasılıkla AH’ye bağlı
olduğunu düşündüğünü gösterir biçimde “Muhtemel Alzheimer
Hastalığı” (bundan böyle İngilizce
karşılığı olan Probable Alzheimer’s Disease
kısaltması PRAD olarak kullanılacak) tanısı
koyabilecektir. Aşağıda Alzheimer Hastalığı
bölümünde ayrıntıyla gözden geçirileceği gibi, DAT terimi
DSM-IV, PRAD ise NINCDS-ADRDA tanı kriterleri olarak
kullanıldığında seçilen özgül tanı etiketleridir.
Aralarındaki ufak nüanslarla birlikte DAT ve PRAD büyük ölçüde örtüşürler.
Bu bölümde PRAD ve DAT kavramları özgül tanı kriterlerinin
karşılıkları olarak bırakılacak, günlük
kullanımda AH’yle ilintili demans için ise “Alzheimer tipi demans” (ATD)
terimi kullanılacaktır. Bu durum ilk bakışta demansla karşılaşan
klinisyenler açısından ciddi bir zorluk gibi görünse de, geniş
nekropsi serilerinde, klinik olarak PRAD ya da DAT tanısı konulan
olguların %90’ının kesin AH patolojik tanısı
aldıkları görülmüştür. Bir başka deyişle, ATD
sendromu %10 gibi küçük bir olasılıkla başka demans etmenleriyle
de ortaya çıkabilmektedir. Günlük pratikte, ilk adımda demans
sendromunu zihinsel bozuklukla kendini gösteren diğer sendromlardan
ayırabilmek, demansa karar verildiği takdirde ikinci adımda
demanslar içinde AH’nin ayrıcalıklı yeri nedeniyle, AH’ye özgü
tipik sendromu, diğer demans nedenlerinin klinik nüanslarından
ayırdedebilmek esastır.
Bu bölümde önce demans sendromu kavramsal olarak
tanımlanacak, normal yaşlılıktan ve diğer zihinsel
bozukluklardan farkı ortaya konulacak ve sonrasında demans muayenesi,
demansların ayırıcı tanısı üzerinde
durulacaktır. Ardından özgül demans nedenlerine geçilecek ve AH
ile AH-dışı demanslar incelenecektir. AH
kısmında klinik tablo, tanı kriterleri, seyir, epidemiyoloji,
genetik, patoloji-patogenez, ve tedavi, alt kısımlarda
ayrıntıyla ele alınacaktır. AH-dışı
demanslar kısmında AH dışı nörodejeneratif demans
hastalıkları arasında olan Lewy cisimcikli demans (LCD), fronto-temporal
Demans (FTD) ve Pick Kompleksi kavramı, prion
hastalıkları ve özellikle Creutzfeldt-Jacob
hastalığı (CJH), hareket bozukluklarıyla birlikte
demans, sekonder demanslar arasında özellikle vasküler demans (VaD)
incelenecek, diğer nadir demans nedenleri kısaca
özetlenecektir. Tıp öğrencileri için büyük puntolarla
yazılmış olan kısımlar esasken, küçük punto ile
yazılmış kısımlar ileri okuma olarak
tasarlanmıştır.
DEMANS SENDROMU
BIR KLİNİK KAVRAM OLARAK DEMANS
Dilimizde popüler kullanımıyla bunama
adı verilen demans kelimesi, etimolojik olarak Latince zihin anlamına
gelen mens kelimesinden türemiştir ve demens zihnin yitirilmesi
anlamına gelir. Latince’deki kullanım biçimiyle
“yerleşmiş, varolan, edinilmiş olan zihnin sonradan yitirilmesi”
anlamını taşımaktadır. Bu biçimiyle edinilecek
olanın, yani zihnin “hiç edinilememesi” durumundan
farklıdır. Böylelikle, demansı zihinsel gerilikler ya da
Yunancasıyla oligofreni (yetersiz zihin), Latincesiyle amensiyadan (zihin
yokluğu) etimolojik ve semantik olarak da ayırabiliriz.
Herhangi bir zaman kesitinde bir demanslı hasta ile bir oligofrenik
hastanın zihinsel özürlülük, ya da yeterlilik düzeyleri işlevsel
olarak benzerlik gösterebilir, ancak demanslının edinsel nedenlerle
(genetik ya da çevresel, sıklıkla da ikisinin birlikte etkileriyle)
kendinde mevcut olan kapasiteyi yitirdiğini, oligofrenin ise
gelişimsel nedenlerle bu kapasiteyi yetersiz edindiğini
söyleyebiliriz. Öyleyse demansın tanımına, “erişkin
merkezi sinir sisteminin hasarlanması sonucu gelişen zihinsel
yeteneklerde bozulma” şeklinde başlayabiliriz.
Zihinsel yeteneklerde hangi tarz bozulma demans
kavramını karşılar? Zihinsel yeteneklerde demansa
özgü tarzda bozulma içinde, bozulan zihinsel yeteneklerin (ya da teknik
terminolojiyle kognitif işlevlerin) sayısı, bu bozulmanın
başlangıç tarzı, şiddeti, süresi ve doğal seyri, onu
diğer bozulma tarzlarından ayırdedici olmaktadır.
Demans olarak nitelendirilebilecek bir zihinsel
bozulma, tanımı gereği, öncelikle birden fazla kognitif
işlevi bozmalıdır. Bu tanım uyarınca,
örneğin serebro-vasküler olaylar sonucu gelişen sol ya da sağ
hemisfer hasarlarına eşlik eden izole kognitif bozukluklar (afazi,
ihmal, amnezi, vb.) demans çerçevesinde değerlendirilmezler.
Oysa ki, orta ve büyük çaplı arterlerin birbirini izleyen
tıkanmaları sonucunda gelişen serebral infarktüsler demans düzeyinde
birden fazla kognitif alan bozukluğuna neden olabilir (bu tür bir
demans VaD altında Multi-infarkt Demans – MID olarak
sınıflanır).
Demans olarak nitelendirilebilecek bir zihinsel
bozulma, tanımı gereği, mesleki performans, sokakta, mali
işlerde bağımsızlık, sıradan aygıtların
kullanımı, hobiler, ev işleri, kendine bakım ile
özetlenebilecek günlük yaşam aktivitelerinde (GYA’lar) kayda değer
bir bozulmaya yol açacak şiddette olmalıdır.
Örneğin, AH’li bir hastanın GYA’ları, erken evrede mesleki
performansındaki bozulma iş arkadaşları tarafından
farkedilir, kendine yabancı mekanlarda yolunu kaybedebilir, evde
yemeğini sık sık yakabilir düzeyde bozulabilecekken, ileri
evrede bu bozulma giyinme, yıkanma gibi temel aktivitelerde yardıma
gereksinecek düzeye ulaşabilir. Merkez sinir sisteminin birçok
hastalığına, bu arada örneğin Parkinson
hastalığı ya da multipl skleroza da sıklıkla kognitif
bozulmalar eşlik eder. Bu hastalar, hastalıkları öncesine
göre daha unutkan, zihinsel tepkileri açısından daha yavaş, yeni
problemlere daha güç çözüm üretir durumda olabilirler. Nöropsikolojik
muayeneleri yapıldığında, bu yakınmalarını
yansıtır düzeyde, bellek, dikkat ve yürütücü işlevler
alanlarında bozulmalar saptanabilir. Ancak, yine sıklıkla
bu hastaların GYA’ları, sözkonusu kognitif sorunları nedeniyle
kayda değer biçimde kısıtlanmamış (parezi ve
bradikinezi gibi motor sorunlara bağlı kısıtlanma ihmal
edilmelidir) ve bağımsızlıklarını sürdürüyor
olacaklardır (anılan iki hastalıkta da bazen kognitif
bozulmanın şiddeti demans düzeyine ulaşabilir). Bu
durumda söz konusu olan demans değil, fakat frontal yürütücü
bozukluktur. Yaşlılıkla ilintili unutkanlık
(age associated memory impairment – AAMI) ve hafif kognitif bozukluk
(mild cognitive impairment – MCI) kavramları demansla ayrılması
gereken diğer durumlardır ve ayrı alt kısımlarda daha
ayrıntılı ele alınacaklardır. AH’nin patogenezi
kısmında ayrıntısıyla gözden geçirileceği gibi,
AH’nin patogenetik başlangıcından, kendisini ATD şeklinde
klinik olarak ortaya koyana kadar 20 yıllık bir süre gerekebileceği
hesaplanmaktadır. Hastalık limbik sistemde başlayacak ve
uzun yıllar buraya sınırlı kaldıktan sonra neokortikal
asosiasyon alanlarına geçecek ve giderek bu bölgedeki hasarın
şiddetini arttıracaktır. Nörodejenerasyon
limbik-paralimbik alanlara sınırlı kaldığı sürece
bu alanlarda temsil edilen epizodik bellek sistemi giderek
işlevsizleşeceğinden bu durum bir ilerleyici, izole
unutkanlık olarak yansıyacaktır. Ancak, nörodejenerasyon
neokortikal heteromodal kortekslere sıçradığında, bu
alanlarda temsil edilen dil ve görsel-mekansal işlevler de bozulmaya
başlayacağından demansın asgari koşulu olan “birden
fazla kognitif bozukluk” kriteri doldurulmuş olacaktır.
İşte AAMI’ler ve MCI’ların bir bölümü, muhtemelen limbik ve
paralimbik dönemlere karşılık gelecek şekilde, AH’nin
demans öncesi pre-klinik ve prodromal evrelerini oluşturmakta, ATD ise bu
süreklilikte demans evresine karşılık gelmektedir.
Demans olarak nitelendirilebilecek bir zihinsel
bozulma, tanımı gereği, doğal seyri açısından
kalıcı ve sıklıkla da ilerleyici olmalıdır.
Öykünün başlangıç ve seyir özellikleri göz önüne
alınmaksızın, salt zihinsel durum açısından
yapılacak bir karşılaştırmada, örneğin akut
konfüzyonel durum (psikiyatrik terminolojide sıklıkla delirium) ile
demans yüzeysel bir benzerlik taşıyabilir; hatta demansın ileri
evreleri dikkatin ileri derecede bozulduğu bir kronik konfüzyonel durum
özelliği gösterebilir. Oysa ki, akut konfüzyonel durumun
(AKD) akut başlangıç ve günler, bazen de haftalar sürüp, yine bazen
kendiliğinden tümüyle düzelme tarzındaki seyri onu demansın
tarzından kolaylıkla ayırır. Demanslar, özellikle de
AH, sıklıkla sinsi başlangıçlı ve yıllar içinde
yavaş ve sürekli ilerleyici seyirlidir. Akut olarak demans
şiddetinde bir zihinsel bozukluğa neden olabilecek etmenler de
vardır: yeterince şiddetli bir kafa travması, kognitif
işlevler açısından stratejik bir konumdaki inme, global serebral
iskemi ya da anoksi, klinik profil açısından demans özellikleri
gösteren bir zihinsel bozukluğa neden olabilirler. Bu durumlarda,
akut başlangıç sonrası zihinsel bozukluğun şiddeti en
fazla olarak en erken evrede görülecek, hasta izleyen aylarda, serebral
plastisitenin devreye girmesiyle, 1. yıl sonuna kadar sürebilecek
bir düzelme eğilimi gösterecek, sonrasında ise düzelme tam
olmamışsa arta kalan klinik tablo sekel olarak kalıcı
olacaktır. Bu sekel halen demans şiddetinde olabilir, ancak
AH’den farklı olarak, klinik ve nöropsikolojik olarak yıldan
yıla farklılık göstermeyen bir statik demans özelliği
gösterecektir. Nöroloji literatürü ve pratik kullanımda bu tür
statik tablolara demans yerine ensefalopati (iskemik/anoksik) ya da kafa
travmasına bağlı olanlarda “travmatik beyin hasarı”
şeklinde adlandırma eğilimi vardır. Bunlara,
zamanında müdahale edilmemiş ya da yetersiz tedavi edilmiş,
başta Herpes simpleks ensefaliti ve tbc meningoensefaliti gibi MSS
infeksiyonlarının bu tür statik demanslarla sonlanacağı
eklenebilir. VaD’lar arasında MID ya da stratejik infarkt demansları,
AH’den farklı olarak akut başlarlar. AH’nin sürekli ilerleyici
seyrine karşın VaD’ın mutad seyri basamaksı ilerleme
şeklindedir (iki inme arasında değişmeden kalan bir plato
dönemi, sonrasında inmeyi izleyen ani kötüleşme). AH’nin
yıllar içinde ilerleme şeklindeki yavaş seyrine
karşılık örneğin CJH 1 yıl içinde yatağa
bağımlı nihai evreye ulaşma ve ölüm şeklinde çok
hızlı bir seyir gösterir. Bir başka deyişle, sinsi
başlangıç, kronik yavaş seyirli demansların prototipi, böyle
bir seyre sahip çok sayıda demans hastalığı arasında
nasıl AH ise, “hızlı seyirli demanslar”, prototipi CJH olan ve
çok sayıda hastalıktan oluşan ayrı bir alt başlık
olarak sınıflandırılabilir. Bu tarz bir
yaklaşımla, ilk durumda AH’yi altın standart olarak alarak
mevcut durumda görülen olgunun ne ölçüde standart bir AH tablosuyla
örtüştüğü, ne ölçüde ayrıldığı temelinde bir
ayırıcı tanı akıl yürütmesi yapılacakken, ikinci
durumda bu kez altın standart tipik CJH özellikleri olacaktır.
Demans tanısı bilinç
bulanıklığı olmayan hastada konulur. AKD’lere yukarda
söz edilen seyirleri yanısıra sıklıkla bilinç
bulanıklığı da eşlik eder. AKD’ler patogenetik
olarak, uyanıklık, farkındalık ve global dikkatin nöral
karşılığı olan retiküler formasyonun işlevini
bozarak ortaya çıkarlar. Dolayısıyla, klinik tablo bilinç
dalgalanmaları yanısıra, nöropsikolojik olarak başlıca
global dikkat bozukluğu ve tüm kognitif işlevlerde buna sekonder
bozulmalardır. AKD’yi demansa yüzeysel olarak benzer kılan
özellik bu global bozulma durumudur. Demans durumlarının
toksik-metabolik ensefalopatilerin AKD’sinden ayırmak ve muayene
bulgularının dikkat bozukluğuna sekonder geçici nitelikte
bulgular değil, fakat o işlevin nöral altyapısının
primer bozukluğunu yansıttığını söyleyebilmek
için tanı muayenesinin bilinç dalgalanması olmayan bir hastada
yapılması koşulu demans tanımına giren bir
ölçüttür. Ancak, klinik pratikte demans ve AKD durumlarının ak
ve kara gibi kutupsallıklar olmadığı ve seyrek olmayarak iç
içe geçebileceklerini unutmamak gerekir. Bunun başlıca nedeni
demans patogenezinin yarattığı nöronal rezervdeki
aşırı kısıtlanma sonucu demans hastasının
toksik-metabolik durumlar ya da sistemik infeksiyonlar sırasında
AKD’ye çok daha yatkın olmasıdır. Böyle bir durumda, AKD
demans seyrinin üzerine binerek tabloyu bir süre için akut olarak
ağırlaştıracaktır. Titiz alınmış
bir öykü, sinsi seyirli bir zihinsel yıkımın seyrinde aniden
gelişen ve GYA’ları hızla bozan bir tabloyu ifade
edecektir. Ayrıca, LCD’de dikkat dalgalanmalarının,
genellikle bir AKD düzeyinde olmasa da, daha başlangıçtan itibaren klinik
tablonun asli bileşenlerinden biri olduğu
hatırlanmalıdır.
Bu kez, anılan ölçütleri de katarak daha tam
bir tanıma girişelim:
“Demans, erişkin MSS’nin hasarlanması
sonucu, bilinç bulanıklığı olmaksızın, birden
fazla kognitif alanın bozulması, bununla ilintili olarak günlük
yaşam aktivitelerinin eski düzeyinde sürdürülememesine neden olan,
doğal seyri açısından kalıcı, sıklıkla da
ilerleyici bir klinik tablodur”
DEMANSLARIN SINIFLANDIRILMASI
Demanslar öncelikle primer ve
sekonder olarak sınıflanırlar (Tablo 1).
AH’nin de dahil olduğu ve en büyük bölümü
oluşturan primer demanslar, demansa neden olan MSS’nin nörodejeneratif
hastalıklarını içerir. Nörodejeneratif hastalık,
zihinsel işlevlerin alt yapısını oluşturan limbik ve
asosiasyon alanlarında, sıklıkla kendine özgü patolojik izi
bırakarak (örneğin, AH’de senil plak ve nörofibriler yumaklar, LCD’de
Lewy cisimcikleri) buralarda nöron ve sinaps kaybıyla dejenerasyona yol
açar ve işlevini bozar. Bu patogenez belli bir yayılım
aşamasında kliniğe demans olarak yansır.
Nörodejenerasyon, AH’de olduğu gibi anılan alanlara
sınırlı kalma eğilimindeyse, demans izole ya da
ağırlıklı klinik tablo olarak kalır; oysa ki, motor sistem
de dejenerasyona dahil olursa LCD’de olduğu gibi parkinsonizm, Huntington
hastalığında olduğu gibi kore, demansla birlikte, bazen
daha da önünde klinik tablonun ağırlıklı bir
parçasıdır. Dolayısıyla, primer demanslar klinik
görünüşlerine göre, kendi içlerinde primer izole demanslar ve motor
bozuklukla birlikte olan primer demanslar olarak
sınıflanabilirler.
Önceki kısımdaki nihai tanımı
hatırlayalım: “Demans...birden fazla kognitif
alanın..bozulmasına yolaçan.... bir klinik tablodur”. Bu
tanım uyarınca, bozulan ve salim kalan kognitif alanların tarzına
göre de sınıflamayı ilerletebiliriz. Nörodejenerasyon,
yukarda anılan alanlarda eşzamanlı başlayıp süregiden
bir seyir izlemez, fakat patogenezin kendine özgü bir tarzı
vardır. Örneğin, AH, daha önce de değinildiği gibi
yıllarca limbik alanlara sınırlı kaldıktan sonra neokortikal
alanlara geçer. Ancak, neokortikal asosiasyon alanlarına geçiş
de eşzamanlı bir yayılım değildir, fakat yine özgül
bir zamansal seyir gösterir: posterior heteromodal alanlar prefrontal
korteksten nispeten daha önce tutulur. Bu zamansal tarz, erken ve orta
evrelerinde AH’nin kendine özgü nöropsikolojik profilini belirler: tipik AH
sendromu (ATD) yakın bellek (limbik), dilsel (sol hemisfer posterior
heteromodal) ve görsel-mekansal (sağ hemisfer posterior heteromodal)
işlevlerin bozulduğu, prefrontal kortekse özgü yürütücü işlevler
(planlama, soyutlama, zihinsel esneklik, vb.) ve kişiliğin göreli
salim kaldığı bir profile sahiptir. Buna
karşılık fronto-temporal demans (örneğin, Pick hastalığı)
anılan seyrin tersine, öncelikle ve bazen de yalnızca prefrontal
korteksi tutar, limbik ve posterior neokortikal alanlar salim kalır.
FTD bu patogenezi yansıtır bir klinik profile sahiptir: erken dönemde
yürütücü işlevlerde bozulma, kişilik değişikliği, buna
karşılık yakın bellek ve mekan oryantasyonu, navigasyonel
işlevlerde (görsel-mekansal işlevler) göreli korunma.
Sistemik, nörolojik ya da psikiyatrik bir
hastalığın seyri sırasında, mutad klinik
gösterilerinin yanı sıra demansa da neden olması durumunda, söz
konusu demans sekonder demanslar altında sınıflanır.
Sekonder demansların en sık nedeni VaD’dir. Gerek orta ve büyük
çaplı serebral arterlerin birbirini izleyen tıkanmaları (MID),
gerek hipertansif serebral küçük damar hastalığı (Binswanger hastalığı)
ve gerekse de stratejik lokalizasyonlu tek infarktlar VaD nedeni
olabilir. Normal nöronal işlevin gereksindiği, ekstraselüler
ortamın glukoz içeriği, oksijenizasyon düzeyi, hormonal durum ve
elektrolit dengesini bozabilecek herhangi bir sistemik veya metabolik bozukluk
(diyabet, kalp yetmezliği, hipotiroidi, kronik obstrüktif akciğer
hastalığı, böbrek yetmezliği, karaciğer
yetmezliği) ya da normal sinaptik iletimi bozabilecek herhangi bir
terapötik amaçlı ilaç, ağır metallere maruz kalma gibi durumlarda
toksik-metabolik ensefalopati ortaya çıkar. Toksik-metabolik
ensefalopatiler, büyük sıklıkla AKD olarak görünseler de bazen daha
yavaş seyirli bir tabloyla demansa da benzeyebilirler.
İnflamatuar süreçler (Nöro-Behçet sendromu, primer MSS vasküliti,
granülomatöz anjiitis, sistemik lupus, paraneoplastik limbik ensefalit),
infeksiyonlar (Tbc meningoensefaliti, Herpes simpleks ensefaliti, HIV, Lyme,
Whipple, nörosifilis, vb), nörolojik hastalıklar içinde multipl skleroz,
primer ya da sekonder beyin tümörleri, kronik subdural hematom, normal basınçlı
hidrosefali gibi yer kaplayıcı lezyonlar demansa da neden
olabilirler. Psikiyatrik hastalıklar içinde özellikle yaşlılık
depresyonu, afektif bulgularını gizleyen,
ağırlıklı biçimde motivasyonel ve kognitif bulgulardan
oluşan bir demans (bazı yazarlara göre psödo-demans) tablosu
şeklinde kendini gösterebilir.
Primer demansların, bu arada AH’nin klinik
tanısı için sekonder demansların ekarte edilebiliyor olması
gerekir. Nitekim, AH için yayınlanmış olan klinik
tanı kriterleri içinde, gerek PRAD ve gerekse DAT tanıları için
bu koşulun yerine getiriliyor olması esastır.
Tablo 1. Demans
Hastalıklarının Sınıflandırılması
|
Primer
(Dejeneratif) |
Sekonder |
|
Alzheimer
hastalığı Lewy cisimcikli demans Fronto-temporal demans FTD-davranışsal varyant İlerleyici tutuk afazi Semantik demans FTD-ALS Hareket bozukluğuyla
birlikte Parkinson hastalığı demansı Kortiko-bazal dejenerasyon Progresif supranükleer paralizi Huntington hastalığı Multi-sistem atrofiler Wilson hastalığı Nöroakantositoz Prion hastalıkları Creutzfeldt-Jacob hastalığı Gerstmann-Sträussler-Scheinker hastalığı Fatal familyal insomni Çeşitli pediyatrik
demanslar Kufs hastalığı Metakromatik lökodistrofi Gaucher hastalığı Niemann-Pick hastalığı Diğer ender demanslar Limbik demans Poliglukozan cisimcik hastalığı Arjirofilik tahıl hastalığı |
Vasküler
demans Multi-infarkt demans Binswanger hastalığı Stratejik infarkt demansı CADASIL Normal basınçlı
hidrosefali Toksik-metabolik demanslar Wernicke-Korsakoff hastalığı B12 vitamin eksikliği Hipotiroidi Kronik karaciğer hastalığı Organik çözücülere maruz kalma İlaçlar İnfeksiyonlar Herpes simpleks ensefaliti Nörosifilis Kronik menenjitler HIV-demans kompleksi Whipple hastalığı Kafa içi yer
kaplayıcı hastalıklar Neoplastik durumlar Subdural hematom Otoimmun-inflamatuar
hastalıklar Multipl skleroz Behçet hastalığı Paraneoplastik limbik ensefalit VGKC ve NMDAR kanalopatileri Granülomatöz anjitis Primer sinir sistemi vasküliti NAIM sendromu |
Bu tabloda demansa neden olan tüm nedenlerin
sıralanması amaçlanmamıştır. Büyük bir
kısmının adı geçmekle birlikte asıl amaç primer ve
sekonder demans kavramlarına açıklık getirmektir.
Kısaltmalar: VGKC: Voltaj kapılı potasyum kanalı; NMDAR:
N-metil D-aspartat reseptörü; NAIM: Non-vaskülitik otoimmun meningoensefalit.
DEMANSTA ÖYKÜ ÖZELLİKLERİ
Bir zihinsel yıkım tablosu olarak
tanımladığımız demans sendromunu oluşturan
hastalıklar, esas olarak primer sensoryel-motor alanlarla ilintili
bölgeleri değil, fakat zihinsel işlevlerin alt yapısı olan
bölgeleri (limbik ve asosiasyon alanları) hasarlar.
Dolayısıyla, demansa özgü belirtiler güçsüzlük, hissizlik, görme
alanı kayıpları gibi klasik nörolojik muayeneyle
bulgularını ortaya koymaya alıştığımız
belirtiler değil; unutkanlık, konuşma bozukluğu, yön bulma
güçlüğü, yargılama-problem çözme güçlükleri, tanıma
bozukluğu, el becerilerinde bozukluk, kişilik
değişiklikleri, anksiyete, disfori, hezeyan ve halüsinasyonlar gibi
belirtiler olacaktır.
Demans sendromunun semptomatolojisi, üç ana (kardinal)
kategoride sınıflanabilir:
1. Kognitif,
2. Davranışsal,
3. İşlevsel
(GYA’lar).
Demans öyküsü, bu üç kardinal alana özgü
yakınmaların bir sistematik içinde sorgulanması ile
alınır. Bunların yanısıra motor, otonom
sistemler ve uyku bozukluklarına ilişkin yakınma ve
belirtilerin sorgulanması da bazı demans sendromları
açısından önem taşır ve kardinal alanların
yanısıra bu alanlar da ikincil alanlar olarak
düşünülebilir.
Öykü mümkünse hastadan birinci derecede sorumlu
bir hasta yakınının da varlığında, paralel olarak
alınmalıdır.
Yakınmaların başlangıç (sinsi,
akut veya subakut) ve ilerleme tarzı (statik, kronik sürekli veya
basamaksı, hızlı) belirlenip kaydedilmelidir.
Kognitif belirtiler arasında mutlak olmasa da
en sık rastlanılan belirti bellek alanına aittir. Hasta ya
da yakını aynı soruların, aynı konuların
tekrarlanması, özel eşyanın kaybedilmesi, randevuların
unutulması, yemeğin ocakta, ocağın açık
unutulmasından yakınmaktadır. Görsel-mekansal
işlevlere ait bozulma kendini önce yabancı mekanlarda, giderek bildik
mekanlarda yön bulma güçlüğü ve bazen kaybolma şeklinde
gösterir. Dil bozukluğu erken dönemde adlandırma güçlüğü,
kelime hazinesinde daralmayla başlayıp, giderek AH’de daha mutad
olduğu gibi anlamanın da bozulduğu bir akıcı afaziye,
ya da ilerleyici tutuk afazide daha mutad olduğu gibi gramer
yapısının da bozulduğu bir tutuk afaziye
dönüşür. Semantik demansta ise sıradan nesnelerin
anlamlarının kaybolduğu bir tek kelime anlama bozukluğu söz
konusudur. İlerleyici afazisi olan bir hastanın kelime bulma
güçlüğü olan temel yakınmasının da sıklıkla hasta
ve hasta yakını tarafından “unutkanlık” olarak ifade
edileceği göz önünde bulundurulmalıdır. Öykü derinleştirildikçe
bu türden bir unutkanlık yakınmasının adresinin limbik
yakın bellek sistemi değil de sol hemisferik dil sistemi
olduğunun altı çizilir. Praktik bozukluklar (apraksiler) nedeniyle,
basit güncel aygıtların (tarak, makas, diş fırçası,
sofra aygıtları, vb.) manipulasyonları bozulabilir.
Gnostik bozukluklar (agnoziler), nesnelerin ve yüzlerin
tanınmasını, mekanda bir nesnenin, diğer nesnelere göre
pozisyonunun belirlenmesini bozabilir. Yürütücü işlevler
alanındaki bozukluk nedeniyle hasta zihinsel esnekliğini kaybetme
eğilimindedir; metaforların soyut anlamlarını kavramakta,
davranışlarını planlamakta, bireysel ve toplumsal sorunlar
üzerine akıl yürütmekte, günlük yaşamda
karşılaştığı problemlere uygun çözümler üretmekte
zorlanır.
Davranışsal belirtiler arasında, apatiye
varacak şekilde kendiliğindenliğin kaybı, disinhibisyona
varacak şekilde dürtü kontrolünde bozukluklar (hiperseksüalite, hiperfaji)
sayılabilir. Apati spektrumundaki hasta giderek daha az spontanite
örnekleri sergiler. İnisiyatif göstermez, kendiliğinden bir
şey talep etmez, kendisine yönelinmediğinde konuşmaz.
Çevresinde olup biten hiçbir şey ilgisini çekmez gibidir.
Disinhibisyon kendini öncelikle sosyal uygunsuz davranış olarak
adlandırılan sosyal konumla bağdaşmayan rahat
davranışlar, alışılmadık girişkenlik,
şakacılık, çocuksulukla gösterir; hasta yakınları bu
değişiklikleri sıklıkla aile için bir utanç vesilesi olarak
aktarırlar. Hiperseksüalite hastanın o zamana kadar
edindiği sosyal normlar dışında cinsel içerikli imalar taşıyan
söz ve davranışlar sergilemesinden uygunsuz cinsel taleplere kadar
varan şiddetlerde görülebilir. Hiperfaji hastanın
alışılagelen ağız tadının
değişmesi, oburlaşması ve özellikle şekerlemeye
düşkünleşmesi şeklinde başlar ve çay poşeti, kendi
feçesi gibi yenilmeyecek nesnelerin dahi ağza
tıkıştırılabildiği şiddetlere ulaşabilir.
Psikotik belirtiler düşünce ve algı bozuklukları şeklinde
ortaya çıkar. Düşünce bozuklukları özellikle
hırsızlık (“bakıcı paramı çalıyor”),
sadakatsizlik (“eşim beni bir başkasıyla aldatıyor”) ve
terkedilme (“beni bakımevine atacaksınız”) hezeyanları,
misidentifikasyon “burası benim evim değil, eve gidelim” ve Capgras
hezeyanı (tanış olması gereken kişi onun yerine
geçmiş bir taklidi) şeklinde ortaya çıkar. Algı
bozuklukları ise tüm duysal modalitelerde olabilecekleri gibi özellikle
görsel halüsinasyonlar şeklindedir. Bunlar çevresel uyaranların
yanlış yorumlandığı illüzyonlar, mekanda bir
varlığın mevcudiyeti hissi gibi forme olmayan halüsinasyonlar
veya canlı rüyalar gibi çok hafif algı bozukluklarından
nesneler, insan ve hayvanlar gibi forme halüsinasyonların olduğu daha
ağır durumlara değişir. Algı bozuklukları
ağırlaştıkça içgörü kaybolur ve geceye
sınırlıyken giderek gündüzleri de görülmeye
başlarlar. Duygudurum bozuklukları arasında depresyon
özellikle sık olabilir ve afektif-motivasyonel yönleri
sorgulanmalıdır. Anksiyete, huzursuzluk, yerinde duramama,
sürekli yer ve mekan değiştirme, çok çabuk sıkılma
şeklinde ortaya çıkabilir; özel bir çeşidi de yaklaşan
randevularla artan anksiyetedir. Mani, klasik psikiyatrik tablosundan çok
uygunsuz neşe ve/veya öfke, duygudurum dalgalanmaları, grandiozite,
saldırganlık şeklindeki gösterilerin bir veya
birkaçının toplamı şeklinde görülebilir. Fobiler,
hastaya özgü çeşitlilikler gösterebileceği gibi, özellikle de
eşin göz önünden uzaklaşması endişesi ve bu nedenle
eşin peşinden ayrılmama veya yalnız kalma korkusu
şeklindedir. Fiziksel veya verbal şiddeti de içeren ajitasyon,
amaçsız-tekrarlayıcı hareketler (amaçsız
dolaşma-adımlama, dolapları açıp-kapama, çarşafı
katlayıp-açma gibi aynı hareketi tekrarlama, uygunsuz yerlere,
toplama, istif etme) gözlenebilir davranışsal sorunlar
arasındadır. Gözlenebilir davranışsal bozukluklar da
şiddetlendikçe başlangıçta “güneş batma fenomeni” adı
verilen geceye sınırlı olmaktan, giderek güne yayılma
eğilimi gösterirler.
İşlevsel alana ait belirtiler, işini sürdürmek,
ev dışında yolculuk, alışveriş, mali
işleri (fatura ödemeleri, banka işleri, vb.) çekip çevirmek,
günlük aygıtları kullanmak, hobilerini sürdürmek, ev işlerini
yürütmek, kendine bakım veya hijyene (giyinmek, yıkanmak, beslenmek,
tuvalet, vb.) muktedir olmak şeklinde örneklenebilecek özellikleri
bozabilir (işlevsel alana ait belirtilerin AH’de bozulma hiyerarşisi
için ilerdeki “Alzheimer Hastalığının Klinik Evreleri”
kısmına bakınız).
Motor bozukluklar arasında başta
yürüyüş bozukluğu olmak üzere ekstrapiramidal bulgulara atfedilecek
yakınmaların sorgulanması (küçük adımlarla yürüme, donma,
istemeden hızlanma, düşme, dengesizlik, hareketlerde yavaşlama,
tremor, konuşma ve yutma bozukluğu) önemlidir. Vasküler demansa
parezi sekellerinin, fronto-temporal demansa amiyotrofinin eşlik
edebileceği akılda tutulmalı ve zaaf, kas erimesi gibi temel
belirtileri sorgulanmalıdır.
Otonom bozukluklar arasında ise üriner
inkontinansın bulunup bulunmaması önem taşır.
İdrara yetişme güçlüğü (urgency) düzeyinde hafif bir
inkontinansın dahi kaydedilmesi gerekir. Ortostatizm
yakınmaları, senkopal eğilim, kronik konstipasyon, impotans
demans sendromlarının ayırt edilmesinde önem taşıyan
diğer otonomik yakınmalar arasındadır.
Parasomniler arasında özellikle REM
uykusu sırasında tonik inhibitör aktivitenin çözülmesi sonucu ortaya
çıkan rüyaların yatakta sıçrama, dövünme, düşme,
konuşma, haykırma şeklinde dışavurumu olan REM uykusu
davranış bozukluğu (RUDB) belirtileri önem
taşır. Uykuya dalma güçlüğü, kesintili uyuma, sabah çok
erken uyanma gibi insomnia belirtileri ve insomnianın veya uyku apnesinin
sonucu olarak veya bağımsız bir fenomen olarak
“aşırı gündüz uyuklaması”, gerek ayırıcı
tanıdaki önemleri ve gerekse de tüm demans hastalarının zihinsel
performanslarına belirgin olumsuz etkileri ve göreli olarak da kolay
tedavi hedefleri olma özellikleriyle titizlikle sorgulanmalı ve
kaydedilmelidirler. Demansta öykü özellikleri için Tablo 2’ye
bakınız.
Öykü sonunda klinisyen temel sorunu saptayarak
çekirdek kognitif bulguyu başlangıç ve seyriyle belirlemiş
olmalı, buna kognitif alandan varsa diğer katılımları,
çekirdek ve ikincil kognitif bozuklukların GYA’ları etkileme
düzeyini, davranışsal ve diğer (ikincil) alanların
katılım zamanlamalarını ve şiddetlerini
canlandırabilmelidir. İyi alınmış bir öykü özellikle
tipik olgularda ayırıcı tanıyı daha bu aşamada
büyük ölçüde çözeceği ve hastalık şiddetini aşağı
yukarı belirleyeceği gibi, mental durum muayenesi için
kullanılacak standart testlerin yanısıra özellikle
odaklanılması gereken kognitif alanlar için özgül testlerin
esneklikle seçimine de rehberlik edecektir. Dolayısıyla, sinsi
başlayıp, yıllar içinde ilerlemiş, zaman içinde kelime
bulma ve dış mekanda yönünü bulma güçlüklerinin eklendiği, bu
nedenle hastanın son zamanlarda artık yalnız başına
sokağa çıkamadığı, ancak evinde yıkanma, giyinme
gibi temel GYA’larını halledebildiği bir öykü
karşısında klinisyen orta evrede bir AH’li hasta ile
karşı karşıya olduğunu rahatlıkla öngörüp muayene
sonuçlarının da bu öngörüsünü destekleyeceğini tahmin
edebilirken, temel sorunu bellek değil de mekanda yönünü bulmak, gözünün
önündeki nesneleri bulamamak olan bir hasta karşısında bu kez
çok daha ender bir nörodejeneratif antite olan “posterior kortikal atrofi”nin
ilerleyici simultanagnozisinin ifade edilmekte olduğunu ayırdedip
normalde standart muayenesinin içinde yeralmayabilecek simultanagnozi
testlerini seçmeye karar verebilmelidir.
DEMANSTA ÖZ VE SOYGEÇMİŞ ÖZELLİKLERİ
Demanslı hastanın öyküsü
sırasında özgeçmişi içinde belirli genel medikal, nörolojik,
psikiyatrik ve toksik özelliklerin soruşturulması önemlidir.
Genel medikal özgeçmiş içinde vasküler risk
faktörleri (hipertansiyon, iskemik kalp hastalığı, diyabet),
endokrin-metabolik bozukluklar (özellikle hipotirodi, B12 vitamini
yetmezliği), kronik infeksiyonlar (tüberküloz, sifilis, AIDS), sistemik
otoimmun-inflamatuar hastalıklar (Sistemik lupus, Sjögren, romatoid
artrit, Behçet), sistemik neoplazi, genel anestezi kullanılmış
cerrahi girişimler, özellikle kardiyak girişimler
kaydedilmelidir. Bunlardan bir bölümü sekonder demansların nedenleri
olabileceği gibi, B12 eksikliği, hipotiroidi, vasküler risk faktörleri,
genel anestezi gibi durumlar AH’yi komplike eden ve kontrol edilebilecek
komorbid durumlar da olabilirler.
Nörolojik özgeçmişte gelişimsel
bozukluklar, geçirilmiş hemorajik-iskemik inme ya da geçici iskemik
ataklar, özellikle şuur kayıplı kafa travması, subdural
hematom, MSS infeksiyonları (menenjit, ensefalit), epilepsi, beyin tümörü,
hidrosefali gibi durumların varlığı
sorgulanmalıdır.
Geçirilmiş depresyon, psikoz, eski
psikiyatrik hospitalizasyonlar sorgulanır.
Toksik özgeçmiş özellikleri arasında
ağır metallere maruz kalma kronik toksik ensefalopatilerin nedenleri
arasındadır. Kronik alkolizm ve/veya madde kullanımı
kaydedilmelidir. Geriyatrik popülasyon özellikle polifarmasinin
yaygın olduğu bir gruptur. Benzodiazepinler, trisiklik antidepresanlar
ve diğer anti-kolinerjik ajanlar, alfa-metil-dopa, reserpin gibi
antihipertansif ilaçlar kognitif bozukluğun tek başına nedeni ya
da ağırlaştırıcı etkeni olarak bulunabilirler.
Soygeçmiş özellikleri sorgulanırken
özellikle primer dejeneratif demansların büyük ölçüde genetik ve çevresel
faktörlerin karmaşık bir etkileşiminin ürünü oldukları göz
önünde bulundurulmalıdır. Nöro-genetikteki büyük ilerlemelere
rağmen AH ve FTD gibi ailede demans varlığının önemli
bir risk faktörü olduğu durumlarda hala keşfedilmeyi bekleyen
Mendelyen geçiş ve yatkınlık genleri bulunmaktadır.
Örneğin, bu iki durum için hala gösterilmiş bir resesif geçiş
geni yoktur ve akraba evliliğinin göreli sıklığıyla
ülkemizdeki demans pratiği bu açıdan gelecekteki böyle bir buluş
için avantajlı olabilir. Birinci derece akrabalar arasında demans,
Parkinson hastalığı, motor nöron hastalığı gibi
nörolojik, depresyon, psikoz, alkolizm gibi psikiyatrik hastalıkların
mevcudiyeti özellikle sorgulanmalıdır. Ana-babanın akraba veya
aynı köyden olup olmadığı, ana-baba ve
kardeşlerin sağ olup olmadıkları, öldülerse
yaşları ve ölüm nedenleri ayrı ayrı kaydedilmelidir.
Tablo 2. Demansta 3 kardinal ve 3 ikincil alanın
sorgulanması
|
Kognitif |
Bellek Dikkat Dil Görsel-mekansal işlevler Yürütücü işlevler Praksis Gnosis |
Yakın:
yakın geçmişe ait kişisel ve aktüel olaylar; Uzak: ilkokul öğretmeni,
okuduğu okullar, evlilik, emeklilik tarihleri, vb. Dalgalanma, konsantrasyon,
çelinebilirlik Kelime bulma, anlama, okuma,
yazma, hesaplama güçlükleri Yabancı/tanıdık
mekanlarda dolaşabilme, yazı karakterinde (ortografik)
değişiklik Problem çözme,
yargılama, soyutlama bozuklukları Alet kullanma, giyinme,
oturma-yürüme güçlükleri Nesneleri tanıma,
mekanda birbirinden ayırma |
|
Davranışsal |
Kişilik
değişiklikleri Duygudurum bozuklukları Algı bozuklukları Düşünce
bozuklukları |
Apati,
disinhibisyon, sosyal uygunsuzluk Keder, isteksizlik,
huzursuzluk, yerinde duramama, sinirlilik, uygunsuz neşe, eşin
peşinden ayrılmama Görsel ve diğer
halüsinasyonlar Hırsızlık,
sadakatsizlik, Capgras ve diğer türden hezeyanlar |
|
İşlevsel |
Sokakta
GYA’lar Evde GYA’lar Kendine bakım |
İş
yaşamı, yolculuk, mali işler, alışveriş, sosyal
ilişkiler Hobiler, ev
aygıtlarını kullanma, yemek pişirme, diğer ev
işleri, küçük tamirat, gazete-TV ilgisi Yemek yeme, yıkanma,
giyinme, makyaj, traş olma, tuvalet mekaniği, sfinkter kontrolü |
|
Motor |
Yürüyüş
bozukluğu, düşmeler, donmalar, dengesizlik, hareket
yavaşlığı, güçsüzlük, erime, seyirme |
|
|
Otonom |
İnkontinans,
empotans, ortostatizm, konstipasyon, terleme |
|
|
Uyku |
REM-davranış
bozukluğu, aşırı gündüz uykusu, uyku apne sendromu |
|
DEMANS MUAYENESİ
Kognitif ve/veya davranışsal
yakınmalarını ileten hastanın muayenesi demans sendromunun
mevcut olup olmadığını, mevcutsa demans sendromları
içinde tipik bir tabloya uyup uymadığını saptamaya yönelik
olmalıdır. Demans muayenesi:
1. Davranışsal
gözlemler,
2. Mental durum muayenesi
(MDM),
3. Somatik nörolojik muayene
alt başlıklarıyla kaydedilebilir.
Davranışsal Gözlemler
Hastanın muayene sırasındaki
davranışları gerek MDM’den alınacak sonuçların
değerlendirilmesinde önem taşırken, gerekse de bazı
durumlarda tanı koydurucu olabilir. Bu alt başlık
altında apati, sosyal uygunluk gibi ölçülmesi zor ya da mümkün olmayan
bulgular üzerine muayene edenin yargıları belirtilir.
Motivasyonun optimal olması MDM
sonuçlarının uygun değerlendirilebilmesi için gerek
şartların başında gelir. Muayene edenin tecrübe ve
mahareti, muayene tekniklerini iyi uygulayabilmesinin yanısıra
hastanın muayene boyunca motivasyonunu korumasını
sağlayabilmesini de gerektirir. Motivasyonsuzluk, gerek
dürtüsellik/çelinebilirlik, ve gerekse de apati şekillerinde muayeneye
işbirliğini bozacak ve ağırlığı ölçüsünde
MDM’yi istenildiği düzeyde uygulanamaz kılacaktır.
Motivasyonsuzluk, apati yönünde muayene sırasında hastadan
beklenen normal inisiyatif düzeyinin gözlenememesi şeklinde bir
kendiliğindenlik azalmasından, oturduğu yerde çevresine tümden
ilgisizliğe kadar değişebilir. Diğer yandan
dürtüsellik ve dikkatin çelinebilirliği de, MDM’de optimal performans için
gereken sebatlılığı bozacak perseveratif cevapları
arttıracaktır. AH’de erken evrelerde motivasyonun korunmuş
olması ya da muayene performansını bozmayacak düzeyde bir
kendiliğinden davranışta azalma beklenirken, ileri evrede apati
ya da çelinebilirlik nedeniyle formel muayene mümkün olmayabilir. FTD daha
erken evrelerden itibaren apati ve/veya dürtüsellik ile karakterizedir.
Bu nedenle bazı FTD hastaları başlangıçtan itibaren
muayeneye izin vermeyebilirler. Yine motivasyon bozukluğunun,
özellikle geriyatrik popülasyonda kendi başına ya da dejeneratif
demanslarla birlikte olduğunda, depresyonun başlıca belirtisi
olduğu söylenebilir.
Muayene eden, duygudurum ve afekt üzerine
gözlemlerini de kaydetmelidir. Kendiliğinden gözlenebilen
parmaklarıyla ya da giysileriyle oynamaktan, koltukta oturamayıp
sürekli dolaşma ya da odayı terketme isteğine kadar
değişen huzursuz, anksiyöz davranış bizzat muayenedeki
performans bozukluğuyla da ortaya çıkabilir. İritabilite,
kolay sinirlenme, negativizm, disfori eğilimi, kederli ruh hali, kolay
ağlamayla kendini gösterecektir.
İçgörü bozukluğu inkar eğilimiyle
başlayabilir.
Sosyal uygunluk, muayene edenin hastanın
genel davranışının geldiği sosyal çevrenin
normlarından beklediği davranışa ne derecede uygun
düştüğü üzerine yargısıdır. Disinhibisyonun
bir parçası olan uygunsuz davranışlar alaycılık
eğiliminden saldırganlığa kadar değişebilir.
AH’de sosyal uygunluk ileri evrelere kadar korunurken FTD’de en erken dönemden
itibaren bozulan tanı koydurucu özelliklerdendir. “Kullanma
davranışı” yine başlıca FTD’ye özgü bir disinhibisyon
özelliğidir. Kullanma davranışı sergileyen hasta muayene edenin
masasını karıştıracak, kalemlerini alma,
dosyalarını düzenleme, kağıtlarını okuma gibi
davranışlar sergileyecektir; masa üstünde bulunabilecek refleks
çekici, gözlük gibi nesneler yoklanıp kullanılmaya
çalışılacaktır.
Mental Durum Muayenesi
Mental durum muayenesi (MDM) kendi içinde “yatak
başı testleri” ve “tarama testleri” olarak
sınıflanabilir. Gerektiğinde davranışsal
belirtilerin ağırlığını saptayacak, işlevsel
durumu nicelleştirecek, demans şiddetini belirleyecek ölçekler de
kullanılabilir.
Yatak Başı Testleri
MDM, yatak başı testlerinden
oluşan bir kısa nöropsikolojik muayene olarak
düşünülebilir. Nöropsikoloji laboratuarında standart normlara
göre uygulanan ve değerlendirilen nöropsikolojik muayenenin tersine, hastanın
öykü, sosyokültürel durum ve davranışsal özelliklerine göre muayene
edenin esnek biçimde değiştirebileceği genellikle basit, nicel
skorlardan çok nitel performans değerlendirmelerine dayanan testlerden
oluşur. Amaç, limbik-paralimbik ve heteromodal alanlarda temsil edilen
kognitif işlevler ve unimodal alanlarda temsil edilen karmaşık
algısal ve motor işlevlerin etkilenme düzeyleri üzerine yorum
yapılabilecek kadar test edebilmektir.
Bu işlevler, 1. Dikkat, 2.Dil, 3.
Görsel-mekansal işlevler, 4. Bellek,
5. Yürütücü işlevler, 6. Gnosis, 7. Praksis
alt başlıklarında sıralanabilir.
Nörolojik muayenede normal ve anormal bulgular
oldukça kesindir. Bireysel ve kültürel varyasyonlar genellikle ihmal
edilebilir düzeydedir. Oysa MDM sonuçlarını değerlendirirken
kognitif işlevlerin birbirinden ayrıldığı
sınırların o kadar aşikar olmadığı,
testlerin eğitim düzeyi ve yaştan büyük ölçüde etkilendiği
hesaba katılmalıdır. Ağır dikkat bozukluğu
nedeniyle kayıt güçlüğü olan hastanın yakın bellek
testlerindeki gerçek performansını değerlendirmek mümkün
olmayabilir. Yine adlandırmayı bozacak bir dil bozukluğu,
genellikle bir kelime listesi öğrenmek tarzında olan yakın
bellek muayenesini verbal olmayan yollara uyarlamayı zorunlu kılar.
Eğitimsiz bir kişide saptanan bir performans bozukluğu o
eğitim düzeyi için normal sınırlarda kalabilecekken, yüksek
eğitimli bir kişinin bozukluğunu ortalama testler ortaya
koymayabilir.
Muayenenin sonunda ön planda ve ikincil düzeyde
bozulan alanlarla korunan alanların bildirileceği bir kognitif profil
belirlenir.
Tarama Testleri
Tarama testleri, özellikle AH’de kognitif
yıkımın ağırlığını saptamak,
yıkımın zaman içindeki ilerleme hızını ve ilaca
cevabı izlemekte kullanılan kısa global kognitif muayene
araçlarıdır. Epidemiyolojik çalışmalar gibi
geniş saha çalışmalarında duyarlılık ve özgüllük
oranları uyarınca normalleri demanslılardan ayırmak için
kullanılsalar da, ofiste tek bir hastanın muayenesinde, hiç bir zaman
asıl MDM’nin yerine tanı amaçlı olarak kullanılmamalı,
asıl işlevlerinin uzunlamasına izlemede tek bir hastadaki
değişimi nicel olarak belgelemek oldukları
unutulmamalıdır.
Bu tür birçok test geliştirilmişse de,
uluslararası literatürde en fazla adı geçen ve yurdumuzda da öteden
beri yaygın olarak kullanılanların başında Mini Mental
Durum Muayenesi (MMSE), Blessed Oryantasyon Bellek Konsantrasyon Testi (BOMC),
Kısa Mental Durum Testi (STMS) sayılabilir. Montreal Kognitif
Değerlendirme Testi (MoCA) ve Addenbrook Kognitif
Muayenesi-Yenilenmiş (ACE-R) yukarda anılan nispeten eski testlerin
zaaflarının giderilmesine yönelik olarak tasarlanmış,
yakın tarihlerde kullanılmaya başlanan tarama testleridir. Bu
yeni testlerin demans öncesi kognitif bozukluğu (yani, MCI) normal
kognisyondan ayırma yetenekleri olduğu ileri sürülmektedir.
ACE-R’ın 40 yıla yaklaşan bir süredir en yaygın olarak
kullanılan test olmasıyla bütün zaaflarına rağmen
terkedilemeyen MMSE maddelerini içinde barındırması ve bir alt
skor olarak MMSE skorunu da belirlemesinin bir avantajı olduğu
söylenebilir.
Davranışsal Ölçekler
Davranışsal belirtilerin
ağırlığını saptamak, zaman içindeki seyirlerini
ve ilaca cevaplarını izlemekte davranışsal ölçekler de
kullanılabilir.
Alzheimer Hastalığında
Davranışsal Bozukluklar Ölçeği (BEHAVE-AD), AH’de bu amaca
hizmet eden kullanılışlı bir testtir. Nöropsikiyatrik
Envanter (NPI) tüm demanslarda kullanılabilecek şekilde
geliştirilmiş son şekliyle 12 davranışsal ekseni
sorgulayan bir ölçektir. Frontal Davranış Envanteri (FBI)
özellikle FTD'li hastaların davranışsal bozukluklarının
ölçümü için geliştirilmiştir. Geriyatrik Depresyon Ölçeği
(Geriatric DS), demanslı popülasyonda diğer depresyon ölçüm
araçlarına göre daha güvenilir bir araç olabilir. Orijinal şekli olan
30 madde, 15 maddeye kısaltılarak (GDS-15) yoğun bir pratikte
daha da kullanışlı kılınmıştır.
İşlevsel Ölçekler
Kognitif yıkıma bağlı olarak
Tablo 2’de gösterilen GYA’lardaki bozulmayı nicel olarak saptamayı
hedefleyen ölçeklerdir. Bunlar arasında, Günlük yaşam
Aktiviteleri/Enstrümental Günlük Yaşam Aktiviteleri Ölçeği
(ADL/IADL), Blessed Demans Derecelendirme Ölçeği–CERAD versiyonu
(BDRS-CERAD) ve Alzheimer Hastalığı İşbirliği
Çalışması–Günlük Yaşam Aktiviteleri Ölçeği (ADCS-ADL)
sayılabilir.
Parasomni Ölçekleri
Gerektiğinde RUDB ve insomniaya neden olan huzursuz
bacak ve uyku apne gibi durumları nicelleştiren Mayo Uyku
Soruşturusu (MSQ) ve aşırı gündüz uykululuğunu
nicelleştiren Epworth Uykululuk Ölçeği (ESS) kullanılabilir.
Nörolojik Muayene
Diğer nörolojik hastalıklarda
olduğu gibi, doğal olarak demans hastalıklarının
muayenesine de tam bir somatik nörolojik muayenenin eşlik etmesi
beklenir. Bununla birlikte bazı demans sendromlarıyla özellikle
bazı nörolojik bulgular birlikte gittiği için söz konusu bulguların
bulunup bulunmaması önem taşır. Bunlar arasında
ekstrapiramidal sistem bulgularının (EPS) özel bir yeri
vardır. Göz hareketleri muayenesinde ortaya konabilecek
aşağı bakış felci, yürüyüş bozukluğu varsa
bunun Parkinsonyen ve/veya ataktik niteliği, geçirilmiş inmelere ait
fokal nörolojik bulguların tümü de demans sendromlarının
ayırt edilmesinde kullanılacaklardır. Özellikle EPS’nin
nicelleştirilmesi amacıyla Parkinson hastalığının
(PH) belirti ve bulguları için geliştirilen UPDRS isimli ölçeğin
Kısım III alt bölümü kullanılabilir.
DEMANS SENDROMUNUN FENOMENOLOJİK AYIRICI
TANISI
Demans sendromunun tanısında, daha önce
de belirtildiği gibi, üç kardinal semptomatolojik kategori arasında
işlevsel alanda bozulma şarttır. Demansın
ayırıcı tanısı, işlevsel bozulmaya yol açan
kognitif alana ait bozulmanın profili, davranışsal alana ait
bozukluğun olup olmaması, mevcut ise tabloya katılma
zamanlaması ve ağırlığı, yine motor
bozukluğun mevcudiyeti ve tabloya katılma zamanlaması,
ağırlığı değerlendirilerek
yapılır. Örneğin, ATD öyküsü ağırlıklı
olarak, izole kognitif demans olarak başlar; davranışsal ve
motor bulgular tabloya ileri evrelerde katılır.
Nöropsikolojik Profil
Demansın nöropsikolojik profili patogenezin
nöral coğrafyadaki
yatkınlığını yansıtır ve
dolayısıyla hastalığı yüksek olasılıkla
tahmin ettirir. Daha önce de değinildiği gibi, demans daha
yakın zamana kadar tasarlandığı gibi, MSS’nin yaygın
hasarıyla gelişen global bir zihinsel yıkım değildir.
Farklı nörodejeneratif etyopatogenetik süreçler farklı nöroanatomik
yatkınlıklar gösterirler. Nöropsikolojik profilleri de, bu
yatkınlığı yansıtacak şekilde, kognitif ve
davranışsal alanların bazen izole, ama sıklıkla da
kendine özgü bileşimlerinden oluşan bozukluklarının
ifadesidir. Nöropsikolojik profil, bozulan ve göreli korunan alanları
belgeleyerek, nöroanatomiyi dolaylı olarak sergiler. Hasarlı
olduğu varsayılan anatomik alan daima tek bir etyopatogeneze
karşılık gelmeyebilir. Farklı süreçler, benzer bir
anatomik yatkınlık gösterebilirler. Ancak, günümüze kadar
biriken nöropatolojik veriler, farklı nöropsikolojik profillerin,
dolayısıyla anatomik yatkınlıkların belli
hastalık süreçlerine karşılık gelme ya da tümüyle
dışlama olasılıklarını güvenilir biçimde ortaya
koyabilmektedir.
Farklı nörodejeneratif
süreçler, genellikle bir çeşit nihai ortak yola sahiptirler.
Dolayısıyla, hastalığın çok ilerlediği,
ağır ve yaygın yıkımın gerçekleştiği
son evrede nöropsikolojik olarak farklı profiller ayırmak mümkün
olmayabilir. Nöropsikolojik profiller, özellikle erken evrede anlamlı
farklılıklar sergileyeceklerdir. Bu profiller
sıklıkla, en azından başlangıçta simetrik ya da
asimetrik olarak fokal kalacak olan dejenerasyonu yansıtırlar.
Altta yatan patolojik sürecin niteliğine göre, hastalığın
nihai evrelerinde dejenerasyon fokal özelliğini bırakıp
yaygınlaşırken, klinik tablo da global bir yıkım
özelliği edinecektir. Pick hastalığı (PiH) gibi
bazı süreçler ise bütün evrelerinde daima fokal kalabilirler.
Mesulam ve Weintraub, özellikle erken evre profillerini vurgulayarak, demanslar
için 4 farklı nöropsikolojik profil tanımlar:
1. Progresif amnestik disfonksiyon (PAD)
2. Primer progresif afazi (PPA)
3. Progresif görsel-mekansal Bozukluk (PGMB)
4. Progresif davranışsal/yürütücü bozukluk
(PDB/PYB) ya da progresif frontal şebeke sendromu
Mesulam ve Weintraub’un bu 4 tablosuna progresif
apraksiyi de mümkün bir 5. tablo olarak eklemek listeyi daha
kapsayıcı bir hale getirecektir. Yazarların da işaret
ettiği gibi, bunlar demansın mümkün başlangıç
gösterilerinin tümü değildir ve bazı olgular bu 4 ya da 5 tabloya
göre sınıflanamayabilir. Bazı patolojik süreçlerde biri ön
planda olmak üzere bu tablolar bir arada görünebilir (örneğin FTD’de 2 ve
4 ya da FTD’nin semantik demans varyantında 2, 4 ve 3’den özellikler, KBD’de
4 ve 5). Yine de, günlük demans pratiğinde büyük ölçüde
kapsayıcı özelliğiyle bu yaklaşım, hem
ayırıcı tanıda pratik bir yol olmakta, hem de hasta ve
hasta yakınlarına yapılacak non-farmakolojik eğitici müdahalelerrde,
bozulan ve korunan niteliklerinin belirlenmesiyle yol göstermekte
yardımcı olmaktadır.
Mesulam, tanımladığı
tabloların tanı kriterlerinin ortak özellikleri olarak kısaca,
tabloya adını veren bozukluğun en az iki yıl boyunca
ilerleyici (progresif) izole bozukluk olarak kalmasını (primer) ve bu
süre içinde GYA’ları bozan yegane neden olmasını ileri
sürer. Bu iki yıllık süre sonrasında diğer kognitif
alanlardaki bozulmalar da tabloya katılabilir. Aşağıda
sıralanan nöropsikolojik profillerde Mesulam ve Weintraub’un önerileri
temel alınmakla birlikte, adı geçen öneride “primer” sıfatı
için gerekli koşul olan “en az 2 yıl izole bozukluk” kriteri
sıkı bir şekilde korunmamış, bunun yerine “primer”
sıfatı izole olsun ya da olmasın, en ağır olarak bozulan
veya profilin çekirdeğinde bulunan kognitif bozukluğu nitelemek amacıyla
kullanılmıştır. Dolayısıyla, burada
sıralanan profiller ayrı ayrı antiteler olarak değil fakat
ayırıcı tanı amaçlı, en güçlü ihtimalle belli
nörodejeneratif hastalıklara karşılık gelmesi beklenen
klinik sendromlar olarak anlaşılmalıdır.
Progresif Amnestik Disfonksiyon (PAD)
Başka bir kognitif bozukluk
olmaksızın yakın bellekte saptanan ilerleyici karakterli
bozukluk GYA’ları da bozmaktadır.
Tipik bir hasta, genellikle 65
yaşının üzerindedir. Unutkanlıktan yakınan hasta
ve/veya yakını bunu özel eşyalarını kaybetmek,
aynı soruları ve konuları tekrarlamak, okuduklarını
aklında tutamamak, randevularını, alışveriş
listesini, yeni gördüğü mekan ve yüzleri unutmak şeklinde
örneklendirir. Unutkanlığı genellikle iş
arkadaşları ve ailesi tarafından da farkedilmektedir.
Bunlar dışında, sokakta ve sosyal ortamlarda
bağımsızlığı
kısıtlanmamıştır. Dış mekanda eskisi
gibi kaybolmadan dolaşır, mali işlerini ve
alışverişi ancak listeyle eskisi gibi halleder. Ev
işlerinde zaman zaman yemek yakıyor olmak dışında
bağımsızlığını sürdürür, hobilerini devam
ettirir. Nöropsikolojik olarak kayda değer bir verbal ve görsel
yakın bellek bozukluğu saptanırken, diğer alanlardaki
performans korunmuş ya da normal sınıra yakın
düzeylerdedir. İkinci bir kognitif alanın da bozulması ve
bunun günlük yaşama yansımasıyla birlikte artık PRAD ya da
DAT kriterleri doldurulur hale gelir. Nöropsikolojik olarak en erken,
yakın bellek bozukluğuna eklenen demansa ilişkin bulgular sözel
akıcılıkta kategori akıcılığının
(örneğin 1 dakikada mümkün olduğunca fazla hayvan ismi söylemek),
leksikal akıcılığa (örneğin 1’er dakikada K, A ve S
harfleriyle başlayan mümkün olduğunca fazla cins isim söylemek) göre
daha erken ve ağır bozulması ve karmaşık
görsel-mekansal işlevlerde bozulma olmaktadır. Zaman içinde
uzunlamasına izlenen bir hastada önceleri bu işlevler korunurken
sonradan bozulduklarının gösterilmesi, artık nörodejenerasyonun
limbik sistemin dışına, sol ve sağ temporo-parietal
heteromodal neokortekse geçtiği anlamına gelir.
Bu tablo demansların erken evreleri içinde en
sık rastlanılan tablodur. PAD'ın ilerde daha
ayrıntılı tartışılacak olan MCI tablosu ile
pratik olarak aynı anlama geldiği söylenebilir. Nadiren çok
uzun yıllar boyunca, ya da bütün seyri boyunca aynı kalsa da büyük
sıklıkla AH’nin henüz PRAD/DAT kriterlerinin
doldurulamadığı erken veya prodromal evresine
karşılık gelmektedir. Görüntüleme çalışmalarında,
özellikle MRG ile volümetrik olarak hippokampal atrofi saptanabilir. (Şekil
1) Son yıllarda geliştirilen PET ile amiloid görüntüleme
tekniği, PAD sergileyen hastalar arasında yoğun serebral amiloid
yükü taşıyanları ayırt ederek ilerde ATD geliştirecek,
bir diğer deyişle pre-demans veya prodromal AH’yi öngörebilir. Yine,
BOS’ta total tau ve fosfo-tau proteini düzeyleri ile amiloid-beta fragmanı
düzeyi benzer bir işlev görebilir. Hippokampus volümetrisi, PET ile
amiloid görüntüleme ve BOS’ta anılan protein düzeyleri ilerde AH’nin
potansiyel biyoişaretleyicileri bölümünde ayrıntıyla tartışılacaktır.
Nöropatoloji simetrik olarak limbik sistemde ve ağırlıkla da,
hippokampal-entorinal yapılardadır. Anılan bölgelere
yatkınlığı dolayısıyla, tahmin edilebileceği
gibi, beklenen hastalık yüksek ihtimalle AH'dir. Non-spesifik
dejenerasyon, PiH, LCD, arjirofilik tahıl hastalığı veya
sadece yumak demansı diğer nadir asosiasyonlardır.
Şekil 1. Hippokampus Görüntülemesi,
Şekil 1a. 73
yaşında normal yaşlı hippokampusları;
sağ hippokampusun normalde biraz daha büyük göründüğüne dikkat ediniz
(Hippokampus başından geçen T1 ağırlıklı koronal
kesit).
Şekil 1b. 75
yaşında hafif demanslı kadın hasta; hippokampus
başlarındaki hacim kaybına dikkat ediniz (Hippokampus
başından geçen T2 ağırlıklı kesit).
Şekil 1c. Aynı olgunun 5 yıl sonraki MRG’si;
demans artık ağır evrede ve hippokampal atrofi çok daha belirgin
hala gelmiş (Şekil 1b ile aynı özellikte kesit.
Progresif Afazi (PA)
İlerleyici dil bozukluğunun, izole olarak
veya diğer kognitif ve/veya davranışsal bozukluklarla birlikte
çekirdek bozukluk olduğu tablolardır.
Tipik bir hasta, genellikle 65
yaşının altındadır. Konuşma
bozukluğundan yakınan hasta, bunu kelime bulma, nesneleri ve
kişileri adlandırma, bazen düşüncelerini yazılı ve
sözlü ifade etme, bazen de yazılı ve sözlü ifadeleri anlama, okuma
güçlüğü şeklinde örneklendirir. Özel bir alt tipi olan semantik
demansta (SD) tek tek kelimelerin anlamsal içerikleri
boşalmıştır. Hastalar sıradan nesnelerin isimlerini
ilk kez duyuyorlarmış gibi davranırlar. Dil kullanmak
gerekmediği durumlarda sokakta ve evde
bağımsızlığını kısıtlayacak günlük
olayları hatırlama, dış mekanda yolunu bulma, günlük
olaylar üzerine akıl yürütme, hobilerini sürdürme gibi sorunları yoktur.
Diğer bir alt tip olan logopenik PA’lı (LPA) hastalar hesaplama
güçlüğünden yakınırlar. SD’li hastalar progresif afazilerinin
yanısıra aşina yüzleri tanıma güçlüğü ve disinhibisyon
belirtileri sergileyebilirler.
Nöropsikolojik profil, tutuk ya da akıcı
afazi tarzındadır.
Yerleşmiş bir tutuk afazi profili,
klasik Broca afazisini andıran tarzda agramatik spontan konuşma ve
tekrarlama bozukluğu ile birlikte anlamanın göreli korunduğu bir
tarzdır. Bununla birlikte, klasik afazik sendromların, tablonun akut
olarak yerleştiği ve zaman içinde düzelmeye yüz tuttuğu
biçimlerinin tersine ilerleyici afazilerin sinsi başlangıçlı,
tedrici ilerleyici seyirleri göz önüne alınmalıdır.
Dolayısıyla, erken dönemdeki bir ilerleyici tutuk afaziğin
klasik afazi muayenesiyle ayırt edilemeyebileceği, klasik afazik
sendromların olmazsa olmaz koşullarından konfrontasyon
adlandırmasının dahi salim kalabileceği kaydedilmelidir. Bu
erken dönem hastalar spontan konuşmalarında kelime bulma güçlükleri,
fonemik parafaziler, dizartrik, bazen kekelemeli konuşma tarzı,
prozodilerinde sanki bir yabancının aksanlı
konuşmasıymışcasına değişme ve kelime
akıcılıklarında, özellikle de leksikal akıcılıklarında
bozulma ile ayırt edileceklerdir. İlerleyici tutuk afazi (progressive
non-fluent aphasia-PNFA) bir FTD alt tipidir. Hastalık seyri
sıklıkla yıllarca izole dil bozukluğu şeklinde
kalabilirken (yani, Mesulam PPA’sı), daha erken dönemde yürütücü bozukluk
özelikleri de eşlik edebilir. KBD tarzı bir asimetrik sağ
hemiparkinsonizm ve amiyotrofik lateral skleroz (ALS) muhtemel motor
eşlikçilerdendir.
İlerleyici akıcı afazi SD veya LPA
şekillerinde sunulur. SD’de ağır konfrontasyon
adlandırması bozukluğu bir nesne agnozisini andırır
özellik taşır. Hasta için kelimeler anlamını
yitirmiştir. Nesneleri adlandıramayan (anomi) hasta bir
Wernicke afaziğinin aksine bir bölümünün ne işe
yaradığını da söyleyemez (agnozi). Bununla birlikte,
karşılıklı iletişim genellikle mümkünken, karmaşık
gramerin kullanıldığı emirlerle de anlamanın
korunduğu görülür. SD’li hastanın semantik bellek testleri
(örneğin, Piramidler ve Palmiye Ağaçları Testi) ve meşhur
yüzleri tanıma testleri de bozulmuştur. LPA bir ilerleyici anomi
olarak düşünülebilir. Spontan konuşma kelime bulma güçlükleriyle
giderek yoksullaşır. Anlama korunmuştur. Anomik hasta
ayrıca nesne agnozisi, semantik bellek ve yüz tanıma
bozuklukları gibi SD özellikleri sergilemez. Akalkuli başta olmak
üzere, sağ-sol disoryantasyonu ve parmak agnozisi gibi Gerstmann sendromu
özellikleri görülebilir.
Gerek PNFA’da ve gerekse de akıcı
türlerde verbal dolayımlı testler, özellikle de kelime listesi
öğrenme gibi klasik bellek testleri, dil bozukluğuna sekonder olarak
bozuk bulunabilir. Ancak dil dolayımına dayanmayan görsel
bellek, sağ hemisfer görsel-mekansal işlevleri iyi
korunmuştur. Bununla birlikte, LPA’nın ileri
aşamalarında simultanagnoziye varan ağır görsel-mekansal
bozukluklar görülebilir.
PNFA’da BT ve MRG ile asimetrik sol peri-sylvien
genişleme, (Şekil 2) bazen sol fronto-temporal atrofi, SPECT
ve PET ile adı geçen bölgede hippoperfüzyon veya hipometabolizma, EEG ile
sol temporal yavaşlama saptanabilir. SD’nin ayırıcı
özelliği MRG’de büyük sıklıkla solda baskın, özellikle
koronal T1 kesitlerde rahatlıkla ayırt edilebilen temporopolar (TP)
atrofidir (Şekil 3). rTV’de atrofi tam tersine sağda
baskındır (Şekil 4). LPA’nın nöropatolojik
karşılığı olan sol anguler girus atrofisi yapısal
görüntülemede gözden kaçabilirse de SPECT veya PET ile fonksiyonel görüntüleme
asimetrik bir sol parietal hipoperfüzyon/metabolizmayı ortaya
koyacaktır (Şekil 5).
PNFA sıklıkla KBD, PSP ve FTD
spektrumuna dahil taupatilerle ilişkiliyken, ALS eşlik ettiği
durumlarda beklenen nöropatoloji bir tau-/ubikutin+ (T-/Ub+) proteinopati veya
ubikutinize proteinin adıyla bir TDP-43 proteinopatisidir. SD hemen daima
bir TDP-43 proteinopatidir. LPA’nın AH’nin atipik sunumlarından biri
olduğuna dair deliller birikmektedir. PNFA, dvFTD, parkinsonizm ve ALS
bileşimlerinin ailevi biçimlerinin genetik karşılıkları
aşağıda “progresif davranışsal bozukluk” içinde
tartışılmıştır. SD’nin ailevi biçimi
bildirilmemiştir.
Wernicke afazisine benzer anlamanın da erken
dönemde bozulduğu bir ilerleyici tutuk afazi izole biçimde değil
fakat sıklıkla, bellek ve diğer kognitif bozukluk bulguları
arasında dil bozukluğunun ön planda olduğu tablolar bir atipik
sunum olarak AH ile ilintili olabilir.
Şekil 2. İlerleyici tutuk afazide
asimetrik sol peri-sylvien atrofi
Sol frontal
operkulum ve sol anterior insula atrofisine bağlanabilecek asimetrik sol
peri-sylvien genişleme (üst sıra, sağ), bir üst kesitte de
hafifçe daha geniş sol ventrikül frontal boynuzuyla birlikte (üst
sıra, sol) görülüyor. Sol lateral ventrikül de sağa kıyasla hafifçe
daha geniş (alt sıra).
Şekil 3. Semantik demansta asimetrik sol
baskın temporopolar atrofi
T2
ağırlıklı koronal kesitlerde sol frontopolar
ağır, sağda daha hafif atrofi (üst sıra), mezial temporal
bölgeden geçen kesitlerde 2 yanlı amigdala atrofisi (alt sıra).
Şekil 4. Sağ temporal varyant
semantik demans görüntülemesi.
T12
ağırlıklı aksiyel ve koronal kesitlerde sağda
baskın, 2 yanlı temporopolar atrofi
Şekil 5. Logopenik
progresif afazide SPECT.
Tc99 HMPAO-SPECT ile MRG’sinde ayırt
edilebilir bir bulgu olmayan logopenik progresif afazili bir olguda asimetrik
ağır sol parietal hipoperfüzyon.
Progresif Görsel-Mekansal Bozukluk (PGMB)
Bu profilin başlıca özellikleri görsel
işleme bozukluklarıdır. Nörodejenerasyon iki yanlı
oksipitoparietal bölgededir. İlk belirtiler sıklıkla,
bildik yerler dahil dış mekanda yolunu bulamama,
aradığı bir nesneyi gözünün önünde olduğu halde diğer
nesnelerden ayırıp bulamama, okurken satırların
birbirine karışması, yazı karakterinin bozulması ve
giyinme güçlüğüdür.
Muayenede Balint sendromunun bileşenleri
(ağırlıkla simultanagnozi ve daha silik olarak optik ataksi,
oküler apraksi) ile giyinme apraksisi saptanır. Nöropsikolojik
olarak görsel-mekansal testlerde ağır bozukluk görülür ve spesifik
testlerle bakıldığında simultanagnozi ortaya konulur.
Bellek korunmuştur. Yürütücü işlevler ve dilsel
işlevlerde hafif bozukluklar eşlik edebilir. Nörodejenerasyon
inferior parietal lobülleri de tuttuğunda sol mekan ihmali ve Gerstmann
sendromu özellikleri de (sağ-sol disoryantasyonu, parmak agnozisi, agrafi,
aleksi, akalkuli) tabloya katılabilir ve LPA’yı andıran bir
anomi yerleşebilir.
Görüntüleme çalışmalarında bu
profili yansıtır şekilde iki yanlı oksipito-parietal atrofi
(özellikle, MRG koronal T1 kesitlerde ayırt edilen), hipoperfüzyon ve
hipometabolizma saptanır (Şekil 6 ve 7).
Yayınlanmış az sayıdaki otopsi
olgularında non-spesifik dejenerasyonun yanısıra
sıradışı olarak parietal neokortekste
yoğunlaşmış nörofibriller yumaklarla AH
sayılabilir. Fokal atrofiler içinde en az “Pick kompleksi”
patolojileri, tersine en fazla AH ile ilintili olandır. Hızlı
seyirli olduğunda tipik bir CJH sunumu olabilir. Ailevi biçimleri
bilinmemektedir.
Şekil 6. Posterior kortikal atrofide
MRG.
Posterior
kortikal atrofili koronal T1 kesitlerde simetrik dorsal parietal atrofi
Şekil 7. Posterior kortikal atrofide
SPECT.
Posterior kortikal atrofili bir olguda Tc99
HMPAO-SPECT incelemesinin koronal kesitleri. Üst sıra frontal kesitlerden
alt sıra oksipital kesitlere doğru önden arkaya doğru
izlendiğinde, 2. sıra 1. kesitte sağ frontoparietal olarak
asimetrik başlayan hipoperfüzyon aynı sıradaki 4. kesitte
simetrikleşiyor ve 3. sıradaki dorsal parietal ve alt sıra 1.
kesitteki oksipitoparietal kesitlerde ağırlaşıyor
Progresif Davranışsal/Yürütücü Bozukluk
(PDB/PYB)
Prefrontal sisteme atfedilen işlevlerin
ilerleyici bozukluğu bu tablonun temel özelliğidir. Tipik bir
hasta 65 yaşının altındadır. Toplumsal konumuyla
bağdaşmayan tarzda davranışlar, normlara
aldırışsızlık, çocuksuluk, uygunsuz cinsel imalar,
beslenme alışkanlığında değişiklik ya da
oburluk, dikkatin kolayca çelinebilir olması, içgörü kaybı,
inisiyatif kullanımının azalması erken belirtilerdir.
Hasta unutkan gibi görünebilir, fakat muayene sonucu bunun dikkat
bozukluğuna ikincil olduğu ortaya konacaktır. Sokakta
kaybolmadan dolaşabilir. Bazı durumlarda sokaktaki
bağımsızlığını yitirmiş gibi
görünebilir, fakat uygun bir sorgulamayla bunun mekan oryantasyonuyla ilgili
değil, fakat sosyal uygunsuz davranışların sonucu olarak
hasta yakınlarının dışarı çıkmayı
kısıtlaması nedeniyle olduğu görülecektir. Bu tablo,
izole ilerleyici bir tablo gibi seyredebileceği gibi, bazen PNFA tipi, bir
dil bozukluğu da eklenebilir. Bu tablo PNFA ve SD ile birlikte FTLD
tanı kriterlerinde bir üçüncü alt tip olarak belirlenmiştir ve
davranışsal varyant olarak da adlandırılır (dvFTLD).
Sağ temporal varyant olarak adlandırılan SD (RTV-SD) tipinde
tablo dvFTLD’den ayırt edilemeyen bir davranışsal bozuklukla
başlayabilir fakat hemen daima daha başlangıçtan itibaren veya
kısa zaman içinde eklenen bir aşina yüzleri tanıma güçlüğü
ile daha geri planda SD tipi bir dil bozukluğu bulunacaktır. dvFTLD’ye
eşlik edebilecek motor bozukluklar ise asimetrik akinetik-rijid veya
aksiyel postüral bozukluklarla parkinsonizm veya ALS olabilir.
Çelinebilirlik (distraktibilite), dürtüsellik
(impulsivite) ve motivasyon güçlüğü (apati) nedeniyle bu hastaların
nöropsikolojik muayeneleri son derece güç olabilir. Bu nedenlerle
performansı değerlendirmek mümkün olamayabileceği gibi, gerçek
yaşamda özellikle karşılaşılan yeni problemler
karşısında çok ciddi sorunları olan bir hastanın “ofis
performansı” dolayısıyla bunlar nöropsikolojik testlerle ortaya
konulamayabilir ve testler tümüyle normal sınırlarda olabilir.
Ancak, tipik bir hastada yürütücü işlevlerle ilgili testlerde akıl
yürütme, planlama, soyutlama, zihinsel esneklik ve sebatlılıkta
bozukluk, uygunsuz cevabı baskılayamama, perseverasyonlar gibi
bulgular saptanır. Görsel-mekansal işlevler ve PA
eşlik etmiyorsa dilsel işlevler korunmuştur. Yakın
bellek testlerinde kayıt ve serbest hatırlamada güçlüklere
karşın tanımanın korunduğu görülür. Bu hastalarda
limbik sistem salim, dolayısıyla uzun süreli bellek
kayıtları mümkün, ancak prefrontal “arşivleyici” sistem bozuk
olduğundan bu kayıtlara kendiliğinden etkin bir biçimde
ulaşmak güç olmaktadır. Bazen ekstrapiramidal motor bulgular
asimetrik veya aksiyel baskın, asimetrik apraksi, ALS
varlığı durumunda 1. ve 2. motor nöron bulguları (el
intrensek kaslarında atrofi, bulber zaaf, fasikülasyonlar, piramidal
bulgular) saptanabilir. Çok nadiren inklüzyon cisimciği miyoziti ve Paget
hastalığı (IBM/Paget) bulguları saptanabilir.
MRG’de simetrik veya asimetrik, frontal loblara
sınırlı veya birlikte anterior temporal lobları da içeren
atrofi saptanır. RTV-SD’de, klasik SD’nin tam tersine sağda
baskın temporopolar atrofi izlenir. Asimetrik parkinsonizm eşlik
ettiğinde parkinsonizmin kontrlateralindeki hemisferde baskın
asimetrik fronto-parietal atrofi daha muhtemeldir. SD ve RTV-SD’de MRG
özellikle koronal T1 kesitler içerdiğinde hemen daima tek başına
yeterli olurken, izole PDB ve asimetrik parkinsonizmli PDB bazen MRG ile
ayırt edilemeyebilir. Böyle durumlarda SPECT veya PET ile izole PDB’de
frontal loblara sınırlı, parkinsonizm durumunda asimetrik
fronto-parietal perfüzyon ya da metabolizma azalmasını göstermek
yararlı olabilir.
Bu klinik tablonun altında en sık
rastlanılan nöropatoloji eşit oranlarda denebilecek şekilde
taupati ve TDP-43 proteinopatidir. ALS ve SD eşliği hemen daima
TDP-43 ile ilişkiliyken PNFA veya parkinsonizm eşliği hemen
daima taupati ilişkilidir. Saf davranışsal varyantın geriye
kalan %10’luk bir hasta grubunda ubikutinize protein yakın zamanlara kadar
tanımlanamamıştı ve atipik ubikuitin pozitif FTLD
(aFTLD-U), aksonal sferoidli nöronal orta boy (“intermediate”) filaman
hastalığı (NIFID) ve bazofilik inklüzyon cisimciği
hastalığı (BIBD) olarak üçe ayrılmaktaydı. Bu
tanımlanamayan proteinin de yakın tarihlerde ”fused in” sarkoma (FUS)
proteini olduğu bulundu. Davranışsal varyantın şimdiye
kadar AH patolojisiyle ilişkisi bildirilmemiştir. Hızlı
seyirli biçimleri, CJH ve FTD-ALS akla getirmelidir. Ailevi taupatiler
17. kromozomda kodlanan tau proteini genindeki (17q21 MAPT) mutasyonlar
sonucuyken, ailevi TDP-43 proteinopatileri (sıklıkla parkinsonizmli
davranışsal varyant şeklinde sunulurlar) yine 17. kromozomda
kodlanan progranulin proteini genindeki (17q24 PGRN) mutasyonlarla
ilişkilidir. FTD-ALS, 9. kromozomda p21 lokusundaki henüz bilinmeyen bir
gendeki ve 3. kromozomda kodlanan kromatin modifiye edici protein genindeki (3p
CHMP2B) mutasyonlarla ilişkilidir. Nihayet, IBM/Paget ve dvFTLD 9.
kromozomda kodlanan valozin içeren protein genindeki (9p13 VCP)
mutasyonlar sonucu gelişir. 16. kromozomda kodlanan FUS ve 1. kromozomda
kodlanan TDP-43 gen mutasyonlarının (TARDP)
ağırlıkla saf ailevi ALS’ye neden oldukları
gösterilmiştir (FALS). Sadece bir TARDP mutasyonlu olgu ALS
olmaksızın PDB fenotipine eşlik eden vertikal bakış
paralizisi ve koreyle sunulmuştur. FUS mutasyonlu bir olguda ise
FTD-ALS fenotipi bildirilmiştir
Progresif Apraksi
Sıklıkla asimetrik olarak tek bir
ekstremiteye ait ilerleyici beceriksizlik, sakarlık bu tablonun temel
özelliğidir. 65 yaşın altında pre-senil
başlangıç daha mutaddır. Hasta elini eskisi gibi
kullanamadığından, eline aldığı nesneleri
düşürebildiğinden yakınmaktadır.
Çatal-kaşık kullanmak, düğme iliklemek gibi iki elin
koordinasyonunu gerektiren eylemler güçleşmiştir. Dominan el
apraktik olduğunda doğal olarak zorluklar daha da artmaktadır:
yazı yazmak, alet kullanmak öncelikle bozulanlardır. Aksiyel kaslar
katıldığında yatma, oturma, yürüme ve giyinme gibi
eylemlerin, beden eksenini nesnelere göre ayarlayamamaktan kaynaklanan
bozuklukları ortaya çıkar. Yatakta dönmek
zorlaşmıştır. Hasta koltuğa iğreti,
düşecekmiş gibi oturur. Yürüyüş sırasında düz yoldaki
basamak, tümsek gibi düzensizliklere adımlarını
ayarlayamaz. Düşebilir ya da basamakları ikişer, üçer
adımlayabilir. Kapılara, engellere çarpabilir. Giyinmek
güçleşmiştir. Ters dönmüş bir giysiyi düzeltemez,
kollarını bulmakta, düğmelerini iliklemekte güçlük
çeker. İlerlemiş olgularda apraktik el yabancı el
sendromunda olduğu gibi tümüyle denetimden çıkabilir.
İleri aşamadaki bir başka görünümde tutulan elin sürekli bir
distonik postürde “kullanışsız el” durumuna gelmesidir. Bu tablo
saf ilerleyici bir tarzda seyredebileceği gibi, daha sıklıkla
kişilik değişikliği, depresyon ve PNFA tablonun bir
parçası olabilir.
Muayenede genellikle asimetrik taraf apraksisi ve
gövde apraksisi tek başına ya da tutuk afazi, yürütücü bozukluk ve
bazen de görsel-mekansal bozukluk bulgularıyla birliktedir. Bellek
göreli olarak korunmuş veya dikkate sekonder olarak
bozulmuştur. Ekstrapiramidal sistem tutulumunu işaretleyen
motor bulgular da sıklıkla apraktik taraftadır. Bazen iki
nokta ayrımında bozukluk, agrafestezi, astereognozi gibi kortikal
duyu kusurları da mevcuttur. Bu tabloya “kotikobazal sendrom (KBS)”
adı da verilir.
Görüntüleme çalışmalarında
fronto-parietal, sıklıkla asimetrik bir atrofi ve/veya
hipoperfüzyon-hipometabolizma saptanır.
Bu klinik tablo, tipik şekliyle kortiko-basal
dejenerasyonun (KBD) en iyi bilinen klinik sendromudur.
Ağırlık aksiyel tutulumda olduğunda bazen progresif
supranükleer paralizi (PSP) de böyle bir tablonun nedeni olabilir.
Sporadik olgular büyük ölçüde KBD tarzı bir taupati olmakla birlikte
ailevi olgular Kr-17 PGRN mutasyonu ile ilişkili TDP-43
proteinopatisi sonucu olabilirler.
Andrew Kertesz davranışsal,
dilsel ve motor varyantlardan biriyle başlayan ve sıklıkla zaman
içinde ön planda olan özelliğe diğerlerinin de eklenmesiyle ilerleyen
bu tabloların tümünü içermek üzere “Pick Kompleksi” kavramını
önermiştir.
Tablo 3: Çekirdek Nöropsikolojik
Profillerin Ayırıcı Tanısı
|
Çekirdek Progresif Sendrom |
Amnezi |
Afazi |
Davranışsal |
Agnozi |
Apraksi |
||
|
Ana semptom |
Unutkanlık |
Kelime bulma, konuşma |
Kelime anlamının kaybı, meşhur yüzlerin
tanınamaması |
Kelime bulma |
Apati, disinhibisyon |
Yön bulma, gözünün önündeki nesneleri ayırt edememe |
Sakarlık |
|
Kognitif tarz |
Bozuk tanıma belleği, semantik akıcılık |
Agramatizm, anomi |
Anomi, görsel agnozi, prosopagnozi |
Anomi, akalkuli |
Yürütücü |
Simultanagnozi |
Apraksi, yürütücü |
|
Nörolojik muayene |
Normal |
Normal, bazen asimetrik parkinsonizm |
Normal |
Normal |
Normal, bazen parkinsonizm/ALS |
Normal |
Asimetrik parkinsonizm |
|
MRG |
Hippokampal atrofi |
Sol peri-sylvien genişleme |
Asimetrik anterior temporal atrofi |
Sol parietal atrofi |
Frontal, fronto-temporal atrofi |
Simetrik dorsal parietal atrofi |
Asimetrik fronto-parietal atrofi |
|
Anatomik yatkınlık |
Limbik |
Anterior peri-sylvien |
Sol temporo-polar |
Sol inferiyor parietal |
Prefrontal |
Bilateral oksipito-parietal |
Asimetrik fronto-parietal, basal ganglia |
|
Hastalık |
Alzheimer |
FTLD-PNFA |
FTLD-SD |
Logopenik PA (Atipik AH) |
FTLD-dv (KBD, PSP, ALS) |
Posterior kortikal atrofi (Atipik AH, LCD, CJH) |
KBD |
|
Nöropatoloji |
AP, NFY |
T+NI, GCI, |
Ub+/T- DN |
NFY (?) |
T+NI, Ub+/T- NCI, NII, DN, Ub+T-TDP- NCI |
AP, NFY, DLDH, PrP+, LC |
GCI |
|
Proteinopati |
Aβ-Tau |
Tau |
TDP-43 |
Aβ-Tau |
Tau, TDP-43 veya FUS |
Aβ-Tau, α-SN, PrPSc |
Tau |
|
Genetik |
PS1, PS2, APP |
MAPT, PGRN, CHMP2B, VCP |
? |
? |
MAPT, PGRN, CHMP2B, VCP, 9p, TARDP, FUS |
?, PRNP |
MAPT, PGRN, CHMP2B, VCP |
Kısaltmalar: Aβ:
amiloid beta protein fragmanı; AH: Alzheimer hastalığı;
ALS: amiyotrofik lateral skleroz; AP: amiloid plak; APP: amiloid prekürsör
protein geni;
CHMP2B: kromatin modifiye edici protein 2B geni; CJH: Creutzfeldt-Jacob
hastalığı; dv: davranışsal varyant; DN: distrofik
nöritler; FTLD: fronto-temporolobar dejenerasyon;
FUS: “fused in” sarkoma;
GCI: glial sitoplazmik inklüzyon; KBD: kortiko-basal dejenerasyon; LCD: Lewy
cisimcikli demans; MAPT: mikrotübül asosiye tau geni; NCI: nöronal sitoplazmik
inklüzyonlar; NII: nöronal intranükleer inklüzyonlar; NFY: nöro-fibriller
yumak; NI: nöronal inklüzyon; PGRN: progranulin geni; PNFA: ilerleyici tutuk
afazi; PRNP: prion proteini geni;
PrP: prion proteini; PS1: presenilin 1 proteini geni; PS2: presenilin 2
proteini geni; PSP: progresif supranükleer paralizi; T: tau proteini; TARDP:
TDP- 43 geni; Ub: ubikutin proteini; VCP: valosin içeren protein
geni
Demansta Davranışsal Özellikler
Demans sendromunun kognitif veya motor olmayan ve
semiyolojik olarak genel psikiyatrinin alanına giren semptomatolojisini
adlandırma konusunda yaşlılık ve demans alanında
terminolojik bir güçlük izlenmektedir. Bazı yazarlar “demansın
non-kognitif belirtileri” tanımını tercih etseler de kognitif
olmayan özellikler yalnızca psikopatoloijk değil aynı zamanda da
motor, otonom ve uyku bozukluklarını da kapsadığı için
bu terim kastını aşmaktadır. Daha fazla
rastlanılan terim “demansın davranışsal özellikleri” şeklindedir.
Bu bölümde de sözkonusu terim tercih edilmiş olmakla birlikte, bu
literatürle bir uzlaşım ihtiyacı gereğidir.
“Davranışsal” terimi genel psikolojide insan
davranışının gözlenebilir özelliklerini
karşılamak üzere kullanılır ve doğal olarak algılama
ve düşünce süreçlerine ilişkin içsel yaşantılamaları
dışta bırakır. Oysa ki bizim
kullanımımızda “demansın davranışsal özellikleri”
gözlenebilir davranışsal bozuklukların yanısıra
duygudurum, algı ve düşünce bozukluklarını da içerecek
şekildedir.
Duygudurum bozuklukları anksiyete, depresyon,
mani, fobiler gibi majör bozukluklar ve bunlara eşlik edebildikleri gibi
yalnız başına da görülebilen emosyonel labilite, iritabilite,
disfori, öfori, ajitasyon gibi duygudurum halleridir. İlgili bölümde
daha ayrıntılı söz edileceği gibi demans sendromu
tanısı konulmadan 10 yıl kadar önceye giden depresyon AH için
başlı başına bir risk faktörüdür. Depresyon AH’de
sıklıkla erken evrede görülür. Depresyon kaybolmakta olan
zihinsel yeteneklere karşı psişik reaksiyon olarak ortaya
çıkabileceği gibi depresif ve depresif olmayan AH’li olguların
otopsilerinin karşılaştırılmasının
gösterdiği üzere nörotransmitter kayıplarına ikincil biyolojik
nedenli de olabilir. Anılan çalışmalarda depresif
olguların beyinsapında noradrenerjik locus ceruleus, serotonerjik
dorsal raphe ve dopaminerjik substantia nigra ve ventral tegmental Tsai
çekirdeklerinde depresif olmayan olgulara kıyasla nöron kaybının
anlamlı düzeyde fazla olduğu görülmüştür. Genç depresif
hastalardaki keder, ölüm düşünceleri, suçluluk ve değersizlik
hisleri, karamsarlık ile karakterize afektif, isteksizlik, hevessizlik,
kendiliğindenlikte azalma, sosyal çekilme ile karakterize motivasyonel
bileşenleri demanslı hastalarda alışagelen biçimde
ayırt etmek her zaman kolay olmayabilir. Bazen motivasyonel
özellikler affektif olanları gölgeleyip depresyon tanısını
geciktirebileceği gibi kendine özgü nöral alt yapısı ile
demansların apati spektrumunda apayrı bir davranışsal
gösterisi olabilen motivasyonel bozukluklar depresyonla
karıştırılabilir. Saf motivasyonel bozukluklar AH’de
orta-ileri evrelerde görülürken FTD’nin erken döneminin başlıca
özelliklerindendir. İnsomni ve gündüz uyuklamaları
şeklindeki diurnal ritm bozuklukları AH’de erken evrelerden itibaren
depresyon çerçevesinde veya izole olarak görülebilir ve performans
bozukluğuna ciddi katkıda bulunabilir. Anksiyete
depresyon çerçevesinde izlenebileceği gibi AH’de orta evreden
itibaren primer olarak da görülür. Kognitif bozukluk arttıkça
şiddeti de artar. Genellikle huzursuzlukla birlikte amaçsız
dolanma, bulunulan mekanı terketme isteğiyle birlikte görülür.
Öfori, iritabilite gibi duygudurum halleri yine orta evreden itibaren
baskın hale gelir. Bazen uykusuzluk, grandiozite,
saldırganlık eşliği ile aşikar bir mani halini
alabilir. Ajitasyon terimi iritabilite ve genellikle verbal ve fiziksel
şiddet gösterilerinin eklenebildiği
saldırganlığın eşlik ettiği mani benzeri tablolar
için kullanılır. AH’de fobiler yine orta evrenin özellikleri
arasındadır. Yalnız kalma/bırakılma, banyo gibi
temalar içerebilir.
Hezeyanlar AH’de duygudurum bozukluklarından
sonra 2. sıklıkta gözlenen düşünce bozukluklarına
ilişkin gösterilerdir. Hezeyanlar da AH’de orta evrenin
özelliklerindendir. Buna karşılık LCD’de destekleyici
tanı kriterleri arasında yeralmalarından
anlaşılacağı gibi ilk evrede görülmeleri
mutaddır. Daha önce de sözedildiği gibi
hırsızlık hezeyanı en sıktır. Bununla birlikte
eşin sadakatsizliği, kendisine kötülük yapılacağı,
misidentifikasyon/Capgras, terkedilme hezeyanları da izlenir.
Halüsinasyonlar algı bozukluklarına
ilişkin gösterilerdir. AH’de yine orta evreden itibaren
görülürler. Şizofrenide en sık işitsel halüsinasyonlar
kaydedilirken, demans sendromlarında görsel halüsinasyonlar daha
sıktır, işitsel modalite onu izler. Olfaktor, gustatuar ve
taktil halüsinasyonlar da izlenebilir. LCD’de ise görsel halüsinasyonlar
temel tanı kriterleri, diğer modalitelerdeki halüsinasyonlar ise
destekleyici özellikler arasında yeralır.
Dolayısıyla, LCD’de hem erken evrenin özelliklerindendirler, hem de
AH’ye göre çok daha sık görülürler.
Kişilik değişikliği yukarda sözedilen
motivasyonel bozukluk spektrumunda apati şeklinde görülebileceği gibi
hiperseksüalite ve oburluk tarzı dürtü kontrol bozuklukları
şeklinde de görülebilir. FTD’de klinik tablonun temel
özelliğini oluşturan kişilik değişikliği, bu iki
uçtan biri ya da diğerinin izole olarak ortaya çıkması
şeklinde görülebileceği gibi sıklıkla birinin baskın
olduğu her ikisinden de özellikler taşıyan tablolar
şeklinde izlenirler. AH’de dürtü kontrol bozuklukları apatiye
kıyasla daha seyrektir. Hiperseksüalitenin de oburluğa
kıyasla daha seyrek olduğu söylenebilir. LCD/PHD’de dürtü kontrol
kusurları dopaminerjik replasman tedavisinin (DRT) bir komplikasyonu
olarak dopamin disregülasyon sendromu (DDS) çerçevesinde görülür. Patolojik
kumar DDS’in bir parçası olarak veya izole şekilde bir dürtü kontrol
kusuru olarak ortaya çıkabilir.
Gözlenebilir davranışsal bozukluklar
AH’nin orta-ileri evrelerinin özellikleridir. Amaçsız
adımlama/dolanma şeklinde hiperaktivite en sık görülendir.
Amaçsız tekrarlayıcı hareketler de sık izlenir.
Demans Eşlikçisi Motor ve Diğer Bulgular
Üç temel alanın yanısıra, motor
belirtiler de bazı demansların ana bileşenlerinden
olabilirler. Yukarıda sözedildiği gibi, motor bulgularla
birilikte olan demanslar, demans sınıflamasının temel
kategorilerindendir. Bu tür demansların klinik tablosunda motor
bulgular, sıklıkla demansa göre daha ağırlıklı ya
da eşdeğer nitelikte olurlar. Motor bulgular
ağırlıkla ekstrapiramidal sisteme ait hipokinetik bir hareket
bozukluğu özelliği gösterirler: bradikinezi, rijidite, parkinsonyen
yürüyüş, postüral denge bozukluğu. multi-sistem atrofilerde
(MSA’lar), özellikle de parkinsonyen formunda (MSA-P), simetrik akinetik-rijid
sendrom, ağırlık ve zamanlama olarak demansın önündedir;
serebellar ve otonom bulgular eşlik edebilir. Kortiko-basal
ganglionik dejenerasyonda (KBD) belirgin bir asimetrik akinezi söz
konusudur. Akinetik taraf sıklıkla apraksiden, ağır
distoni ve “yabancı el” fenomenine kadar değişen özellikler
gösterir ve aynı tarafta kortikal duyu kusuru saptanabilir. KBD
bazen demans, bazen de hareket bozukluğu şeklinde başlayabilir.
Motor nöron hastalığına (MNH) bazen FTD şeklinde ve
hızlı seyirli bir demans da eşlik edebilir (FTD-MNH). Bu
durumda, MNH’ye özgü intrensek el kaslarında atrofi, hafif yutma
güçlüğü şeklinde bulgular demansın gerisinde gizlenebilir.
NPH’da yürüme güçlüğü ve sfinkter kusuru demansla
eşzamanlıdır. Binswanger sendromunda yine NPH’ya çok
benzer bir triad (demans, yürüme güçlüğü, sfinkter kusuru) görülür.
Buradaki yürüme güçlüğü “alt beden yarısı parkinsonizmi” ya da
vasküler parkinsonizm adı verilen şeklindedir ve derin küçük
infarktlara bağlıdır. Gerek parkinsonizm ve gerekse de
sfinkter kusuru AH’ye hastalığın ileri evrelerinden itibaren
katılır. Multi-infarkt demansta hemiparezi sekelleri, görme
alanı kusurları saptanabilir.
Demans sendromunda motor bulguların
varlığı büyük sıklıkla subkortikal işlev
bozukluğunu işaret eder. Bu işlev bozukluğunun nedeni
subkortikal gri (laküner durum) ya da beyaz madde (Binswanger
Hastalığı, CADASIL) yapılarında çok sayıda
iskemik vasküler hasar sonucu olabileceği gibi doğrudan bazal
ganglia-serebellum sistemlerinin nörodejenerasyonu sonucu da olabilir.
Gerçekten de, primer (nörodejeneratif) demanslar motor bulgularla
birlikte olduğunda bir kaç istisna dışında bu bulgular
hemen daima bir ekstrapiramidal sendrom (EPS) niteliğini
taşır. MSA’nın serebellar formunda (MSA-C), başta
gövde ve ekstremite ataksisi ve dizartri olmak üzere serebellar bulgular
ağırlıklı bulgulardır.
Daha önce de değinildiği gibi, patolojik
değişikliğin kortikal ya da subkortikal
ağırlığına göre tabloda motor bulgular kognitif
bulgulara göre, geri ya da ön planda, bazen de eşit düzeyde olurlar.
Bazı hastalıklar bu ağırlık açısından
uniform nitelikte olurlarken (örneğin, AH, PSP, Huntington), diğer
bazı hastalıkların kendi içinde, dejenerasyonun
yatkınlığı bazen kortikal, bazen ise subkortikal yönde
bozulabilir (örneğin, LCD, KBD).
DEMANS TANISINDA YARDIMCI LABORATUAR
YÖNTEMLERİ
Tanı ve ayırıcı tanıda
kognitif, davranışsal ve işlevsel alanları sorgulayan titiz
alınmış bir öykünün yanısıra mental durum muayenesi ve
özellikle fokal bulgular, yürüyüş bozuklukları ve ekstrapiramidal
bulgulara dikkat edilecek nörolojik muayeneden oluşan klinik yöntem esas
olmakla birlikte, asgari düzeyde laboratuar incelemeleri de
duyarlılığı arttıracaktır. Amerikan
Nöroloji Akademisi’nin (AAN) 1995’te yayınladığı Uygulama
Elkitabı’nın “Demans Tanı ve Değerlendirmesi” bölümünde
aşağıdaki biyokimyasal testlerin rutin olarak
yapılması gerektiği söylenir: tam kan sayımı,
elektrolitler, kalsiyum, glukoz, BUN/kreatinin, karaciğer testleri, tiroid
testleri, serum B12 vitamin düzeyi ve sifilis serolojisi.
Kliniğimizdeki rutin uygulamada sifilis serolojisine de ihtiyari olarak
başvurulmaktadır (Tablo 4). Elkitabı’nda diğer
biyokimyasal testler, görüntüleme (BT/MRG), nöropsikolojik muayene, EEG ve
lomber ponksiyon gibi inceleme yöntemlerinin, rutin dışında
kalan özel durumlarda yapılabileceği belirtilmektedir. BT ya da
MRG’nin, klinik değerlendirme tecrübeli bir klinisyen elinde
yapıldığında veya hasta tipik olarak 60
yaşının üzerinde, sinsi başlangıçlı, fokal bulgular,
nöbetler ya da yürüyüş bozuklukları göstermeyen bir demans
tablosu sergilediğinde nadiren gerekeceği kaydedilir. Yine de,
çok erken olgularda alışkın bir göz MRG’nin koronal T1
kesitlerinde gördüğü hippokampal atrofiyle klinik olarak arada
kaldığı bir olguda DAT/PRAD kararı verebilir. Yukarda
Çekirdek Nöropsikolojik Sendromlar bölümünde değinilen ve
aşağıda Alzheimer Dışı Demanslar bölümünde daha
ayrıntılı gözden geçirilecek olan özgül atrofi tarzlarının
saptanması Alzheimer dışı demansların
tanısında değerli olabilir. Öyküde bir ya da daha fazla lober
hematomla birlikte MRG’de gradyan eko tekniğiyle saptanan mikro-kanamalar
demansı serebral amiloid anjiyopatiyle (CAA) ilişkilendirebilir.
Yine, hızlı seyirli bir demansta başta difüzyon
ağırlıklı görüntülemede (DWI) olmak üzere FLAIR’de de
görülebilen kortikal ve bazal ganglionik-talamik yüksek sinyal CJH’yi
işaretleyebilir. Ancak, sıklıkla rutin bir MRG taramasına
dahil edilmedikleri için atrofi değerlendirilmek istendiğinde koronal
T1 kesitlerin, CAA veya CJH’dan kuşkulanıldığında
gradyan eko veya DWI teknikli incelemelerin dahil edilmesi radyoloji
laboratuarından özel olarak talep edilmelidir. Nöropsikolojik muayene
temel olarak erken demansı değerlendirmekte, demans ve depresyonu
ayırmakta, hukuki ehliyetin dokümantasyonunda yararlı
olacaktır. EEG’ye, nöbetlerin varlığında,
Creutzfeldt-Jacob hastalığı kuşkusunda
başvurulabilir. Lomber ponksiyon, metastatik kanser, MSS infeksiyonu
kuşkusu, reaktif sifilis serolojisi, hidrosefali, genç demans,
hızlı seyirli ve atipik demans, immunosupresyon ve MSS vasküliti
kuşkusu durumlarında rutin olarak yapılmalıdır.
Rutin ve tek olgu incelemesi
dışında kalan, özellikle AH’de araştırma amaçlı
bir dizi yöntemden de sözedilebilir. MRG-volümetri ile hippokampus, entorhinal
korteks ve superior temporal sulkus volümetrisi yöntemi, özellikle normal
yaşlanma-MCI-AH devamlılığının
ayrımında giderek rafine edilmektedir (Şekil 1).
MR-Spektroskopi (MRS) yöntemi ile bir nöronal işaretleyici olan N-asetil
aspartat miktarında meziyal temporal bölgelerden yapılan
ölçümlerde saptanan azalmanın yine hippokampal volüm ile korele
ettiği gösterilmiştir. MRS ile NAA/Cr düzeyleri nöron
kaybıyla karakterize olan AH, FTLD ve VaD gibi demanslarda düşer.
MI/Cr düzeyleri ise AH ve FTLD gibi gliozis ile karakterize demanslarda
yükselir. Nihayet, Cho/Cr düzeyleri AH ve LCD gibi ağır kolinerjik
kayıp ile karakterize demanslarda yükselir. İşaretli glukoz
kullanan klasik FDG-PET ile pariyeto-temporal hipometabolizma, SPECT ile
aynı bölgelerde hipoperfüzyon bulgularının AH’de
duyarlılığı yüksek olsa da yeterince özgül
değildirler. FDG-PET ile posterior singulat
hipometabolizmasının AH’nin en erken bulgularıyla korele
ettiğine ilişkin bildirimler mevcuttur. Yeni gelişmekte
olan PET-amiloid görüntülemenin potansiyeline daha önce PAD bölümünde
değinilmişti. BOS’ta bazı Aβ fragmanları düzeylerinde
azalma, yine total ve fosfo-tau proteini düzeyinde artma ümit vaadeden
laboratuar yöntemleri arasında aynı bölümde söz edilmişti.
Genetik inceleme ile AH’de bir risk faktörü olan APO-E e4 alellerinin
saptanması artık atipik olgularda düşünülebilir. Familyal
gibi duran olgularda genetik analizin yararı aşikardır. Kognitif
elektrofizyoloji özel olarak tasarlanmış ve analiz edilmiş olaya
ilişkin potansiyeller (OİP) biçiminde AH’nin erken döneminde
ayırt edici bir yöntem olarak kullanım alanı bulabilir.
Başlangıçta ucuz ve basit bir yöntem olarak umut
vaadeden anti-kolinerjik damla (tropicamide) ile anormal pupiller cevabın
özgüllüğü kabul edilir bulunmamıştır. Günümüzde
sadece araştırmalara sınırlı gibi görünen bu
yöntemlerden bazıları gelecekte AH rutininde kullanılabilecek
potansiyel biyo-işaretleyiciler arasındadırlar.
Tablo 4. Demans
tanısında rutin laboratuvar
ALZHEIMER HASTALIĞI
Demans antik çağlardan beri bilinen bir
hastalıktır. Ancak, Alzheimer hastalığının
(AH) ilk olgusu olma onuru bayan Auguste D.’ye aittir. Auguste
D 1850 Mayıs’ında doğmuştu. 1890’ların sonlarına
doğru bellek ve dil bozuklukları şeklinde tanımlanan demans
semptomları geliştirmeye başladı. Kısa zamanda ciddi
psikotik özellikler ve ajitasyon eklendi ve Kasım 1901’de, Alois Alzheimer
tarafından muayene edileceği Frankfurt Akıl Hastanesi’ne
getirildi, 8 Nisan 1906’da bu hastanede öldü. Alzheimer 1902’de Kraepelin’in
daveti üzerine önce Heidelberg’e gitmiş, 1 yıl sonra da yine
Kraepelin ile birlikte Münih’e geçmişti. Auguste D.’nin ölümünü duyan
Alzheimer eski çalışma arkadaşlarından klinik kayıtlarını
ve beyin dokusunu rica etti. 3 Kasım 1906’da, Tübingen’deki Güneydoğu
Alman Psikiyatri Derneği’nin 37. Toplantısı’nda Auguste D.’nin
klinik ve nöropatolojik bulgularını "Über einen eigenartigen,
schweren Erkrankungsprozess der Hirnrinde" isimli 11. bildiriyle sundu.
Hastanın yaşının genç olması dolayısıyla
hastalık bir pre-senil demans biçimi olarak değerlendirilecektir.
Beynin nöropatolojik incelemesinde serebral korteksin normale göre
incelmiş olduğunu gördü. Ayrıca, daha sonra amiloid plaklar
olarak adlandırılacak miliyer odakları ve yine daha sonra
nörofibriler yumaklar adını alacak intraselüler fibril
birikintilerini tanımladı. Auguste D. ile ilk kez
tanımlanmış olan klinik olarak demans ile nöropatolojik olarak
NFY ve AP bugün hala AH’nin ana diyagnostik triadını
oluşturmaktadır. Hocası ve çalışma
arkadaşı, şizofreninin de isim babası Emil Kraepelin Psychiatrie:
Ein Lehrbuch für Studierende und Aerzte adlı döneminin en ünlü ders
kitabının 1913’teki 8. baskısında bu tür olgulara
Alzheimer’in adını verdi. Tuhaf olan bunca yaygın bir
hastalığın “Havva” olgusu olarak adlandırılabilecek
olan ilk olgunun atipik bir olgu olmasıdır. Gerek erken
başlangıç ve gerekse de, aslında erken
başlangıçlı AH olgularında aykırı olmayacak
şekilde, davranışsal belirtilerin ön planda olduğu klinik
tablo ATD’nin tipik tablosu değildir. 1970’ler ortalarına kadar
AH’nin “Pre-senil demanslar” kategorisi altında PiH’le birlikte nadir
hastalıklardan biri olarak sınıflandırılması
büyük ölçüde bu nedenledir. Bir asırlık tarihinin son
çeyreğinde aslında çok daha sık görülen ve “kronik organik beyin
sendromu” ya da “serebral skleroz” gibi adlar da verilen “senil
demansın” AH ile aynı patolojik değişiklikleri
paylaşan bir ve aynı antite olduğu
kavranmıştır. Robert Katzman’ın 1976’da “senil demans
AH’dir demesiyle AH tarihinin moderan çağına geöilmiş olur.
Günümüzde, özellikle ortalama yaşam beklentisinin giderek
arttığı gelişmiş ülkelerde, henüz etyolojisinin
aydınlatılamamış ve kesin tedavisinin bulunamamış
olması nedenleriyle de, en önde gelen morbidite ve mortalite nedenlerinden
biri olmaktadır. Alzheimer anahtar kelimesi ile Pub Med’de bir
araştırma yapılarak son yarım yüzyılın 10’ar
yıllık dilimlerindeki yayın sayısına
bakıldığında bulunan nerdeyse logaritmik artış
çok çarpıcıdır: 1960-69: 28, 1970-79: 327, 1980-89: 5464,
1990-99: 17328, 2000-09: 29738.
KLİNİK TABLO
Tanı Kriterleri
Yukarıda yeri geldiğince
tartışıldığı gibi ATD, merkezini limbik sistem
dejenerasyonuna bağlı yakın bellek bozukluğunun
oluşturduğu, sinsi başlangıçlı, yavaş seyirli bir
tempoyla neokortikal tutulumun da katılmasıyla diğer kognitif
işlevlerin de bozulduğu bir demans sendromudur (GYA’ları bozan
sinsi başlangıç, yavaş seyirli en az iki kognitif
bozukluk). Bu tanımdan anlaşılacağı gibi demans
AH nörodejenerasyonunun göreli geç bir gösterisidir. AH’nin klinik
tanısı için yayınlanmış olan ve bugün yaygın
biçimde kullanılan NINCDS-ADRDA (National Institute of Neurological and
Communicative Disorders and Stroke-Alzheimer’s Disease and Related Disorders
Association) ve DSM-IV (Diagnostic and Statistical Manual of Mental Disorders,
4th edition) kriterlerinin her ikisi de (Tablo 5 ve Tablo 6) bir
takım nüanslarına karşın özetle yukardaki
koşulların doldurulmasını AH’ye özgü tipik sendromun
tanısı için zorunlu tutmaktadırlar. Böyle bir tabloya yolaçabilecek
sekonder nedenlerin (sistemik, nörolojik, psikiyatrik) ekarte edilebiliyor
olması da her ikisi için zorunlu dışlama kriteridir. Bu
koşulların gerçekleştirildiği tipik tabloya DSM-IV
“Alzheimer Tipi Demans” (DAT) adını vermektedir. NINCDS-ADRDA
için ise aynı tablonun adı “Muhtemel Alzheimer
Hastalığı” (PRAD) olmaktadır. Her iki sisteme
göre de son kliniko-patolojik serilerdeki tanı özgüllüğü %90’a
yaklaşmaktadır. Bir başka deyişle PRAD/DAT
tanısı %10 ihtimalle AH dışı durumlara
yanlış olarak konulmakta veya AH dışı nedenler
PRAD/DAT tablosunu taklit ederek sunulmaktadırlar. Tipik klinik
tablonun duyarlılığı tüm AH olgularının yine %90
kadarını kapsamaktadır. Bir başka deyişle AH
hastalarının %10’u atipik klinik tablolar gösterdikleri için PRAD/DAT
tanısı alamamaktadırlar. NINCDS-ADRDA, başlangıç ve
seyirde beklenmedik özellikler (akut başlangıç ve/veya
hızlı seyir), demansa neden olabilecek fakat klinisyenin demans
nedeni olarak düşünmediği 2. bir durumun varlığı
(örneğin, inme, hipotiroidi, vb.) ve izole tek bir ilerleyici kognitif
bozukluk olarak sıraladığı atipik tablolar için “Mümkün
Alzheimer Hastalığı” (Possible AD-PosAD) adını
kullanır. PosAD tanısıyla duyarlılık artar, yani
tanı konulmayan olguların sayıları azalırken özgüllük
azalmakta, yani yanlış tanıların sayısı
artmaktadır.
Her iki yaklaşım da AH’nin demans
aşamasının tanınması için özellikle
duyarlılık ve bir ölçüde de özgüllük açısından kabul
edilebilir düzeyler sağlasalar da tanısal kesinlikten uzak oldukları
da açıktır. Nitekim, her 2 kriter dizisi de AH’nin göreli geç
bir aşamasına karşılık gelen bir klinik tablo, sendrom
tanımlamaktadır. NINCDS-ADRDA kriterlerinde açıkça ifade
edildiği gibi kesin tanı nöropatologa bırakılmaktadır.
Bir başka deyişle mevcut durumda klinisyenler değil, sadece
patologlar Alzheimer hastalığı tanısı koyabilirler.
Hastalığın seyrine müdahale olanaklarının
sınırlı olduğu yakın tarihlere kadar bu durumun pek de
sakınca yarattığı söylenemezdi. Bununla birlikte,
hastalığın nörobiyolojisinin kavranmasındaki baş
döndürücü gelişmeler doğrudan etyopatogenetik mekanizmaları
hedefleyen nedene yönelik tedavilerin araştırılabilir
olmasına olanak sağladı. Aşağıda daha
ayrıntılı gözden geçirileceği gibi, bu amaçla sınanan
ve büyük maddi kaynaklar ayrılarak klinik çalışmaların
olgun aşamalarına kadar gelebilen çok sayıda molekül nihai
engelde takılarak ipi göğüsleyemedi. Bu üst üste hayal
kırıklarının nedenleri arasında mutad
çalışma tasarımından kaynaklanan nedenler ve etkisizlik
yanısıra kuvvetle muhtemel bir diğeri ise bu klinik
çalışmaların anılan kriterlerle tanı konmuş
demans evresindeki hastaları toplaması gibi görünüyor; oysa ki
doğrudan bir etyopatogenetik mekanizma basamağını
hedefleyen bir ajan için yerleşmiş demans ajanın gerçek
potansiyelini sergileyebilmesi için çok geç kalınmış bir
aşama olabilir. AH’nin demans aşamasını kanserin metastatik
aşamasına benzetip tedavi başarısı beklentisi
açısından bir analoji kurabiliriz. Bu nedenle AH’nin daha demans
öncesi aşamada tanıya yönelik biyo-işaretleyicilerin de
yardımıyla güvenilir bir şekilde ayırt edilmesi, AH
tanısının bizzat klinisyen tarafından konulması
giderek artan bir önem arzetmektedir. Aşağıda, AH’de
yardımcı tanı yöntemleri ve potansiyel biyo-işaretleyiciler
gözden geçirildikten sonra yakınlarda bir öneri olarak sunulan böylesi bir
yeni tanı kriterleri dizisi tanıtılacaktır. Ayrıca
benzer bir perspektifle her 2 kriter dizisinin gözden geçirilmekte olduğu,
NINCDS-ADRDA’in NIA-AA (Ulusal Yaşlılık Enstitüsü – Alzheimer
Derneği) şekliyle 2010 sonlarında DSM-IV2ün DSM-V olarak 2011
içinde yenilenmesinin beklendiği kaydedilmelidir.
Tablo 5. NINCDS-ADRDA Alzheimer
Hastalığının Klinik Tanı Kriterleri (McKhann et al.
1984)
I.
MUHTEMEL Alzheimer Hastalığı klinik tanı kriterleri
şunları içerir:
· Klinik muayene ile saptanan,
Mini-Mental Test, Blessed Demans Ölçeği ya da benzer bir test ile
dokümante edilen ve nöropsikolojik testlerle de doğrulanan demans tablosu;
· İki ya da daha fazla
kognitif süreçte bozulma;
· Bilinç bozukluğu yok;
· Başlangıç 40-90
yaşları arasında, büyük sıklıkla da 65
yaşından sonra;
· Bellek ya da diğer
bilişsel süreçlerde ilerleyici bozukluğa yolaçabilecek sistemik ya da
beyne ait başka bir hastalık yok.
II.
MUHTEMEL Alzheimer Hastalığı tanısı şunlarla
desteklenir:
· Dil (afazi), motor yetenekler
(apraksi) ve algı (agnozi) gibi özgül kognitif işlevlerde ilerleyici
bozulma;
· Günlük yaşam
aktivitelerinde bozulma ve davranış biçiminde değişme;
· Ailede benzer bozukluk öyküsü
(özellikle patolojik olarak kanıtlanmışsa);
· Laboratuarda:
Standart tekniklerle normal lomber ponksiyon,
EEG’nin normal olması yada yavaş dalga aktivitesinde artış
gibi non-spesifik değişiklikler,
BT’de serebral atrofiye ilişkin bulgular ve seri incelemelerde bu
bulguların ilerleyişi.
III.
Alzheimer hastalığı dışındaki nedenler
dışlandıktan sonra, MUHTEMEL Alzheimer Hastalığı
tanısı ile uyumlu olabilecek diğer klinik özellikler
şunlardır:
· Hastalığın
seyrinde platolar;
· Depresyon, uykusuzluk,
inkontinans, hezeyan, illüzyon ve halüsinasyonlar, verbal, emosyonel ya da
fiziksel katastrofik patlamalar, cinsel bozukluklar ve kilo kaybı gibi
eşlikçi bulgular;
· Bazı hastalarda, özellikle
hastalığın ileri dönemlerinde, kas tonusunda artış,
miyoklonus ya da yürüme güçlüğü gibi diğer nörolojik bozukluklar;
· Hastalığın ileri
evresinde nöbetler;
· Yaş için normal BT.
IV. MUHTEMEL
Alzheimer Hastalığı tanısını
belirsizleştiren ya da ihtimal dışına çıkaran
özellikler şunlardır:
· İnme tarzında ani
başlangıç;
· Hemiparezi, duysal kayıp,
görme alanı defektleri ve inkoordinasyon gibi fokal nörolojik
bulguların hastalığın erken evrelerinde bulunması;
· Nöbetler ya da yürüyüş
bozukluklarının, daha başlangıçta ya da
hastalığın çok erken evrelerinde bulunması;
V. MÜMKÜN
Alzheimer Hastalığı tanı kriterleri şunlardır:
· Demansa neden olabilecek
diğer nörolojik, psikiyatrik ya da sistemik bozukluklar
olmaksızın, başlangıç, prezantasyon ya da klinik seyirde
varyasyonların bulunması durumunda konulabilir;
· Demansa neden olabilecek, ancak
demansın nedeni gibi görünmeyen ikinci bir sistemik ya da beyin
hastalığının bulunması durumunda konulabilir;
· Diğer belirlenebilir
nedenlerinin dışlandığı, tek ve yavaş ilerleyici
bir bilişsel bozukluğun bulunması durumunda, araştırma
çalışması amaçlı olarak kullanılabilir.
VI. KESİN
Alzheimer Hastalığı tanısı kriterleri
şunlardır:
· Muhtemel Alzheimer
Hastalığı klinik kriterleri;
· Biyopsi ya da otopsiyle elde
edilen histopatolojik kanıtlar.
Tablo 6. DSM-IV Alzheimer Tipi Demans
İçin Tanı Kriterleri (1995)
A.
Birden fazla kognitif alanı içeren bozukluk kendini
aşağıdaki iki maddeyi de kapsayacak şeklinde ortaya
çıkar:
(1)
Bellek bozukluğu (yeni bir bilgi öğrenme ve öğrenilmiş eski
bir bilgiyi hatırlama yeteneğinin bozulması)
(2) Aşağıda
sıralanan kognitif bozuklardan en az biri:
(a) Afazi (dil
bozukluğu)
(b) Apraksi
(motor işlevlerin normal olmasına karşın belirli motor
eylemlerin yerine getirilmesi yeteneğinde bozulma)
(c) Agnozi
(duysal işlevlerin salim olmasına karşın
nesneleri
tanımakta güçlük)
(d) Yürütücü
işlevlerde bozulma (planlama, organize etme, sıralama, soyutlama)
B.
A1 ve A2 kriterlerinde tanımlanan bilişsel bozukluklar toplumsal ve
mesleki işlevselliği ciddi biçimde bozmakta ve eski işlevsellik
düzeyine göre anlamlı bir gerilemeyi temsil etmektedir.
C.
Seyir, sinsi başlangıç ve yavaş ilerleyici kognitif
yıkım özelliklerindedir.
D. A1
ve A2 kriterlerinde tanımlanan bilişsel bozukluklar
aşağıda sıralanan nedenlerden herhangi birine
bağlı değildir:
(1)
Bellek ve diğer bilişsel işlevlerde ilerleyici bozulmaya neden
olabilecek merkezi sinir sistemine ait diğer durumlar (örneğin
serebrovasküler hastalık, Parkinson hastalığı, Huntington
hastalığı, subdural hematom, normal basınçlı
hidrosefali, beyin tümörü)
(2)
Demansa neden olabileceği bilinen sistemik durumlar (örneğin
hipotiroidizm, B12 vitamini ya da folik asid eksikliği, niasin
eksikliği, hiperkalsemi, nörosifilis, HIV infeksiyonu)
(3)
İlaçlar ve madde kullanımı ile ilgili durumlar
E.
Bozukluklar delirium seyri dışında ortaya
çıkmıştır.
F. Bozukluk
başka bir Eksen I hastalığı ile açıklanabilir
nitelikte değildir.
Alzheimer
Hastalığının Pre-Demans Evreleri: Normal
Yaşlanma-Yaşla İlintili Unutkanlık-Hafif Kognitif
Bozukluk-Demans Devamlılığı
İlerleyici hastalıklar patogenetik süreçleri
içinde belli bir eşiği geçtikleri aşamada klinik olarak
saptanabilir olurlar. Bu eşik de hemen daima normal kabul
edilemeyecek bir belirtidir. Örneğin, glioblastoma yeterince büyüyüp
kafa içi basıncını arttırarak
başağrısına neden olana kadar sub-klinik olarak
ilerler. Başağrısı kadar aşikar bir anormal
belirti olmayan unutkanlık durumunda sub-klinik dönemle klinik dönem daha
kalın çizgilerle içiçe girer. Alzheimer
hastalığının zamansal evrimi bunun en güzel
örneklerindendir. AH demans aşamasına ulaşmadan önce uzun
yıllar (bazı hesaplamalara göre 15-20 yıl) kliniğe
ilerleyici unutkanlık şeklinde yansıyan bir demans öncesi veya
“prodromal” evreden geçecektir. Bununla birlikte, unutkanlık yaşlılıkta
son derece sıklıkla duyulmaya alışılmış,
gayet normal kabul edilen bir yakınmadır. Bellek de dahil olmak
üzere bazı kognitif yeteneklerde yaşlanmayla birlikte beklenen
“normal” azalma nöropsikolojik standardizasyonda eğitimle birlikte göz
önüne alınması gereken temel özelliklerindendir. Bir yakın
bellek testinde yetmişli yaşların normal değeri yirmili
yaşların normalinin yarısı olabilir. Normal
yaşlanma kavramı bellek de dahil kognitif kapasitelerini hiç
yitirmeyen küçük bir azınlığı da içerse de,
ağırlıkla nöropsikolojik testlerde yaşa göre uyarlanmış
düzeylerde bir performansa karşılık gelir. MRG/BT’de
kortikal sulkuslar ve ventrikül sistemi genişlemeye
başlamıştır. Bu tür görüntülemelerin
raporlarının sonuç bölümünde sıklıkla “yaşla ilintili
atrofi” yazacaktır. Kognitif açıdan normal kişilerin otopsileri,
tümüyle normal olabileceği gibi 50’li yaşların
başlarından itibaren neokortekste gevşek nitelikli amiloid
plaklar ve bazen bunlara eşlik eden entorhinal kortekste NFY’lerin
görülebileceğini ortaya koymaktadır. Sağduyu bu 2. gruba ait
beyinlerde AH nörodejenerasyonun başladığı fakat henüz
klinik belirti eşiğini geçmediğini düşündürmelidir.
Başka bir deyişle, 2. grup değişiklikleri göstermeye
başlayan beynin sahibi yeterince uzun yaşasaydı,
yakınmalarının zaman içinde görünür olup sonunda AH klinik
kriterlerini dolduracağı ileri sürülebilir.
Şekil 8. Normal yaşlanmadan
ağır demansa bellek bozukluğu sürekliliği
Sosyal yaşamda başka açılardan
sağlıklı bir yaşlının özel isimleri unutuyor,
özel eşyalarını kolaylıkla bulamıyor olması
anormal kabul edilmez. Aynı soruları tekrarlar olması
yakınları arasında kuşku uyandırmaya
başlar. İşini sürdüremez olması, ya da yabancı
bir mekanda kaybolması ise artık normal olarak kabul edilemez
olur.
“Yaşla İlintili Bellek Bozukluğu
(AAMI)” kavramı tam da yukarıdaki paragrafta anormal bulunmayan
unutkanlık durumuna karşılık gelmektedir. Daha önceki
yıllarda “Selim Yaşlılık Unutkanlığı”
kavramıyla karşılanmaya çalışılan ve isminin de
çağrıştırdığı gibi demansa ilerlemeyecek iyi
huylu unutkanlığa karşılık gelecek şekilde
kullanılan söz konusu durum için Amerikan Ulusal Zihinsel Sağlık
Enstitüsü (NIMH) 1986 yılında araştırma amaçlı
tanı kriterleri yayınladı (Tablo 7). AAMI
tanımına uygun düşen yakınmaları olan bir
yaşlı günlük yaşamında tümüyle
bağımsızdır. Bu kişinin nöropsikolojik
muayenesinde özellikle yakın bellek testlerinde yaşa göre normal
sınırlarda, fakat genç erişkinlere göre ortalama değerlerin
1 standart sapma (SD) altında yer aldığı
saptanacaktır. Hangi yaşlı “selim” yaşlılık
değişikliklerini göstermekte, hangisi ise AH’nin en erken izlerini
taşımaktadır sorularının cevabını
şimdiki durumda ne salt klinik fenomenoloji ne de mevcut nöropatolojik
kavrayış ile vermek mümkün değildir. AAMI tanısı alan
bir kişi yakınmaları karakter değiştirmeden
öldüğü takdirde beyni tümüyle normal bulunabileceği gibi, “Nöropatoloji”
kısmında ayrıntısıyla görüleceği gibi, limbik
sisteminde az sayıda nörofibriler yumak ve neokorteksinde
gevşek karakterli amiloid plaklar taşıyor olabilir. PET tekniklerinde
yeni gelişmelerle sadece amiloid plaklara (PIB-PET) ve hem plaklara hem
yumaklara (FDDNP-PET) bağlanan ligandlarla bu tür patolojik
değişiklikleri taşıyan ve taşımayan kognitif
olarak normal yaşlı kişileri saptamak mümkün olmaktadır.
Uzunlamasına izlenen kognitif olarak normal veya AAMI tanısı
alan yaşlıların PIB/FDDNP+ olanları, PIB/FDDNP- olanlara
göre anlamlı olarak daha fazla demans geliştirdikleri
gösterildiği takdirde bu yöntem prodromal/pre-klinik AH’nin bir
işaretleyicisi olarak yerleşebilir.
Yaşlılıkta pre-demans kognitif
bozukluğu tanımlama gayretiyle bir dizi daha kavram ve kriterler
dizisi ileri sürülmüş olsa da tümü de demansa ilerleyen bir bellek
bozukluğu kavrayışından ziyade demans kadar şiddetli
olmayan bir “hafif kognitif bozukluk”u çeşitli isimlerle
tanımlamışlardır. Bunlar arasında 1990’lar boyunca
tanımlanan ICD-10’un “minimal kognitif bozukluk – MCD”, DSM-IV’ün “hafif
nöro-kognitif bozukluk – MNCD”, Uluslararası Psikogeriyatri Birliği’nin
“Yaşla İlintili Kognitif Bozukluk – AACD” ve Kanada
Sağlıklı Yaşlanma Çalışması (CSHA)
çerçevesinde kullanılan “demans olmayan kognitif bozukluk – CIND”
kavramları sayılabilir. Tüm bu tanımlamalar, belli
nüanslarına rağmen, tanımladıkları demans
şiddetinde olmayan kognitif bozukluk durumunu esas olarak statik özerk
antiteler olarak görmektedirler; öyle ki, bunlar arasından bir
kısım hasta düzelip normale dönebileceği gibi, bir
kısmı ise ilerleyip demans geliştirdiğinde nitelik
değiştirip başka bir antiteye dönüşmüş gibi kabul
edilmektedir.
Tablo 7. NIMH-AAMI Tanı
Kriterleri
|
Yaş
> 50. Günlük yaşamda
belleğe ilişkin yakınmalar. Genç erişkinlere göre 1
standart sapma daha düşük nesnel bellek performansı. Demans mevcut değil. Normal ya da normalin üstü
zeka seviyesi. |
Gerçekte, AAMI tanısı alan
yaşlıların AH nörodejenerasyonuna sahip bir bölümü daha da
kötüleşecek ve bellek yakınmaları yakınlarının da
dikkatini çekmeye başlayacaktır. Gene de böyle bir kişi
anılan bellek problemlerinin yarattığı, ancak listeyle
alışverişe çıkar olmak gibi genellikle üstesinden
gelebildiği güçlükler dışında, günlük yaşamında
halen bağımsızdır. Nöropsikolojik muayenesinde
ağırlıkla bazen de tek başına bellek
alanında, anlamlı düzeyde düşük performans gösterir.
Bu tabloya “Hafif Kognitif Bozukluk” (MCI) adı verilir (Tablo 8).
Orijinal biçimiyle bu tanım, prodromal AH olması muhtemel, ATD’ye
dönüşme riski taşıyan bir alt grubu araştırma ve
mümkünse tedavi hedefi yapmak üzere belirlenmişse de sonradan revize
edilen biçimiyle amnestik olmayan demanssız kognitif bozukluklar da dahil
edilerek prodromal AH’ye olan vurgu çabası kapsayıcılık
adına zayıflamış görünmektedir (Tablo 9).
İlgili tablodan da görüleceği gibi, sınıflama tümüyle
bellek bozukluğunun bulunup bulunmamasına ve mevcut kognitif bozukluğun
tek veya çoğul olma durumuna göredir. Bu farklı tabloların
ilerledikleri takdirde en mutad olarak belli demans hastalıklarına
dönüşecekleri beklenir. İlk bakışta bu sınıflama
yukarda tanımlanan “çekirdek sendromlar” yaklaşımının
bir benzeri gibi düşünülebilirse de burdaki “mevcut MCI tablosu-ilerdeki
demans sendromu” ilişkisi çok daha indirgemeci bir akıl yürütmedir.
“Çekirdek sendromların anatomik adreslerinden bu anatomik adrese
yatkınlık sergileyen en muhtemel proteinopati” tarzı bir
akıl yürütme esnekliği sergileyemez. Örneğin, progresif
simultanagnozi-simetrik dorsal parietal alanlar-amiloidopati silsilesi
progresif simultanagnozinin bu sınıflamadaki
karşılığı olan izole non-amnestik MCI ile, bu MCI alt
sınıfının AH’ye dönüşmesi beklenmediğinden
sağlanamaz. Dahası, çekirdek sendrom yaklaşımıyla,
ilerde demansa dönüşüp dönüşmeyeceğine
bakılmaksızın anatomik yatkınlık ve muhtemel
proteinopatiden yola çıkarak hastalık tanısına varmak
mümkünken, MCI tanımı ile ancak demans aşamasında bir
hastalıktan sözedilebilecek durumla normallik arasında
kalmış bir geçiş aşaması tasarlanmış
olmaktadır.
Uzunlamasına izlenen büyük serilerde MCI’ın demansa dönüşme
hızı yıllık %8-14’ken 5 yıllık izlemlerde %50
kadardır. Oysa ki, normal popülasyonda demans insidansı
%1-2’dir.
Görüntüleme çalışmalarında mezyal temporal patoloji bu
hastaları normallerden ayırmaktadır. MRG-volümetride
bazı çalışmalarda hippokampus, diğer bazılarında
ise entorhinal korteks atrofisi MCI’lı serilerde yaşla
eşlenmiş normal kontrollere göre anlamlı düzeyde
görülmektedir. FDG-PET’le yapılan çalışmalarda
başlangıç noktasında başlıca posterior singulat
hipometabolizmaya sahip olmanın izlemde dönüşmenin anlamlı bir
öngörücüsü olduğu ortaya konmuştur. PIB-PET ile yapılan
çalışmalarda ATD’lilerin hemen tümü PIB tutulumu gösterirken
MCI’ların yarısından fazlasında ve enteresan olanı
normal yaşlıların da küçük bir bölümünde görülmektedir.
Dahası gerek MCI’lılar ve gerekse de normallerde PIB tutulumu
gösteren grupta PIB yükü ile bellek skorları korele etmektedir. Bu bulgu
MCI ve normal olarak sınıflanmış olan PIB tutulumuna sahip
bu bireylerin gerçekte sırasıyla prodromal ve pre-klinik AH
taşıdıkları şeklinde yorumlanabilir. Ayırt edici
başka bir bulgu ise nöritik plaklara bağlanan PIB ligandı ile ancak
demans aşamasında mezyal temporal tutulum görülürken MCI ve
normallerde görülmemesidir. Buna karşılık NFY’lere de
bağlanan FDDNP-PET ile normal yaşlılarda ve MCI’lılarda
bellek skorlarıyla korele eden bulgular saptanmaktadır. FDDNP-PET ile
bu bulgu da PIB-PET ile yukarıdaki yoruma benzer bir şekilde
yorumlanabilir.
MCI otopsilerinde normalden hafif PRAD patolojilerine kadar
değişen bir spektrum izlenmekle birlikte, NFY’nin halen
limbik-paralimbik alanlara sınırlı ancak sayısal olarak
normallere göre anlamlı düzeyde artmış olması en
tutarlı bulgu gibi görünmektedir. Nöritik plaklar henüz limbik
sistemde görülmemektedir. APOE-ε4 alel sıklığı ve
BOS’ta total tau ve fosfo-tau proteini düzeylerinde artış ile
β-amiloid42 düzeylerinde düşüşün, entorhinal ve/veya
hippokampal atrofi yanısıra demansa dönüşecekleri
dönüşmeyenlerden ayıran öngörücüler olduğu ileri
sürülmektedir. MCI’ın bir klinik antite olarak yani prodromal AH
olarak daha iyi sınırlarının çizilmesi, bu evrede etkili
olacak tedavilerle demansa dönüşmenin geciktirilmesi ya da önüne
geçilmesi, günümüzde AH’nin yükünün azaltılmasında temel bir hedef
gibi görünmektedir.
Tablo 8. Petersen-Mayo MCI
Kriterleri
|
1. Hasta
yakını tarafından da doğrulanan bellek
yakınması. 2. Genel kognitif
işlevlerde bozulma yok. 3. Günlük yaşam
aktivitelerinde bozulma yok. 4. Yaş ve
eğitim normlarına göre saptanan bellek bozukluğu. 5. Demans mevcut
değil. |
Yukarıdaki paragrafta tanımlanan,
artık sağduyunun da normal kabul edemediği, günlük yaşamda
bağımsızlığını sürdüremeyecek düzeyde
bozulma durumunda artık demanstan sözedilebilir. Bu duruma
karşılık gelen otopsilerde NFY’ler artık neokortekse
yayılmış, gevşek biçimden nöritik biçime
değişmiş AP’ler limbik sistemde görülür olmuş durumdadır.
Tablo 9. Revize MCI
Kriterleri ve Muhtemel Demans İzdüşümleri
Alzheimer Tipi Demansın Evreleri
İlerleyici bir hastalıktan
sözettiğimize göre klinik tabloyu statik bir profille tarif
edemeyiz. Yine sağduyu ile amatör bir yaklaşım dahi
ilerleyen bir tablonun hafif, orta ve ağır olarak
sınıflandırılması gerektiğini öngörebilir.
Formel evreleme ölçeklerinin de taklit ettiği yöntem bundan farklı
değildir. Zihinsel yıkımın demans düzeyine
ulaşması günlük aktivitelerde bozulma ile belirlendiğine göre,
evrelemeyi de GYA’lara göre kabaca yapabiliriz: AAMI’yı sübjektif bellek
yakınmaları, MCI’yı GYA’ların korunduğu ama
yakın kişilerin farkında olduğu bir bellek bozulması
olarak tarif etmiştik; bu durumda devamlılık ölçütünü gözönünde
tutarak hafif demansı işte ve ev dışında
bağımsızlığın bozulmaya
başladığı, orta demansı, bunlar tümüyle
bağımlılaşırken ev yaşamı ve kendine
bakımda sorunların başladığı, ağır
demansı ise sürekli bakım gereken, hastanın tümüyle
bağımlı olduğu bir durum olarak tarif edebiliriz.
Bugün yaygın olarak kullanılan iki
evreleme sisteminden biri olan “Global Bozulma Ölçeği” (Global
Deterioration Scale-GDS)’nin de geliştiricisi olan Reisberg, AH’deki
ilerleyici yıkım sürecini, bebeklik-erken ve geç çocukluk ve ergenlik
şeklindeki insanın ilerleyici
bireyselleşme-bağımsızlaşma gelişimsel sürecinin
tam tersine çevrilmesi olduğunu ileri sürer ve bu ilerleyici
yıkımı retrogenez olarak adlandırır. Buna göre
AAMI-MCI’lı yaşlı büyük ölçüde bağımsız olsa da
bazı kararları için erişkin gözetimine gerek duyan ergene, hafif
demanslı, evinde ve ev dışında tanıdık mekanlarda
belli bir bağımsızlığı kazanmış, ancak
sosyal ilişkiler, muhakeme gerektiren karmaşık işlevlerde
halen denetim gereksinen 7-12 yaşlarındaki okul çocuğunu
andırır. Orta demanslı ise kabaca, ev yaşamı ve
giyinme, yıkanma, yemek yeme gibi temel GYA’larda henüz gözetim gereken
2-6 yaşları arasındaki okul öncesi çocuğu gibidir.
Ağır demanslı, yaşamını sürdürmek için 24 saat
ana-babaya (bakıcıya) tümüyle bağımlı 0-2 yaş
bebeğine benzer. Ağır evrenin kendisi de, yani giderek tüm
motor ve verbal yeteneklerin kaybedildiği yatağa tam
bağımlı nihai döneme doğru yıkım, oturma, yürüme,
konuşma, sfinkter kontrolünün geliştiği bebekliğin dinamik
gelişiminin tam tersi olarak kavranıp alt evrelere
ayrılabilir.
GDS sözedildiği gibi AH’yi evrelemeye yarayan
ölçeklerden birisidir. AH dışı demanslarda AH’ye
özgüllüğü dolayısıyla kullanılamaz. GDS evreleri 1
ila 7 arasında değişir. Kabaca, GDS1 hiç yakınması
ve bulgusu olmayan normal yaşlıya karşılık
gelirken GDS2’ye AAMI, GDS3’e ise MCI evreleri denilebilir. GDS
4-5-6-7 ise hafif, orta, ağır ve çok ağır olmak üzere
AH’nin klinik evreleridir. Demansın Klinik Evrelendirilmesi
(Clinical Dementia Rating Scale-CDR) ise yaygın olarak kullanılan 2.
ölçektir. CDR’da bellek yine merkezi önemde olmakla birlikte, çok eksenli
tasarımıyla diğer demansların evrelenmesinde de
kullanılabilir. CDR evreleri 0-0,5-1-2-3 olarak
sıralanırlar. CDR 0, AAMI’ı da içerecek şekilde
normal yaşlılığa karşılık gelir. CDR
0,5 MCI’ya karşılık gelir ve “kuşkulu demans” evresi
adını alır. CDR 1, 2 ve 3 sırasıyla hafif, orta
ve ağır evrelerdir (Tablo 10, 11).
Hafif evredeki demanslı hasta halen
çalışmaktaysa artık işinde verimliliğini
yitirmiştir. Yaratıcılık gerektirmeyen tekdüze
işler başlangıçta sürdürülebilse de, iş
arkadaşları performans düşüklüğünün
farkındadırlar ve kısa süre içinde emeklilik
kaçınılmaz olur. Yakın geçmişe ait olayların
hatırlanmasındaki güçlük, aynı soruların
tekrarlanması, kelime bulma güçlükleri yakınların dikkatini
çeken başlıca özelliklerdir. Halen bildik mekanlarda
dolaşabilse ve yolculuk yapabilse de, yabancı mekanlarda
kaybolabilir. Araba kullanırken sinyalizasyona dikkatsizlik,
tepkilerde yavaşlama, yönleri karıştırma gibi güçlükler
başlamıştır. Banka işleri, fatura ödemeleri gibi
mali işlerde hatalar olmaktadır. Banka kartı, cep telefonu
gibi yenilikleri öğrenip kullanmayı başaramaz. Hobiler
(dikiş-nakış, bahçecilik, sanatsal uğraşılar,
yetenek oyunları, vb.) sürdürülemez olmuştur. Yemek lezzetinde
bozulmalar gibi mutfak işlerinde güçlükler
başlamıştır. Çamaşır, bulaşık
gibi ev işlerini sürdürebilse de, bunlarda eski özenini bir ölçüde
yitirmiştir. Okumak ve gazete-TV aracılığıyla
aktüaliteye ilgi azalmıştır. Giyinmek, yıkanmak,
sofra alışkanlıkları ve temel hijyende henüz bir sorun
yoktur. İritabilite, duygulanımda küntleşme ve inkar
eğilimi ile kendiliğindenliğin azalması
dışında davranışsal belirtiler yoktur ve sosyal
uygunluk iyi korunmuştur. Uyku kalitesi bozulmaya başlar.
Cinsel ilgi ve iştah bozulur. Eksikliklerin farkedilmesinin de
katkısıyla bazı olgularda depresyon belirtileri ön planda
olabilir. Ancak depresyon sıklıkla keder ifadesi gibi afektif
belirtilerden çok, isteksizlik gibi motivasyonel belirtilerle kendini
gösterir. Muayenede yakın bellek ön planda olmak üzere,
görsel-mekansal bozukluk, uzak bellekte bozulmalar, adlandırma güçlükleri,
dikkat ve soyutlama-planlamada bozulmalar saptanır. Praksis
muayenesinde “beden-parçası-nesne-gibi” cevaplar alınır.
Henüz gnostik bir kusur saptanmaz. Somatik nörolojik muayene
normaldir. MMSE skoru kabaca 20-26 arasında olabilir.
Sıklıkla GDS 4, CDR 1 olarak evrelenirler. Bu hastaların
otopsilerinde heteromodal kortekste NFY’ler, limbik sistemde nöritik SP’ler
saptanır.
Orta demans evresine
ulaşıldığında, hasta ev dışındaki
bağımsızlığını artık tümüyle
yitirmiştir. Gözetimle sokağa çıkabilse de, yalnız
kaldığı takdirde yolunu bulamaz.
Başkalarının evinde odaları karıştırabilir.
Yeni öğrenme artık hemen hiç mümkün olamamaktadır. Anlama,
okuma ve yazma giderek bozulur; evrenin sonlarına doğru imzası
tanınmaz olabilir. Birinci derece yakınları
hakkındaki bilgiyi genellikle korusa da, torunlarının
sayısı, isimleri, okulları gibi bilgileri
karıştırmaktadır. Evdeki işlevselliği son
derece yüzeyselleşmiştir. Ancak sofrayı toplamaya
veya sebze doğramaya yardım düzeyinde olabilir. Giyinme
sırasında mevsime ya da günün saatine uygun giysiyi seçmede zorlanma,
giysilerin sırasını karıştırma (gömleğin
üzerine iç çamaşırı gibi), düğmeleri yanlış
ilikleme gibi güçlükler başlar. Sofrada öncelikle
bıçağı kullanamaz olduğunda yemeklerinin önceden kesilmesi
gerekir. Giderek döküp saçarak yemek belirginleşir.
Çatal-bıçağı karıştırmak, sıvıları
çatalla almaya çalışmak gibi hatalar görülebilir.
Yıkanmakta öncelikle sıcağı soğuğu ayarlamakla
başlayan yardım gereksinimi ortaya çıkar.
Henüz sfinkter kontrolü seyrek gece kaçırmaları
dışında sorunsuzdur. Tuvalet mekaniği, elini yüzünü
yıkamak gibi işlevleri kendi başına yapabilir.
Davranışsal belirtiler artık vurgulanmaya
başlamıştır. Hırsızlık, terkedilme ve
sadakatsizlik hezeyanları olabilir. Yalnız kalmaktan ürker ve
yakınını (eşi, çocuğu) sürekli gözünün önünde
ister. Hekim vizitleri gibi yaklaşan randevular aşikar bir
beklenti anksiyetesine yolaçabilir. Uyku-uyanıklık ritiminde
bozulma artık belirginleşmiştir. Gece sık uyanmalar
ve gündüz sık uyuklamalarla geçer. Muayenede hafif evre
bulguları biraz daha ağırlaşmıştır.
Dilsel işlevlere ait bulguların ağırlaşması dile
dayanan testlerin yapılamaz olmasına neden olabilir. Praksis
bozukluğu taraf apraksisi düzeyine ulaşabilir. Gnostik
bozukluklar, özellikle sofrada göz önündeki nesneyi diğerleri
arasından ayırıp bulamamak (simultanagnozi) şeklinde
olabilir. Temel nörolojik muayenede hafif parkinsonyen
değişiklikler saptanabilir. MMSE skoru 10-19 arasında
değişir. GDS 5, CDR 2 olarak evrelenirler. Bu hastaların
otopsilerinde NFY’ler unimodal asosiasyon kortekslerine de
yayılmış olabilir.
Ağır demans evresinde bellekte
artık sadece parçacıklar söz konusudur. Yakınını
(eşi, çocuğu) ana-babasıyla karıştırabilir,
aynadaki kendi yüzünü tanıyamayabilir. Giyinmek, yıkanmak,
yemek gibi temel GYA’larda artık tam bir gözetim gerekmektedir.
Evrenin sonlarında yutma güçlüğü de ortaya çıkar. Kelime
hazinesi son derece fakirleşmiştir. Evrenin sonlarında tüm
verbal yetenekler yitirilir. Ambulasyon giderek zorlaşır ve
sonlara doğru giderek oturmak dahi mümkün olmaz hale gelir.
Televizyondaki kişileri ev içindeymiş gibi sanıp konuşmak,
aynadaki kendi hayaliyle yabancıymış gibi konuşmak
gözlenebilir. Ambulasyonun korunduğu sırada amaçsız
dolaşma, istifçilik, amaçsız tekrarlayıcı hareketler
izlenebilir. Tuvalet mekaniğinde bozulmalar (idrar ya da gaita
sonrası uygun biçimde temizlenme, sifonu çekme sorunları), idrar
kaçırma giderek belirginleşir. Epileptik nöbetler ortaya
çıkabilir. Ağır evredeki hastaların formel muayenesi
son derece güçtür ve mümkün olamayabilir. Yapılabildiğinde
global bir yıkım saptanır. Temel nörolojik muayenede
tonus değişiklikleri, yürüyüş bozuklukları şeklinde
parkinsonyen bulgular biraz daha ortaya çıkmıştır.
MMSE 0-9 arasındadır. GDS 6-7, CDR 3 olarak evrelenirler.
Şekil 9’da AH’nin klinik ve bir dizi
laboratuar ölçütünün pre-klinik evreden başlayarak ağır demans
evresine kadar gösterdiği değişkenlik gösterilmektedir.
Şekil 9. Alzheimer
hastalığının evreleri boyunca klinik ve laboratuar
göstergelerinin değişimi
BOS Aβ düzeyi daha preklinik evrede PIB-PET ile
amiloid yükünün saptanabilmesinden önce düşmeye başlar; klinik
evrelerden itibaren PIB-PET ile amiloid yükü fazlaca değişmez.
AAMI’dan itibaren kognisyon bozulmaya, BOS tau düzeyleri artmaya başlar.
MCI evresinde FDG-PET ile bölgesel metabolizma azalması izlenir olur ve
sonrasında demans evreleri boyunca yaygınlaşarak şiddetlenir.
Hippokampus hacmi AAMI’dan itibaren küçülmeye başlar. İşlevsel
bozukluk (GYA’lar) tanı gereği demans aşamasından itibaren
başlar ve ilerleyici olarak kötüleşir.
Kısaltmalar: AAMI: yaşla ilintili bellek bozukluğu; BOS:
beyin-omurilik sıvısı; FDG-PET: işaretli glukoz pozitron
emisyon tomografisi; GYA’lar: günlük yaşam aktiviteleri; MCI: hafif
kognitif bozukluk; PIB: Pittsburgh bileşeni
Tablo 10. Demansın Klinik
Evrelendirilmesi Ölçeği -CDR (Morris 1997)
|
CDR |
1.
Bellek |
2.
Oryantasyon |
3.
Yargılama – Problem çözme |
|
0 |
Bellek
kaybı yok ya da hafif ve belirsiz unutkanlık |
Tümüyle
oryante |
Günlük problemler ve
çalışma hayatı ve mali işlerle ilgili problemleri
iyi çözer; yargılama iyidir |
|
0,5 |
Hafif
fakat aşikar unutkanlık; olayların kısmen
hatırlanabilmesi; "selim"
unutkanlık |
Zaman
ilişkilerinde hafif güçlük dışında tümüyle oryante |
Problem çözme, benzerlik ve
farklılıkları kavramakta hafif bozukluk |
|
1 |
Orta
düzeyde unutkanlık, yakın dönem olayları için daha
belirgin; unutkanlık günlük aktiviteleri etkiliyor |
Zamanda
orta düzeyde güçlük; muayene sırasında mekana
oryante, dışarıda coğrafi disoryantasyonu olabilir |
Orta
düzeyde bozukluk; toplumsal yargılama genellikle
korunmuştur |
|
2 |
Ağır düzeyde
unutkanlık; yalnızca çok iyi öğrenilmiş materyel
tutulabilir; yeni materyel hızla yitirilir |
Zaman
ilişkilerinde ağır düzeyde güçlük; genellikle zamana,
sıklıkla da mekana disoryante |
Ağır
düzeyde bozukluk; genellikle toplumsal yargılama da
bozuktur |
|
3 |
Ağır
düzeyde unutkanlık; yalnızca parçacıklar kalır |
Yalnızca
kişilere oryante |
Yargılama
ve problem çözme tümüyle bozuk |
|
CDR |
4.
Ev dışında işlevsellik |
5.
Ev yaşamı – Hobiler |
6.
Kişisel bakım |
|
0 |
İşte,
alışverişte, gönüllü gruplar ve toplumsal gruplar içinde her
zamanki düzeyde bağımsız işlevsellik |
Ev
yaşamı, hobiler ve entelektüel ilgiler iyi korunmuş |
Kendine
bakıma tümüyle muktedir |
|
0,5 |
Anılan
aktivitelerde hafif bozulma |
Ev
yaşamı, hobiler ve entelektüel ilgilerde hafif bozulma |
|
|
1 |
Anılan
aktivitelerden bazılarını halen sürdürse de,
bağımsız işlev görememe; yüzeysel bir bakışla
hala normal görünebilir |
Evdeki
işlevlerde hafif fakat aşikar bozulma; güç ev işleri,
karmaşık hobiler ve ilgiler
terkedilmiş durumda |
Gayrete
getirilmesi gerekiyor |
|
2 |
Ev
dışında bağımsızlığını tümüyle
yitirmiş / Ev dışında aktivitelere götürülebilecek
kadar iyi görünür |
Yalnızca
basit işler yapılabiliyor; ilgiler son derece
sınırlı |
Giyinme,
hijyen ve diğer kişisel bakım için yardım gerekiyor |
|
3 |
Ev
dışında bağımsızlığını tümüyle
yitirmiş / Ev dışında aktivitelere götürülemeyecek kadar
hasta görünür |
Evde
kayda değer bir işlevselliği yok |
Kişisel
bakım için önemli ölçüde yardım gerekir; genellikle
inkontinandır |
Evrelendirme: Bellek ekseninin dışındaki eksenlerden
en az üçü bellek ekseninden farklı değilse, evre bellek ekseniyle
aynıdır. Farklı olduğu durumda ise, evre bellek
ekseninin üstünde ya da altında kalan bu 3 eksenin derecesidir. Bu
kuralın tek istisnası bellek ekseni dışında kalan 5
eksenden üçünün belleğin bir tarafında geri kalan ikisinin
diğer tarafında olmasıdır ki bu durumda da evre bellek
ekseniyle aynıdır. Bellek ekseni 0,5 ise evre 0 olamaz;
diğer eksenlerin derecesine bağlı olarak 0,5 ya da 1
olmalıdır. Bellek 0, fakat en az iki eksen 1 ya da daha fazla
ise, evre 0,5 olmalıdır.
Tablo 11. Global Bozulma
Ölçeği –GDS (Reisberg et al. 1998)
|
1 |
Bellek
kusuruna ilişkin yakınma yok. Klinik görüşme ile
bellek kusuru saptanmıyor. |
|
2 |
Bellek
kusuruna ait, özellikle aşağıda sıralanan alanlarda
yakınmalar var:
(a)
eşyalarını koyduğu yerleri unutuyor;
(b) önceden iyi
bildiği isimleri unutuyor.
Klinik görüşmede bellek kusuruna ait nesnel bir kanıt yok.
İş ve toplumsal ortamlarda nesnel bir bozukluk yok.
Semptomatolojiye yönelik uygun düzeyde endişe
taşıyor. |
|
3 |
En
erken gösterilebilir bozukluk bulguları.
Aşağıdaki alanlarda birden fazla bulgu:
(a) iyi
bilmediği çevrelere gittiğinde kaybolabiliyor.
(b) iş
arkadaşları, hastanın bozulmaya yüztutan çalışma
performansının farkındalar.
(c) kelime
ve isim bulma güçlükleri yakınları tarafından fark ediliyor.
(d) bir kitap
yada yazıyı okuduğunda eskisi gibi hatırında
kalmıyor.
(e) yeni
tanıştığı insanların isimlerini
hatırlamakta güçlüğü var.
(f)
değerli bir nesne kaybedilmiş yada konulmaması gereken bir
yere konmuş.
(g) konsantrasyon
eksikliği klinik testler sırasında aşikar.
Bellek bozukluğuna ilişkin, ancak yoğun bir görüşmeden
sonra ortaya konulabilen nesnel bulgular.
Uğraşı gerektiren iş koşulları yada toplumsal
ortamlarda düşük performans.
Hastada inkar mekanizması belirgin hale gelir olmuş. Belirtilere
ılımlı yada orta düzeyde bir anksiyete eşlik
edebilir. |
|
4 |
Dikkatli
bir klinik görüşme sonrasında ortaya konulan aşikar bozukluk
bulguları.
Bozukluk aşağıdaki alanlarda ortaya konuyor:
(a) günlük ve
yakın geçmişe ait olaylara ilişkin bilgide azalma.
(b) kişisel
geçmişe ait bazı bellek problemleri.
(c)
çıkarma dizileriyle ortaya konulan konsantrasyon bozukluğu.
(d) yolculuk
yapma, para işleriyle uğraşma gibi yeteneklerde azalma.
Aşağıdaki alanlar genellikle sorunsuz:
(a) yer ve zaman
oryantasyonu
(b) bildik
kişi ve yüzlerin tanınması.
(c) bilinen
yerlere yolculuk yapabilme.
Karmaşık işlevlerin yerine getirilemez olması.
Baskın savunma mekanizması olarak inkar kullanılıyor.
Duygulanımda küntleşme ve
sıkıntı yaratan durumlardan kaçınma. |
|
5 |
Yaşamlarını
sürdürebilmeleri için yardım gerekmektedir.
Hasta güncel yaşamına ilişkin temel özelliklerden birini
hatırlayamıyor. Örneğin:
(a)
yıllardır kullanmakta olduğu adres yada telefon
numarasını.
(b) yakın
aile üyelerinin isimlerini (torunlar gibi).
(c) mezun
olduğu lise yada yüksek okulun adını.
Zaman (gün, haftanın günü, mevsim, v.b) yada mekan oryantasyonunda
bozulmalar.
Eğitimli bir kişi, 40'tan geriye 4er, yada 20'den geriye 2'şer
saymakta güçlük çekebilir.
Bu evredeki kişiler kendilerine ve diğerlerine ait temel gerçeklere
ilişkin bilgiyi korurlar.
Kendi isimlerini daima, eş ve çocuklarınınkileri genellikle
bilirler. Temizlenmek
ve yemek yemekte yardım gerekmez, ancak uygun giysiyi seçmekte
güçlükleri olabilir. |
|
6 |
Bazen,
yaşamlarını sürdürmek için tümüyle bağımlı
oldukları eşlerinin ismini unutabilirler.
Yaşamlarındaki yakın geçmişe ilişkin olay ve
deneyimlerin tümünden büyük ölçüde habersizdirler.
Çevreye ilişkin bazı bilgiler korunabilir; yıl, mevsim, v.b.
10'dan geriye, bazen de ileriye doğru 1'er saymakta güçlükleri olabilir.
Günlük yaşam aktivitelerinde yardım gerekir:
(a) idrar
inkontinansı olabilir.
(b) yolculuk için
yardım gerekir, fakat bazen bildik yerlere gidebilirler.
Diurnal ritm sıklıkla bozulmuştur.
Hemen daima kendi isimlerini hatırlarlar.
Genellikle, çevrelerindeki tanıdık kişileri yabancılardan
ayırabilirler.
Kişilik ve emosyon değişiklikleri görülür. Bunlar
oldukça değişkendir ve şunları içerir:
(a) hezeyan davranışı, örn.,
eşlerini taklit olmakla suçlayabilirler; çevredeki
hayali kişilerle, yada
aynadaki kendi imgeleriyle konuşabilirler. (b) obsesif
belirtiler olabilir, örn., hasta sürekli olarak basit bir temizlik hareketini
tekrarlayabilir.
(c) anksiyete belirtileri, ajitasyon
ve daha önce mevcut olmayan tarzda bir saldırganlık
görülebilir.
(d) kognitif abuli, örn., amaca yönelik bir
davranışın uygulanması için gerekli düşüncenin
yeterli süre taşınamaması nedeniyle irade gücünün kaybı. |
|
7 |
Bu
evre sürecinde tüm verbal yetenekler kaybedilir. Bu evrenin erken döneminde
bazı kelime ve cümleler söylenebilse de konuşma son derece
sınırlanmıştır. Evrenin ilerlemesiyle,
homurdanmak dışında, konuşma tümüyle yitirilir. İdrar inkontinansı;
temizlik ve yemek yemek için yardım gerekir. Temel psikomotor yetenekler
(örn. yürümek) evrenin ilerlemesiyle kaybedilir. Beyin bedene ne yapması
gerektiğini söyleme yeteneğini artık yitirmiş gibidir. Genel ve kortikal nörolojik
bulgu ve belirtiler bu evrede genellikle mevcuttur. |
EPİDEMİYOLOJİ
Prevalans ve İnsidans
Dünyada şimdiye kadar yapılan prevalans
(belli bir zaman kesitinde bulunan tüm olguların nüfusa oranı)
çalışmalarının metaanalizi 65-70 yaşları
arasında %4-5 kadar olan AH’nin her 5 yılda bir katlanarak artıp
90’lı yaşlarda %50’ye kadar ulaştığını
ortaya koymaktadır. Türkiye’den ilk çalışma
diyebileceğimiz Istanbul, Kadıköy’de gerçekleştirilen TAPS
isimli prevalans çalışmasının sonuçları Tablo 12’de
gösterilmiştir. 90’lı yaşlardan sonraki prevalans konusunda
henüz bir uzlaşım olmamakla birlikte artışın devam
etmeyip bu oranın plato çizdiği (“AH yaşla ilintilidir”)
görüşü ile sürekli yükseldiği (“AH yaşlanmayla ilintilidir)
görüşü arasındaki tartışma tam olarak
çözümlenmemiştir.
Yaşa-özgül insidans, ya da belli bir zaman
aralığında ortaya çıkan yeni olguların
sayısı da yaş aralıkları arttıkça hızla
artar. Jorm ve Jolley’in 1998 tarihli bir meta-analizlerinde 65-69 ile
başlayıp 85-89 ile sonlanan 5’er yıllık 5 yaş
katmanında insidans %1.6, %3.5, %7.8, %14.8 ve %26.0 şeklinde yine
katlanarak artmaktadır.
Tablo 12. TAPS
Çalışması’nda yaşa ve cinsiyete göre
katmanlandırılmış prevelans oranları
|
|
PRAD |
Toplam
AH |
Toplam
demans |
|
Yaş
grupları bir arada Tümü |
%11 |
%16 |
%20 |
|
Erkek |
%9 |
%13 |
%17 |
|
Kadın |
%13 |
%17 |
%22 |
|
Cinsiyet
grupları bir arada 70-74 |
%9 |
%14 |
%18 |
|
75-79 |
%14 |
%19 |
%22 |
|
80+ |
%13 |
%17 |
%23 |
|
Erkek 70-74
|
%8 |
%12 |
%16 |
|
75-79 |
%9 |
%12 |
%15 |
|
80+ |
%11 |
%16 |
%20 |
|
Kadın 70-74 |
%10 |
%15 |
%19 |
|
75-79 |
%18 |
%23 |
%26 |
|
80+ |
%15 |
%20 |
%25 |
PRAD: muhtemel Alzheimer
hastalığı; toplam AH: PRAD artı mümkün AH; toplam demans:
toplam AH artı non-AH demanslar. (Kaynak 15’den
uyarlanmıştır.)
Risk Faktörleri (Tablo 13)
Günümüzde yaşam beklentisindeki dramatik
artış global bir fenomendir. Batılı ülkeler
başta olmak üzere, tüm dünyada yaşlı nüfusun genel nüfusa
oranının giderek artması bir toplumsal sorun olarak AH’nin
önemini de arttırmaktadır. Hastalığın toplumsal
maliyetinin demans şiddetiyle doğru orantılı olduğunu
gösteren çalışmalar mevcuttur. TAPS’ta da yaş bir risk
faktörü olarak ortaya konmuştur.
Bir risk faktörü olarak cinsiyet
tartışmalıdır. Bir çok çalışma AH
prevalansının kadınlarda erkeklere göre daha fazla olduğunu
göstermektedir. Ancak bu prevalans farklılığı
genellikle kadınlarda yaşam beklentisinin daha uzun olmasıyla
açıklanmaktadır. Bununla birlikte östrojen beyinde bir
nörotrofik faktör olarak işlev görmekte, erkeklerde ömür boyu mevcut olan
testosteron beyinde östrojene çevrilip işlevini sürdürürken, kadınlar
menopoz sonrası östrojensiz kalmaktadırlar. Bu durum
yaşamın ikinci yarısında bir nörotrofik faktörden yoksun
kalan kadınların neden demans için daha fazla risk
taşıdıklarının açıklamalarından biri
olabilir. Nitekim, epidemiyolojik çalışmalarda post-menopozal dönemde
östrojen replasmanı kullanan kadınlarda kullanmayanlara göre demans
prevalansının daha düşük olması bu varsayımı
destekler niteliktedir. Cinsiyetin gerçek anlamda bir risk olup
olmadığına daha iyi bir cevap verebilecek olan insidans
çalışmalarının sonuçları ise çelişkilidir.
Bir çalışma 85 yaşının üzerindeki erkeklerde AH
insidansını yılda %2.7, buna karşılık
kadınlarda %8.9 olduğunu bulmuştur. Başka bir
çalışmada ise bir AH’linin 1. derece kadın
akrabalarının 1. derece erkek akrabalarına göre yaşam boyu
hastalık riskinin daha fazla olduğu gösterilmiştir. Ancak çok
sayıda başka çalışmacı grubu bu farklılığı
doğrulamayarak herhangi bir farklılık
bulmamışlardır. TAPS çalışmasında da
kadınlarda prevalans daha yüksek olsa da cinsiyetin bir risk faktörü
olmadığı görülmüştür. Bununla birlikte, dört
ayrı prospektif Avrupa çalışmasının
havuzlanmış verilerini analiz eden EURODEM Insidens
Araştırma Grubu kadın cinsiyetin 85 yaş ile birlikte bir
risk faktörü olarak belirdiğini, 90 yaş üzerinde bu riskin daha da
arttığını bildirmişlerdir. TAPS çalışmasında
85 ve 90 yaş üzeri tabakalardaki birey sayısının azlığı
bizim çalışmamızdaki negatif sonucun açıklaması
olabilir.
Yorumu yine tartışmalı olsa da
düşük eğitimin başlı başına bir risk faktörü
olduğu artık yerleşmiş bir bilgidir. Eğitim
deneyimindeki artışın bireyin kognitif rezervinin
genişlemesine yolaçarak hastalığın ortaya
çıkış eşiğini yükseltmesi çekici bir açıklama
gibi durmaktadır. TAPS çalışmasında da düşük
eğitim demans için bir risk faktörü olarak belirlenmiştir.
TAPS çalışmasında elektro-manyetik
alan (EMF) maruziyeti de AH için bir risk faktörü olarak ortaya konmuştur.
Elektrikçiler, tamirciler, santral operatörleri, teknisyenleri,
kaynakçılar, marangozlar, terziler gibi elektrikli aletlerle
çalışanlar aşırı düşük frekanslı (ELF) EMF
maruziyeti için risk altında meslek gruplarıdır. Bizim
çalışmamızı da içeren ELF-EMF maruziyeti ve AH
ilişkisi ile ilgili 12 çalışmanın meta-analizi olan bir
çalışmada bu ilişkinin bağımsız bir risk
olduğu kabullenilmekle birlikte mevcut çalışmalarda maruziyet
dozu ile risk artışı ve maruziyet ile cinsiyetlerin ilişkisinin
belirlenememiş olması dolayısıyla daha fazla
çalışmanın gerektiğinin altı çizilmiştir. Maruz
kalınan diğer toksik ve zararlı durumlar arasında organik
çözücüler, alüminyum ve genel anestezi ön plana çıkar gibi
görünmektedir. Ancak bunların yerleşmiş
tutarlılıkta bulgular olduğu söylenemez. Sigara içenlerde,
kolinerjik nikotinik reseptörlerin üst-regülasyonu ve bunun sonucu olarak
Aβ düzeylerinin düşmesi ile açıklanan biçimde AH’nin daha az
görüldüğü bulgusu, başlangıçta sigara içenlerin ileri
yaşlara gelmeden diğer hastalıklar nedeniyle ölmeleri ve
vasküler demansa daha yatkın olmaları şeklinde metodolojik
olarak eleştirilmişken, son zamanlardaki Rotterdam ve New York
çalışmalarında tam tersine AH riskini bir kaç kez
arttırdığı yönünde bulgular elde edilmiştir.
Ayrı bir alt başlıkta
tartışılacak olan otozomal dominant geçişli familyal AH
olguları yanısıra, ailede demans öyküsü AH için kendi
başına bir risk faktörüdür. AH’li hastaların kardeşlerinde
yaşam boyu hastalık riski beklentisi ikiye katlanarak %23’ten %48’e
çıkar. Monozigotik ikizlerde dizigot ikizlere oranla AH
birlikteliği anlamlı oranda fazladır. APOE-ε4 aleli
ile AH ilişkisi "genetik" altbaşlığı
altında tartışılacaktır.
Yine genetik başlığı
altında sözedilecek olan Down sendromu ve AH ilişkisi
yanısıra, Down sendromu aile öyküsü AH riskini 2 ila 3 misli
arttırmaktadır. Ayrıca enteresan bir bulgu, Down sendromlu
çocuk doğuran annelerin AH riskinin, diğer tiplerde mental retarde
çocuklar doğuran annelere göre 5 misli artmasıdır. Bu
farklılık anne yaşının doğumda 35’in altında
olduğu durumlarda ortaya çıkmaktadır. Down
babalarında AH için ayrıca bir risk söz konusu değildir.
Bu bulgu, AH geliştirmek ve 35 yaşının altında Down
sendromlu çocuk doğurmak arasında paylaşılan bir genetik
yatkınlık olasılığı yönünde
tartışılmıştır.
Kafa travması öyküsü ile AH arasında
ilişki bildiren önemli çalışmalar olmakla birlikte
(örneğin, EURODEM) bunu reddenler de vardır (örneğin, Rotterdam
Çalışması ve Kanada Sağlık ve Yaşlanma
Çalışması). Bilinç kaybına neden olmuş şiddetteki
kafa travmalarının bir risk faktörü olarak değerlendirilmesi
mevcut çalışmalardaki çelişik sonuçları aşabilir.
APOE-ε4 yükü ile kafa travmasının AH’nin başlangıç
yaşı üzerine birlikte etkisi olduğunu ileri süren
çalışmalar mevcuttur. Bir çalışmada ise tek
başına etkisiz görünürken, normalde iki misli artmış olan
APOE-ε4 heterozigotlarının riskini 10 misline
arttırdığı ortaya konmuştur. Kafa travmasının
etkisinin β-amiloid metabolizması üzerinden olması muhtemel
görünmektedir. Dementia pugilistica geliştiren boksörlerin otopsilerinde
saptanan gevşek neokortikal amiloid plaklar bu varsayımı
destekler niteliktedir.
Öz geçmişte 10 yıla kadar geriye giden
tedavi gerektirmiş bir depresyon öyküsü AH riskini 3 kat
arttırır. Depresyonun hastalığın erken bir
gösterisi olduğunu iddia eden çalışmalar vardır. Jorm
tarafından yapılan 2001 tarihli meta-analizde
yaşlılıkta depresyon özgeçmişinin demans geliştirme
riskini ikiye katladığı bildirilmiştir.
Kısıtlı sosyal ilişkiler
Kungsholmen, İsveç ve Kuzey Manhattan, ABD gibi önemli toplum temelli
kesitsel ve uzunlamasına gözlem çalışmalarında
bağımsız bir risk faktörü olarak ortaya konmuştur. Sosyal
ilişkiler akraba ve arkadaş ilişkilerinin zenginliği,
sinema, lokanta, klüp, dernek faaliyetleri amaçlı sokağa
çıkmalar olarak özetlenebilir. Honolulu-Asya Yaşlanma
Çalışması’nda bu risk orta yaşlarından itibaren sosyal
aktiviteleri düşen bireylerle sınırlı bulunmuştur.
Vasküler olaylar arasında miyokard iskemisi,
hipertansiyon, diyabet ve inme öyküsü ile metabolik sendrom mevcudiyetinin AH
riskini anlamlı biçimde arttırdığı bir çok
çalışmada gösterilmiştir. Bu bulgular o denli güçlüdür ki,
daha yakın zamanlara kadar 2. sıklıkta demans nedeni olarak
gösterilen vasküler demansın AH’den bağımsız bir antite
olarak varlığı bile son yıllarda tartışma konusu
olmuştur. Aynı zamanda bir inme risk faktörü olan yüksek plazma
homosistein düzeyinin AH için de bağımsız bir risk faktörü
olduğu bildirilmiştir.
Küçük kafa çevresi ile AH arasında bazı
çalışmalarda gösterilen ilişki, eğitim ile olan
ilişkide olduğu gibi kognitif rezerv teorisiyle açıklanmaya
çalışılmaktadır.
Hipotirodinin gerek genel olarak demans ve gerekse
de AH ile ilişkisi ortaya konmuştur. AH ile ilişkisinin
mekanizması aşikar olmasa da potansiyel olarak düzeltilebilir bir
durum olması bakımından önemlidir.
Yorumu yine tartışmalı olsa da
düşük eğitimin başlı başına bir risk faktörü
olduğu artık yerleşmiş bir bilgidir. Eğitim
deneyimindeki artışın bireyin kognitif rezervinin
genişlemesine yol açarak hastalığın ortaya
çıkış eşiğini yükseltmesi çekici bir açıklama
gibi durmaktadır. TAPS çalışmasında da düşük
eğitim demans için bir risk faktörü olarak belirlenmiştir.
TAPS çalışmasında elektro-manyetik alan
(EMF) maruziyeti de AH için bir risk faktörü olarak ortaya konmuştur.
Elektrikçiler, tamirciler, santral operatörleri, teknisyenleri,
kaynakçılar, marangozlar, terziler gibi elektrikli aletlerle
çalışanlar aşırı düşük frekanslı (ELF) EMF
maruziyeti için risk altında meslek gruplarıdır. Bizim
çalışmamızı da içeren ELF-EMF maruziyeti ve AH
ilişkisi ile ilgili 12 çalışmanın meta-analizi olan Garcia
ve arkadaşlarının 2008 tarihli makalelerinde bu ilişkinin
bağımsız bir risk olduğu kabullenilmekle birlikte mevcut
çalışmalarda maruziyet dozu ile risk artışı ve
maruziyet ile cinsiyetlerin ilişkisinin belirlenememiş olması
dolayısıyla daha fazla çalışmanın öneminin altı
çizilmiştir. Maruz kalınan diğer toksik ve zararlı durumlar
arasında organik çözücüler, aluminyum ve genel anestezi ön plana
çıkar gibi görünmektedir. Ancak bunların yerleşmiş
tutarlılıkta bulgular olduğu söylenemez. Sigara içenlerde,
kolinerjik nikotinik reseptörlerin üst regülasyonu ve bunun sonucu olarak
Aβ düzeylerinin düşmesi ile açıklanan biçimde AH’nin daha az
görüldüğü bulgusu, başlangıçta sigara içenlerin ileri
yaşlara gelmeden diğer hastalıklar nedeniyle ölmeleri ve
vasküler demansa daha yatkın olmaları şeklinde metodolojik
olarak eleştirilmişken, son zamanlardaki Rotterdam ve New York çalışmalarında
tam tersine AH riskini bir kaç kez arttırdığı yönünde
bulgular elde edilmiştir.
Ayrı bir alt başlıkta
tartışılacak olan otozomal dominan geçişli familyal AH
olguları yanısıra, ailede demans öyküsü AH için kendi
başına bir risk faktörüdür. AH’li hastaların
kardeşlerinde yaşam boyu hastalık riski beklentisi ikiye
katlanarak %23’ten %48’e çıkar. Monozigotik ikizlerde dizigot
ikizlere oranla AH birlikteliği anlamlı oranda
fazladır. APOE-ε4 aleli ile AH ilişkisi "genetik"
altbaşlığı altında tartışılacaktır.
Yine genetik başlığı
altında söz edilecek olan Down sendromu ve AH ilişkisi
yanısıra, Down sendromu aile öyküsü AH riskini 2 ila 3 misli
arttırmaktadır. Ayrıca enteresan bir bulgu olarak, Down sendromlu
çocuk doğuran annelerin AH riskinin, diğer tiplerde mental retarde
çocuklar doğuran annelere göre 5 misli artmasıdır. Bu
farklılık anne yaşının doğumda 35’in altında
olduğu durumlarda ortaya çıkmaktadır. Down
babalarında AH için ayrıca bir risk söz konusu değildir.
Bu bulgu, AH geliştirmek ve 35 yaşının altında Down
sendromlu çocuk doğurmak arasında paylaşılan bir genetik
yatkınlık olasılığı yönünde
tartışılmıştır.
Kafa travması öyküsü ile AH arasında
ilişki bildiren önemli çalışmalar olmakla birlikte (örn.,
EURODEM) bunu reddedenler de vardır (örn., Rotterdam
Çalışması ve Kanada Sağlık ve Yaşlanma
Çalışması). Bilinç kaybına neden olmuş şiddetteki
kafa travmalarının bir risk faktörü olarak değerlendirilmesi mevcut
çalışmalardaki çelişik sonuçları aşabilir.
APOE-ε4 yükü ile kafa travmasının AH’nin başlangıç
yaşı üzerine birlikte etkisi olduğunu ileri süren
çalışmalar mevcuttur. Bir çalışmada ise tek
başına etkisiz görünürken, normalde iki misli artmış olan
APOE-ε4 heterozigotlarının riskini 10 misline
arttırdığı ortaya konmuştur. Kafa travmasının
etkisinin β-amiloid metabolizması üzerinden olması muhtemel
görünmektedir. Dementia pugilistica geliştiren boksörlerin otopsilerinde
saptanan gevşek neokortikal amiloid plaklar bu varsayımı
destekler niteliktedir.
Öz geçmişte 10 yıla kadar geriye giden
tedavi gerektirmiş bir depresyon öyküsü AH riskini 3 kat
arttırır. Depresyonun hastalığın erken bir
gösterisi olduğunu iddia eden çalışmalar vardır. Jorm
tarafından yapılan 2001 tarihli meta-analizde
yaşlılıkta depresyon özgeçmişinin demans geliştirme
riskini ikiye katladığı bildirilmiştir.
Kısıtlı sosyal ilişkiler
Kungsholmen, İsveç ve Kuzey Manhattan, ABD gibi önemli toplum temelli
kesitsel ve uzunlamasına gözlem çalışmalarında
bağımsız bir risk faktörü olarak ortaya konmuştur. Sosyal
ilişkiler akraba ve arkadaş ilişkilerinin zenginliği,
sinema, lokanta, klüp, dernek faaliyetleri amaçlı sokağa
çıkmalar olarak özetlenebilir. Honolulu-Asya Yaşlanma
Çalışması’nda bu risk orta yaşlarından itibaren sosyal
aktiviteleri düşen bireylerle sınırlı bulunmuştur.
Vasküler olaylar arasında miyokard iskemisi,
hipertansiyon, diyabet ve inme öyküsü ile metabolik sendrom mevcudiyetinin AH
riskini anlamlı biçimde arttırdığı bir çok
çalışmada gösterilmiştir. Bu bulgular o denli güçlüdür ki,
daha yakın zamanlara kadar 2. sıklıkta demans nedeni olarak
gösterilen vasküler demansın AH’den bağımsız bir antite
olarak varlığı bile son yıllarda tartışma konusu
olmuştur. Aynı zamanda bir inme risk faktörü olan yüksek plazma
homosistein düzeyinin AH için de bağımsız bir risk faktörü
olduğu bildirilmiştir.
Küçük kafa çevresi ile AH arasında bazı
çalışmalarda gösterilen ilişki, eğitim ile olan
ilişkide olduğu gibi kognitif rezerv teorisiyle açıklanmaya
çalışılmaktadır.
Hipotirodinin gerek genel olarak demans ve gerekse
de AH ile ilişkisi ortaya konmuştur. AH ile ilişkisinin
mekanizması aşikar olmasa da potansiyel olarak düzeltilebilir bir
durum olması bakımından önemlidir.
Koruyucu faktörler
Epidemiyolojik çalışmalar bir dizi
faktörle AH arasında, bu faktörlerin “koruyucu” olabileceğini
düşündürecek şekilde ters bir ilişki göstermiştir.
Düşük eğitim düzeyi AH için bir riskken,
doğal olarak yüksek eğitim düzeyi ve profesyonel aktivite bir
koruyucu olarak ortaya çıkmaktadır. Benzer biçimde APOE-ε4
aleli risk iken ε2 aleli bazı çalışmalarda koruyucu gibi
görünmekteyse de ε2’nin bu olumlu rolü, ε4’ün olumsuz rolü kadar
sağlam bir bulgu olarak yerleşmemiştir. Birinci durum
yukarda sözedilen “kognitif rezerv” kavramı ile açıklanırken, 2.
durumda ε2’nin β-amiloid fragmanının temizlenmesinde etkin
bir alternatifi temsil ettiği düşünülmektedir.
Özellikle dejeneratif hastalıklarda oksidatif
gerilim ve eksitotoksisite mekanizmalarının açıklayıcı
olarak devreye girmesinden sonra, bu mekanizmaları tersine çevirdiği
iddia edilen terapötik ajanların popülaritesi de
artmıştır. Anti-oksidanlar arasında özellikle de E
vitamininin koruyucu etkisi üzerinde durulmuş, Rotterdam ve Kanada
Yaşlanma gibi önemli çalışmalarda C vitaminiyle birlikte bir
koruyucu etkileri varmış gibi görünse de Washington Heights-Inwood
Columbia Üniversitesi Yaşlanma Çalışması ve Honolulu
Çalışması gibi diğer önemli çalışmalarda ise bir
etki gösterilememiştir. Önceden Sano ve arkadaşlarının
(bir klinik çalışmasında (1997) 2000 IU/gün dozunda E vitamininin
AH’lilerin hastalık seyirlerini yavaşlattığının
gösterilmesiyle AH tedavisinin bir parçası olarak önerilmekte olan E
vitaminin popülaritesi daha yakın tarihli bir çalışmada
(Petersen ve ark. 2005) MCI’dan demansa dönüşmeyi engelleyemediğinin
görülmesiyle kaybolmuş gibi durmaktadır.
Anti-inflamatuar ajan kullananlar arasında AH
sıklığının daha az olduğu bir çok gözlemsel
çalışmada gösterilmiştir. Bu bulgu, söz konusu
ajanların AH’deki inflamatuar süreci tersine çevirmeleri şeklinde
yorumlanmıştır. Ancak, özgül olarak sağlıklı
yaşlılarda demans gelişimini engellemek veya AH’lilerde
hastalık seyrini yavaşlatmak amaçlı tasarlanan anti-inflamatuar
klinik çalışmaları başarılı olamamıştır.
Östrojen kullanan post-menapozal kadınlarda AH’nin daha az görülmesi,
östrojenin koruyuculuğu üzerine yoğun bir tartışma
başlatmıştır. Ancak, Kadın
Sağlığı İnisiyatifi çalışmasında
yaşlı kadınlarda östrojen replasmanının jinekolojik
kanserlerde artmanın yanısıra, ayrıca demans riskini
düşürmek bir yana tam tersine arttırdığı
bulunmuştur. Yine, kolesterol düşürücü ilaçlar, özellikle de statin
grubu kullananlarda AH prevalansı kullanmayanlara göre daha
düşüktür. Son zamanlarda bu etkinin kolesterol düşürücü etkinin
ötesinde, statinlerin doğrudan α-sekretaz proteolitik yolunu
uyararak, sAPP oluşumunu arttırmalarıyla ilintili olduğuna
ilişkin deneysel deliller bulunmuştur. Yine de, AH’lilerde
klinik çalışmalarda sınanan statin tedavisinin faydası
gösterilememiştir. Anti-hipertansif ve anti-diabetiklerin vasküler risk
faktörlerinin kontrolü aracılığıyla demansa karşı
koruyucu olabileceği sağduyuyla da çıkarımlanabilir.
Nitekim, bu öngörü anti-hipertansifler açısından bir kaç prospektif
çalışmada doğrulanmıştır. İnsülin degrade
edici enzimin (IDE) Aβ klirensinde bir şaperon olarak işlev
gördüğü bilinmektedir. İnsuline dirençli tip II diyabetiklerde, IDE
beyinde işlev görmek yerine periferde artan insülini parçalamak için
kullanılmaktadır. Dolayısıyla, özgül olarak insülin
direncini düşürerek işlev gören peroksizom proliferatörü ile aktive
reseptör gamma (PPAR-γ) agonistleri sınıfı
anti-diyabetikler vasküler risk kontrolü yanısıra potansiyel olarak
özgül anti-Alzheimer etki de gösterebilirler.
Birçok çalışmada hafif-orta alkol
tüketiminin koruyuculuğu bildirilmiş, giderek bu etkinin bir U
eğrisi sergilediği yüksek tüketimde kaybolduğu ileri
sürülmüştür. Alkollü içkiler içinde özellikle kırmızı
şarap üzerinde durulmaktadır. Kırmızı
şarabın içerdiği polifenoller ve özellikle de bir uzun ömür
faktörü olarak Aβ’nın etkilerini tersine çeviren sirtuin
sinyallemesinde rol oynayan resveratrol son zamanların ilgi
odağıdır.
Akdeniz diyeti bir koruyucu faktör olarak son
yıllardaki en popüler çevresel faktör araştırma
konularından biridir. Akdeniz diyeti (MeDi) ile tahıllar, sebzeler,
meyvalar, peynir, süt, özellikle balık, zeytinyağı ve
kırmızı şaraptan zengin bir diyet kastedilmektedir.
Columbia Üniversitesi MeDi Çalışması, New York’ta MeDi uyumuna
göre düşük, orta ve yüksek olarak
sınıflandırdığı 1393 normal
yaşlının MCI ve 482 MCI’lının demans geliştirme
sonlanma noktalarında yüksek uyum grubunun düşük uyum grubuna göre
avantajlı olduğunu göstermiştir. Rotterdam ve Chicago
Sağlık ve Yaşlanma Projesi gibi 2 büyük çalışmada
balık tüketiminin daha yavaş kognitif bozulmayla ilişkisi
gösterilmiştir.
Son olarak, zihinsel ve fiziksel aktivitenin
koruyuculuğundan sözedilebilir. Osteoporotik Fraktürler
Çalışması’nda uzunlamasına izlenen 6000 65 yaş üzeri
kadın arasında en fazla yürüyen üst çeyrek, en az yürüyen alt
çeyrekle karşılaştırıldığında, 6-8
yıl sonra daha az kognitif kayıp sergilemekteydiler. Colcombe ve
Kramer’in meta-analizinde (2003) egzersize tabi tutulan sedanter
yaşlıların tutulmayanlarla karşılaştırıldığında
kısa süreli kognitif düzelme sergiledikleri ortaya konmuştur.
Fiziksel aktivite bir yandan vasküler riskin kontrolu
aracılığıyla, diğer yandan da eğitim için
olduğu gibi zengin uyarana maruz kalmanın kognitif rezervi
arttırması yoluyla etki ediyor olabilir. Çok sayıda
çalışma, okumak, sanatsal faaliyette bulunmak, oyun oynamak gibi
zihinsel aktivite gösteren yaşlıların AH geliştirme
risklerinin daha düşük olduğunu ileri sürmektedir. Wilson ve
arkadaşları Chicago’da ortalama 5.3 yıl izledikleri 4000
yaşlı arasında, kognitif olarak stimüle edici faaliyetlere daha
sık katılan yaşlıların daha düşük ihtimalle
kognitif bozulma sergilediklerini gösterdiler. Zihinsel aktivite de daha zengin
kognitif rezerv oluşturarak etki ediyor olabilir.
Tablo 13. Alzheimer
Hastalığı’nda Rol Oynayan Faktörler
|
Risk faktörleri |
Koruyucu faktörler |
|
· Yaş · Kadın cinsiyet · Düşük eğitim · Ailede demans öyküsü · Genetik etkenler (APOE-ε4) · Bilinç kayıplı kafa
travması · Down sendromu · Majör depresyon öyküsü · Vasküler olaylar · Plazma homosistein düzeyi · Küçük kafa çevresi · Hipotiroidi · Bazı toksik ve zararlı
durumlara maruz kalma |
·
Yüksek eğitim · APOE-ε2 · Anti-oksidan
kullanımı (?) ·
Anti-inflammatuar kullanımı (?) · Östrojen
kullanımı (?) · Statin
kullanımı (?) ·
Kırmızı şarap · Akdeniz diyeti · Fiziksel ve
zihinsel aktivite |
Genetik
AH’de genetik faktörler büyük oranda
hastalığın gelişimi için çevresel faktörlere bir
yatkınlık zemini yaratacak şekilde birer risk faktörü niteliğindedirler.
AH bu özelliğiyle Huntington hastalığında olduğu gibi
Mendelyen yasaların etkisinde basit genetik belirlenimli değil
de, genetik ve çevresel faktörlerin karmaşık bir etkileşimi
sonucu ortaya çıkan kompleks nörolojik hastalıklardandır.
Yine de, tüm AH olgularının uluslararası literatürde %5’ine
yaklaşan bir oranı basit Mendelyen otozomal dominant geçişle
hastalığa yakalanırlar.
Genetik açıdan genel olarak kompleks
nörolojik bir hastalık olarak nitelenmesinin yanısıra, AH
Mendelyen geçiş açısından da heterojenite gösteren
polijenik/multialelik bir hastalıktır: birden fazla kromozomdaki gen
lokuslarının çok sayıda farklı mutasyonları aynı
hastalığa yolaçar. Otozomal dominant (OD) geçişten sorumlu
olan şimdiye kadar 3 ayrı gen bulunmuştur: amiloid prekürsör
protein (APP) geni (APP, 21. kromozom), presenilin 1 geni (PSEN1, 14.
kromozom) ve presenilin 2 geni (PSEN2, 1. kromozom). Bu
genlerin kodladığı 3 protein de normal işlevleri çok iyi
bilinmeyen, nöronal plastisitede rol oynadıkları yönünde
varsayımlar ileri sürülen transmembran proteinlerdir. Anılan
genlerdeki mutasyonlar her durumda APP proteolizini Aβ
fragmanının üretiminin artışına neden olacak yola
kaydırırlar. APP mutasyonu taşıyan aileler çok
az sayıda olsa da, bu mutasyonlar bir yandan amiloid metabolizması
üzerinden hastalığın patogenezine ışık tutarken,
bir yandan da Down sendromu ile AH arasındaki ilişkiyi de
aydınlatmıştır. Bilindiği gibi Down sendromlular
21. kromozomun 3 kopyasını taşırlar (21 trizomisi).
Dolayısıyla, ömürleri boyunca APP proteolizinden üretilecek daha
fazla Aβ’yı temizlemek zorundadırlar. Bu hastalar 30’lu
yaşlardan itibaren hemen daima AH’nin nöropatolojik
değişikliklerini göstermeye başlarlar.
OD-AH’liler tipik olarak erken
başlangıçlı (60 yaş öncesi) hastalığa
sahiptirler. OD-AH ile ilişkilendirilen ilk mutasyon John Hardy
tarafından 1991 yılında bildirilen APP’nin
yanlış anlamlı (missense) bir mutasyonudur (V717I).
Enteresan biçimde, aynı gendeki bir başka mutasyon bir yıl önce
Hollandalı tipi amiloidozlu serebral hemoraji ile
ilişkilendirilmişti (G693Q). Biriken veriler serebral amiloid
anjiyopatiye neden olan mutasyonların APP’nin β-amiloid
bölgesinin içinde kümelenirken, OD-AH’ye neden olanların biri
dışında tümünün γ-sekretaz
kesim bölgesini tutarak Aβ42
üretimini arttırdıkları, bir mutasyonun ise β-sekretaz
kesim bölgesinde hem Aβ40, hem
de Aβ42 üretimini
arttırdığını göstermektedir. St. George Hyslop
1995 yılında PSEN1 genini klonlayarak bu gendeki AH’ye neden
olan mutasyonu tanımladı. Bu genin kodladığı
Presenilin 1 proteininin (PS1) APP’nin γ-sekretaz
proteolizine ve aynı zamanda embriyogenez sırasında nöral
gelişimde temel sinyalleme sistemi olan NOTCH sinyalizasyonuna
aracılık eden bir transmembran protein olduğu
anlaşıldı. Aynı yıl içinde bu kez Schellenberg grubu
tarafından PS1 ile %80 homoloji taşıyan Presenilin 2 (PS2)
proteinini kodlayan PSEN2 geninde OD-AH’ye neden olan mutasyon
tanımlandı. Günümüze kadar 76 ailede 28 ayrı APP, 361
ailede 164 ayrı PSEN1 ve 18 ailede 10 ayrı PSEN2 mutasyonu
bildirilmiştir. Her 2 gendeki mutasyonlar da γ-sekretaz
aktivitesini etkileyerek Aβ’nın daha toksik formu olan Aβ42
üretiminin artmasına neden olmaktadırlar. OH-AH aileleri genç
yaşta ortaya çıkmaları (PS1, 25-60 yaş; APP,
40-65 yaş; PSEN2, 45-84 yaş) yanısıra, bellek
dışı bir çekirdek sendromla ve/veya erken davranışsal
özellikler, miyoklonus ve nöbetler şeklinde atipik klinik tablolarla da
çıkabilirler. AH’nin ilk bildirilen olgusunun erken
başlangıçlı, davranışsal belirtilerin ön planda olduğu
bir tabloya sahip olduğu hatırlandığında Auguste
D.’nin böylesi bir genetik varyasyona sahip olması ihtimali üzerinde
durulmakla birlikte bu olgunun saklanan dokusundan yakın tarihlerde
yapılan bir çalışmada PSEN1 mutasyonu
taşımadığı ve APOE genotipinin ε3/ε3
olduğu bulunmuştur. Genel olarak PSEN1 aileleri APP ailelerine
göre daha atipik ve daha kısa süreli ve ağır demans
sergilerlerken, PSEN2 aileleri daha fazla sporadik AH’ye benzeyen ve
daha yavaş seyirli hastalık tablolarına sahiptirler.
Şimdiye kadar tanımlanan
mutasyonlar familyal AH’nin %50’si kadarını oluştururlar ve PSEN2’nin
tanımlanması üzerinden geçen 15 yıla ve yoğun
araştırmalara rağmen yeni bir Mendelyen geçiş geninin
bulunamaması dikkat çekicidir. Anılan 3 genle
bağlantılandırılamayan AH aileleri olduğuna göre henüz
bulunmayan genler ve otozomal dominant olmayan geçiş biçimleri ileri
sürülebilir. Özellikle henüz hiç otozomal resesif geçiş
bildirilmemiş olan bu hastalıkta akraba evliliklerinin sık
görüldüğü ülkemiz böylesi bir buluş için potansiyel
taşımaktadır.
ApoE proteinini kodlayan gen (APOE,
19. kromozom) mutasyonlarıyla doğrudan belirleyici değil fakat
polimorfizmiyle sporadik AH’de risk faktörü olarak ortaya konmuştur.
Kolesterolün taşınmasında rol oynayan bir protein olan ApoE,
toplam 299 rezidüsünün ikisinde arjinin ve/veya sistein bulunmasıyla
farklılaşan üç ayrı alelik forma sahiptir: ε2, ε3 ve ε4.
Kuzey Amerika ve Kuzey Avrupa’dan normal popülasyonlarda en sık ε3
(%70-80) görülürken, ε4’ün
sıklığı %10-20, ε2’ninki ise
%5-10’dur. AH’de ε4 sıklığı
ikiye katlanarak %40-50’ye ulaşır. ε4 AH
riskini doza bağlı bir şekilde arttırır ve
hastalık başlangıç yaşını azaltır.
AlzGene veritabanında (2007) beyazlar ve Japonlarda ε4
heterozigotlarında AH riski, ε3
homozigotlarına göre ihtimaliyet oranları (OR’ler)
sırasıyla 3 ila 4 olacak şekilde artmıştır. ε4
homozigotlarında hastalık heterozigotlarına göre daha erken
başlar ve risk, OR’ler aynı gruplarda bu kez sırasıyla 11.8
ve 21.8 olacak şekilde artar. Ayrıca, OR-AH’de de ε4 varlığı
hastalık başlangıç yaşını erkene
alır. Bir yatkınlık geni olan ApoE’nin ε4’ünün
varlığı hastalık başlangıcı için ne zorunlu
ne de yeterlidir. Bununla birlikte ε4
homozigotlarının yaşam boyu kümülatif risklerinin %90
olduğu hesaplanmıştır. ε4
sıklığı ve patolojik rolü için coğrafi
farklılıklar ileri sürülmüştür. Bizim de
katıldığımız bir görüşe göre Avrupa’da kuzeyden
güneye sıklığı azalacak şekilde bir gradyan mevcuttur.
Örneğin, ε4 sıklığı
normal popülasyonda kuzeyde Finlandiya’da %24 bulunurken, güneyde Yunanistan’da
%5.6 gibi küçük bir orana kadar inmektedir. Biz TAPS
çalışmasının olgu-kontrollü kısmında ε4
sıklığını normal kontrollerde %8.2, buna
karşılık AH grubunda %14.7 bulduk. ε4 OR,
2.21 gibi AlzGene’de bildirilenlerden daha düşük olmakla birlikte yine de
anlamlı bulundu. ApoE alel sıklığını dünya
çapında bir meta-analizde ele alan Corbo ve Scacchi (1999) bu gradyan ve
ε4’ün bir AH riski oluşturmasında gözlenen etnik ve coğrafi
farklılıklar için akla yakın bir açıklamada bulundular:
ε3, Akdeniz Havzası gibi kadim bir tarım
uygarlığına sahip insan topluluklarında en sık alel
gibi durmaktadır (%84.9-%89.8). Ataların aleli denebilecek olan
ε4, Pigmeler (%40.7), Bantular (%37.0), Malezya ve Avustralya aborjinleri
(%24.0 ve %26.0), Papualılar (%36.8) ve Lapplar (%31.0) gibi
avcı-toplayıcı ekonominin hala mevcut, besin
kaynaklarının göreli olarak kısıtlı olduğu
halklarda sık olsa da başta Sahra-altı halklarda olmak üzere bir
AH riski değildir. Yazarlar, ε4’ün Batılı diyet, daha uzun
yaşam süreleri gibi çağdaş çevresel koşullara
maruziyetinin, bazı işlevsel özelliklerine dayanarak onu AH için bir
yatkınlık aleli haline dönüştürmüş olabileceğini ileri
sürdüler. ε4’ün hastalığın patogenezindeki rolü tam
anlamıyla ortaya konamamış olsa da, gerek Aβ
klirensinde ve nöronal plastisitede rejenerasyon ve onarımda rol oynayan
APOE’nin, bu rolünde ε4’ün, ε2 ve ε3’e
göre daha başarısız olduğuna ilişkin deliller
vardır. ApoE nakavt farelerde sinaptik yoğunlukta
yaşa bağımlı bir azalma görülür. Kafa travması,
serebro-vasküler hastalık gibi beyin hasarlarından sonra ε4
taşıyıcıları daha kötü prognoz
gösterirler.
Şimdiye kadar incelenen APOE-ε4
dışı hiç bir polimorfizmin etki büyüklüğü ε4
OR’lerine yaklaşamamıştır. Bertram ve
arkadaşlarının AlzGene veritabanını kullanarak
yaptıkları bir meta-analizde (2007) bağlantı analiziyle o
zamana kadar 69 gende bildirilen 127 polimorfizm incelenerek 13 gendeki 20
polimorfizm pozitif olarak değerlendirilmiş ve ortalama risk OR’leri
1.25, ortalama koruyucu OR’leri 0.85 olarak bulunmuştur. Bu 20 polimorfizm
arasından özellikle anjiyo-tensin converting enzim (ACE), GRB2
asosiye bağlayıcı protein 2 (GAB2) ve transferrin (TF)
genlerindeki polimorfizmler küçük etkilerine rağmen en az 10
çalışmada doğrulanmış göreli olarak büyük örneklem
sayılarıyla dikkate değer ek risk faktörleri olabilirler. Son
zamanlarda üstünde durulan başka bir yatkınlık geni de sortilin
ilişkili reseptör genidir (SORL1). SORL1 APP’nin hücre yüzeyinden
Golgi-endoplazmik retikulum kompleksine naklinde rol oynadığı
bilinmektedir. SORL1 ifadesinin azalması Aβ
artışına neden olmaktadır. Lee ve
arkadaşlarının gözden geçirdiği üzere büyük örneklemlerdeki
alelik asosiasyon çalışmasında 1.4 ila 2.2 arasında OR’ler
saptanmıştır. Genom Boyu Asosiasyon Çalışması
(GWAS) bağlantı analizinden farklı olarak bir genetik lokus ile
hastalığın ilişkisini önceden teorik olarak varsaymak
gerekmeksizin, binlerce hastalık büyük hasta örneklemlerini
kontrollerle tüm genomun analizi aracılığıyla
karşılaştırıp hastalık grubunda anlamlı olarak
daha sık görülen varyasyonları ortaya koymaya dayanıyor. 2009
yılında tamamlanan 2 büyük GWAS çalışmasının
ilkinde 5000’i aşkın hasta ve 10000’i aşkın kontrolün
analizinde Harold ve arkadaşları clusterin (önceden APOJ
olarak bilinen) geni (CLU) ve 11. kromozomda bulunan
phosphatidylinositol bağlayan clathrin asamble proteini geni (PICALM)
ile anlamlı ilişkiler bildirdiler. Nature Genetics’in aynı
sayısında yayınlanan 2. GWAS çalışmasında
5000’den fazla hasta ve kontrol bireylerinde Lambert ve arkadaşları
8. kromozomda bulunan CLU’daki lokusu doğrularken, bu kez 1.
kromozomdaki kompleman bileşeni reseptörü 1 geninde (CR1) bir lokus
daha gösterdiler. Bu 3 genin kodladığı proteinlerin de Aβ
klirensinde rol oynadığı düşünülmektedir. Öyle görünüyor
ki, her şeye rağmen yeni yatkınlık genlerinin
araştırılması ve bulunanların doğrulanmasına
yönelik yoğun çabalar sürecektir. Bu bölümün bir özeti için Tablo 14’e
bakınız.
Tablo 14. Alzheimer
hastalığının genetiği
|
Gen
ve Proteini |
Kromozom |
Mekanizma |
Etki |
Risk |
|
APP – Amiloid prekürsör protein |
21 |
Mutasyon/ Duplikasyon |
Aβ40 ve Aβ42 üretiminde artış |
Tam penetrans |
|
PSEN1 – Presenilin 1 |
14 |
Mutasyon |
Aβ42 üretiminde artış |
Tam penetrans |
|
PSEN2 – Presenilin 2 |
1 |
Mutasyon |
Aβ42 üretiminde artış |
Tam penetrans |
|
APOE – Apolipoprotein E |
19 |
Polimorfizm ε2, ε3, ε4 |
Aβ klirensinde bozulma |
ε4: 3.68 (3.30-4.11) ε3: 0.62 (0.45-0.85) |
|
ACE – Anjiotensin I converting enzim |
17 |
Polimorfizm C, T |
Aβ klirensinde bozulma |
0.83 (0.72-0.95) |
|
GAB2 – GRB2 asosiye bağlayıcı protein 2 |
11 |
Polimorfizm T, G |
Aβ klirensinde bozulma |
0.85 (0.76-0.94) |
|
TF – Transferrin |
3 |
Polimorfizm C2, C1 |
Aβ klirensinde bozulma |
1.18 (1.06-1.31) |
|
SORL1 – Sortilin ilişkili reseptör |
11 |
Polimorfizm T, C |
Aβ üretiminde artma |
1.10 (1.03-1.17) |
|
CLU – Clusterin |
8 |
Polimorfizm T, C |
Aβ klirensinde bozulma |
0.86 (0.82-0.89) |
|
PICALM – Phosphatydinilinositol bağlayıcı klatrin asamble
proteini |
11 |
Polimorfizm C, T |
Aβ klirensinde bozulma |
0.87 (0.83-0.91) |
|
CR1 – Kompleman bileşeni (3b/4b) reseptörü 1 |
1 |
Polimorfizm T, C |
Aβ klirensinde bozulma |
1.18 (1.09-1.28) |
Alzheimer
hastalığının otozomal dominan geçişli
formlarından sorumlu 3 gen ile yatkınlık geni APOE yanısıra
2010 Nisan ayı itibariyle AlzGene veritabanındaki (www.alzgene.org) pozitif
çalışmalar arasında en az 10 çalışma ve 10000’e
yaklaşan örneklem büyüklüklerine sahip olan aday yatkınlık
genleri (ACE, GAB2, TF, SORL1) ile genom boyu asosiasyon
çalışmalarında yakın tarihlerde ortaya konulan aday
yatkınlık genleri (CLU, PICALM, CR1) gösterilmiştir.
Risk, ihtimaliyet oranları (OR’ler) ve parantez içlerinde %95 güvenilirlik
aralıkları ile gösterilmiştir.
NÖROPATOLOJİ
AH’nin klinik tanıda başvurulabilecek
patognomik bir işaretleyicisi olmadığı gibi patolojik
tanısı da benzer bir güçlük taşır. Pick
cisimciklerinin varlığı PiH kesin tanısı için gerekli
ve yeterli koşuldur. Oysa ki, AH kesin tanısı için
patolojik olarak nörofibriler yumaklar (NFY) ve senil ya da amiloid
plakların (AP) saptanması gerekli ancak yeterli değildir.
Her iki lezyon da normal yaşlanmada olduğu gibi bir dizi başka
dejeneratif hastalıkda da görülebilir. AH’nin kesin tanısı
için patolog bu iki lezyonun varlığını saptamanın
yanısıra belli bir nöroanatomik dağılımda ve
belli miktarlarda olduklarını da göstermelidir.
İlerde demansa dönüşecek olsun ya da
olmasın normal insanlar 60 yaşlarından itibaren neokortikal
gevşek plaklar ve bazen limbik NFY’ler geliştirmeye
başlarlar. Ancak titiz kliniko-anatomik korelasyon
çalışmaları NFY’lerin neokorteks, AP’lerin ise nöritik veya sert
plak biçimiyle limbik sistemde görünür olmalarının AH için %100’e
yaklaşan duyarlılık ve özgüllük gösterdiğini ortaya
koymaktadır. Aşağıda daha iyi görüleceği gibi,
NFY’lerin limbik sistemde birikip neokortekse geçişi ve giderek
neokortekste yaygınlık göstermeye başlaması, normal yaşlanma-MCI-giderek
ağırlaşan demans devamlılığının
patolojik korelatı gibi durmaktadır. Yine klinik evreyle
sayısal ve bölgesel açıdan korelasyon göstermemekle birlikte
gevşekten nöritike doğru biçimini değiştirip limbik
sistemde görülür hale gelen AP hemen daima AH’yi MCI’dan ayıran özelliktir
denebilir.
NFY ve AP’lerin yanısıra
gliozis-inflamasyon, nöron ve sinaps kayıpları, aksonal ve dendritik
morfolojik değişiklikler, kortikal kolinerjik innervasyon ve
diğer nörotransmitter sistemlerinde kayıplar AH nöropatolojisinin bileşenlerini
oluştururlar (Tablo 15).
Tablo 15. AH Nöropatolojisi
|
Nörofibriler
yumaklar (NFY) Amiloid plaklar (AP) Nöron kaybı Dendritik ve aksonal
değişiklikler Sinaps kaybı Gliozis - inflamasyon Kolinerjik innervasyonun
kaybı Diğer nörotransmitter
kayıpları |
Nörofibriler yumaklar
NFY’lerin (Şekil 10) temel
bileşeni hiperfosforile t (tau) proteinidir. Tau 17. kromozom
tarafından kodlanan mikrotübül asosiye proteinler (MAP) ailesinden bir
proteindir. Mikrotübüller özellikle aksonlarda yerleşir.
Mikrotübüllerin stabilizasyonu, sitoskeletal bütünlük ve kargo veziküllerinin
aksonal nakliyesinde önemli roller oynar. Kargo vezikülleri eskiyen
yapıların replasmanı ve sinaptik aktivite için gerekli protein
ve enzimleri içerir. Mikrotübüller tau-tubulin etkileşimi ile stabilize
olabilen labil yapılardır. Bu etkileşim tau proteininin
izoformuna ve fosforilizasyon durumuna bağlıdır. Tau proteini
karboksi ucunda mikrotübüllere bağlanma bölgesi sayısına göre
üçü 3 tekrar (3R), üçü ise dört tekrar (4R) taşıyan toplam 6 izoforma
sahiptir. 4R tau mikrotübül stabilizasyonunda daha etkindir. İkincil
mesajcıların uyarımı post-sinaptik nörondaki tüm
intraselüler proteinleri olduğu gibi tauyu da protein kinazların
aktivitesiyle fosforile eder. Fosforile tau mikrotübülden ayrılır. Yine
bütün fosforile intraselüler proteinler gibi tau da işlev bittikten sonra
fosfatazlar aracılığıyla defosforile edilmelidir ki yeniden
mikrotübüle bağlanabilsin. Fosforile taunun defosforilasyonundan
başlıca sorumlu fosfataz protein fosfataz 2A’dır (PP2A). AH
patogenezinde hiperaktif kinazlar ve/veya hipoaktif fosfatazlar tau proteininin
hiperfosforilizasyonuna yolaçarak mikrotübüllere bağlanma yeteneğini
bozarlar. Bu kinazlar arasında özellikle glikojen sentetaz kinaz 3β’nın (GSK-3β)
yalnız AH’de değil, diğer taupatilerde de patolojik
hiperfosforilasyondan sorumlu olan enzim olduğu üzerinde
durulmaktadır. Bağlanmamış hiperfosforile tau monomerleri
fiziksel konformasyonlarını değiştirerek kendi üzerlerine
katlanırlar (patolojik hatalı katlanma) ve toksik oligomerlere
dönüşürler. Tau oligomerleri bir sonraki aşamada yine toksik
çözülemeyen çift sarmallı filamanlara (PHF) polimerize olurlar.
PHF’ler ısı şoku proteinleri ve başlıca ubikutin
proteozom sistemlerinin devreye girmesiyle giderek intranöronal NFY’ler
şeklinde kondanse olurlar (Şekil 11). NFY’lerin toksik
PHF’lerin etkisini sınırlamaya çalışan bir savunma
mekanizması olduğu giderek geçerlilik kazanan bir görüştür.
Yetersiz bir savunma olan NFY ile sonunda hücre iskeleti bütünlüğü ve
aksonal nakliye bozularak hücre ölümü meydana gelir. Hücre ölümüyle
ortaya çıkan ekstraselüler NFY’ye “hayalet yumak” adı verilir. NFY’den hayalet
yumağa geçişin yılları kapsayan uzun bir süreç olduğu
düşünülmektedir.
Şekil 10. İntraselüler
Nörofibriler Yumak (Kaynak: http://www-edlib.med.utah.edu/WebPath/CNSHTML/CNS094.html)
NFY’ler beyinde rasgele değil
fakat belli bir bölgesel yatkınlığı yansıtacak şekilde
yerleşirler. Normal yaşlılık sürecinde limbik
(entorhinal ve hippokampal) NFY sayısıyla kronolojik yaş
arasında korelasyon gösterilmiştir. Braak ve Braak’ın I ve II.
evrelerine karşılık gelen bu aşamanın
sağlıklı yaşlanmadan yaşla ilintili unutkanlığa
(AAMI) geçişin nöral altyapısı olduğu ileri sürülebilir. En
erken değişiklikler entorhinal korteksin (ECx) II. moleküler
tabakasında oluşmaktadır. ECx limbik sistemin dış
dünyayla arayüzü olarak değerlendirilebilir. Hatırlanmak üzere
kaydedilmesi gereken her türlü yeni bilgi limbik sisteme ECx II
aracılığıyla girer ve bu tabakadaki nöronların
aksonları perforan yolu oluşturarak ECx’i hippokampal formasyonun
giriş kapısı denilebilecek olan dentat girusunun (DG) granüler
tabakasına bağlarlar. DG epizodik (otobiyografik) bellek sistemindeki
bilgi işlemenin nöral altyapısı olan 2 kapalı devrenin
yolaklarını taşır: lokal intrensek hippokampal devre ve
geniş boyutlu Papez devresi (Şekil 12). Her iki devre de
nihayetinde subikulum aracılığıyla ECx alt tabakalarına
(IV-VI) bağlanarak kapanacak ve ECx IV-VI işlenmiş limbik
bilgiyi multimodal asosiasyon kortekslerine geri bildirebilecektir.
İntrensek hippokampal devre içinde DG yosunsu lifler
aracılığıyla Ammon boynuzunun 3. sektörünün (CA3) piramidal
tabakasına bağlanır. Shaffer kollateralleri bu kez CA3’ü CA1’e
bağlarlar. CA1 ise subikuluma bağlanır. Subikulum hippokampal
formasyonun bu kez çıkış kapısı konumundadır ve
yukarda değinildiği gibi her 2 devrenin de sonlandığı
yapıdır. Papez devresi ise DG’den fimbria-fornix
aracılığıyla hipotalamusun mamiller cisimciklerine (CM)
olan bağlantıyla başlar. CM, mamillo-talamik traktus (MTT)
aracığıyla talamusun limbik çekirdekleri de denilen anterior
grup çekirdeklerine bağlanır. Talamus anterior grubu posterior
singulat girusla (pCG), pCG ise retrosplenial girusla (rSG)
bağlantılıdır. rSG’den subikuluma olan
bağlantılarla Papez devresi kapanır. AH’de nörodejenerasyonun
ağırlığı ve süresiyle paralel biçimde NFY’nin önceden
görüldüğü bir bölgedeki sayısı artarken o bölgeyle
bağlantılı olan komşu bölgeye transsinaptik olarak
yayılır. Dolayısıyla, ECx II dejenerasyonuyla DG
diskoneksiyonu ve bunun sonucunda meydana gelen ve sadece CA3’ün sahip
olduğu ve girdilerinin %95’ini olıuşturan oto-asosiyatif
liflerince sağlanan kompansatuar CA3 aşırı aktivitesinin
sonucu olan değişerek reorganize olan intrensek hippokampal devre
AAMI’daki bellek bozukluğunun nöral temeli olabilir. Braak ve Braak, I’den
VI’ya sıraladıkları evreler boyunca NFY’lerin entorhinal
kortekste görülmeye başlayıp önce hippokampus ve sonra da paralimbik
alanlara, sonunda da heteromodal neokortikal alanlara yayılıp
sayılarının artışını izleyen bir evreleme
sistemi geliştirdiler. Mesulam, Braak ve Braak sistemine paralel bir
klinokopatolojik korelasyon evrelemesi önererek (Şekil 13) NFY
yayılımını normal yaşlanmadan MCI, hafif ve
ağır demans evreleri sürekliliği içinde izler. Bu evreler
boyunca intrensek hippokampal devrenin ve Papez devresi
yapılarının giderek daha fazla NFY ile yüklendikleri ilerleyici
süreç AH’nin AAMI-MCI-demans sürekliliği boyunca merkezi ilerleyici bellek
bozukluğu fenotipinin de açıklamasıdır.
AH’nin yerleşik tedavisi içinde
yeralan ajanlardan biri olan memantinin in vitro olarak PP2A agonizması
yaptığına dair deliller AH’lilerle yapılan küçük bir
çalışmada çalışma sonunda BOS fosfo-tau düzeylerinde
gösterilen düşme ile desteklenmiştir. GSK3β
inhibitörleri (örneğin, lityum, valproat, thiadiazolidindionlar [TDZD’ler:
NP-13]) ve mikrotübül stabilizatörleri (örneğin, paclitaxel,
davunetide [AL-108]) erken klinik gelişim aşamasındaki anti-tau
stratejiler arasındadır. Fosfo-tau immunizasyonuna ilişkin
başarılı bir pre-klinik çalışma mevcuttur .
Şekil 11. Mikrotübül
asosiye tau proteinin NFY’ye evrimi
Üç veya dört tekrar bölgesinden mikrotübüle
bağlı olan tau proteini hiperaktif kinazlar (özellikle GSK3β)
ve/veya hipoaktif fosfatazların (özellikle PP2A) varlığında
hiperfosforile olarak mikrotübüllerden serbestler. Serbest tau patolojik olarak
katlanarak oligomerleşir. Oligomerler çift sarmallı filamanlara (PHF)
kondanse olurlar. PHF ise ısı şoku proteinleri (özellikle HSP90)
ve proteozomal ubikutinizasyon sistemi aracılığıyla
NFY’lere hapsolunurlar.
Kısaltmalar:
GSK3β: glikojen sentetaz kinaz 3β; HSP: ısı şoku
proteini; MAP: mikrotübül asosiye protein; NFY: nörofibriler yumak; P: fosfor;
PHF: çift sarmallı filaman; PP2A: protein fosfataz 2A
Şekil 12.
İntrensek hippokampal ve Papez devreleri
Multimodal
asosiasyon alanları ECx’in üst tabakaları ile ileri besleme
bağlantılarına sahiptir. ECx II moleküler tabaka perforan yol
aracılığıyla hippokampal formasyonun giriş
kapısı DG’nin granüler tabakasına bağlanır. DG fornix
aracılığıyla Papez, yosunsu lifler
aracılığıyla intrensek hippokampal devreyi
başlatır. Her 2 devre de hippokampal formasyonun çıkış
kapısı olan subikulumun üzerine döner. Subikulum ECx alt
tabakaları aracılığıyla multimodal asosiasyon
kortekslerine limbik geri besleme yapar. CA3’ün %95 girdisi kendi
oto-asosiyatif lifleri aracılığıyladır. Perforan yol
CA3’e bir kollateral verir. ECx III’den CA1 ve subikuluma doğrudan
bağlantılar mevcuttur.
Kısaltmalar:
Ant. Th: talamus anterior grup çekirdekler; CA1 ve 3: cornu ammonis
1 ve 3; CM: corpus mamillare; DG: dentat girus; ECx: entorhinal
korteks; pCG: posterior singulat girus; rSG:
retrosplenial girus.
Şekil 13. Nörofibriler yumakların zaman ve uzamsal
ilerleyişi. Kaynak 29’dan modifiye edilmiştir. + sayısı ve giderek koyulaşan renk gradyanı
artan NFY yükünü temsil etmektedir. AAMI: Yaşla ilintili bellek bozukluğu;
MCI: Hafif kognitif bozukluk.
Amiloid plaklar ve Amiloid-β
Fizyopatolojisi
AH’deki ikinci temel nöropatolojik
değişiklik olan ve NFY’lerin tersine yükleri hastalık şiddetiyle
korele etmeyen senil plakların (Şekil 14) amiloid maddelere
yüksek afiniteli Kongo kırmızısı ile
boyandığı 1927’de Divry tarafından gösterilmiş, bir
dizi farklı morfolojik yapıda olsalar da ana bileşenlerinin
amiloid beta peptidi (Aβ) olduğu 1984’te Glenner
ve Wong tarafından bildirilmiştir (yani, senil plak = amiloid
plak). Yukarda genetik bölümünde sözedildiği gibi Aβ, 21.
kromozomda kodlanan ve işlevi tam olarak
anlaşılamamışsa da bir sinaptik adhezyon proteini
olduğu düşünülen bir transmembran protein olan amiloid prekürsör
proteininin (APP) metabolizma ürünlerindendir. APP geninin
yokedildiği transjenik farelerde (APP nakavt) nöral plastisitede rolü
olabileceğini düşündüren ılımlı bulgular mevcuttur.
APP yokluğu onun gibi davranan ve “amiloid prekürsör benzeri proteinler”
adı verilen APLP1 ve APLP2 tarafından kompanse ediliyor olabilir.
Nitekim, APP/APLP2 çifte nakavtlar sağ kalamamaktadır.
Bütün transmembran proteinlerde
olduğu gibi APP’nin de bir hücreiçi karboksi ucu (CT), bir membran kateden
parçası, bir de hücredışı amino ucu (NT) vardır (Şekil
15). 770, 751 ve 695 rezidülük 3 alternatif bağlanmış
izoformu olan APP’nin intramembranöz parçası 700-723. rezidülere rastlar. β-amiloid,
671 ila 711-713. rezidüler arasında yeralan 40 veya 42 rezidülük distal
kısmı ekstraselüler, proksimal kısmı intramembranöz
yerleşmiş bir parçasıdır. APP bir dizi proteolitik
enzimle kesilerek metabolize edilir. Bu enzimlere α, β ve γ-sekretaz
adları verilir. α-sekretaz
APP’yi tam da Aβ’nın ortalarına raslayan 687. ekstraselüler
bölgede keser. Bu kesim sonunda distalde parenkime atılan çözülebilir
APP ya da sAPPα adı verilen ekstraselüler
parça oluşurken geride hala membrana bağlı 83 rezidülük
C-teminal fragmanı-α (CTF-α)
kalır. sAPPα’nın hücre kültürlerinde nörotrofik etkileri
bulunmuş ve APP nakavt transjeniklerdeki plastisite defisitlerini geri
çevirdikleri gösterilmiştir. Oysa ki, alternatif distal kesimi yapan
β-sekretaz enzimi APP’yi Aβ’yı salim bırakacak
şekilde, ekstraselüler bölgede α-sekretazın
daha distalinden 671. pozisyondan keserek akson budanması ve
dejenerasyonuna neden olduğu düşünülen distal parça sAPPβ’yı parenkime atar ve geride salim
Aβ fragmanını da içeren 99 rezidülük membrana
bağlı CTF-β’yı bırakır. Proksimal kesme ise
intramembranöz bölgede ağırlıkla 711, daha az olarak 713.
pozisyondan γ-sekretaz enzimi
aracılığıyladır. γ-sekretaz CTF-α’yı kestiğinde bu kez
parenkime Aβ’nın
budanmış ve zararsız p3 adı verilen
parçacığı atılırken, geride kalan CTF-γ’nın γ-sekretaz
tarafından biraz daha proksimalde, ε pozisyonundan
(720/721) bir kez daha kesilmesiyle nükleer sinyallemede rol
oynadığı bulunan amiloid intraselüler domeni (AICD) isimli
fragman sitoplazmaya salınır. Buna karşılık, γ-sekretaz
CTF-β’yı
kestiğinde 711 veya 713. pozisyonlardan kesilmeye göre daha
ağırlıkla Aβ40, daha
az oranda da, daha toksik formu olan Aβ42 fragmanı,
yani klirensi başarıyla sağlanamazsa amiloid plaklarda birikecek
materyel ortaya çıkar. Normal yaşlanmada α-sekretaz
aktivitesi baskınken, AH’de denge β-sekretaz
alternatifine kaymaktadır. Yine yukarda genetik bölümünde sözedildiği
gibi OD-AH özellikle Aβ42 üretimini
arttırırken başta APOE olmak üzere bütün yatkınlık
genleri Aβ klirensini etkileyerek risk oluşturmakta veya
koruyucu olmaktadırlar. α-sekretaz
aktivitesini çok sayıda transmembran proteinin proteolizinde görevli olan
bir disintegrin ve metalloproteinaz (ADAM) ailesinden bir dizi
metalloproteinazın yerine getirdiği bilinmektedir. Bunlar
arasında en aktiflerinden biri ADAM17 olarak da bilinen tümör nekrotizan
faktör-α çevirici enzim (TACE), diğeri ADAM10’dur. β-sekretazın
son yıllarda 11. kromozomdaki geni de bulunarak karakterize edilmiş
ve BACE (beta-site APP cleaving enzyme) adı verilmiştir. BACE
nakavt farelerde kayda değer bir değişiklik olmamakla birlikte
APP/PS1 gibi son derece virulan bir AH transjeniği ile
çaprazlaştırılan BACE nakavt farelerin soyunda, plak birikimi
için BACE’in temel önemde olduğunu gösterecek şekilde plak birikimi
olmamaktadır. Bu da β-sekretaz inhibisyonunu çekici
bir tedavi hedefi kılmaktadır. γ-sekretazın
ise aktif bölgesinde presenilini de içeren ayrıca nikastrin, APH-1 ve
PEN-2 isimli proteinlerden oluşan bir proteaz kompleksi olduğu
bilinmektedir. Daha önce de sözedildiği gibi PS1 aynı zamanda
nöro-gelişimde temel olan NOTCH sinyalizasyonunda da rol oynamaktadır
ve PS1 nakavt fareler ağır gelişimsel kusurlar sergileyerek
sağ kalamamaktadırlar.
Parenkimdeki Aβ
monomerleri kendi başına zararsızdır ve mikroglia
tarafından doğrudan fagositoza maruz bırakıldıkları
gibi APOE alelik formları, insülin degrade edici enzim (IDE), α2-makroglobulin
(α2-MG), neprilizin ve matriks metalloproteinaz (MMP) gibi
proteinleri içeren bir dizi “şaperon moleküle” bağlanıp,
düşük dansiteli lipoprotein reseptörü ilişkili protein (LRP-1)
aracılığıyla astrosit ve nöronlara internalize edilerek
otofaji ve proteozomal degradasyona maruz bırakılırlar. LRP1
farklı hücre tiplerinde Aβ’nın internalizasyonu için bir
kapıcı olarak düşünülebilir ve nitekim vasküler endotelinden
sirkülasyona aktarılırak temizlenmeye de aracılık eder.
Degradasyon ve klirens aracılığıyla başa
çıkılamayan miktarlardaki monomerler bir araya gelerek dimer ve
trimerler şeklinde çözülebilir Aβ
oligomerlerini oluşturacaklardır. Oligomerler parçalanarak “amiloid
betadan türeyen difüze olabilen ligandlara” (ADDL’ler) değişebilirler
veya toplanmayı sürdürerek bir sonraki aşamada artık çözülemeyen
protofibrilleri ve giderek toksik fibrilleri oluştururlar. ADDL’ler
arasında NT’de ilk 2 residüsü budanmış ve piroglutamatla
değiştirilmiş Aβ [Aβ
(3(pE)-42)] toksik potansiyeli ve birikime yatkınlığı en
fazla biçim olarak ilgi çekmeye başlamıştır. Aβ
fragmanının önce oligomerizasyonu, sonra da fibrilizasyonuna amino
ucuna yakın bir konumda bulunan ağır metal
bağlayıcı bölgelerinin aracılık ettiği
düşünülmektedir. Buna göre, Cu++ bağlanması
oligomerizasyon, Zn++ bağlanması fibrilizasyon için
gereklidir. Zn++ yanısıra metal bağlayıcı
bölgenin yakınlarındaki proteoglikan polisakkarid makromolekülleri
bağlayan başka bir bölgenin de fibrilojenezde etkili olduğu
bildirilmiştir. Fibriler Aβ plaklara
“hapsedilecektir”.
İlk birikim NFY’lerin tersine
limbik sistemde değil, neokortekste ve gevşek (difüz) plaklar
şeklindedir. Bu plaklardaki amiloidin büyük çoğunlukla fibriler
değil granüler Aβ oligomerlerini içerdikleri
bulunmuştur. Gevşek plaklar kognitif bozukluk geliştirmeden
ölmüş olan yaşlı beyinlerinde de büyük miktarlarda görülebilir.
Ayrıca AH’lilerde hastalığın semptomatolojisine katkıda
bulunduğu düşünülmeyen striatum, talamus ve serebellum gibi
bölgelerde çevrelerinde gliozis ve nöritik sitopatolojik
değişiklikler olmaksızın görülebilirler. Ayrıca, çok
sayıda immunositokimyasal çalışma ile gevşek plak
içeriğinin daha toksik ve birikime yatkın başlıca Aβ42 formu
olduğunu görülmüştür. Dolayısıyla, plak oluşumu Aβ
monomerinden birikime doğru giden bir patogenetik mekanizmanın daha
olgun bir evresinden çok hastalığın demans öncesi
aşamasındaki bir savunma mekanizması olarak düşünülebilir:
degradasyon ve klirens mekanizmalarıyla Aβ40 ile
başa çıkılmakta bu mekanizmalar Aβ42 için
yetersiz kaldığında plak formasyonu yoluna gidilmektedir. Buna
karşılık diğer plak formu olan katı (kompakt) plaklar
çekirdeğini beta katlanmış fibriler Aβ’nın oluşturduğu
plaklardır, Bu aşamadan sonra dejenere nöritler içeren katı
plaklara nöritik plaklar adı verilir. Nöritik plaklar immun
cevabı tetikleyerek mikroglia ve astrosit aktivasyonu ile TNF-α ve
IL-β gibi sitokinlerin salınımı, kompleman aktivasyonu ve
serbest radikal oluşumuna neden olurlar. Bu immun aktivasyon
nörotoksisitenin başlıca etmenlerindendir gibi durmaktadır.
Nöritik plaklar yalnızca demanslı beyinlerde görülür. Nöritik
bileşen sinaptik artık ve nörofilamanlardan oluşur ve çoğu
aynı zamanda t-PHF için de pozitif immunoreaksiyon gösterir. PHF(+)
plaklarda nöritik bileşenin NFY içeren akson kalıntıları
olduğu söylenebilir. Böylelikle bölgesel olarak farklı
başlangıç yatkınlıklarına sahip olan NFY ve AP’ler
hastalığın seyri içinde PHF(+) plaklarla birlikte yan yana
gelmiş olurlar. Nöritik plaklar tam boy Aβ40 ve Aβ42’nin
her ikisini de içerdikleri gibi ADDL’leri de içerirler. Bunlar arasında
yukarda en toksik olduğu söylenilen amino ucundan budanmış Aβ
(3(pE)-42 formu demans şiddetine korele edecek şekilde Braak ve Braak
evre IV-VI boyunca nöritik plak kompozisyonundaki oranı giderek artar. Bu
bulgu da yalnızca demanslı beyinlerde görülen bu daha olgun plak
formunun da bizzat kendisinin bir tetikçi olmasından çok alternatif
savunma mekanizmalarının çok yetersiz kaldığı
nörodejenerasyonun bu ileri aşamalarına özgü bir ek savunma
girişimi olduğunu düşündürmektedir. Son zamanlarda
Aβ’nın özellikle Aβ (3(pE)-42
formu ile agrezom adı verilen intranöronal birikintilerde biriktiğine
dair çalışmalar artmaktadır. Bu çalışmalar sonucunda
intranöronal birikimin sinaps ve nöron kaybına neden olan, ekstraselüler
plak birikimini önceleyen erken bir fenomen olduğu ileri sürülmüştür.
Bu yeni bulgular ışığında Aβ monomerinden nöritik
plağa doğru evrim eski perspektifle olduğu gibi giderek daha
zararlı bir forma doğru çizgisel bir evrilme şeklinde değil
de toksik Aβ oligomeri ile farklı aşamalarda başa
çıkma stratejileri olarak görülmelidir. Degrade edilemeyen toksik
Aβ’nın fibrilizasyonu ve sonrasında da plaklara hapsolunması
toksisitesinin giderilmesine yönelik önlemler olmakla birlikte, bu çözülemeyen
birikintilerle çözünür oligomerik formlar arasında sürekli bir dinamik
dengenin mevcudiyeti aslında bu formların toksik biçimler için
sürekli bir rezervuar işlevi görmeleri anlamına gelmektedir. Aβ
oligomerinin yolaçtığı sinaptik disfonksiyon ve giderek nöron
kaybını önceleyen ve aşan sinaps kaybı mekanizmalarına
aşağıda değinilecektir.
AP yükünün AH’de demans
şiddetiyle korele etmediğini gösteren nöropatolojik
çalışmaların ötesinde, PIB’den başlayarak PET amiloid ligandlarının
geliştirilmesinden sonra AP yükünün bir kısım kognitif
yakınması olmayan yaşlılar ve bir kısım
MCI’lılarda AH tanısı almış demanslılar düzeyine
yaklaştığı, hatta AP yükü açısından AH’ye
benzeyen MCI’lıların
uzunlamasına izlemlerinde demansa dönüştükleri, bir kez demans
geliştikten sonra ise AP yükünün fazlaca değişmediği ortaya
konmuştur. PET ile ölçülen amiloid yükü esas olarak MCI’dan da önce
pre-klinik aşamada ilerleyici olarak artıyor, pre-demans MCI ve
demans aşamalarında ise büyük ölçüde değişmeyecek denge
durumuna ulaşıyor gibi durmaktadır.
Aβ monomerinin
üretiminin engellenmesinden başlayarak tüm çözülebilir ve çözülemez
formlarının klirensini desteklemek geleceğin temel tedavi
stratejisi gibi durmaktadır. APP proteolizini non-amiloidojenik yöne
kaydırmak amaçlı bir spesifik α-sekretaz
aktivatörü (NP-17) ilk kez klinik çalışma evresine kadar
ulaşmıştır. β-sekretaz
inhibisyonu yukarda sözedilen nedenlerle daha çekici bir hedef gibi görünse de
spesifik küçük moleküllü ajanların geliştirilmesi açısından
geride kalmıştır. Yakın tarihlerde bir ajan (CTS-21166) ilk
kez bir faz I çalışmada başarıyla sınanmış,
bir diğeri için de (HPP854) faz I çalışması için başvuruda
bulunulduğu bildirilmiştir. γ-sekretaz
inhibisyonu hedefi, yine yukarda sıralanan nedenlerle problemli gibi
görünse de sınanan böyle bir ajan (flurizan) faz III’e kadar gelip bu
noktada takılmış, diğeri (semagacestat) ise halen oldukça
büyük boyutlu bir faz III’te sınanmaktadır. Doğal klirens
mekanizmaları yetersiz kaldığında immunizasyon
aracılığıyla başa çıkma çabası çekici bir
hedeftir. Nitekim, tam boy Aβ peptidine
karşı geliştirilen antijenle başlatılan bir faz IIa
çalışması (AN1792) hastaların bir bölümünde gelişen
meningoensefalit nedeniyle durduruldu. Ölen bir olgunun otopsisinde
anti-amiloid etki için tetiklenen hümoral immunite cevabı
yanısıra inflamasyonun nedeninin ayrıca tetiklenen T-hücre
cevabı olduğu ileri sürüldü. Hücresel immuniteyi tetiklemeden sadece
hümoral immuniteyi tetikleyecek bir yöntem üzerinde çalışan
araştırıcılar sonunda hücresel cevabın Aβ’nin C-terminaline karşı
geliştiğini, N-terminalini içeren 5-6 rezidülük budanmış
epitopların terapötik olarak yeterli hümoral cevaba neden olduğunu
buldular. Sonrasında humanize monoklonal Aβ
antikorlarıyla pasif immunizasyonun da başarı şansı
yüksek bir müdahele biçimi olduğu ortaya kondu. Halen çok sayıda
aktif (örneğin, ACC-001, CAD106, V-950) ve pasif (örneğin,
bapineuzumab, LY2062430) immunizasyon ajanı klinik çalışmaların
çeşitli evrelerinde sınanmaktadır. Sekretaz modülasyonu ve
immunizasyonun yanısıra Aβ monomerinin
toksik olgun biçimlerine dönüşmesini engelleme stratejileri de
denenmektedir. Anti-oligomerizatörler arasında sınıflanan
tramiprosat isimli molekül büyük umutlarla geldiği faz III’ü
aşamamıştır. Diğer bir
anti-oligomerizasyon/fibrillizasyon yöntemi β-katlanmanın
aracılarından ağır metal bağlayıcı bölgenin
inhibisyonudur. Metalloprotein zayıflatıcı ajanlar (MPAC’ler)
böylesi ajanlardır. Bunlar arasında clioquinol (PBT-1) toksik
metaboliti bertaraf edilemediği için gelişimi durdurulduysa da benzer
bir molekül olan PBT-2’nin faz II çalışmaları sürdürülmekteydi.
Exebryl ve scyllo-inositol (ELND005/AZD-103) isimli moleküller klinik
çalışmalarda sınanmakta olan diğer küçük moleküllü
anti-fibrillizatörler arasındadırlar.
Şekil 14.
Amiloid Plak (Kaynak: http://www-edlib.med.utah.edu/WebPath/CNSHTML/CNS090.html)
Şekil 15. APP proteolizi
ve Aβ birikimi
Amiloid
prekürsör protein (APP) 770 aminoasidlik (aa) bir transmembran (TM) proteindir.
700 ve 723. aa’lar membranı kateden parçasına rasgelir. Aβ
karboksi ucuna (CT) yakın 40-42 aa’lık bir fragmanıdır.
Aβ’nın 700 ila 711-713. aa’lara karşılık gelen
kısmı membran içinde (transmembran domen – TMD), 671 ila 699. aa’lar
arasında kalan kısmı ise ekstraselüler (jukstamembran domen –
JMD) yerleşimlidir. APP proteolizi distalde ekstraselüler parçada
amiloidojenik veya non-amiloidojenik olarak yapılır. Amiloidojenik
proteoliz Aβ fragmanını salim bırakacak şekilde 671.
pozisyondan β-sekretaz kesimiyle başlatılır. Bu kesim amino
ucunda (NT) akson budanmasında rol oynadığı bulunan
sAPPβ ve CT’de membrana bağlı 99 aa’lık (C99) fragmanı
oluşturur. Non-amiloidojenik proteoliz ise APP’yi Aβ-JMD içinden,
687. pozisyondan kesen α-sekretaz aracılığıyla
başlar. Bu kesim de NT’de nörotrofik etkileri bilinen sAPPα ve CT’de
yine membrana bağlı 83 aa’lık (C83) fragmanı
oluşturur. C99 ve C83 intramembranöz parçada γ-sekretaz kompleksi
(GS) ile bir dizi ardışık kesimden geçer. İlk kesim ε
kesimi adı verilen 720 veya 721. pozisyonda meydana gelir. Oluşan
49-50 aa’lık APP intraselüler domeni (AICD) isimli fragmanın hücre
çekirdeğine transloke olarak protein sentezini desteklediği
bilinmektedir. İkinci kesim ζ kesimidir ve 717 veya 718. pozisyonda
ve nihayet 3. kesim olan γ kesimi 711 veya 713. pozisyondadır. Bu son
kesim non-amiloidojenik C83’te ise ortama 24-26 aa’lık inert bir
budanmış Aβ parçacığı olan p3, amiloidojenik
C99’da ise 40-42 aa’lık salim bir Aβ fragmanı üretir. GS APP’nin
yanısıra bir dizi TM proteinin daha proteolizinden sorumlu olan bir
aspartil proteaz kompleksidir. Aktif katalitik bölgesi presenilin 1 veya 2
(PSEN) olmak üzere 4 ayrı proteinin bileşimidir (Pen-2, Nicastrin,
Aph1B). GS’nin diğer önemli proteolitik hedefi nöro-gelişimde önemli
rol oynayan NOTCH proteinidir. GS kesimi sonrası NOTCH intraselüler domeni
(NICD) adı verilen fragman, gen ifadesini ve hücresel farklılaşmayı
başlatmak üzere nukleusa transloke olur. Ortama salınan Aβ
monomerli mikroglial fagositozla temizlenebilecekleri gibi şaperon
moleküller tarafından vasküler endotelden klirensi sağlanabilir veya
yine şaperon moleküller tarafından lizozomal veya proteozomal
degradasyona maruz kalacakları nöronlar ve astrositler içine sokulur. Bu
yöntemlerin her hangi biriyle ortamdan uzaklaştırılamayan,
özellikle de potansiyel olarak daha toksik form olan Aβ42 gevşek
plaklarda birikir. Bu son savunma mekanizması da başarısız
kaldığı takdirde Aβ monomeri çözülebilir oligomerler
halinde bir araya gelir. Çözülebilir oligomer bir dizi sinaptik disfonksiyon
mekanizmasını tetikleyerek AH’nin en erken fizyopatolojik
özelliği olan LTP depresyonuna neden olur. Oligomerler daha toksik
ADDL’lere budanabilecekleri gibi çözülemeyen Aβ fibrillerine doğru da
evrilebilirler. Aβ fibrilleri de hücre yıkıntı ürünleri
olan distrofik nöritlerle birlikte nöritik plaklarda hapsedilirler. Fibriler
Aβ gibi nöritik plaklar da kararlı yapılar olmaktan çok toksik
oligomerler için bir tür rezervuar işlevi görmektedirler. Nöritik plaklar
çevrelerinde oksidatif gerilim yanısıra aktive mikroglia
aracılığıyla inflammasyona yolaçarlar.
Nöron kaybı
AH’de nöron kaybı entorhinal
korteksten başlar ve limbik sistemi terkettiğinde superior temporal
sulkusta tespit edilebilir. Nöron kaybının zaman içinde
ilerleme ve anatomik yatkınlık tarzı genel anlamda NFY’nin
tarzına benzese ve NFY ile nöron sayılarının
arasında anlamlı negatif korelasyonlar saptansa da, nöron
ölümünden tek başına NFY’ler sorumlu tutulamaz. Subkortikal
çekirdekler gibi NFY’lerin bulunduğu bölgelerde ille de nöron
kaybının olması gerekmediği gibi, NFY’lerin az sayıda
olduğu ya da hiç bulunmadığı bölgelerde de ağır
nöron kaybı görülebilir. Amiloid nörotoksisitesinin neden
olduğu sinaps kaybı ve transsinaptik dejenerasyon hücre ölümünde rol
oynadığı düşünülen diğer etmenlerdir.
Kolinerjik kayıp
Bütün korteksin kolinerjik
innervasyonu temel limbik yapılardan biri olan bazal önbeyindeki Meynert
çekirdeğinden sağlanır. Bu innervasyon dikkat ve bellek
işlevlerinin optimal sürdürülebilmesi açısından büyük önem
taşır. Tau hiperfosforilazasyonun ilk görüldüğü alanlardan
biri de Meynert çekirdeğidir. AH’de limbik alanlardaki yaygın
NFY formasyonu ve nöron kaybı Meynert çekirdeğini de
etkiler. Kolinerjik aksonların kaybı diğer patolojik
özellikler gibi bir bölgesel yatkınlık gösterir. Lokal internöronlardan
sağlanan striatal kolinerjik innervasyon ve talamusun beyinsapı
pedinkülopontin çekirdek kaynaklı kolinerjik innervasyonu
etkilenmediği gibi, etkilenen Meynert kaynaklı kolinerjik innervasyon
da aynı tarzı yansıtır: kortekste en fazla etkilenen
bölgeler limbik ve asosiasyon korteksleri iken, primer sensoryal ve motor
korteksler göreli olarak salim kalırlar.
Kolinerjik buton da denilen
pre-sinaptik kolinerjik terminalde kolin asetil transferaz enzimi (ChAT) bir
yandan mitokondrilerden asetil koenzim A aracılığı ile
serbestlenen kolinden, diğer yandan da yüksek afiniteli kolin geri
alınım sistemi aracılığıyla alınan kolinden
asetil kolin (ACh) üretir. ACh molekülleri veziküllere paketlenip
depolarizasyonu izleyerek sinaptik aralığa salgılanır.
Sinaptik aralıkta difüzyonla ilerleyen ACh post-sinaptik membranda
nikotinik reseptörlere bağlanarak doğrudan, muskarinik reseptörlere
bağlanarak ise G-proteini ilişkili ikincil mesajcılar üzerinden
etkisini gösterir. Nikotinik etkilerin hücrenin
uyarılabilirliğini arttırarak dikkat tonusunun
sağlanmasında rol oynadığı, muskarinik etkilerin ise
kalıcı sinaptik değişikliklerle yeni bilginin
depolanması şeklindeki nöroplastisite mekanizmalarının
unsuru olduğu bilinmektedir. Beyinde α7 nikotinik
reseptörler (α7nAChR) pre-sinaptik
yerleşimlidir ve uyarılmaları ACh’nin pre-sinaptik akson
terminalinden salınımını arttırırken yine
pre-sinaptik muskarinik 2 ACh reseptörleri (M2AChR) tam tersine ACh
salınımı inhibe ederler. AH’nin erken döneminde α7nAChR’de kompansatuar bir artış
söz konusuyken pre-sinaptik denge giderek nikotinik aleyhine ve muskarinik
lehine dönmeye başlar. Post-sinaptik membranda bulunan ve her ikisi de AH
sürecinde erken dönemde kaybolmaya başlayan M1AChR ve hem pre- hem de
post-sinaptik yerleşmiş olan α4β2nAChR’lerin
uyarılması yukarda değinilen hızlı (nikotinik) ve
yavaş (muskarinik) kolinerjik nörotransmisyona aracılık eder.
Hem nikotinik, hem de muskarinik stimülasyon kaybının gerek α-sekretaz
etkinliğinin azalmasıyla Aβ
oluşumunun artması ve gerekse de GSK-3β
etkinliğinin artmasıyla taunun patolojik fosforilizasyonunun da
artması şeklinde in vitro etkileri gösterilmiş, diğer
yandan Aβ’nın α7nAChR
blokajı üzerinden ACh’nın sentez, salınım ve post-sinaptik
etkinliğini azaltabileceği de ortaya konmuştur. Bu durum Aβ ve
kolinerjik kaybın birbirlerini pozitif geri besledikleri bir
kısır döngü oluşturmaktadır. Muskarinik kolinerjik
nörotransmisyonun tetiklediği fosfoinositol sistemi üzerinden
anti-Alzheimer mekanizmalar Şekil 16 ve 17 de gösterilmiştir.
Bu mekanizmalar α4β2 ve α7
nikotinik ile M1 muskarinik agonizmanın bir tedavi hedefi olarak önemini
göstermektedir ve bunların tümüne ilişkin terapötik ajanlar, doz
kısıtlayan gastro-intestinal yan etkilerinin sunduğu büyük güçlüklere
rağmen klinik çalışmalarda ilerlemeye
çalışmaktadırlar (örneğin, sırasıyla TC-1734, MEM
3454, AF267B). M2 antagonizmasının hayvan modellerinde M1
agonizmasına benzer anti-AH etkileri gösterilmiştir.
Asetilkolinesteraz (AChE) enzimi katalitik iyonik kısmının
aktivitesiyle ACh’yi asetat ve koline hidrolize ederek ACh’nin post-sinaptik
aktivitesini durdurur. AChE’nin diğer yandan periferik aniyonik
kısmı aracılığıyla Aβ
oligomerleriyle oluşturdukları fibriler kompleksin de tek
başına oligomerinkini aşan toksik etkileri gösterilmiştir.
AChE inhibitörleri AH tedavisinin ilk yerleşik ajanları olarak
terapötik cephanede çoktan yerlerini almışlardır. Klasik
katalitik bölge inhibisyonu yanısıra periferik bölge inhibisyonu da
yapabilen ajanlar (örneğin, NP-61) erken klinik gelişim
aşamasındadırlar. Östrojen limbik nöronlarda sinaptogeneze
katkıda bulunup nöroplastisitede rol oynarken, Meynert’te ACh üretimine de
katkıda bulunur. Post-menapozal kadınlarda östrojen replasmanının
koruyucu etkisi anılan işlevleri dolayısıyla olabilir.
Şekil 16. Kolinerjik
sinaps
Kolinerjik Sinaps.Pre-sinaptik akson terminalinde ChAT,
asetil CoA aracılığıyla kolini asetilleyerek ACh oluşturur.
ACh veziküller içine paketlenerek sinaps aralığına salınır.
Presinaptik nikotinik α7
reseptörlerinin uyarılması ACh salınımını stimüle
ederken (yeşil ok), muskarinik M2 reseptörlerinin uyarılması
inhibe eder (kırmızı çubuk). Sinaptik aralığa salınan
ACh post-sinaptik nikotinik α4β2
reseptörleriyle bağlandığında Na4 kanalı açılır
ve içeriye Na+ girerek dikkatten sorumlu hızlı kolinerjik
nörotransmisyonu başlatır. Muskarinik M1 reseptörlerine bağlanan
ACh, reseptöre bağlı Gq proteinini aktive ederek PLC’yi fosforiller.
Fosforile PLC bir yandan ikincil mesajcılardan membrana bağlı
diasil gliserolü (DAG) aktive ederek PKC’yi doğrudan fosforillerken, diğer
yandan da fosfoinositol difosfattan (PIP2) inositol trifosfat (IP3)
üreterek endoplazmik retikulumdaki (ER) kendi reseptörüne bağlanacak diğer
ikincil mesajcıyı hücre içine salar. IP3, ER’den
Ca++ salınımını
sağlar ve artan intraselüler Ca++ böylece daha fazla PKC’yi
dolaylı yoldan fosforillemiş
olur. Fosforile PKC, mitojen aktive protein kinaz (MAPK) aracılığıyla
nükleer transkripsiyonu devreye sokar ve öğrenmenin temeli olan yeni
sinaps yapımına aracılık eder. Sinaps aralığında
işlevi biten ACh, AChE aracılığıyla asetat ve koline
parçalanır. Kolin pre-sinaptik terminalden kolin taşıyıcısı
aracığıyla geri alınır
Şekil
17. AH’li sinapsta Aβ’nın anti-glutamaterjik ve
anti-plastisite etkiler
Şekil 17a. Glutamaterjik
sinaps, GABA-erjik olanın tersine AH’de erken dönemde saldırı altındadır. APP’nin
amiloidojenik proteolizi sonrası ortamdan temizlenemeyen çözünür Aβ oligomeri
post-sinaptik membranda bir dizi patolojik zinciri tetikler. Bunların
sonucunda, önce NMDA, sonra da AMPA olmak üzere glutamaterjik (GluR) sinapslar
endositozla kaybedilir. Sonuçta glutamaterjik aktiviteye dayanan ve
öğrenmenin moleküler mekanizması olan uzun süreli potansiasyon (LTP)
giderek azalırken, uzun süreli depresyon (LTD) artar. Bu etkinin GluR ile
doğrudan etkileşimle değil fakat başka bir transmembran
protein (TMP) olan bir aracı üstünden olduğuna dair deliller
mevcuttur. Bu aracılardan biri olarak normal TMP prion proteini (PrPc)
gösterilmiştir. Diğeri ise yalnızca glutamaterjik sinapslarda bulunan ve GluR stabilitesini
sağlayan neuroglin 1’dir. Neuroglin 1-Aβ kompleksinin Wnt sinyalizasyonunu
başlatacak ligandın reseptörüne bağlanmasını engellediği,
bunun sonucunda da GSK3β-Axin-APC’den
oluşan “yıkım kompleksi”nin inhibe edilemeyerek bir nükleer
transkripsiyon faktörü olan β-kateninin işlevini göremeden
yıkıldığı bilinmektedir. Çözünür Aβ oligomeri
ileri glikasyon son ürünleri reseptörü (RAGE) ile de etkleşir. Nöronal
membranda etkileşim temel inflammatuar sinyalizasyon olan NFκB
sinyalizasyonunu tetikler ve böylelikle ortama inflammatuar sitokinler
salınır. Aβ
fagositozu için aktive olmuş mikroglia da RAGE-Aβ
etkileşimi ile salınan inflammatuar sitokinlerin bir diğer
kaynağıdır. Bir TMP olan RAGE, bu tür proteinlerin
proteolizinden sorumlu olan bir metalloproteinaz olan ADAM10
aracılığıyla kesilerek parenkime salınan çözünür RAGE
(sRAGE) oluşur. sRAGE de Aβ bağlar, ancak artık membrana bağlı
olmadığından bir patolojik zinciri başlatmanın tam
tersine Aβ’yı
nötralize eden bir işlev görür. Aβ düşük yoğunluklu
lipoprotein reseptörü 1 (LRP1) aracılığıyla kan-beyin
bariyerinden dolaşıma verilir. Vasküler endotel membranındaki
RAGE Aβ’yı
yeniden beyne alırken, dolaşımdaki sRAGE Aβ
bağlayarak buna karşıt çalışır. Dolayısıyla,
RAGE antagonistleri ve sRAGE agonistlerinin potansiyel anti-AH farmakolojik
stratejiler olduğu söylenebilir. Yerleşik tedavi stratejilerinden
biri olan memantinin bilinen etki mekanizması NMDAr’da Mg++ tıkacı
işlevi görerek mevcut Ca++ sızıntısı
kaynaklarından birini engellemek iken hücre içinde de hiperfosforile tauyu
parçalayacak fosfataz agonizması yaptığına dair de deliller
vardır.
Şekil 17b. Kolinerjik
sinaps da AH’de erken tehdit altiındadır. Çözünür Aβ oligomerleri
pre-sinaptik membranda asetil kolin (ACh) salınımını inhibe
eden M2 muskarinik reseptörlere değil de, tam tersine
salınımı arttıran α7 ve α4β2 nikotinik reseptörlere
bağlanarak kolinerjik nörotransmisyonu azaltırlar. M2 antagonizması
ve/veya α7 ve α4β2
agonizması yapan ajanlar bu dengeyi tersine çevirebilirler. Post-sinaptik
M1 kolinerjik aktivite gibi insulin sinyalizasyonu da protein kinaz C (PKC)
fosforillenmesi üzerinden anti-AH etkiler (ADAM17 aktivasyonu, β-sekretaz ve
GSK3β
inhibisyonu) gösterir. Dolayısıyla,
M1 agonistleri ve insulin potansiyel anti-AH etkilere sahiptirler. Asetil kolin
esterazın (AChE) periferik kısmına bağlanan Aβ ile
oluşturduğu kompleksin oligomerin toksisite aracılarından
biri olduğu gösterilmiştir. Dolayısıyla, katalitik
kısma bağlanan klasik inhibitörlerden farklı olarak ayrıca
periferik kısmı da bloke edecek “dual inhibitörler” daha güçlü bir
anti-AH etki gösterebilirler. DNA onarımıyla görevli sirtuin
sinyalizasyonunun Aβ ile tam tersi etkilere sahip olduğu
bilinmektedir. İntraselüler Aβ sirtuin sinyalizasyonunda rol oynayan
başlıca moleküllerden biri olan SIRT1’i inhibe ederken, bir TMP olan
fraktalcine’nin (CX3CL1), örneğin kırmızı şarapta da
bulunan bir polifenol olan rosveratrol ile uyarılması CX3CL1’in,
aynı zamanda APP’nin non-amiloidojenik proteolizini sağlayan
ADAM17’yi aktive etmesini sağlamakta, bunun sonucunda aktive olan SIRT1
ise RAGE’i kesecek ADAM10’u aktive ederek pro-inflammatuar NFκB
sinyalizasyonunu durdururken, diğer yandan da pro-plastisite nükleer
transkripsiyon faktörlerini harekete geçirmektedir.
Sinaps kaybı
Sinaps kaybı kortikal biyopsi
örneklerinde klinik demans ağırlığıyla, nöron
kaybından dahi daha kuvvetli en yüksek korelasyon gösteren yapısal
değişikliklerin başında gelir. Sinaptofizin gibi
sinaptik proteinlerin miktarlarının da demans
ağırlığıyla korele ettiği gösterilmiştir.
Sinaps kaybı başlangıçta NFY ve nöron ölümünün anterograd
Wallerian dejenerasyon sonucu sekonder etkisi ile açıklansa da, primer
hasarın özellikle de intraselüler çözülebilir Aβ
oligomeri aracılığıyla sinapslara olması ve
bozukluğun retrograd olarak hücre gövdesine taşınarak, NFY
oluşumu ve nihayetinde hücre ölümüne yolaçması artık daha
muhtemel görünmektedir.
Çözülebilir Aβ oligomerinin
toksisitesi ve neden olduğu erken sinaptik disfonksiyon üzerine odaklanan
son çalışmalar çözülemez fibriler formun veya daha olgun
plakların parenkimde oynadığı rol yerine toksik çözülebilir
oligomerin hücre yüzeyinde uygun bir reseptör bulup bağlandıktan
sonra veya membrandan intraselüler mesafeye geçtikten sonra başlattığı
bir dizi zincirleme reaksiyonun doğasını aydınlatmaya
başlamıştır. Bu mekanizmaların
basamaklarının aynı zamanda nedene yönelik tedavi hedefleri de
oluşturmaları heyecan vericidir. Bu çalışmaların
ortaklaştığı bir nokta Aβ oligomerinin
başlangıçta inhibitör GABA-erjik sinapsları salim bırakırken
eksitatör glutamaterjik sinapslarda değişikliklere neden olması,
buna bağlı olarak öğrenmenin temelini teşkil eden moleküler
mekanizma olan uzun süreli potansiasyonu (LTP) baskılarken, uzun süreli
depresyonu (LDP) teşvik etmesidir. Inestrosa grubu oligomerin eksitatör
sinapslarda NMDA reseptörü stabilitesini sağlayan neurogilin 1 ile
etkileşirken inhibitör sinapslardaki neurogilin 2 ile
etkileşmediğini gösterdiler. Aynı
araştırıcılar bu etkileşimin embryogenez gibi bir dizi
diğer işlevleri yanısıra akson rehberliğinden sorumlu
Wnt sinyalizasyonunu bozduğu ve AH’de bu sinyalizasyonun son ürünü olan
nükleer transkripsiyon faktörü β-katenin miktarlarının
azaldığı buldular. Bu etkileşim sonunda NMDA ve AMPA reseptörlerinin
postsinaptik membrandan endositozla kaybına yolaçar. Reseptörlerin
kaybıyla sinaps atrofiye uğrar. Wnt sinyalizasyonunu aktive eden
ligandlar Aβ’nın tetiklediği sinaptik değişiklikleri
geri çevirebilmektedir. Wnt sinyalizasyonu sinaptik öz-savunma
mekanizmalarından biridir. AH için de başlıca risk faktörü olan
bu sinaptik öz-savunma mekanizmaları yaşlanmayla birlikte
zayıflamaya başlarlar. Şekil 17a’dan A β’nın neden
olduğu glutamaterjik sinaptik disfonksiyon ve Wnt sinyalizasyonu şematik
olarak gösterilmiştir. Bu
sinyalizasyon mekanizmalarından bir diğeri de sirtuin
sinyalizasyonudur. Sirtuinler proteinlerin asetil grubunu kaldırarak
(deasetilaz) ve ADP-riboziltransferaz işlevleri gören bir uzun ömür
faktörüdür. SIRT1 ve SIRT2 isimli sirtuinlerin DNA onarımında rol
aldığı, aktivitelerinin yaşlanmayla birlikte
zayıfladığı gösterilmiştir. Aβ oligomerlerinin
SIRT1 aktivitesini bozarken tersine kırmızı şarapta da
bulunan bir polifenol olan resveratrol gibi maddelerle SIRT1 geninin aktivasyonu
mümkün olduğu gibi bir sitokin proteini olan fraktalkinin (CX3CL1)
aktivitesinin arttırılması AH’de azalan SIRT1 aktivitesini
arttırmakta ve böylelikle aktive olmuş NF-κB sinyalizasyonu
inhibe edilerek tetiklenen inflamatuar süreç tersine çevrilebilmektedir. Bu
gözlemler AH’de birer terapötik ajan olarak resveratrol ve CX3CL1
agonizmasını gündeme getirmiştir. NF-κB sinyalizasyonu
üzerinden inflamatuar değişikliklere aşağıdaki ilgili
bölümde daha ayrıntıyla değinilecektir. Serebral insülin sinyallemesi
ve Aβ oligomerinin hücre yüzeyine bağlandıktan sonra tetiklediği
mekanizmalar da birbirinin tam tersi gibi görünüyor. Serebral insülin
sinyallemesi sinaptik enerji homeostazından sorumludur. Nöronlarda bir
tirosin kinaz reseptörü gibi işlev gören insülin reseptörlerine (IR)
bağlanan insülin fosfadidilinositol-3-kinaz (PI3K) üzerinden
nihayetinde protein kinaz C’yi aktive ederek aşağıda “kolinerjik
kayıp” bölümünde daha ayrıntıyla sözedilen ve Şekil 17b’de
şematik olarak gösterilen anti-AH mekanizmaları tetikler. AH
hastalarının bir bölümü yüksek açlık insülin düzeylerine
sahiptir (periferik direnç). FDG-PET ile de gösterilen nöronal glukoz
kullanımı bozukluğu (hipometabolizma AH’de göreli erken bir
özelliktir ve demans şiddetiyle korele eder. AH’li hastaların bir
bölümünde nöronal IR’ler, glukoz taşıyıcı proteinler ve
diğer insülin yolağı ilintili bileşenler azalır
(santral direnç). Aβ dendritik IR’leri azaltırken insülin oligomerin
tetiklediği LTP baskılanması ve sinaptik kaybı tersine
çevirir. Bu bulgular insülin sinyallemesini etkileyen ajanları AH’nin
potansiyel terapötik ajanları durumuna getirmiştir. Periferik insülin
direnci düşürerek IDE’nin periferiden ziyade MSS’de bir şaperon
molekül olarak kullanılmasını amaçlayan varsayımların
sınanması PPAR-γ agonistlerini (rosiglitazon ve pioglitazon)
klinik çalışmaların olgun evrelerine kadar getirmiştir. Son
olarak, doğrudan insülinin intranazal yoldan MSS’ye verilmesi halen
sürmekte olan bir faz II çalışmada (SNIFF çalışması)
sınanmaktadır. Aβ’nın LTP depresyonu yapacak şekilde
NMDA reseptörlerine bağlanması ve nihayet önce glutamaterjik NMDA
sonra da glutamaterjik AMPA reseptörlerinin endositozla kaybolması
bilinmekle birlikte bunun doğrudan Aβ’nın reseptöre
bağlanmasıyla değil fakat bilinmeyen bir transmembran proteine
bağlanıp bir kompleks oluşturduktan sonra gerçekleştiği
ileri sürülmekteydi. Yakın tarihte bu bilinmeyen transmembran proteinin
prion proteini (PrPc) olduğu ileri sürüldü.
Araştırıcılar Aβ’nın 350 transmembran protein
arasından baskın olarak PrPc’ye
bağlandığını gösterdiler. İlk bakışta
bu durum şaşırtıcı çünkü bu işlevi bilinmeyen
proteinin proteolitik kesimi ve sonrasında hatalı katlanarak fiziksel
konformasyonunu değiştirmesi (PrPSc) ve birikmesi bir
başka nörodejeneratif hastalıklar grubuna, yani içlerinde
Creutzfeldt-Jacob hastalığının da bulunduğu prion
hastalıklarına yolaçmakta. PrPc’ye AH patogenezinde
atfedilen bu temel rol doğrulandığı takdirde 2 büyük
nörodejeneratif hastalığın aydınlatılmasında
önemli bir dönüm noktası olabilir.
Araştırıcıların çalışmaları oldukça
ikna edici görünmektedir. PrP nakavt transjenik farelerde Aβ ile LTP
supresyonu gerçekleşmiyor. Son derece virulan bir transjenik model olan
APP/PSEN mutantların PrP nakavtlarla çaprazlanan soyunda
sağkalım artıyor, APP/PSEN Tg’lerdeki tipik pasif kaçınma
öğrenme bozuklukları görünmüyor ve mekansal öğrenmeleri
doğal tip farelere benzer düzeyde normal oluyor. Buna
karşılık Aβ-Prpc kompleksi NMDA reseptörlerinin
tipik endositozuna yol açtıkları gibi bu farelerde dentritik diken
kaybı, nöritik distrofi ve sonunda hücre kaybı gibi AH’nin diğer
tipik sinaptik patolojisinin tümü görülüyor. Son olarak, intraselüler
Aβ’nın mitokondriyal stabiliteyi de bozup apoptozu tetikleyecek
reaktif oksijen türlerinin (ROS) oluşumuna neden olduğu bilinmekte.
AH’li beyinlerde mitokondriyal membran stabilitesinden sorumlu füzyon
proteinleri dynaminlerin ve bunların arasında özellikle de DLP1’in
düzeyi düşmekte, bunun sonucunda intraselüler mitokondri
dağılımı bozulmakta, sayıları azalmakta ve bu
durum sonunda dentritik diken sayısını azaltıp sinaps
kaybına yolaçmakta. Tersine, deneysel modellerde DLP1 aşırı
ifadesi ADDL ile tetiklenen mitokondriyal değişiklikleri tersine
çevirmekte. Dimebon isimli eski bir anti-histaminik molekülün AH’de
işlevsiz kaldığı gösterilen mitokondriyal delikleri
kapatarak işlev gördüğüne dair pre-klinik deliller sonrası halen
olgun klinik çalışmalarda etkinliği sınanmaktadır.
Gliozis ve İnflamasyon
Gliozis AH nöropatolojisinin bir
başka özelliğidir. Aβ’nın nöritik plaklarda birikiminin mikroglial
ve astroglial hücreleri aktive ettiği AH patogenezinde göreli geç bir olay
olarak öteden beri iyi bilinmektedir. Mikroglial aktivasyon sitokinlerin
salgılanması, akut faz reaktanları ve komplemanın aktive
edilmesiyle inflamasyonu harete geçirir. Son zamanlarda plaklardan
bağımsız olarak doğrudan Aβ
oligomerinin ileri glikasyon son ürünleri reseptörü (RAGE) ile bağlanarak
MSS’deki başlıca pro-inflamatuar sinyalleme sistemi olan aktive B
hücrelerinin kappa hafif zincir güçlendiricisi nükleer faktörünü (NF-κB)
tetiklemesinin AH’de erken bir fenomen olarak inflamasyona
yolaçmasının üzerinde durulmaktadır. NF-κB infeksiyona
immun cevapta DNA transkripsiyonunu düzenleyen bir protein kompleksidir.
Sinaptik plastisite ve bellek işlevlerinde de rolü gösterilmiştir.
Kanser, çeşitli inflamatuar ve otoimmun hastalıklar ve AH’de
NF-κB sinyallemesi yolundan sapar. RAGE immunoglobulin süper
familyasına dahil bir transmembran reseptör proteindir. İleri
glikasyon son ürünleri (AGE’ler) yaşlanma yanısıra diabetik hiperglisemi
gibi patolojik durumlarda glikozillenme ile başlayan bir dizi kimyasal
reaksiyonun nihai ürünleri olan ligandlardır. RAGE, AGE’ler
yanısıra Aβ, HMGB1, s100, Mac-1 gibi bir
dizi başka ligandı bağlayarak diabet komplikasyonlarıyla
birlikte, AH, multipl skleroz, inme gibi nörolojik hastalıklar, romatoid
artrit, ateroskleroz, koroner kalp hastalığı gibi sistemik
hastalıklarda rol oynadığı bulunmuştur. Tam uzunlukta
RAGE (flRAGE) bir transmembran protein iken alternatif düğümlenme ile üretilen
şekli olan endojen sekretuar RAGE (esRAGE) ve RAGE’in proteoliziyle ortaya
çıkan enzimatik kesilmiş RAGE (ecRAGE) RAGE’in çözünür biçimleridir
(sRAGE). sRAGE de flRAGE gibi ligand bağlar, ancak hücre yüzeyine
bağlı olmadığı için NF-κB gibi
sinyalizasyon tetiklenmesine neden olmadığı gibi flRAGE’e bağlanarak
pro-inflamatuar zinciri başlatacak olan ligandları kendine
bağlayarak bir tampon işlevi görür. Yaşlanma süreci gibi AH’nin
de flRAGE düzeylerinin artıp sRAGE düzeylerinin düştüğü süreçler
olduğu düşünülebilir. sRAGE aynı zamanda sirkülasyonda bulunur
ve artmış düzeyleri daha fazla Aβ’nın LRP1 aracılığıyla
kan-beyin bariyerinden dolaşıma aktarıldığı bir
küvet (sink) etkisiyle Aβ’nın klirensine karşılık gelir. Bu
gözlemler bir tedavi hedefi olarak flRAGE inhibitörleri (örneğin, TTP488)
ve/veya sRAGE-mimetiklerini (TTP4000) gündeme getirmiştir. NF-κB
sinyalizasyonunun Aβ-RAGE etkileşimi ile
tetiklenmesinin yanısıra, yukarda değinildiği gibi Aβ’nın yolaçtığı
SIRT1 inaktivasyonu da devreye giren NF-κB
sinyalizasyonunun üzerindeki denetimi kaldırarak bir pozitif geribildirim
etkisi yapmaktadır. RAGE etkileri Şekil 17a ve 17b’de şematik
olarak gösterilmiştir. İnflamasyonun, serbest radikallerin ortaya
çıkışı, oksidatif gerilim, kalsiyum homeostazı ve
mitokondriyal membranda bozulmalar ile birlikte gittiği
düşünülmektedir. Astrositler butiril kolin esteraz (BChE) gibi
normalde aktivitesi ihmal edilecek düzeyde olan bir dizi molekülün de
kaynağıdır ve dolayıyla da gliozis asetil kolinin
parçalanmasında da alternatif yolu devreye sokmaktadır.
Diğer nörotransmitter
kayıpları
Serotoninerjik kayıp
AH’de serotoninerjik (5-HT-erjik)
dorsal raphe çekirdeğinde Meynert düzeyinde olmasa bile kayda değer
nöron kaybı ve kalan nöronlarda NFY’ler görülür. Kortikal 5-HT-erjik
akson terminallerinde 5-HT’nin salınımı ve geri
alınımı ciddi düzeyde bozulmuştur. AH’de 5-HT-erjik
kayıpla depresyon ve saldırgan davranışın korelasyonu
bildirilmektedir. 5-HT geri alınım blokerlerinin (SSRI) AH’deki
depresyon ve agresyonun tedavisindeki rasyoneli bu gözlemlere
dayanmaktadır.
Dorsal raphede bildirilen bu
kayıp ve pre-sinaptik serotonerjik aktivitedeki bu eksikliklere
rağmen AH’de serotonerjik sistemin hiperaktif olabileceğine dair
görüşler mevcuttur. Bu sağduyuya karşıt gibi görünen
durumun nedeni farklı post-sinaptik 5-HT reseptörlerinin işlevsel
farklılıkları ve yerleşimleri ile ilintildir. Bunlar
arasında 5-HT1a ve 5-HT6 reseptörler siklik AMP’nin
intraselülerdüzeylerini azaltarak ve dolayısıyla plastisite
karşıtı bir mekanizmayla işlev görürler. Her iki reseptör
tipi de limbik sistemde zengin bulunur. 5-HT6 özellikle glutamaterjik ve
kolinerjik nöronları doğrudan inhibe edecek pozisyonda bulunan
GABA-erjik nöronlarda zengindir. Bu bulgulardan yola çıkarak 5-HT1A ve
5-HT6 antagonistleri AH için klinik çalışmalarda sınanmaya
başlanmıştır (örn., ilki için SRA333 ve WAY100635, ikincisi
için SB-742457 ve SAM-351).
Spesifik 5-HT antagonizması
aracılığıyla cAMP intraselüler düzeylerinin
azaltılmasına karşı durmanın pro-plastisie etkilerinin
gösterilmesinden sonra cAMP’yı yıkan enzim olan fosfodiesterazın
(PDE) inhibisyonunun terapötik poyansiyeli üzerinde durulmuştur.
Pre-klinik çalışmalarda güçlü pro-kognitif ve pro-plastisite etkileri
görülen PDE4 inhibitörleri erken klinik çalışmalara
ulaşmış durumdadır (örn., AVE8112 ve MEM 1414).
Noradrenerjik kayıp
Beyinsapındaki noradrenerjik
(NA-erjik) çekirdek olan locus ceruleus’ta (LC) da dorsal raphe benzeri bir
nöronal kayıp ve NFY oluşumu gözlenir. LC’deki patoloji seçici
yatkınlığa uygun biçimde LC’nin kortikal projeksiyonları
olan anterior ve medyal bölümlerini etkilerken spinal ve serebellar
projeksiyonları içeren kaudal ve lateral bölümleri salim
kalır. NA-erjik kaybın klinik karşılığı
iyi belirlenmemiştir.
Dopaminerjik kayıp
AH ileri evrelerinde parkinsonizm
sıradışı bir olgu değildir. Bu olguların
patolojik karşılığı büyük sıklıkla
substantia nigra pars compacta (SNc) dopaminerjik (DA-erjik) nöronlarında
kayıp ve Lewy cisimcikleridir (LC). LCD kendine özgü bir antite olarak
ortaya çıkmışken LCD ve AH’nin içiçe geçtiği durumlar olan
hem NFY-AP hem de kortikal Lewy cisimciklerinin sonucu olan klinik
durumların LCD mi yoksa AH’nin Lewy varyantı (AH-LV) olarak mı
adlandırılacağı tartışmalıdır. LC’ler
ve NFY-AP birlikteliği için 2 ayrı nöropatolojik örüntü
sınıflandırmaya yardımcı olabilir. LC’ler gerek
sporadik ve gerekse de familyal AH’de en sık amigdalada ve %60’a
yaklaşan bir oranda görülürler. Diğer limbik alanlar, özellikle de
periamigdaloid ve entorhinal korteks amigdaladan sonra en sık görüldükleri
yerlerdir. LCD’ye özgü neokortikal ve beyinsapı yerleşimi çok daha
seyrektir. Buna karşılık LCD’de yaygın neokortikal ve
beyinsapı yerleşimine değişken miktarlarda AH tipi patoloji
eşlik edebilir.
Alzheimer Hastalığının Nedenselliği
Üzerine Son Değerlendirmeler
AH araştırmacıları plaklar ve
yumaklar temelinde, hangisinin birincil rolü oynadığı üzerine
nerdeyse uzlaşmaz iki kampa büyük ölçüde ayrılmış
durumdadırlar. Bu kamplaşmanın dinsel fanatizme
benzerliğine ince bir eleştiriyle de, tau patolojisini
(yumakları) ön plana alanlara tauistler, β-amiloid patolojisini
alanlara ise baptistler dendiği olmuştur.
Gerçekten de, ancak demans evresinde
tanınabilen hastalığın klinik evreleri NFY
yayılımıyla çok iyi korele ederken, AP ile korelasyonun
olmaması tauistlerin temel tezi olurken, gösterilen bütün genetik
bozuklukların Aβ üzerinden etkisini göstermesi, buna
karşılık tau mutasyonlarının AH dışı
dejenerasyonlara neden olması baptistlerce tezlerinin
kanıtı gibi sunulmuştur.
Günümüzde ilk bakışta baptizm
tartışmasız bir zafer kazanmış görünmekte ve amiloidin
hastalığın tetikçisi olduğu, buna karşılık
tau patolojisinin bir epifenomen olduğu artık egemen görüş
olarak yerleşmiş durumda. Tetikçi amiloid olduğuna göre bunun
doğal sonucu olarak neredeyse tüm nedene yönelik tedavi çabaları
anti-amiloid stratejilere vakfedilmiş durumda. Bu çabanın temel
rasyoneli, tetikçi ortadan kaldırıldığı takdirde ona
sekonder olarak gelişen fizyopatolojik değişiklikler, tau da
dahil olmak üzere düzelecektir şeklinde özetlenebilir. Yeni
yüzyılın ilk onyılında bu çabaların
yarattığı büyük heyecan ve iyimserlik, klinik
çalışmalarda nihai gelişim aşamasına kadar gelen hiç
bir anti-amiloid aday molekülün ipi göğüsleyememesiyle, büyük maddi ve
moral kayıplarla birlikte birbiri ardına hayal
kırıklıklarına dönüşmüştür. Bunlar arasında
özellikle de Aβ antijeniyle ilk aktif immunizasyon
çalışması olan ve katılımcılar arasında
gelişen meningoensefalit olguları nedeniyle durdurulan AN1792
çalışması temel varsayımın sınanması
açısından öğretici olmuştur. Katılımcılar
arasından ölen olguların otopsilerinde serebral plak yükünde (hem
gevşek, hem de sert plaklar) kayda değer bir azalmaya karşın
NFY yükünde bir farklılık izlenmemiştir. Dahası, bu
çalışmaya katılıp da yeterli antikor geliştiren
hastaların uzun süreli izlemleri de dahil olmak üzere, çok sayıda
farklı anti-amiloid tedavi çalışmalarına katılan hiç
bir hasta grubunda bildirilen yararlanmanın büyüklüğü amiloid
hipotezinden yola çıkarak beklenileceği gibi dramatik
olmamış, plaseboya karşı farklı sonlanım
ölçütleri açısından ancak marjinal yararlar ifade edilmiştir.
İlk onyılın sonunda bu durum en militan baptistler arasında
dahi orijinal görüşe yönelik bir skeptisizmle birlikte görüşün
revizyonu taleplerinin ileri sürülmesine neden olmuştur.
Bu kafa
karışıklığının nedeni AH’nin yukarda
özetlenmeye çalışılan sürekli değişen dinamik
doğasına karşın hastalığı tanıma,
„tetikçilik“ kavramını anlama gibi bir dizi etmene indirgenebilir.
Her şeyden önce şimdiki durumda Alzheimer hastası
dendiğinde demanslı bir kişi anlaşılmakta ve bu türden
çalışmalar demanslı hastalar toplanarak yapılmaktadır.
Daha önce gözden geçirildiği gibi demans AH’nin oldukça ileri bir evresine
karşılık gelmektedir ve bu ileri evrede amiloidle başa
çıkmak yeterli olmamaktadır. Alzheimerli beyin, orduların
sürekli yeniden mevzilendiği bir savaş alanı gibi
düşünülebilir. Demans aşaması bu savaşın artık
büyük ölçüde kaybedildiği bir evreye karşılık gelmektedir.
Öyle görünüyor ki bu savaşın başlangıcında
savunmanın henüz başarılı olduğu dönemde kolay degrade
edilebilen Aβ40’ın güvenlik kuvvetleri olarak
düşünülebilecek şaperon moleküller aracılığıyla
klirensini sağlamak ve kolay başa çıkılamayan daha
saldırgan Aβ42’yi gevşek plaklarda hapsetmek temel
savunma stratejisidir. Böylesi bir „çatışmanın“ henüz hiç bir
kognitif yakınması olmayan bir yaşlının beyninde
sürmekte olduğu ileri sürülebilir (AH’nin pre-klinik evresi). İlk
savunma mevzilerinin düşmeye başlamasıyla saldırganın
Aβ (3(pE)-42 gibi daha elit birlikleri hücre içine girip sinapsları
öldürmeye başlayacak ve bu kez savunma bunları ölü sinaps
artıklarını da içeren daha maliyetli ekstraselüler nöritik
plaklara ve intraselüler agrezomlara hapsetmeye çalışacaktır. Bu
aşamada savunma da insülin, sirtuin, WnT, sRAGE gibi kendi elit
birliklerini devreye sokarak saldırıya göğüs germeye
çalışacaktır. İlk savunmanın böylesi çökmesinin
karşılığı AH’nin pre-demans veya MCI aşaması
olarak düşünülebilir. Bu ikinci savunma mevzisi de çökecek olursa bu kez
tau da saldırganın 2. elit birliği olarak devreye girecek ve en
başarılı olduğu savaş alanı olan limbik sistemden
başlayarak nöronları öldürmeye başlayacaktır. Bu üçüncü
aşama AH’nin demans evresidir ve tau yeni mevziler kazanıp
yayıldıkça savunma da giderek şiddetlenen demans maliyetini
ödemek pahasına, sonuna kadar direnebileceği en tahkim edilmiş
bölgeleri olan primer sensori-motor alanlara doğru geri çekilecektir. Bu
senaryoya göre bir „tetikçi“ tanımlamak gerekiyorsa bu Aβ değil
fakat nöronun başlıca katili olan taudur gibi durmaktadır.
Aβ’yı ise tetikçiyi tutan sponsor olarak düşünebiliriz. Bu
senaryoya göre AH’den korunmanın yolu sponsorun tetikçiyi bulmasına
engel olmak gibi durmaktadır. Öyleyse, hastalığı demans
öncesi aşamasında, hatta pre-klinik döneminde tanımak artık
bir zorunluluk olmuştur. Öyle görünüyor ki AH’nin bu demans öncesi
evreleri yeterli duyarlılık ve özgüllükte belirlenip
tanındıktan sonra çeşitli anti-amiloid stratejileri bu erken
aşamalarda denemek, bu ilk savunma kaybedildiğinde veya zaten
hastalık gecikmiş demans aşamasında
tanındığında bu kez başlıca anti-tau stratejilere
yönelmek gerekecektir. Önümüzdeki onyılın genetik ve çevresel olarak
AH riski taşıyan normal yaşlı bireylerin ve gerçekte
prodromal AH olan AAMI ve MCI’lı bireylerin biyo-işaretleyici
kombinezonlarıyla hassasiyetle ayırtedilip anti-amiloid tedavi klinik
çalışmalarına dahil edileceği ve AH demansı için
başarılı anti-tau ajanların geliştirileceği bir
dönem olacağını ileri sürmek bir kehanet
olmayacaktır.
Mesulam, AH’nin nedenselliği için baptizm ve
tauizm dışında kalan ve hastalığın
yaşlılık riskiyle nöroanatomik yatkınlık
tarzını gayet iyi açıklayan “nöroplastisite yetmezliği” teorisini
ileri sürmüştür (Mesulam, 2000, Mesulam, 1999). Bu hipoteze göre AH özetle,
genetik ve çevresel etkilerle yaşam boyunca artarak biriken plastisite
yükünün artık kaldırılamaz olması ile normal
yaşlanmadan sapmanın sonuçlarıdır.
Nöroplastisite, MSS’nin yeni yaşantılama
ve doku hasarına karşı verdiği yapısal tepkileri
içerir. Nöronun post-mitotik bir hücre olması dolayısıyla,
hücre yenilenmesi özelliği olmayan MSS’de ontogenezin intra-uterin ve
doğum sonrası ergenlik boyunca da sürecek l dönemlerinde büyük
bir dinamizm görülür. Genetik olarak belirlenen ve sınırlanan
bir çerçevede, daha intra-uterin dönemden başlayarak çevresel
uyaranların modülatuar etkisi altında nöral gelişimin temel
mekanizmaları olan nörogenez, nöral migrasyon, nöral eliminasyon ve
stabilizasyon, sinaptogenez ve aksonal miyelinizasyon ilerleyici bir şekilde
tamamlanır. Erişkin MSS’ye ait mimari tamamlanmadan önce,
nöroplastisitenin nerdeyse sınırsız potansiyeli ile, genetik
olarak verilenin çerçevesi içine, o bireyin maruz kaldığı, hiç
bir zaman başka bir bireyle tam anlamıyla örtüşmeyecek olan,
emsalsiz yaşantı parçalarının temsili, yukarda anılan
mekanizmalarla, kişiliği de oluşturmak üzere
yerleşir. Erişkin MSS’nin plastik dinamizmi gelişimsel
aşamadaki düzeyini yitirir. Gelişimsel dönemde her kritik yeni
yaşantılama kişiliği oluşturacak yeni bir yapıtaşı
olarak MSS’ye kaydedilirken, erişkin dönemde artık kritik yaşam
deneyimleri bu denli büyük değişikliklere yolaçmaz. Gelişimsel
dönemde bir hemisferektomiyi tolere edebilecek durumda olan MSS, erişkinliğe
ulaştıktan sonra bir hemisferin çok daha sınırlı bir
alanındaki hasar sonucu ağır düzeyde sakatlanabilir.
Yine de, nöroplastisite erişkin dönemle birlikte tümden durmaz. Yeni
öğrenme, aşınma ve hasar onarımı amaçlı olarak
MSS’nin belli bölgelerinde daha belirgin olmak üzere devam eder. Bunlar
aksonal tomurcuklanma, dendritik dallanma, yeni sinaps oluşumu
(sinaptogenez) ve varolan sinapsın yeniden şekillenmesi (sinaptik
“remodeling”) ve uzun süreli potensiasyon (LTP) olarak
sıralanabilir. Bu mekanizmalar yeni yaşantıların
temsili ihtiyacına karşı yaşantılama
bağımlı plastisite ile aşınma, yıpranma ve hasara
cevap olan reaktif veya kompansatuar plastisitenin temel
mekanizmalarıdır.
Mesulam varsayımını bilinen AH ile
ilintili tüm risk faktörleri ve patolojik ürünlerin nöroplastisiteye müdahale
ettiğine ilişkin delillerle desteklemektedir. Nöroplastisite
potansiyeli, AH’de NFY oluşumunun yayılımı ile örtüşür
biçimde en yüksek limbik sistemde iken, azalan sırayla paralimbik
yapılar, asoasiasyon neokorteksi ve en az da primer sensoryel-motor
korteksler şeklinde kendini gösterir. MAP-τ üretimi ve
fosforilizasyonunda artış plastik değişiklikler sürecinin
temel işlemlerindendir. Özellikle plastisite yükünün yüksek
olduğu bölgelerde kinaz-fosfataz döngüsünün dengesinin bozulması
plastik talep karşısında fosforillenmiş taunun görevi
bittiğinde defosforilizasyonunun sağlanamayarak yukarda
sözedildiği gibi patolojik katlanmasına ve birikim eğilimi
gösteren oligomerlere dönüşmesine neden olmaktadır. APP,
daha önce de sözedildiği gibi, erişkin beyninde başlıca sinaptik
bütünlüğün korunması olmak üzere, nöroplastisitedeki rolü deneysel
olarak gösterilmiş bir membran proteinidir. APP proteolizi sonucunda
α-sekretaz ile kesilen ürün olan sAPP’nin de nörit büyümesi ve LTP’deki
rolü bilinmektedir. Buna karşılık, β sekretazla
alternatif kesim sonucu oluşan Aβ fragmanı nörotoksiktir,
aksonal tomurcuklanma ve LTP’yi inhibe eder. β-sekretazın
distal amino terminal ürünü olan sAPPβ’nın da akson budanması
tarzı anti-plastisite etkisinden yukarda sözedilmişti. PS1’in, nörogenezde
çok önemli işlevi olan Notch proteini ile etkileşerek işlev
gördüğüne de değinilmişti. Bu etkileşimin erişkin
beyninde plastisite yükseltici etkileri gösterilmiştir. PS1 ve PS2
mutasyonları APP proteolizini Aβ yoluna kaydırmak yanı
sıra, Notch ile anılan etkileşimi gösteremeyen bozuk ürünlere
yol açarak da plastisite potansiyelini baskılıyor olabilir.
Kolesterol ve fosfolipidlerin transselüler naklinde rol oynayan ve
dolayısıyla aksonal büyüme ve sinaptogeneze katkıda
bulunduğu düşünülebilecek ApoE’nin plastisite ile
ilişkisine daha önce değinilmişti. Buna göre ApoE-ε4
plastisiteyi baskılarken, ApoE-ε3 ve ε2 yükseltici etki
göstermektedir. Yine östrojenin nörotrofik etkilerine
değinilmişti. Yaş AH için temel risk
faktörüdür. Uzun ömür doğal olarak hem yaşantılama
bağımlı plastisite, hem de aşınma, yıpranma ve
hasarlanmanın artmasıyla kompansatuar plastisite ihtiyacını
arttırmaktadır.
Dolayısıyla da yaşla birlikte artan
bu ihtiyacı karşılamak için gerek τ fosforilasyonunda ve
gerekse de APP ekspresyonundaki adaptif artışla birlikte denge ne
kadar plastisite yükseltici faktörler lehine ise o kadar AH patolojisinden
korunulabilmekte, tersine durumda ise bir yandan serbest τ polimerlerinden
NFY oluşumu, diğer yandan da Aβ’nın önce gevşek
plaklarda birikmesi, sonrasında da bunların katı plaklara
dönüşmesi artmaktadır.
Çağdaş uygarlık tek bir bireyin
optimal adaptasyonu için yüksek bir kognitif kapasite, özellikle de bellek
kapasitesi talep etmektedir. Çağdaş insan genetik
yapısını, o dönemin doğal çevresine adaptasyon için
seleksiyona uğramış 30.000 yıl önceki
avcı-toplayıcı uygarlığının Homo
sapiensinden miras almıştır. Avcı-toplayıcı
uygarlığına kıyasla gerek bireysel olarak uzayan ömür ve
gerekse de sosyal olarak nicel ve nitel açılardan olağanüstü
değişen koşullar nöroplastisite yükünü
taşınılamaz hale getirerek AH’yi modern insanlık durumunun
kaçınılmaz bir eşlikçisi yapmaktadır. Bu perspektifle
bakıldığında yukarda resmedilen savaş senaryosunun
tarafları da pro-plastisite ve anti-plastisite güçler olmaktadır.
Dolayısıyla, nöroplastisite yüküne odaklanılmayan uzun ömür veya
AH kesin tedavisi düşleri de anlamsızlaşmaktadır.
TEDAVİ
AH’de mevcut ve deneysel tedavilere toplu bir
bakış için Tablo 16’ ya bakınız.
Tablo 16.
Alzheimer
hastalığının yerleşik ve potansiyel tedavi
stratejileri
Kognitif Semptomatik Tedavi
Halen tedavinin çekirdeğini
kolinesteraz inhibitörleri (ChEİ’ler) ve memantin ile kognitif semptomatik
tedavi oluşturmaktadır.
Kolinesteraz İnhibitörleri
Takrinin (Cognex®) 1993
yılında Amerika Besin ve İlaç İdaresi (FDA) tarafından
Alzheimer hastalığının tedavisi için
ruhsatlandırılması o zamana kadar tedavi edilemez kategoride
kabul edilen bir hastalık için ilk onaylanmış tedavi olması
anlamına geliyordu. AH tedavisinde bir ilacın FDA onayı
alması için yalnızca bir kısım kognitif sonlanım
ölçütünde değil, klinik olarak da gözlenebilir bir etkisinin olması
(GYA’lar ile davranış üzerine ve genel klinik izlenim olarak) ve bu
etkisini en az 6 aylık bir süre içinde korurken güvenilir de olması
gerekmektedir. Bu ruhsat takrinin 1987’de başladığı
klinik çalışmalar sınavlarından sonunda geçtiğini
sembolize ediyordu aynı zamanda. Ancak bir yandan gastro-intestinal
yan etkilerinin şiddeti etkili doza çıkmayı
sınırlarken, bir yandan da hepatotoksisite nedeniyle haftalık
karaciğer enzimleri taraması zorunluluğu kullanımı
iyiden iyiye güçleştirmekteydi. 1990’ların 2. yarısından
itibaren yan etki profili açısından daha kullanışlı
ChEİ’lerin birbiri ardına ruhsatlandırılması takrinin
piyasa ömrünün çok kısa olmasına neden oldu. Yine de AH
tedavisinde “ilk göz ağrısı” olma onuru ebediyyen takrine ait
olacaktır.
Donepezil (Aricept®), rivastigmin
(Exelon®) ve galantamin (Remynil®) sınanan çok
sayıda ChEİ’den şimdiye kadar ruhsatlananlardır.
Bunların hepsi de ülkemizde mevcuttur ve ilk ikisinin çok sayıda
jenerik muadili de çıkmıştır. Şimdiye kadar piyasaya
verilmiş olsun olmasın klinik çalışmalarda düzgün biçimde
sınanan tüm ChEİ’ler başarılı olmuştur. Bütün
CHEİ’ler için klinik çalışmalarda genel olarak söylenebilecek
olan ortak etki plaseboya göre anlamlı farklılığın
yanısıra, sonlanım ölçütlerinin zaman içindeki değişim
eğrileri izlendiğinde 6. ayda plasebo grubu kötüleşmişken,
kötüleşmeksizin hala başlangıç noktasında olma ve 1.
yılda başlangıç noktasına göre kötüleşmeye
karşın ChEİ öncesi dönemin uzunlamasına izlemlerdeki
bozulma hızlarına referansla (”tarihsel plasebo”) daha iyi durumda
olmaktır. Plasebo grubundayken 6. ayda açık etiketli uzatma
evresinde aktif ilaca geçirilen hastalar başlangıçta düzelseler de
sonrasında bozulmaya başlarlar ve eğrileri hiç bir zaman
baştan beri aktif grupta olanların eğrisini kesmeden izlem
boyunca paralel olarak bozulurlar. Dolayısıyla, ChEİ’lerin
hastaya yaklaşık 1 yıl kazandırdığı ve
tanı konulduktan sonra ilaca ne kadar erken başlanırsa o kadar
iyi olduğu söylenebilir. Donepezil (FDA onayı 1996) günde tek
doz (prospektüs bilgisi akşamları önerirken biz birimimizde bazen
uykuyu bozması ve fazik fizyolojik kolinerjik aktiviteyle uyumu nedeniyle
sabahları tercih ediyoruz), 5 ila 10 mg tabletler şeklinde
kullanılan selektif bir ChEİ’dir. Her 2 dozu da etkin olmakla
birlikte yüksek dozun etkinliği de daha yüksektir. Başta GİS’e
ait olanlar olmak üzere, yan etkileriyle baş edebilmek için 4 haftada
titrasyonla yüksek doza çıkmak uygundur. Yakın tarihlerde piyasaya
verilen ağızda eriyen şekli yutma güçlüğü de olan özellikle
ileri evre hastalarda yararlı olmaktadır. Rivastigmin (FDA onayı
1997) 1.5, 3, 4.5 ve 6 mg’lık kapsüller şeklinde
piyasadadır. Bütiril kolin esteraz (BuChE) inhibisyonu etkisi de
olan ve günde iki kez kullanılan rivastigminin en düşük etkili dozu 6
mg/gün olmak üzere, 4’er haftalık titrasyonlarla 12 mg/gün’e yükseltilmesi
hedeflenmektedir. Renksiz ve kokusuz sıvı şekli ilaç
kullanmayı reddeden hastalarda kullanımı
kolaylaştırmıştır. AChEİ’lerin kafa kafaya
karşılaştırıldığı en kaliteli çalışma
olan, büyük boyutlu ve uzun süreli EXCEED isimli çalışmada ikinci
yılın sonunda rivastigmin 2 ikincil sonlanım ölçütünde (demans
evrelendirmesi ve GYA’lar) donepezile üstün bulunmuşsa da donepezil de ilk
4 aylık titrasyon döneminde rivastigmine kıyasla anlamlı ölçüde
daha iyi tolere edilmiştir. Birincil sonlanım ölçütü (kognitif)
açısından her 2 ilacı kullananların da yaklaşık
%35’i 2. yılın sonunda başlangıç noktalarından daha
iyi durumdaydılar. Rivastigmin yakınlarda deriden emilen bant
formunun oral formuna eşdeğer etkinliği ve plaseboya yakın
tolerabilitesini gösterdiği IDEAL çalışmasıyla kayda
değer bir yenilik getirmiş oldu. İlki 6mg/gün, ikincisi 12mg/gün
klasik dozlarına eşit 5 ve 10cm2 dozlarıyla piyasada
olan bant şekli de 4 haftada titre edilmelidir. Galantamin (FDA
onayı 2001) 8, 16 ve 24 mg/gün aralığında günde iki doz
şeklinde kullanılan ve pre-sinaptik allosterik nikotinik reseptör
modülasyonu etkisi de olan bir ChEİ’dir. Önce sıvı
şekli, daha sonra da yavaş emilen kapsül formu piyasaya
verilmiştir. Sıvı form hala günde 2 doz şeklinde 4’er
haftalık titrasyonla önce etkisiz 8mg/gün ile başlanıp, 4 hafta
sonra etkin olduğu ilk doz düzeyi olan 16mg/gün, son olarak da kompliyans
sağlanabildiği takdirde 24mg/gün düzeyine
çıkılmalıdır. Kapsül formu günde tek doz benzer titrasyon
şemasıyla arttırılmalıdır. En düşük dozdan
başlayıp 4 haftalık aralıklarla yükselindiğinde üçü
için de ortak problem olan bulantı-kusma ile başa çıkmak daha
kolay olmaktadır. ChEİ’ler hafif ve orta evre AH’de
kullanım için ruhsatlanmış olsa da ağır evrede
yararlarına ilişkin gerek klinik çalışma ve gerekse de
kendi deneyimizden anekdotal deliller bulunmaktadır.
İlaçların etkisizlik ve kesilme kararlarına ilişkin çok
aşikar göstergeler olmasa da, düzenli kullanılan ilaca
karşın yıl içinde sürekli kötüleşme gözlemleri sonucunda
kesme kararı verilebilir. Yine gerek bizim deneyimimiz ve gerekse
birinden diğerine geçiş verileri (”switch data”), ilaçlardan biri
kesildiğinde diğerinin kullanılmaya başlanmasının
hastaların %50’sinde yararlı olabildiğini göstermektedir
.
ChEİ’lerin non-kognitif ya da
davranışsal belirtiler üzerindeki olumlu etkileri de
bildirilmektedir. Buna bir sonraki bölümde değinilecektir.
ChEİ’ler büyük ölçüde hafif-orta evre hastalarda
çalışılmış ve bu evrelerde kullanım için
endikasyon almışlardır. Bununla birlikte, orta-ağır
evre çalışmaları da genellikle pozitiftir. Günümüze kadar
biriken verilerin kapsamlı bir gözden geçirmesi için Cochrane
veritabanına bakınız.
Memantin
ChEİ’lerden sonra bu kez farklı bir etki
mekanizmasına sahip olan bir ajan daha onay almış, böylece
kolinerjik nörotransmisyonun yanısıra glutamaterjik nörotransmisyonun
da NMDA reseptör modülasyonu aracılığıyla düzenlenmesinin
AH’de bir tedavi stratejisi olabileceği
kanıtlanmıştır. Memantin (FDA onayı 2003) düşük
afiniteli bir NMDA reseptör antagonistidir. Memantinin bozuk Alzheimer
sinapsında post-sinaptik NMDA reseptöründe magnezyum tıkacı
görevi yaparak patolojik intraselüler kalsiyum birikimini engellediği
bilinmektedir. Buna karşılık, NMDA antagonizmini geri
dönüşümsüz değil fakat düşük afiniteyle
yaptığından, NMDA reseptörleri aracılıklı normal
glutamaterjik nörotransmisyon için gerekli olan depolarizan bir uyaranla
magnezyumun yapacağı gibi reseptörü terketmekte ve böylelikle de LTP
mekanizmasına engel olmamaktadır. LTP bozukluğunun AH
fizyopatolojisinin erken ve temel özelliklerinden biri olduğuna yukarda
değinilmişti. Memantinin 10mg’lık tablet şeklinde piyasaya
verilmiştir ve günde 2 doz kullanılacak şekilde, haftada
5mg’lık artışlarla 4. haftada 20mg/gün dozuna ulaşılır.
Sonradan bu günlük dozun gece tek doz verilmesinin de tolerabiliteyi
bozmadığı, buna karşılık bir kısım
hastada görülebilen gündüz sedasyonuyla başa çıkmayı
kolaylaştırdığı görülmüştür. İzleyerek,
memantinin de sıvı formu 10 damla = 5mg olacak şekilde
geliştirilmiş ve ChEİ’lerde olduğu gibi yutma güçlüğü
ve ilaç reddi olan hastalarda kullanım kolaylığı
sağlamıştır. Yine de günlük toplam doza 40 damlayla
ulaşılması bir güçlük nedeniyken bu sorun da yeni piyasaya
verilen pompalı sıvı formuyla aşılmış gibi
görünmektedir. Bu yeni formda bir bardak su içine akacak şekilde pompa
düzeneğine bir kez basış 5 mg eşdeğeri memantin
sağlamaktadır. ChEİ’lerin olduğu gibi memantinin de
non-kognitif, davranışsal etkileri mevcuttur ve bunlara
aşağıda değinilecektir. Günümüze kadar biriken verilerin
son sistematik metaanalizi için Cochrane veritabanına bakınız.
AH’de mevcut tedaviler olan ChEİ’ler ve
memantinin etkileri genel olarak mütevazi görünse de bu tedavilerin
erişilebilir olmasından itibaren hasta ve yakınlarının
yaşam kalitelerine katkıları olduğu kuşkusuzdur. Bu
yönde özellikle de endüstri sponsorluğunda olmayan, ABD’nin 2 ayrı
mükemmeliyet merkezi diyebileceğimiz Alzheimer merkezinden gelen 2
bağımsız çalışmanın verileri oldukça önemlidir.
Bunların ilkinde Boston, Massachusetts General Hospital Alzheimer
Merkezi’nin 1990-2005 yılları arasındaki verileri analiz
edilmiş. Bu 15 yılın başları ilaçsız dönem,
ortaları sadece ChEİ’lere erişilebilen dönem ve sonları ise
ChEİ memantin kombinasyonunun mümkün olduğu dönemdir. Bu
çalışmanın sonuçları, ortalama 30 ay izlenen 382 hastada, kognitif
ve fonksiyonel sonlanım ölçütleri açısından solo ChEİ
kullanan grubun ilaçsız gruptan daha iyi, ChEİ+memantin kombinasyon
tedavisi kullanan grubun ise her ikisinden de daha iyi olduğu yönündedir.
İkincisinde Pittsburgh Üniversitesi Alzheimer Merkezi’nin 1983-2006
arası verileri analiz edilmiştir. Bu 23 yıllık dönem de
benzer şekilde ilaçsız, tek başına ChEİ’li ve
kombinasyon tedavili dönemler olarak ayrılabiliyor. Bu kez bakımevine
yatırılma bir sonlanım ölçütü olarak alınmış ve
ortalama 60 ay izlenen hastalarda solo ChEİ’nin ilaçsız gruba göre bu
süreyi geciktirdiği, kombinasyon tedavisinin ise yine her ikisine göre
daha dageciktirdiği gösterilmiştir .
Non-Kognitif Semptomatik Tedavi
ChEİ’lerin özellikle halüsinasyon ve
hezeyanlar şeklindeki psikotik belirtilere, ajitasyon ve apatiye iyi
gelebildiği gösterilmiştir. Benzer şekilde, memantinin de
psikotik belirtilere ve özellikle de ajitasyona iyi geldiği
gösterilmiştir, Biriken verilerin analizi memantinin mevcut ajitasyonu
kontrol altına alabildiği gibi uzunlamasına izlemde ilaç
kullanan hastalarda daha az ajitasyon ortaya çıktığı
gözlemlenmiştir. Dolayısıyla da, bu anılan belirtilerin
şiddetleri anti-psikotiklerle derhal müdahaleyi gerektirmediği
durumlarda yeni başlanılan bir ChEİ’nin ve/veya memantinin
onların üzerine muhtemel etkisini değerlendirmekle gereksiz ilaç
kullanımının da önüne geçilmiş olur.
Depresyon, mani ve anksiyete gibi duygudurum
bozuklukları AH’de sıktır. Depresyon tedavisinde öncelikle
bu yaş grubunda sık kullanılan ve depresyonu bizzat ortaya
çıkarabilecek veya şiddetlendirebilecek olan reserpin,
α-metil-dopa, β-blokerler gibi anti-hipertansifler kesilerek kalsiyum
kanal blokerleri veya ACE inhibitörleri gibi daha uygun ilaçlara geçilmelidir.
Trisiklik anti-depresanlar anti-kolinerjik yan etkileri nedeniyle seçilmemesi
gereken gruptur. SSRI grubundan ilaçlar, venlafaksin, mirtazapin,
duloksetin gibi yeni kuşak hem serotonin, hem de norepinefrin
gerialım blokeri olan (SNRI) anti-depresanlar güvenle kullanılabilir.
Tam bir mani oldukça seyrek olsa da emosyonel labilite, iritabilite ve öfori
gibi özellikleri bazen tedavi gerektirebilir. Bu durumda lityum,
karbamazepin ve valproat gibi duygudurum stabilizatörleri
kullanılabilir. Lityum ve valproatın GSK3β inhibitörü
olarak potansiyel işlevleri klinik çalışmalarda
sınanmaktadır. Anksiyete tedavisinde çok gerekmedikçe kognitif
depresan etkileri nedeniyle benzodiazepinlerden
kaçınılmalıdır. SSRI’lar ve trazodon bu amaçla
öncelikle seçilebilir. Etkisiz kaldığında lorazepam
(Ativan®) veya alprozalam (Xanax®) gibi kısa etkili
benzodiazepinler seçilebilir.
Hezeyan ve halüsinasyon gibi psikotik belirtilerin
tedavisinde ekstrapiramidal ve anti-kolinerjik yan etkilerden
arınmışlık başlıca tercih nedenidir. Bu
nedenle atipik nöroleptikler öncelikle tercih edilenlerdir. Risperidon
(Risperdal®) ve olanzapinin (Zyprexa®) ekstrapiramidal
yan etki potansiyelleri onları gerçek anlamda birer atipik olarak
sınıflamayı engellemektedir. Risperidonun aktif metaboliti
olan paliperidol (Invega®) yakın zamanlarda piyasaya
verilmiştir ve tek doz kullanılabilen bu ajanın gerçek bir
atipik olduğu, kilo aldırmadığı ve sedasyona neden
olmadığı ileri sürülmektedir. Klozapin (Leponex®)
çok etkili bir atipik olsa da kemik iliği baskılama potansiyeli
nedeniyle haftalık lökosit sayımlarının getirdiği
güçlük kullanımını sınırlamaktadır.
Atipikler arasında ketiapin (Seroquel®) diğerleri için
sıralanan kısıtlamalardan arınmış ve geniş
bir güvenlik aralığında rahatlıkla
kullanılabilmektedir. Diğerleriyle kıyaslanamayacak kadar
ucuz olması düşük dozlarda haloperidolü (Norodol®) tipik
bir nöroleptik olsa da hala bir seçenek olarak korunmasına neden
olmaktadır. Demansta psikoz tedavisinde çok düşük dozlarda
başlayıp optimal dozu olguya göre uyarlamak esastır. Dozun
tümünü ya da büyük bölümünü gündüz sedasyonundan kaçınmak amacıyla
geceleri vermek uygun olur. Ketiapin 50 ila 100 mg’lık dozlarda
genellikle etkin olurken, 400 mg/gün düzeylerine çıkarmak da gerekebilir.
Risperidon 0.5 ila 1 mg, seyrek olarak da 2 mg/gün dozlarında
kulanılır. Paliperidol ile demanslı popülasyonda yeterli tecrübe
olmamakla birlikte en düşük dozu olan 3 mg denebilir. Olanzapin dozu 2.5-5
mg, bazen 10 mg/gün’dür; bir klinik çalışmada 15 mg/gün’den itibaren
etkinliğinin paradoksal olarak düştüğü
gösterilmiştir. Klozapin 6.25 mg ila 50 mg/gün dozlarında
yeterli etkinlik gösterse de bazen 200 mg/gün dozlarına kadar çıkmak
gerekebilmektedir. Haloperidol dozu 1 ila 3 mg/gün arasında
değişebilir ve damla şeklinde kullanımı bir avantaj
olarak düşünülebilir.
Ajitasyon ve saldırganlığın
tedavisinde yine atipik nöroleptikler ilk seçimlerdir. Ajitasyon için bazen tek
başına trazodonla başlayıp cevap alınmazsa
nöroleptikleri tercih etmek bir seçenek olabilir. Karbamazepin, valproat
ve lityum da tek başına ya da ilave tedavi olarak denenebilir.
İmpulsivite ve dürtü kontrol
kusurlarının tedavisinde öncelikle SSRI’lar
denenir. Hiperseksüalite için ayrıca anti-androjen ajanlar
kullanılabilir.
Uykusuzluk tedavisinde ilaçtan önce yatmadan bir
süre önce içilecek ılık süt önerilmelidir. Gerçekte bir
anti-depresan olan trazodon (Desyrel®) 50-200 mg dozları
arasında yatmadan 1 saat önce verildiğinde oldukça etkin bir sedatif
etki gösterir. Mirtazapin (Remeron®) yatmadan önce ½ tablet (15mg)
dozunda benzer etkiye sahiptir.
Non-Farmakolojik Tedavi
Tedaviye non-farmakolojik yaklaşım hasta
ve hasta yakını ile kurulan ve hastalıkla başa çıkmaya
yönelik ilaç dışı etkileşimin bütününü içermektedir.
Bunlar farklı başlıklarda
sıralanabileceği gibi hastalık evrelerine göre de
farklılık gösterir. Evresel perspektif şöyle
özetlenebilir:
Erken evrede hastalık hasta ve hasta
yakınına kültüre uyumlu bir şekilde anlaşılabilir bir
şekilde aktarılır ve hastalığın mümkün
olduğunca ayrıntılı bir şekilde kavranacağı
internet, Alzheimer dernek ve vakıflarının ve benzeri
kaynakların dergi, broşür, kitap gibi bilgilendirici
yayınlarının okunması teşvik edilir. Hastayla
vesayet meselelerini de içerecek şekilde, geleceğin gerçekçi bir
biçimde tasarlanmasına yönelik tartışılmaya gayret
edilir. Hobiler ve sosyal ilişkileri sürdürmenin önemi
aktarılır. Araba kullanmayı sürdürmenin
sakıncalarından sözedilir. Hasta yakınına destek grupları
hakkında bilgi verilir ve dernek aktivitelerinde yer alması
doğrultusunda cesaretlendirilir.
Orta evrede hasta ve hasta
yakınına yalnız sokağa çıkmanın
sakıncaları anlatılır. Mali ve bunun gibi hukuksal
işlerde artık vesayetin zorunluluğu aktarılır.
Ev aygıtlarını kullanmadaki güçlükler ve kaza ihtimalleri gözden
geçirilir. Giyinmek, yıkanmak ve sofra davranışı gibi
güçlükler ele alınır. Artık yalnız
yaşamaması yönünde teşvik edilir ve alternatifler ele
alınır. Psikotik belirtiler mevcutsa çevresel müdahalelerle
başa çıkılmasına gayret edilir. Sfinkter kontrolünü
arttırıcı yöntemler üzerinde durulur. Bu tür
kuruluşlar o çevrede mevcut olduğunda gündüz bakım evlerinin
değerlendirilmesi teşvik edilir. Hasta yakını yükü
gözden geçirilir ve muhtemel bir depresyona karşı önlem
alınır.
İleri evrede hasta yakınıyla
suçluluk duyguları, hastalık yükü ve kayıp üzerine
tartışılır. Evde profesyonel yardım ve bir ötesi
olarak kalıcı bir bakımevine nakil üzerine
tartışılır ve teşvik edilir. Yatak yaraları,
kontraktürler, üriner ve solunum sistemi infeksiyonları riskleri konusunda
uyarılır. Sonda ile beslenme gerekliliği ele
alınır. Nihayet terminal bakım ve ölüm meseleleri üzerinde
durulur.
Spesifik müdahale alanları ise
aşağıdaki şekilde sıralanabilir:
İşlevsel performans açısından
sözel ipuçlarından fiziksel olarak göstermeye kadar değişen
kademeli yardımın demanslı hastanın günlük
performansını arttırdığı
gösterilmiştir. Davranış modifikasyonu ile tuvaletin
uyarılması veya saatlerinin önceden belirlenmesi kuru kalmayı
teşvik eder. Uğraşı rehabilitasyonu kognitif performans,
psikososyal işlevsellik ve emosyonel dengeyi düzeltir.
Işığın azaltılması ve seslerin sükünet verici bir
duruma getirilmesi gibi çevresel manipülasyonların yeme
davranışını arttırdığı
gösterilmiştir.
Davranışsal problemler açısından
banyo ve sofra sırasında müziğin ajitasyon ve
saldırganlığı azalttığı
gösterilmiştir. Mevcudiyetin simülayonu tedavisi adı verilen
aile üyelerinin ses ve görüntü kayıtlarının dinletilmesi veya
izletilmesinin de benzer etkisi olduğuna ilişkin bildirimler
vardır. Yürüyüş ve benzeri hafif egzersizler
gezinme-adımlama davranışının önüne geçebilir.
Psikotik belirtiler saldırganlık, kaçma veya yemeyi reddetme gibi
tehlikeli veya aşırı sıkıntı verici şiddette
değilse yeniden yönlendirme (konuyu değiştirme) en iyi başa
çıkma yoludur. Ajitasyon ve saldırganlık için de yeniden
yönlendirme, dikkatini çelme ve aktivite tedavisi etkili olabilir.
Öncelikle eğer ayırt edilebiliyorsa ajitasyonu tetikleyen olayın
önüne geçilmeye çalışılmalıdır (örneğin, aynadaki
“yabancı” görüntüden ürküp kavga eden hasta için aynaların
kaldırılması). Kahve, çay gibi uyarıcıların
akşam içilmemesine özen gösterilmelidir, onların yerine içilecek
ılık süt uykusuzluğun önüne geçebilir.
Hasta yakınları için hastalık
hakkında kısa ve uzun süreli eğitim programlarının
sıkıntıyı azaltan ve nihai evreyi geciktiren etkileri kesin
bir biçimde gösterilmiştir. Hasta yakını destek
gruplarının emosyonel denge açısından büyük önemi
vardır. Bilgisayar ağları, telefon desteği
eğitim için kullanılacak seçeneklerdir. Gündüz bakım
evlerinin hasta yakını yükü açısından olumlu etkisi
kuşkusuzdur.
ALZHEIMER DIŞI DEMANSLAR
ALZHEIMER DIŞI DEJENERATIF DEMANSLAR
Friedrich Lewy, 1923 yılında parkinsonizmi
olan ve hemen nerdeyse yarısında demansın da var olduğu
küçük bir hasta serisinde daha sonra kendi adıyla anılmaya
başlanan sitoplazmik inklüzyonları tanımlamıştır.
Sonraki 50 yıl içerisinde Lewy cisimciği-demans birlikteliği ile
ilgili sadece tek tek olgu bildirimleri mevcuttur. Ancak, 1980’li
yıllarda, demansı olan bir grup hastada nöropatolojik olarak kortikal
Lewy cisimciklerinin gösterilmesinden sonra, dünyanın farklı
merkezlerinden hasta serileri bildirilir olmuştur.
Lewy cisimcikli demans (LCD) tanımı ilk
kez 1996 yılında şu anki LCD Birliği’nin Birinci
Uluslararası Çalıştayı’nda
kullanılmıştır (McKeith et al. , 1996). Bu
çalıştaydan önce demanstaki Lewy cisimciklerinin
varlığı, diffüz Lewy cisimcik hastalığı,
Alzheimer hastalığının Lewy cisimcikli varyantı
(AHLCV), Lewy cisimcik tipli senil demans ve Alzheimer
hastalığına eşlik eden Lewy cisimcik
hastalığı gibi farklı terimlerle ifade edilmiştir.
Yukarda “Alzheimer Hastalığı” bölümünün “Dopaminerjik
Kayıp” alt başlığında
tartışıldığı gibi AHLCV’yi AH nöropatolojisine
sınırlı limbik LC’lerin (amigdala, peri-amigdalar ve entorhinal
korteksler) eşlik etmesi durumunda kullanıp, neokortikal ve
beyinsapı LC’lere değişen oranlarda AH nöropatolojisinin
(baskın olarak amiloid plaklar) eşlik etmesi durumunda vurgunun LCD
üzerinde olması daha uygun gibi görünmektedir.
Epidemiyoloji
Bazı
merkezlerin demanslar arasında sıklık açısından AH’den
sonra 2. sırayı VaD veya FTD’ye vermelerine karşın LCD’nin
ikinci sırayı aldığı daha yaygın bir kabül görmektedir
(McKeith et al. , 2004). Populasyon temelli çalışmaların
gözden geçirildiği bir meta-analizde LCD prevelansı %0.1-5.0
arasında, demanslar arasında sıklığı ise
%1.7-30.5 arasında değişmektedir. Yukarda AH bölümünde sözedilen
TAPS çalışmasında Kadıköy’de 70 yaş üzeri 1019
yaşlının tarandığı prevelans
çalışmasında saptanan toplam 93 demans olgusunun 70’i PRAD ve
PosAD tanıları alan toplam AH olguları iken LCD (toplam muhtemel
ve mümkün LCD) 9 olgu ile 2. sırada yer almıştı. Otopsi
serilerinde de benzer şekilde bu oran %15 ile 25 arasındadır.
Insidans, Cache County çalışmasında izlenen 65 yaş üzeri
5092 birey arasında yılda 6 yeni LCD olgusu ile %0.1 olarak
saptanmıştı. Cinsiyetin bir risk faktörü olarak niteliği
çok aşikar olmasa da nöropatolojik olarak aynı zamanda kesin AH
tanı kriterlerini dolduran beyinsapı LC’li 85 otopsi olgusunda saf
AH’li 192 olgunun tam tersine erkek baskınlığı
bildirilmiştir. Yukarda söz edilen meta-analizde yaşın LCD için de
bir risk faktörü olduğu, buna karşılık Brezilya ve Sri
Lanka kentleri ile Japonya kırsal alanından aynı prevelans
rakamlarının bildirilmiş olmasına dayanılarak çevresel
faktörlerin rolü olmadığı ifade edilmiştir.
Klinik
Tablo:
LCD de
Alzheimer hastalığına benzer tarzda, senil dönemde sinsi
başlangıçlı, kronik progresif seyirli, genellikle belirgin
görsel-mekansal ve yürütücü fonksiyonlarda bozulmanın ön planda
olduğu bir demans tablosudur. LCD tanısı alan hastaların
kolinesteraz inhibitörlerine yanıtının iyi olması ve
nöroleptiklere aşırı hassasiyetleri nedeniyle farmakolojik
yaklaşım açısından ve aynı zamanda seyrinin ve
prognozunun diğer demans tiplerinden farklı olması nedeniyle
LCD’nin klinik olarak doğru tanınması önemlidir.
Son
yıllarda edinilen bilgilerden sonra, LCD’nin klinik ve patolojik tanı
kriterleri için, McKeith ve arkadaşları tarafından 1996
yılında tanımlanan uzlaşı kriterlerinin kabul
edilebilir bir özgüllüğünün olmasına rağmen
duyarlılığının suboptimal olması ve bununla
beraber temel özelliklerden olan zihinsel işlevlerde dalgalanmanın
tanınmasındaki zorluklar ve temel özelliklerin (görsel
halüsinasyonlar, seyirde dalgalanma ve parkinsonizm) üçünün bir arada
düşük sıklıkta görülüyor olması nedeniyle tanı
kriterlerinde revizyon yapılması ihtiyacı doğmuştur.
McKeith
ve arkadaşları tarafından 2005 yılında yayınlanan
yeni uzlaşı kriterleri Tablo 17’de belirtilmiştir (McKeith et
al. , 2005). Revize edilmiş kriterlerde önceki merkezi, çekirdek ve
destekleyici özellikler kategorilerine klinik ve inceleme bulgularından
oluşan işaretleyici özellikler eklenmiştir.
İşaretleyici özellikler diğer demans tiplerine göre anlamlı
olarak daha sık görülürken, destekleyici özellikler ise sıklıkla
görülür ancak özgüllükleri azdır.
LCD’de
ilerleyici engelliliğin sebebi hem kognitif bozukluğa hem de
nöropsikiyatrik, motor, uyku ve otonomik bozukluklara bağlı
olabilmesine rağmen zihinsel bozulma tanı için gerek
şarttır. LCD demansın kardinal (kognitif ve işlevsel
bozukluk olarak demans ve görsel halüsinasyonlar) ve ikincil alanlarından
(spontan parkinsonizm, REM-uykusu davranış bozukluğu [RUDB],
inkontinans) özelliklerin daha ilk evrelerden itibaren görülebildiği
demanslar arasında en zengin sunuma sahip olanıdır. Merkezi
özelliğe ek olarak üç çekirdek özellikten ikisinin mevcudiyeti “muhtemel
LCD”, birinin mevcudiyeti ise “mümkün LCD” tanısı için esastır
(Bkz. Tablo 17). Bir veya daha fazla çekirdek özelliğin
varlığına, işaretleyici özelliklerden bir veya daha
fazlasının eşlik etmesi durumunda tanı “muhtemel LCD”
olabilir. Buna karşın çekirdek özellliklerden hiçbirisinin
olmaması durumunda, işaretleyici özelliklerden bir veya daha
fazlasının olması ise “mümkün LCD” tanısı için
yeterlidir. Ancak “muhtemel LCD” tanısı tek başına
işaretleyici özelliklere dayanarak konmamalıdır.
Her üç
çekirdek özellik için de yeni kriterlerde fazlaca değişiklik
belirtilmemesine rağmen, tanı ve semptomların şiddetini
değerlendirmek için klinik değerlendirmede daha gelişmiş
metodların kullanımı önerilmiştir.
Merkezi
Özellik
Hastanın
sosyal ya da iş hayatındaki fonksiyonelliğini etkileyen
ilerleyici bir kognitif bozulma olmalıdır. Bu kognitif kayıp
erken evrede özellikle karmaşık dikkat, görsel-mekansal ve yürütücü
işlevlerde bozulma ile karakterizedir. Yürütücü işlevler; planlama,
soyut düşünme yeteneği, yargılama, içgörü, karar verme gibi
amaca yönelik hareketler için önemli olan kognitif işlemleri içerir.
Hastalığın erken evresinde göreli olarak daha az olan bellek ile
ilgili yakınmalar ve muayene bulguları ancak hastalık ilerledikçe
aşikar hale gelir. Episodik bellek ile ilgili testlerdeki
performansları (çoktan seçmeli ile hatırlamaları serbest
hatırlamadan iyidir) AH’si olan hastalara göre daha iyidir. Ayrıca
sözel bellek testlerinde görsel bellek testlerine göre daha iyi performans
gösterirler. Erken ve ön planda görsel-mekansal bozukluk LCD için tipiktir.
MMSE’de AH’ye kıyasla göreli yüksek puan alabilen, hatta demans için kesme
puanının üzerinde kalan bir hastanın kesişen
beşgenlerden puan kaybetmesi özellikle ayıt ettiricidir. Görsel-mekansal
bozukluğun şiddeti erken dönemde posterior kortikal atrofide izlenen
şiddette bir simultanagnoziye neden olabilir.
Çekirdek
Özellikler
Dalgalanma: Klinik pratikte
çekirdek kriterler arasında en zor karakterize edilebileni zihinsel
işlevlerdeki dalgalanmalardır. Bu kriter için
uyanıklık durumu veya kognitif ve işlevsel performansta
dakikalar, saatlerden, günler, haftalara kadar değişen sürelerde,
hasta yakını ve bazen de hekim için aşikar kayda değer
dalgalanmalar geçerli sayılmalıdır. Bununla birlikte dalgalanma
kriterinin varlığı veya yokluğunun saptanmasının
sadece öyküyle değerlendirmeciler arası güvenilirliğinin çok
yüksek olmadığı ve dalgalanmaya özgü geliştirilmiş
ölçeklerin kullanımının önerildiği
unutulmamalıdır.
Görsel
halüsinasyonlar: Hastalığın genellikle erken evrelerinde
görülen tekrarlayıcı canlı, renkli, kompleks görsel
halüsinasyonlar tanı için çekirdek özelliklerden ikincisidir. Görsel
halüsinasyonları olan LCD’li hastaların, halüsinasyonları
olmayan grup ile karşılaştırıldığında
görsel-algısal bozulmaları daha belirgindir. SPECT görüntülemelerinde
oksipital kortekste tutulum azalmıştır. Görsel
halüsinasyonları olan posterior kortikal atrofili (PKA) grubun, olmayan
PKA’lılara göre bunların gerçekte LCD olduklarını düşündürecek
şekilde daha fazla parkinsonizm ve RUDB’ye sahip oldukları
gösterilmiştir. Otopsi çalışmalarında, görsel
halüsinasyonları olan vakalarda, görsel kompleks imajların jeneratörü
olduğu bilinen anterior ve inferior temporal lob ve amigdalada Lewy cisimciklerinin
artmış olduğu bildirilmiştir. Görsel halüsinasyonları
olan hastalarda kortikal asetilkolin defisitleri daha belirgindir ve bu durum
belki de kolinerjik tedaviye daha iyi yanıtın nedeni olabilir.
Parkinsonizm: LCD’de ekstrapiramidal
motor bulguların ağırlığı genellikle demansı
olsun veya olmasın benzer yaş grubundaki Parkinson
hastalarındaki (PH) motor bulguların
ağırlığına benzer şekilde olabileceği gibi
daha geri planda ve hafif de olabilir. PH’nın ayırdedici asimetrik
sunumu belirgin olmayabilir. LCD’de postüral instabilite, yürüyüş
bozukluğu, bradimimi gibi aksiyal tutulum demanssız PH’lılara
göre daha belirgindir. İstirahat tremoru daha az görülür. Parkinsonizmin
L-Dopa cevabı çok iyi olmayabilir. Miyoklonus, tedavi gerektirmeyecek
kadar hafif bir parkinsonizm, tremorun yokluğu, L-Dopa
cevapsızlığı özelliklerinden birinin LCD’de PH’ya göre 10
kat daha muhtemel olduğu bildirilmiştir.
İşaretleyici
Özellikler
REM
uyku davranış bozukluğu: RUDB demans ve
parkinsonizmden çok önce başlamış olabilir. RUDB spesifik bir
sinükleopati (PH, MSA, LCD) eşlikçisidir ve nadiren diğer dejeneratif
hastalıklarda görülür. RUDB LCD’ye eşlik eden aşırı
gündüz uyuklaması gibi diğer uyku bozukluklarıyla birlikte
zihinsel işlevlerdeki dalgalanmalara katkıda bulunuyor olabilir. RUDB
öyküsü polisomnografi ile doğrulanabilir.
Nöroleptik
aşırı duyarlılığı: Nöroleptik
duyarlılık reaksiyonlarıyla birlikte olan yüksek mortalite ve
morbidite nedeniyle, D2 reseptör bloke edici ajanlar tanı veya tedavi
amacıyla kullanılmamalıdır. LCD’li hastalar ve demansı
olsun olmasın PH’lılar özellikle tipik nöroleptiklere
karşı, demansta kullanılmaya alışılan çok
düşük dozlarına dahi aşırı duyarlılık
gösterirler ve ağır, akut bir akinetik-rijid tablo
geliştirebilirler. Bu durum habis nöroleptik sendrom şiddetinde
olabilir ve ketiapin ile dahi bildirilmiştir. Ancak LCD tanısı
alan hastaların yaklaşık %50’sinde tipik veya atipik
antipsikotik ajanlar almalarına rağmen belirgin yan etki
bildirilmemesine dayanarak, nöroleptik duyarlılığının
olmaması LCD tanısını dışlamazken varlığı
ise tanıyı kuvvetle destekler.
Dopamin
taşıyıcısı görüntülemesi: Dopamin
taşıyıcısı fonksiyonel görüntülemesi, nigrostriatal
dopaminerjik sistemin bütünlüğünü tanımlar. FP-CIT, beta-CIT, IPT,
TRODAT gibi spesifik ligandlarla yapılan dopamin
taşıyıcısı görüntülemesi, presinaptik nöronal
dejenerasyon için işaretleyici olabilirler. LCD’de striatal dopamin
taşıyıcısı aktivitesi düşük iken, Alzheimer
hastalığında normaldir. İdiopatik PH, MSA ve PSP’de de
bozuk olabileceği göz önünde tutulmalıdır.
Destekleyici
Özellikler:
Destekleyici
özellikler LCD’de sıklıkla görülür ancak özgüllükleri çekirdek ve
işaretleyici özellikler kadar yüksek değildir.
Hastalığın erken döneminde ortostatik hipotansiyon, üriner
inkontinans, nörokardiyovasküler insitabilite, konstipasyon, impotans gibi
otonomik semptomlar görülebilir. Otonomik disfonksiyon aynı zamanda
bazı hastalarda görülebilen tekrarlayan düşmeler, senkop ve geçici
bilinç kaybının gelişmesine de katkı da bulunuyor olabilir.
Sistematize hezeyanlar ve depresyon hastalığın herhangi bir
döneminde görülebilir. Hezeyanlar içinde misidentifikasyon ve Capgras
hezeyanı özellikle sıktır.
Şu
anki bilgilerin ışığında, LCD tanısını
desteklemek için klinik olarak uygulanabilir genotipik veya beyin omurilik
sıvısı işaretleyicisi yoktur. Beyin manyetik rezonans
görüntüleme (MRG)’de, hippokampal ve medial temporal lob volümlerinin
korunmuş olması, putamende atrofi ve MRG’de oksipital atrofi
olmaksızın SPECT ve PET ile oksipital hipoperfüzyon ve hipometabolizmanın
görülmesi tanı için yardımcıdır. Yukarda söz
edildiği gibi MRG’de PKA tarzı oksipito-parietal atrofi sergileyen
hastalar arasında görsel halüsinasyonlar kuvvetle LCD düşündürse de
rasgele seçilmiş LCD’liler yapısal MRG’de posteriyor atrofi
tarzı açısından ayırt edilemeyebilirler.
[I-123]-metaiodobenzyl guanidine (MIBG) ile yapılan ve postganglionik
sempatik kardiak innervasyon hakkında bilgi veren sintigrafi LCD’de
anormaldir ve AH’den ayırmada özgüllük ve duyarlılığı
yüksektir. Başka bir çalışmada bu ayrımın özellikle de
aşikar ortostatik hipotansiyonu olan hastalarda parkinsonizm
olmaksızın da aşikar olduğu gösterilmiştir.
Sağkalım
AH’ye kıyasla daha kısa gibi durmaktadır. Bu amaçla
yapılmış yakın tarihli büyük bir çalışmada LCD ve
PH demanslı (PHD) otopsi doğrulamalı 243 olgunun retrospektif
analizinde medyan sağkalımın 5 yıl olduğu
bildirilmiştir. PHD’liler LCD’lilere göre daha uzun sağkalıma
sahiptiler. Başlangıçta aşikar dalgalanma kötü sonlanımı
en iyi öngören özellik olarak belirlenmiştir. AH’ye göre sağkalımın
kısalmasının, özellikle motor ve otonom bozukluklar olmak üzere,
LCD’nin zengin klinik sunumunun engelliliği çok daha fazla
arttırması nedeniyle olduğu ileri sürülebilir.
Nöropatoloji
Nöropatolojik olarak tabloya adını veren intraselüler Lewy cisimcikleri
(LC) başlıca özelliktir. LC’ler, intranöronal sitoplazmik,
eozinofilik, sferik, filamantöz inklüzyon cisimcikleridir. LC’lerin temel
bileşeni 4. kromozomda kodlanan ve işlevi çok iyi
aydınlatılamasa da aksonal nakilde rol oynadığı
düşünülen ve ağırlıkla akson sonlanmalarında lokalize
olan, dolayısıyla sinaptik bütünlüğün korunması
şeklinde bir plastisite rolü de olduğu düşünülen
α-sinüklein isimli proteindir. LC’ler, α-sinükleinin anormal
katlanması ve fosforilasyonu sonrasında oluşurlar. α-sinüklein
immün boyaması geliştirildikten sonra hematoksilen-eozin ile
seçilemeyen neokortikal LC’leri de ayırt etmek mümkün olmuştur.
PH’da ağırlıklı olarak subtantia nigranın DA-erjik
hücre gövdelerini içeren pars compactasına sınırlı olan LC
ve Lewy nöritleri, LCD’de beyin sapına ilave olarak limbik-paralimbik
sistem ve neokortekste birikir. Bununla birlikte saf LCD nöropatolojisi
tüm LCD klinik fenotipinin küçük bir bölümünden sorumludur. Klinik olarak
LCD tanısı alan hastaların postmortem
çalışmalarında AH patolojisi de sıklıkla bir
arada görülmekte ve hem nomenklatürü (AH’nin Lewy cisimcikli varyantı,
yaygın Lewy cismciği hastalığı gibi) hem de klinik
fenotipi bulanıklaştırmaktadır. Örneğin, daha belirgin
olarak neokortikal Alzheimer tipi patolojisi olan LCD hastalarında, klasik
LCD klinik bulguları da daha hafiftir. LCD’li hastalarda en sık
görülen Alzheimer patolojisi amiloid plaklar iken NFY daha seyrektir. NFY’ler
görüldüğü takdirde, AH’yle kıyasla daha düşük Braak ve Braak
evrelerinde, ağırlıkla limbik ve paralimbik alanlarda, AP’ler de
nöritik de olsalar, genellikle PHF (-) ve neokortekse sınırlı
olma eğilimi taşımaktadırlar. Ayrıca, LCD’deki
AP’ler çevresinde mikroglial aktivasyon ve inflamasyona da AH’ye kıyasla
çok daha az rastlandığı ileri sürülmektedir. Hippokampal
CA2 ve CA3 bölgelerinde görülen ubikütin pozitif nöritik dejenerasyonun da
AH’de görülmeyen LCD’ye özgü bir patolojik özellik olduğu ileri
sürülmektedir.
LCD nörokimyasal olarak kortikal kolinerjik işaretleyiciler ve
nigrostriatal dopamin kaybı ile karakterizedir. Raphe çekirdeği ve
locus ceruleus’taki nöron kaybı ve gliozis serotonin ve norepinefrin
azalmasına neden olur ve duygu durum, dikkat ve uyanıklıktaki
dalgalanmaları açıklayabilir. Meynert çekirdeğindeki kolinerjik
nöron kaybı da bellek, dikkat, uyanıklığın derecesi ve
belki de dalgalanmaları anlamlı olarak etkileyebilir. Meynert
çekirdeğindeki dejenerasyona ek olarak başlıca işlevi
diurnal ritmin düzenlenmesi olan talamik kolinerjik innervasyonun
kaynağı pedinculo-pontin çekirdek de AH’den farklı olarak
dejenerasyona katılır. Anılan beyin sapı
nörotransmitter kayıplarının LCD’ye özgü parasomni, parkinsonizm
ve otonom sinir sistemi belirtilerinden sorumlu olduğu düşünülmektedir.
ChAT aktivitesi ile belirlenen neokortikal kolinerjik kayıp AH’ye göre çok
daha belirgindir. Bu kaybın genel olarak demans şiddetiyle,
özel olarak temporal lober kaybın ise davranışsal belirtilerle
korelasyonu bildirilmiştir.
Çözünemeyip biriken fibriler α-sinükleinin, LCD’nin yanı
sıra bir dizi dejeneratif hastalıkta daha temel patolojik
işaretleyici olduğunun gösterilmesi sonrasında bu grup α-sinükleopatiler
olarak adlandırılmaya başlanmıştır.
Bunlar arasında LCD’den ayrı bir antite olup olmadığı
tartışmalı olan PH demansı öncelikle sayılabilir.
Kısa süre öncesine kadar bir PH-AH örtüşmesi olduğu
düşünülen idyopatik PH’da sonradan gelişen demansın, artık
beyin sapı tipi LC hastalığının zamanla kortikal tip
LC’ye yaygınlaşması sonucu geliştiğine dair bir
görüş birliği oluşmaktadır. α-sinüklein fibriler
birikimleri MSA’da glial inklüzyonlar şeklinde görülmektedir. Glial
α-sinüklein inklüzyonları LCD ve PH’da geç evrelerde görülürken,
MSA’da klinik fenotipten sorumlu patolojik değişiklik gibi
durmaktadır. Hallervorden-Spatz hastalığında nöronal
ve glial inklüzyonların yanısıra aksonal sferoidler içinde de
görülürler. Bir çalışmada otonomik ganglionlarda ilerleyici
dejenerasyonla giden saf otonomik yetmezlik de bir α-sinükleopati
olarak tarif edilmiştir.
Şekil 18. Lewy
Cisimciği. Solda substantia nigra’da çevresinde halosuyla intranöronal
eosinofilik inklüzyon izlenirken sağda ise kortekste ubikitin (+), halosu
seçilmeyen intranöronal inklüzyon izlenmekte (Kaynak: http://www.path.sunysb.edu/faculty/woz/NPERESS/webclass5.htm)
Tablo 17. Muhtemel ve Mümkün LCD Klinik
Tanısı için Gözden Geçirilmiş Yeni Uzlaşı Kriterleri (McKeith
et al. , 2005)
1)
Merkezi özellik
(Muhtemel veya
mümkün LCD tanısı için gereklidir):
Demans; Normal
sosyal ya da mesleki işlevselliği etkileyecek
ağırlıkta ilerleyici kognitif bozulma. Erken evrelerde
bellek bozukluğu baskın ve kalıcı düzeyde olmayabilir,
fakat ilerlemeyle birlikte aşikar hale gelir. Dikkat,
frontal-subkortikal yetenekler ve vizuo-spasyal işlevleri ölçen
testlerdeki bozulmalar ön planda olabilir.
2)
Çekirdek özellikler
(Üç temel
özellikten ikisinin mevcudiyeti “muhtemel DLB”, birinin mevcudiyeti ise “mümkün
DLB” tanısı için esastır):
a.
Dikkat ve uyanıklıkta ciddi değişikliklerle birlikte giden
zihinsel işlevlerde dalgalanma.
b.
Tipik olarak iyi forme ve ayrıntılı, tekrarlayıcı
görsel halüsinasyonlar
c.
Parkinsonizmin motor özelliklerinin kendiliğinden mevcudiyeti
3)
İşaretleyici özellikler
(Bu
özelliklerden bir veya daha fazlası yine bir veya daha fazla
çekirdek özelliğin varlığında, tanı muhtemel LCD
olabilir. Ancak çekirdek özelliklerden hiçbirisi yoksa, bu özelliklerden bir
veya daha fazlasının olması mümkün LCD tanısı için
yeterlidir. Muhtemel LCD tanısı ise tek başına
işaretleyici özelliklere dayanarak konmamalıdır.)
a.
REM uyku davranış bozukluğu
b.
Ağır nöroleptik duyarlılığı
c.
SPECT veya PET görüntülemeleriyle gösterilebilen bazal gangliada düşük dopamin
taşıyıcı tutulumu
4)
Destekleyici özellikler
(Genellikle vardır fakat tanısal
özgüllüğü kanıtlanmamıştır.):
a.
Tekrarlayan düşmeler ve senkop
b.
Geçici açıklanamayan bilinç kaybı
c. Ağır
otonomik disfonksiyon (ortostatik hipotansiyon, üriner inkontinans gibi)
d.
Diğer modalitelerde halüsinasyonlar
e.
Sistematize hezeyanlar
f. Depresyon
g.
BT/MRG’de medial temporal lob yapılarının göreli olarak
korunmuş olması
h.
SPECT ve PET’de oksipital aktivitede azalma ile birlikte genel olarak
düşük tutulum
i. MIBG
myokard sintigrafisinin bozuk (düşük tutulum) olması
j. EEG’de
belirgin yavaş dalga aktivitesi ile birlikte temporal loblarda geçici
keskin dalgalar
5)
LCD tanısı ihtimali
aşağıdakilerin mevcudiyeti durumunda azalır:
a.
Fokal nörolojik bulgular veya beyin görüntülemesinde serebrovasküler olaya ait
bir delil varsa
b.
Klinik tabloya tamamen ya da kısmen neden olabilecek başka bir
fiziksel hastalık veya beyin bozukluğunun varlığı
c.
Eğer parkinsonizm ilk kez ağır demans evresinde ortaya
çıkarsa
6)
Semptomların zamansal sıralanımı:
LCD
tanısı, demans parkinsonizmden (eğer varsa) önce veya birlikte
ortaya çıkar ise düşünülmelidir. Parkinson hastalığı
demansı (PHD) terimi, yerleşmiş Parkinson
hastalığı bağlamında oluşan demansı
tanımlamak için kullanılmalıdır. Pratik uygulamada, klinik
durum için en uygun terim kullanılmalıdır ve Lewy cisimcik
hastalığı gibi genelleyici terimler sıklıkla
yardımcıdır. LCD ve PHD ayrımı yapılması
gereken araştırma projelerinde ise demansın
başlangıcı ile parkinsonizm arasındaki 1 yıl
kuralı, LCD tanısı için hala önerilmeye devam edilmektedir.
Klinikopatolojik ve tedavi çalışmaları gibi diğer
araştırma projelerinde ise her iki klinik fenotipinin de
alfa-sinükleopati veya Lewy cisimcik hastalığı gibi daha
geniş kategoriler altında toplanmalrı düşünülebilir.
Tedavi
LCD
tedavisi klinisyen için zor ve karmaşıktır. Non-motor semptomlar
için etkili olan tedaviler, motor fonksiyonları
kötüleştirebilir veya benzer şekilde tersi de mümkündür.
Non-farmakolojik
yaklaşım:
Non-farmakolojik
yaklaşımlar, LCD’li hastaların pek çok semptomlarında ve
fonksiyonel kayıplarında yararlı olabilirler, ancak henüz bu konu
sistematik olarak değerlendirilmemiştir. Görsel halüsinasyonlar ve
kognitif kötüleşme, dikkat ve uyanıklığın az
olduğu zamanlarda daha ağır olabilir. Bu nedenle sosyal
ilişkilerin arttırılması gibi yaklaşımlarla
uyanıklığın ve dikkatin arttırılması yararlı
olabilir.
Farmakolojik
yaklaşım:
Motor
bulgular: LCD’de parkinsonizmin L-Dopa cevabının iyi olmaması ve
DA-erjiklerin psikotik belirtileri arttırma riskine karşın
hastanın kendisine optimize edilmiş bir anti-parkinson tedaviden
yararlanıp yararlanmayacağı tetkik edilmelidir. L-dopa
düşük dozlarda başlanıp, yavaş doz artışı
yapılarak psikiyatrik semptomların artmasını engellemek
amacıyla gerekli olan minimum dozda kullanılmalıdır.
Antikolinerjiklerden kaçınılmalıdır.
Nöropsikiyatrik
semptomlar: Görsel halüsinasyonlar en sık görülen psikiyatrik semptomlardır.
Farmakolojik yaklaşımın gerektiği durumlarda atipik
antipsikotikler ve kolinesteraz inhibitörleri (ChEİ) düşünülmelidir.
ChEİ’lerin davranışsal belirtiler üzerindeki olumlu etkisi ilk
kez LCD’li hastalarla yapılan bir klinik çalışmada ortaya
konmuştur. Dolayısıyla, yeni bir hastada kognitif tedavi
amaçlı başlanan ChEİ, nöroleptik tedaviye karşı
gelişebilecek aşırı duyarlılık da göz önüne
alınarak görsel halüsinasyonlar ve diğer psikotik belirtilerin
tedavisi için bir süre tek başına bırakılabilir.
Cevap alınamadığı takdirde atipik nöroleptikler, onlarla
dahi aşırı duyarlılığın
gelişebileceğine dair anekdotal bildiriler de hesaba katılarak
dikkatle kullanılabilir. AH tedavisine benzer dozlarda kullanılmak
koşuluyla ketiapin, olanzapin, klozapin sırasıyla
seçilebilir. Tipik antipsikotiklerin kullanımından
kaçınılmalıdır. Depresyon da sık görülen bir
semptomdur ancak anti-depresan kullanımı ile ilgili sistematize bir
çalışma yoktur. SSRI veya SNRI’lar kullanılabilir. Ancak
antikolinerjik etkileri nedeniyle trisiklik antidepresanlardan
kaçınılmalıdır.
Uyku
bozuklukları: RUDB yatmadan önce verilen 0.25 mg klonazepam, 3 mg
melatonin veya 12.5 mg ketiapin ile başlanacak dozlarla tedavi edilebilir.
Doz artışı her zaman yavaş yapılmalı ve yan
etkiler açısından dikkatli olunmalıdır.
Aşırı gündüz uyuklaması zihinsel performansın
düşmesine katkıda bulunabilir. Tedavide sabah ve öğlen
dozları şeklinde, 100-400mg arası kullanılacak modafinil
ile yarar sağlanır.
Kognitif
semptomlar: LCD tedavisinde ağır kolinerjik yetmezliğin de
düşündüreceği gibi, kolinerjik temelli tedavi, en azından halen
elimizde bulunan ChEİ’lerle AH’ye benzer biçimde yararlı
olabilir. LCD’de Cochrane Kütüphanesi tarafından değerlendirmeye
alınan tek rasgele kontrollü klinik çalışma olan McKeith
çalışmasında rivastigminin bazı kognitif ölçütlerdeki
üstünlüğüne rağmen sonuçlar etkinlik açısından yeterince
ikna edici bulunmamıştır. Buna karşılık
rivastigmin EXPRESS isimli başarılı klinik çalışmasına
dayanarak PHD tedavisi için, FDA onayı almıştır ve
dolayısıyla da LCD için de düşünülebilir. Memantin tek bir
çalışmada (Emre 2010) sadece global ölçüde plaseboya üstün
bulunmuştur. Kullanımı düşünülebilir.
Parkinson
Hastalığı Demansı
Parkinson hastalığı uzun
yıllar için saf bir motor bozukluk olarak tasarlandıktan sonra zaman
içinde non-motor bozukluklar ve bunlar içinde ağırlıklı bir
yeri olan kognitif bozukluklar kaydedilir olmuştur. Giderek yeterince uzun
sürmüş bir PH’da kognitif bozulmanın belli bir şddeti olarak
demansın nerdeyse kaçınılmaz olduğu gözlenmiştir. Bu
klinik gözlemler büyük ölçüde hastalığın nörobiyolojisi ve
patogenezinin kavranmasındaki gelişmeler sayesinde mümkün olmuştur.
Alfa-sinükleinin SNc’deki Lewy cisimcikleri (LC’ler)
içinde biriken ana protein olduğunun bulunması ve alfa-sinüklein
immünhistokimyasının geliştirilmesinden sonra, nigral LC’lerin
çevresindeki karakteristik haloyu taşımadıkları için klasik
boyamayla gösterilemeyen kortikal LC’ler de gösterilebilir hale gelmiştir.
İzleyen çalışmalarda kortikal LC’lerin demans şiddetiyle
korelasyon gösteren ana patolojik bulgu olduğu
saptanmıştır. Braak ve arkadaşlarının
anatomo-klinik korelasyon çalışmalarında (halen
tartışmalı olsa da) alfa-sinüklein patolojisinin kaudo-rostral
bir ilerleyişle beyinsapından limbik ve heteromodal asosiasyon
kortekslerine doğru yükseldiğinin gösterilmesiyle PH’da
demansın neden geç dönemde ortaya çıktığı
sorusunun cevabı da teorik bir temele kavuşmuştur.
Epidemiyoloji
Belirli kriterlere göre seçilmiş 12 prevelans
çalışmasını sistematik olarak gözden geçiren bir
meta-analizde PHD’nın kesitsel nokta prevelansının %25-30
oranında olduğu bildirilmiştir. Toplum-tabanlı prospektif
bir çalışmada, Aarsland ve arkadaşları
başlangıç, 4 yıl ve 8 yıl sonraki PHD kumulatif
prevelansını sırasıyla %26, %51, %78 olarak
bildirmişlerdir. Diğer çalışmalarda başlangıçta
demansı olmayan Parkinson hastalarında 4 yıl sonunda %42, bir
diğerinde ise 15 yıl içinde %48 oranında demans
geliştiği bildirilmiştir. Sağkalımdan
etkilenmediği için prevelans ölçümlerine göre daha sağlıklı
olan insidans hesaplamalarında üç ila beş yıllık izlem
süresinde PH’da insidansın kontrol grubuna göre 4-6 kat daha fazla
arttığı izlenmiştir.
PHD için en önemli risk faktörleri ileri yaş,
uzun hastalık süresi ve ağır motor özürlülüktür (özellikle
bradikinezi-rijidite ve aksiyel semptomlar). Özellikle ileri yaş ve
ağır motor bulguları olan hastalar genç yaş ve daha hafif
motor bulguları olan hastalara kıyasla yaklaşık 10 kat
kadar artmış bir demans riski gösterirler. Değişik
çalışmalarda bildirilen, ancak her zaman doğrulanamayan
diğer risk faktörleri arasında şunlar sayılabilir: PH
başlangıcının ileri yaşta olması, akinetik-dominan
tipte PH, konuşma bozukluğu ve postüral insitabilite gibi aksiyel
tutulum bulgularının belirgin olması, levodopa ile tedavi
esnasında halüsinasyonların ve psikozun erken dönemde ortaya
çıkması, depresyonun varlığı, hastanın ilk
muayenesindeki mini-mental durum muayenesi puanının düşük
olması ve yürütücü işlevler (özellikle sözel
akıcılık), görsel-mekansal işlevler ve bellek
fonksiyonlarında hafif de olsa işlev bozukluklarının
varlığı. İki yıllık prospektif bir
çalışmada postüral insitabilite ve yürüme güçlüğü fenotipi
bulunan hastalarda tremor-dominan gruba kıyasla PHD ortaya çıkma
riskinin daha yüksek ve kognitif bozulma oranının daha
hızlı olduğu bulunmuştur. Bir başka
çalışmada başlangıçta tremor-dominan bir fenotip gösteren
hastaların demans gelişmesinden önce tremor dominansını
kaybedip akinetik-rijid belirtilerin ön plana çıktığı
saptanmıştır.
Klinik Özellikler
Genel seyri itibarıyla PHD sinsi
başlangıçlı ve yavaş ilerleyici bir demans tablosu ile
karakterizedir. Tipik klinik özellikler ön planda bir yürütücü işlev
bozukluğuyla birlikte dikkatte ve görsel mekansal işlevlerde erken
dönemlerden itibaren bozulma (sıklıkla demansın genel
şiddetinden daha ağır ölçüde), genelde tanımanın
korunduğu daha hafif bir episodik bellek bozukluğu, kelime
akıcılığı ve kelime bulma zorluğu
dışında göreli olarak korunmuş temel lingusitik
işlevleri içerir. Bu kognitif profile sıklıkla apati,
halüsinasyon ve hezeyanlar gibi davranışsal semptomlar eşlik
eder. Başlangıçta zihinsel alandaki bozulmalar motor bulgulara
odaklanan hekimin ve hasta yakınlarının dikkatini çekmeyebilir.
Subjektif olarak hastalar sıklıkla dikkat ve konsantrasyon
güçlüklerinden, yakın olayları iyi
hatırlayamadıklarından, algılamanın ve düşüncenin
yavaşlamasından, cümleyi başlatmakta ve doğru kelimeleri
bulmakta güçlüklerden bahsederler. Giderek önce hesap yapma, banka ve finans
işlerini yürütme, karışık ilaç şemalarını
uygulama gibi daha karmaşık işlevlerde, zamanla da daha temel
günlük yaşam işlevlerinde bozulma görülür. Hastalar kendilerine ya da
üstlendikleri görevleri yapmak için uzun zaman harcarlar, ya da hiç yapamazlar.
Karar vermede, işleri başlatmada, ailevi ve sosyal aktivitelere
katılmakta zorluk çekerler, giderek artan apatiden dolayı bu tip
aktivitelere ve hobilerine ilgileri ve spontaniteleri azalır. Sık bir
bulgu güniçi uyuklamaların artması, gündüz-gece ritminin tersine
dönmeye başlamasıdır. Demansın ileri evrelerinde bu
belirtiler iyice belirginleşir, konuşma giderek kötüleşir, ileri
dizartrik ve anlaşılmaz hale gelebilir; hasta konuşmayı kestiğinde
ne anlattığını ve nerede kaldığını
hatırlamayabilir. Son evrelerde hasta sıklıkla inkontinan,
yatağa veya tekerlekli sandalyeye bağımlı haldedir.
Nöropsikolojik profil açısından PHD klasik olarak yürütücü işlev
bozukluğunun ön planda olduğu , “diseksekutif” tipte bir demans
tablosudur. Ancak bazı hastalarda ileri, limbik tipte bellek
bozukluğunun ön planda olduğu Alzheimer tipi bir demans tablosunun
belirtileri tabloya katılabilir ya da ön plana geçebilir. Bu hastalar
büyük olasılıkla her iki tip patolojinin değişik oranlarda
bir arada olduğu karma-patolojili tabloları temsil ederler.
Nöropsikolojik muayenede kompleks,
yöneltilmiş dikkatte bozulma anlamındaki yürütücü işlev
bozukluğu, sözel akıcılıkta azalma ve psikomotor
hızdaki yavaşlama en erken saptanan özelliklerdir. AH ile kıyaslandığında
PHD’de daha fazla apati, daha belirgin kognitif yavaşlama (bradifreni),
dikkatte daha ağır ve daha dalgalanan bir bozulma vardır.
Yürütücü işlev bozukluğu erkenden ortaya çıkar ve hastalık
ilerledikçe daha belirgin hale gelir. PHD’de belleğin tüm öğeleri
(çalışma belleği, açık bellek, verbal ve görsel
modalitelerde örtük hazırlama belleği, prosedürel öğrenme)
bozulmuştur. Öğrenme testlerinde ve günlük hayatta spontan geri
çağırma AH’ye benzer şekilde bozulmuşken seçenekler
arasından tanıma veya ipuçlarından yararlanarak hatırlama
daha iyidir. Erken ve belirgin görsel-mekansal işlev bozukluğu
PHD’nin diğer bir önemli özelliğidir, bu bozukluk .AH’ye kıyasla
daha ağırdır. Temel dil işlevleri PHD’de büyük ölçüde
korunmuştur. Dil bağlamında en sık görülen sorunları
spontan kelime bulma güçlüğü ve sözel akıcılıkta bozulma
oluşturur. Bazı yazarlara göre yürütücü işlev disfonksiyonuna
bağlı, içe dönük dikkat ve arama stratejilerinde bozulma sonucu
ortaya çıkan, böylelikle de dil işlevlerinde gerçek bir
bozulmayı yansıtmayan bu belirtiler AH’dekinden daha ciddi boyutta
olabilirler. AH’ye bağlı demans tablosuyla PHD tablosu
arasındaki bu farklılıklara karşın PHD ile Lewy
Cisimcikli Demans tabloları büyük ölçüde örtüşürler. LCD’nin temel
özelliklerinden biri olan mental performansta günden güne veya gün içinde
ortaya çıkan dalgalanmalar PHD için de oldukça tipiktirler.
PHD’de davranışsal problemler ve
kişilik değişiklikleri oldukça sıktır. NPI
kullanılarak yapılan bir çalışmada PHD olan hastaların
%89’unda bir veya daha fazla psikiyatrik semptom bulunduğu
saptanmıştır. En sık görülen nöropsikiyatrik
semptomlar depresyon, halüsinasyonlar, apati, anksiyete ve hezeyanlardır.
Depresyon ya da depresif belirtiler hastaların en az %50’sinde görülüp
hastalığın seyri esnasında herhangi bir zamanda ortaya
çıkarlar. Halüsinasyonlar genelde görsel nitelikte olup iyi
şekillenmiş insan (sıklıkla çocuklar, bazen
tanıdık bazen da tanınmayan kadın ve erkekler), hayvan veya
bitki görüntülerinden oluşur ve LCD’de görülenlere benzerler. Kapı ya
da zil sesi, veya ismin çağrılması şeklinde duysal
halüsinasyonlar daha nadirdir; multi-modal halüsinasyonlar ise çok nadir
görülürler. İçgörü sıklıkla korunurken bazen bozulur ve
halüsinasyonlar korkutucu bir nitelik kazanırlar. Hezeyanlar en sık
“phantom boarder” (evde bir yabancının olduğu hissi),
hastanın arkasında ya da yanında birisinin durduğu hissi,
bir gölgenin geçtiği düşüncesi, aldatma hezeyanları olarak
ortaya çıkarlar. Hastalar bazen evlerinin kendi evleri
olmadığı, ya da yakınlarının (sıklıkla
eşlerin) kendi yakını olmadığı hissine
kapılabilir ve “eve” gitmek isteyebilirler. Halüsinasyonlar ve hezeyanlar
dopaminerjik ilaçların alımından sonra ortaya çıkabilir ya
da belirginleşebilirler. Bu durum PHD olan ya da demans geliştirmekte
olan hastalarda daha erken, daha kolay ve daha düşük dozlarda ortaya
çıkarlar. Erken dönemlerden itibaren görülen ve PHD’nin önemli bir
özelliği olan apati hastalık ilerledikçe daha da belirginleşir,
ileri evrelerde hastalar spontanite ve motivasyonlarını tamamen
yitirip günün büyük kısmını uyuyarak ya da bir köşede
oturarak geçirirler.
PHD’de motor fenotip sıklıkla simetrik
bradikinezi, rijiidite ve postüral instabilitenin ön planda olduğu
akinetik-rijid formdur. Daha önce tremor-dominansı gösteren hastalarda
zamanla tremor tamamen ortadan kalkabilir. PHD’de levodopa cevabının
giderek azaldığı düşünülür. Ancak bu gözlem en az
kısmen non-dopaminerjik bulguların giderek ön plana geçmesiyle
açıklanabilir. LCD hastalarına benzer şekilde ölümle
sonlanabilecek nöroleptik duyarlılığı PHD
hastalarında da bildirilmiştir. Uyku ile ilgili bozukluklardan
artmış gün uykusu yanında REM uykusu davranış
bozukluğu da PHD’de sık görülen özelliklerden birisidir. Bu belirti
demansı olmayan hastalarda da ortaya çıkabilmekle birlikte demanslı
hastalarda muhtemelen daha sıktır ve bilinen bir Parkinson
hastasında yeni olarak ortaya çıkması kognitif bozulmanın
ön habercisi olabilir. Kapsamlı ve
karşılaştırmalı olarak araştırılmamalarına
karşın ortostatik hipotansiyon, senkop, düşmeler, barsak ve
mesane bozuklukları, kalp atım hızında azalma, seksüel
disfonksiyon gibi otonomik bozuklukların demansı olan hastalarda daha
sık görüldüğü düşünülmektedir.
Nöropatoloji ve Fizyopatoloji
PHD’de asandan modulatör sistemlerin hemen
hepsinde hücre ve innervasyon kaybı bildirilmiştir. Değişik
çalışmalarda kolinerjik, dopaminerjik, noradrenerjik ve
serotoninerjik monoaminerjik sistemlerin etkilendiği bulunmuştur.
Kolinerjik kayıplar dışındaki bulgular ve bunların
demans ile bağlantısı net olarak gösterilmemiş olmakla
birlikte bu kayıpların tümü de demansın değişik
belirtilerinin ortaya çıkmasına katkıda bulunabilirler.
Örneğin, dopaminerjik kayıp frontostriatal
bağlantıların normal çalışmasını bozarak
yürütücü işlevlerde ve çalışma belleğinde bozulma
yapabilir. Ancak PHD’deki kognitif bozukluğun biyokimyasal kökeni olarak
en sistemli şekilde gösterilebilen bulgu kolinerjik innervasyondaki
azalmadır. Bu azalma anatomik, biyokimyasal ve fonksiyonel
çalışmalarda gösterilmiş olup AH’de görülenden daha fazla
orandadır (gözden geçirme için bkz. Emre, 2007). Yapısal olarak bazal
önbeyni innerve eden Meynert’in nükleus bazalisindeki kolinerjik hücrelerin
sayısının azaldığı saptanmıştır.
Biyokimyasal olarak asetilkolin yapılmasını sağlayan enzim
olan kolinasetiltransferaz aktivitesinin PHD olan hastaların korteksinde
demansı olmayan Parkinson hastalarına göre daha fazla oranda
azaldığı, bu azalmanın özellikle frontal ve temporal
kortekslerde daha belirgin olduğu bulunmuştur. Kolinerjik
işaretleyiciler ile yapılan fonksiyonel görüntüleme
çalışmalarında benzer şekilde kolinerjik aktivitenin PHD
olan hastalarda olmayanlara ve AH olanlara kıyasla daha çok
azaldığı, bu azalmanın miktarının kognitif
tutulumun, özellikle de yürütücü işlevlerdeki bozukluğun miktarı
ile orantılı olduğu saptanmıştır. Tüm
biyokimyasal bulgular birlikte ele alındığında dopaminerjik
eksikliğin yürütücü işlev ve çalışma belleği
bozukluğuna, kolinerjik eksikliğin bellek, dikkat ve yürütücü
işlevlerde bozukluğa, noradrenerjik eksikliğin dikkat
bozukluğuna, serotoninerjik eksikliğin ise affektif bozukluğa
katkıda bulunabileceği öne sürülmüştür.
Zamansal gelişim içinde PHD’nin
nöro-patolojik karşılığı olarak üç tip patoloji
sorumlu tutulmuştur. Bunlar a) subkortikal çekirdeklerin, özellikle
mediyal substantia nigranın dejenerasyonu, b) AH-tipi patoloji (amiloid
plaklar ve nörofibriler yumaklar) c) Lewy cisimciği (LC) patolojisidir.
Her bir patoloji için demanstan sorumlu ana mekanizma olduğunu gösteren
farklı çalışmalar vardır. Ancak son 10 yılda
yapılan, değişik patolojilerin eş zamanlı olarak
değerlendirildiği, özellikle de LC patolojisini saptamak için daha
hassas bir yöntem olan alfa-sinüklein immünhistokimyası kullanılan
çalışmalarda altta yatan ana patolojinin kortikal ve limbik alanlarda
hücre kaybı ve LC-tipinde dejenerasyon olduğu gösterilmiştir. Bu
çalışmalarda LC patolojisi, kognitif bozuklukla en yakın
korelasyon gösteren bulgudur; buna sıklıkla AH-tipi patoloji
eşlik etmekte, ancak nadiren patolojik olarak AH tanısını
koydurmaya yetecek miktarda olmaktadır.
Genetik
özellikler
Ailevi PH’nın nadir
nedenlerinden olan alfa-sinüklein geninde triplikasyon mutasyonu
taşıyan ailelerde PH motor semptomlarının yanında,
bazen erken dönemden itibaren demans görülebilirken, gen duplikasyonu olan
ailelerde bu durum ya hiç olmamakta ya da nadir görülmektedir.
Tanı
Kriterleri
PHD’nin klinik tanı
kriterleri (bkz. Tablo 18) “Movement Disorders Society”’nin
oluşturduğu bir Çalışma Grubu tarafından 2007
yılında yayınlanmıştır (Emre et al. ,
2007). Bu grup tarafından tanımlanan PHD’nin klinik özellikleri, bu
özelliklerin varlığı ya da yokluğuna
dayandırılarak tanımlanan “muhtemel PHD” ve “mümkün PHD”
tanı kriterleri Tablo 18’de gösterilmişlerdir.
PHD tanısı temel
prensipleri itibarıyla herhangi bir hastada demans tanısı koymaktan
farklı değildir. İlk aşamada demansı taklit edebilen
diğer durumların ekarte edilmesi gerekir. Bu durumlar demans
seviyesinde olmayan kognitif tutulumu, sistemik hastalıklar ya da ilaç yan
etkisi olarak ortaya çıkabilen akut-uzamış konfüzyonu ve depresyonu
içerir. Bu durumlar ekarte edilp demans tanısı konulduktan sonra
ikinci aşama ayırıcı tanıyı oluşturur. Bu
aşamada demans ve Parkinsonsizm belirtilerinin bir arada olabileceği
diğer hastalıklar (progresif supranükleer felç, kortikobazal dejenerasyon,
vasküler demans, normal basınçlı hidrosefali ve LCD gibi)
düşünülmeli, Parkinson hastalığına eklenebilecek subdural
hematom, B-12 eksiklği gibi diğer nedenler de göz ardı
edilmemelidir. Tüm bu durumlar ekarte edilmesine karşın tanıda
bazen güçlüklerle karşılaşılabilir. Örneğin, ileri
derecede motor ve konuşma bozukluğunun kognitif yıkımla üst
üste binmesi, depressif belirtielerin eşlik etmesi, olası ilaç yan
etkilerinin tabloyu karıştırdığı durumlarda
tanı güçleşebilir. Ayrıntılı bir öykü, tüm
kognitif alanları içeren mental muayene, diğer hastalıkları
dışlayan laboratuar incelemeleri ile yukarıda sözü edilen PHD
klinik tanı kriterlerinin dikkatlice uygulanması tanıyı
kolaylaştırır.
PHD için özgül bir
nörogörüntüleme bulgusu yoktur. BT ve MRG gibi yapısal görüntülemelerde
daha ziyade beynin posterior bölgelerinde atrofi dikkat çeker; hippokampal ve
medyal-temporal atrofi eşlik edebilmesine karşın AH’de görülen
kadar sık ve ağır değildir. Fonksiyonel görüntülemede SPECT
ve PET incelemelerinde frontal ve temporoparietal kortekste hipoperfüzyon
bulunur; ancak bu bulgu AH’de de ortaya çıkabildiği için özgül
değildir ve tek bir hastanın ayırıcı
tanısına katkı sağlamaz. Presinaptik dopaminerjik terminallerin
yoğunluğunu gösteren “dopamin taşıyıcı”
(DAT) SPECT incelemesinde PHD ve LCD hastalarında striatal DAT
tutulumunda azalma görülürken AH’de bu tutulum normaldir. Böylelikle bu yöntem
ekstrapiramidal bulgular geliştiren Alzheimer hastalarının
(örn., nöroleptik yan etkisine bağlı) PHD ve LCD hastalarından
ayırd edilmesinde yardımcı olabilir. Nihayet AH için
geliştirilen PET ile amiloid görüntülemede LCD grupları AH2liler gibi
amiloid pozitif bulunurken PHD grupları demanssız kontroller gibi
amiloid negatif bulunmaktadır.
Tedavi
PHD düşünülen bir hastada
farmakolojik tedaviye başlamadan önce zihinsel bozukluğu
tetikleyebilecek diğer faktörler dışlanmalıdır. Bu
bağlamda sistemik hastalıklar, özellikle yeni başlanan veya dozu
arttırılan ilaçlar gözden geçirilmeli, hasta antikolinerjik,
trisiklik antidepresan veya benzodiazepin grubundan bir ilaç kullanıyorsa
bu ilaçlar kesilmelidir. Dopaminerjik ilaçlar halüsinasyon, hezeyan gibi
psikotik belirtileri ve konfüzyonu arttırabilecekleri için özellikle
agonistlerin dozu azaltılmalı ya da bunlar kesilmeli, tedavi mümkün
olduğu kadar yeterli dozda yalın levodopa preparatlarına
indirgenmelidir.
PHD’de kolinerjik
eksikliğin tanımlanmasından sonra mevcut tüm kolinesteraz
inhibitörleri (rivastigmin, donepezil, galantamin) çoğunlukla küçük
çaplı ve açık çalışmalarda olmak üzere bu grup hastalarda
denenmiş, tüm çalışmalarda belirli ölçüde olumlu etkiler
tanımlanmıştır. Bu çalışmaların en
büyüğü ve kapsamlısı rivastigmin ile yapılan EXPRESS
çalışmasıdır ve pozitif sonuçlanmıştır (Emre
et al. , 2004). Diğer asetilkolinesteraz inhibitörlerden donepezil ile
de büyük çaplı bir çalışma yapılmış, belirli
ölçeklerde plasebodan üstün olduğu bildirilmiş, ancak bu
çalışma henüz yayınlanmamıştır. EXPRESS
çalışmasının sonuçlarına dayanarak rivastigmin PHD’de
ilk özgül tedavi olarak ruhsatlandırılmıştır. LCD
tedavisinde anılan Emre çalışmasında memantin PHD grubunda
da klinik değerlendirme ölçütünde plaseboya üstün bulunmuştur.
Kullanımı düşünülebilir.
Non-kognitif belirtilerin
tedavisi de LCD’ye benzerdir.
Tablo 18:
Parkinson hastalığı demansının özellikleri (Emre et
al. 2007)
I. Temel
Özellikler
1. Tanısı “Queen
Square Beyin Bankası” Kriterlerine göre konulmuş Parkinson
Hastalığının varlığı
2. Sinsi olarak başlayan,
yavaş ilerleyen, tanısı öykü, klinik ve mental muayene
bulguları ile konan, aşağıdaki şekilde
tanımlanmış demans sendromunun varlığı:
II. Eşlik
eden klinik özelikler
1. Kognitif özellikler
2. Davranışsal
özellikler
III. Parkinson Hastalığı
demansını dışlattırmayan, ancak tanıyı
kuşkuda bırakan özellikler
·
Demansın sebebi oldukları düşünülmeyen, ancak kognitif
bozukluğa neden olabilecek diğer anormalliklerin
varlığı, örn. görüntülemede anlamlı vasküler hastalık
varlığı
·
Motor ve kognitif semptomların gelişimi arasındaki zaman
ilişkisinin bilinmemesi
IV. Mental bozukluğun nedeni olabilecek diğer
hastalıkların veya bozuklukların mevcudiyeti, bunların
varlığında Parkinson hastalığı demansı
tanısını koymak mümkün olmaz
·
Akut konfüzyon
·
Sistemik hastalıklar veya anormallikler
·
İlaç intoksikasyonu
·
DSM IV’e göre majör depresyon
· NINDS-ARIEN kriterlerine göre
“Muhtemel Vasküler Demans” ile uyumlu özellikler
Muhtemel
Parkinson Hastalığı Demansı
A. Temel Özellik: Her iki temel
özellik de bulunmalı
B. Eşlik eden klinik
özellikler:
C. Grup III’de yer alan
özelliklerin olmaması
D. Grup IV’de yer alan
özelliklerin olmaması
Mümkün
Parkinson Hastalığı Demansı
A. Temel Özellik: Her iki temel
özellik de olmalı
B. Eşlik eden klinik
özellikler:
VEYA
C. Grup III’de yer alan
özelliklerden bir veya daha fazlasının varlığı
D. Grup IV’de yer alan
özelliklerin olmaması
Frontotemporal Demans
Arnold Pick ilk kez
1892 yılında “Afazi ve Beynin Senil Atrofisi Arasındaki
İlişki Üzerine” isimli makalesinde, Alzheimer’in Auguste
D.’sinden 14 yıl önce, nörodejeneratif hastalıkların 3. büyük
grubunu oluşturan ve günümüzde son olarak (şimdilik)
“frontotemporolobar deenerasyonlar” (FTLD) olarak adlandırılan ancak
bu terimin patolojiye daha yakın olan
çağrışımlarıyla olsa gerek klinikte çoklukla bir
önceki ismi olan “frontotemporal demans” (FTD) olarak anılan fokal
atrofilerin ilk olgusunu bildirdi. O sırada Prag’da üniversite Psikiyatri
Bölümü’nün direktörü olan Pick başka bir August’u, August H. isimli olguyu
özetle şöyle tanımlamıştı: “Öykü konuşma
bozukluğunun yavaş geliştiğini düşündürüyor ...Sol
hemisferin, özellikle de sol temporal lobun giruslarında ağır
bir atrofi mevcuttu. Her hangi bir fokal hastalık saptanmadı…Öyle
görünüyor ki, belli bir zaman parçasında gelişmiş,
aşağı yukarı keskin bir biçimde sınırlanabilen
bir afazi türü mevcuttur ve muhtemelen de basit nitelikte,
sınırları belirlenebilir beyin değişiklikleriyle
ilişkilendirilebilir…”. Bu paragraf günümüzde semantik demans olarak
bilinen sol temporopolar atrofiye bağlı ilerleyici afazi alt tipinin
bundan nerdeyse 120 yıl önce yapılmış mükemmel bir
tanımını içermektedir. “Erişkinlerin Primer Progresif
Demansı Üzerine” isimli, 1904 tarihli bir sonraki yayınında ise
bu kez de davranışsal varyantı ilk kez tarif edecektir: “41
yaşında bir ev hanımı yavaş yavaş
değişti. Özensiz ve sakar bir hale geldi… olağan işlerini
sürdürmüyor, çocuklarının bakımını yapmıyor,
giysilerini ya da yatak çarşaflarını değiştirmiyor,
saçlarını taramıyordu. İşlerini bitirmeden
bırakıyor ve tembel tembel oturuyordu. Her hangi bir sohbet
başlatmıyor, soruları tekrarlıyor, stereotipik
eğilimli cevaplar veriyor ve sıklıkla persevere ediyordu. Tek
kaygısı bedeniydi, bitlenmeden ve açlıktan yakınıyor
ve daima yemek talep ediyordu. Alışılmadık öfke krizleri
oluyor, çocuklarına sözel olarak şiddet gösteriyor veya
çocuklarına veya inekler dahil yakınında ne varsa onlara
vuruyordu…”. Pick’in çalışmalarıyla ilerleyici
davranışsal belirtiler ve ilerleyici afaziden oluşan bu
antitenin nöropatolojisi intranöronal globüler arjirofilik inklüzyonlu
cisimcikler (Pick cisimcikleri-PiC) ve balonlaşmış şiş
nöronlar (Pick hücreleri-PiHc), yüzeyel kortikal spongioz, glioz, nöron
kaybı olarak tanımlanmıştı. PiC terimini ilk Alzheimer
1911 yılında kullanmıştır. Bu antite 1922’de Gans
tarafından “Pick atrofisi” ve nihayet 1926’da Onari ve Spatz
tarafından “Pick hastalığı” olarak
adlandırıldı (kapsamlı bir gözden geçirme için bkz. Binetti
2001). Gans hastalığın baskın ailevi özelliğinden de
ilk kez sözediyordu. Yakın tarihlerin önmli çalışmalarından
birinde 1974’te Cenevre Beyin Bankası’ndan 40 olguluk bir PiH serisi
bildirildi. Klinik olarak davranışsal artı dilsel bir sendrom veya
yürütücü bozukluk artı parkinsonizm olarak özetlenebilecek 2 tarzda
sunulmuş olan bu seride ilk sunum temporo-orbitofrontal, ikincisi frontal
konveksite atrofisi ile ilişkiliydi. İlk atrofi tarzını
sergileyen 10 olguda PiC ve PiHC birlikte görülürken 8 olguda ikisinin birden
olmadığı sadece gliozis mevcuttu. İkinci tarz atrofi
tarzını sergileyen 10 olguda PiC olmaksızın sadece PiHC, 4
olguda ise yine sadece gliozis mevcuttu. Cummings ve Duchen 1981’de bilgisayarlı
tomografinin erken dönemlerinde 5 ağır anterior temporal atrofili PiH
olgusu bildirdiler. Tümünde de atrofi PiC ile ilişkiliydi ve
amigdalayı içine almaktaydı. Davranışsal özellikleri
“Kluver-Bucy sendromu” unsurları olarak özetlediler. Dilsel
bozuklukların da ön planda olduğunu, bellek ve mekansal oryantasyonun
göreli korunmuş olduğunu vurgulayarak bu klinik tablonun PiH’in bu
varyantı için ayırt edici olabileceğini ileri sürdüler.
İzleyen yıllarda, başlıca daha sonra yayınlanacak ilk
kriterler dizisine de isimlerini verecek olan İsveç’ten Lund,
İngiltere’den Manchester gruplarından
araştırıcıların bir dizi çalışmasıyla,
PiC ve PiHc’nin bu antitenin sık görülen bir bileşeni
olmadığı gerekçesiyle, Pick’in ismi kaldırılarak
atrofi tarzı (fronto-temporal) antitenin adı haline geldi. Bu
çalışmalar özetle frontal lob demansının (FLD) AH’den
ayırt edilebilir bir sendrom olduğunu, tüm demansların
%15-20’sini kapsadığını, olguların çoğunda PiC
bulunmadığını ileri sürmekteydi. Geçen yüzyılın
son 10 yılına egemen olan bu 2 grup 1994’te bir araya gelerek “Manchester-Lund
Fronto-temporal Demans (FTD) Kriterleri” olarak anılacak ilk tanı
kriterleri dizisini yayınladılar. Bu kriterlerde
davranışsal özellikler ayrıntılı bir şekilde
tanımlanırken, dilsel özelliklerin oldukça yüzeysel bir
tanımına rastlanır. Parkinsonizm ve ALS bulgularının
görülebileceği vurgulanır. Erken inkontinans, normal EEG ve
görüntülemede frontal, anterior temporal veya bunların kombinasyonunun bu
tanıya özgü olduğunun altı çizilir. Erken başlangıç ve
sık aile öyküsü AH’yle kontrast oluşturan öykü özellikleri olarak
belirlenir. Nihayet, 1998’de Lund ve Manchester gruplarına Fransız,
Kanadalı ve ABD, California’dan araştırıcıların
katılımıyla isim bir kez daha değiştirilip bugünkü
haline getirilir: FTLD (Neary et al. , 1998). Bu yeni kriterler
dizisinde (bkz. Tablo 19), yukarda “demans sendromlarının
fenomenolojik ayrırıcı tanısı” bölümünde “progresif
afazi” ve “progresif yürütücü/davranışsal bozukluk” alt
başlıklarında ayrıntısıyla
tartışılan davranışsal varyant FTD (FTDdv), ilerleyici
tutuk afazi (PNFA) ve semantik demans (SD) biçimlerinde 3 farklı sunumunun
ayrıntılı tanımları sıralanmıştır.
FTLD “Uzlaşım” Kriterleri’nin yazarları arasında bulunan
Kertesz Pick isminin bir klinik tanı olarak ortadan
kaldırılıp sadece PiC’lerin göründüğü, klinik
karşığı müphem ve dolayısıyla tanısı
çok güç ender bir histopatolojik tanıya indirgenmesine baştan beri
itiraz etmekteydi. Kertesz, ilk kez 1994’te “primer progresif afazi (PPA), FLD,
PPA-ALS ve KBD gibi fokal kortikal dejenerasyonlarla karakterize nörodejeneratif
hastalıkları” Pick Kompleksi (PK) terimiyle adlandırarak Pick
eponimini yeniden canlandırmayı önerdi. Bu öneri yaygın kabul
görmese de Kertesz ısrarını sürdürdü. Zaman içinde Kertesz için
PK dilsel ve davranışsal bileşenlerle birlikte, KBD ve PSP’yi
içeren bir motor bileşen de içeren bir klinik spektrum haline geldi.
Klinik sunumlar bu bileşenlerden biriyle başlayıp seyir boyunca
izole kalabildikleri gibi daha sıklıkla diğer bileşenlerden
özelliklerin de katıldığı karma sendromlar haline
dönüşmektedir. PK spektrumunu oluşturan antitelerin öngörülebilir
nöropatolojik ve genetik karşılıkları olduğunu ileri
sürdü (yeni yüzyıldaki bir dizi Kertesz gözden geçirmeleri için bkz.
Kertesz, 2007, Kertesz, 2003, Kertesz).
Epidemiyoloji
Çok sayıda
prevelans çalışması olmasa da Cambridge, İngiltere
çalışmasında FTD’nin pre-senil dönemde (45-64 yaş) AH kadar
sık olabileceğini gösteren bulgular elde edilmiştir. Rochester,
Minnesota, ABD’den gelen tek insidans rakamları (100,000 kişilik
nüfusta yılda yeni olgu) 40-49 yaş aralığında 2.2,
50-59 aralığında 3.3 ve 60-69 aralığında 8.9
şeklindedir. Daha yaşlı aralık dahil edilmemiştir.
Genellikle cinsiyetleri eşit etkilediği görüşü hakimken,
Cambridge çalışmasında örneklemin %82’si erkeklerdi. Ortalama
hastalık süresi AH’ye göre biraz daha kısa, 8 yıldır
(aralık 2-20 yıl). ALS eşliği sağkalımı 3
yıla kadar kısaltır. Hastaların %40’ının 1.
derece akrabalarında benzer hastalık öyküsü vardır ve
bunların en az yarısı otozomal dominan tarzda bir geçiş
sergilerler. Familyal ve sporadik olgular arasında AH için mutad olan
başlangıç yaşı farkı yoktur.
FTLD’nin alt gruplarının prevelans ve
insidansları spesifik olarak
çalışılmamıştır. PNFA’nın tüm FTLD’nin %10
ila 35’ini oluşturduğu ileri sürülmüştür. Bir
çalışmada başlangıç yaşının diğer FTLD
alt gruplarından anlamlı olarak daha geç olduğu fakat
sağkalımın farklı olmadığı bulunmuştur.
SD tüm FTLD’nin %15-25’ini oluşturur ve başlangıç yaşı
ve sağkalım açısından tipik FTD’den farklılık
göstermez. Belirlenmiş ve yerleşmiş risk faktörleri olduğu
söylenemez. Sadece MAPT geninin H1 haplotipine sahip olmak bir genetik
risk olarak ileri sürülmüş ve H1/H1 homozigot genotipi PSP ve KBD için bir
risk oluşturduğu doğrulanmıştır. Genetik olmayan
risk faktörleri arasında da sadece kafa travması öyküsü tek bir
çalışmada FTLD ile anlamlı ilişkili bulunmuştur.
Klinik Tablo
PK perspektifi ile motor vayant da dahil
edildiğinde bir davranışsal varyant (FTDdv), kendi içinde PNFA
ve SD olarak da ayrılan bir dilsel varyant (SD sol ve sağ temporal varyantlar
olarak bir kez daha ayrılabilir) ve yine kendi içinde asimetrik
parkinsonizm-progresif apraksi tarzında KBD sendromu ve vertikal
bakış felci-aksiyel parkinsonizm-ağır postüral imbalans
şeklinde PSP sendromu şeklinde ayrılabilecek bir motor varyant
ayırt edilir. Özellikle FTDdv ve PNFA’ta ALS eşlik ettiğinde
FTD-ALS’den sözedilir. Bu klinik sunumların ayrıntılı
özellikleri yukarda “Progresif Afazi”, “Progresif
Yürütücü/Davranışsal Bozukluk” ve “Progresif Apraksi” bölümlerinde
ayrıntıyla tartışılmıştır.
Genetik
Tau geni (MAPT)
Familyal FTD ile MAPT geni
ilişkisi ilk kez 1994’te bir ailenin kromozom 17’de MAPT genini
içeren q21-22 bölgesine anlamlı bir bağlantının
varlığının bulunmasıyla gösterilmiştir.
Sonrasında 1996’da o zamana kadar aynı bağlantının
saptandığı 13 geniş aileyi tanımlamak üzere
gerçekleştirilen bir uzlaşım toplantısında “Kromozom
17’ye bağlı fronto-temporal demans ve parkinsonizm (FTDP-17)” terimi
önerilmiştir. İki yıl sonra FTDP-17 ailellerinde ilk MAPT
mutasyonları 3 ayrı grup tarafından bildirildi. Geçen
yıllar içinde toplam 132 ailede 44 farklı mutasyon
bildirilmiştir. MAPT mutasyonlu olgular tüm FTLD
olgularının %5-10’unu, familyal FTD’nin ise %10-20’sini
oluşturur.
Faklı MAPT mutasyonlu
aileler PK’nın tüm varyantlarını içeren oldukça geniş bir
fenotipik varyasyon sergilerler. Bu durum bazen aynı mutasyona sahip bir
ailenin farklı üyelerinin fenotipik değişkenliği olarak da
görülür. En sık görülen mutasyon P301L hatalı anlamlı mutasyonu
sıklıkla 45-65 arasında başlar ve başlangıç
yaşı hiç bir zaman 70’i aşmaz. MAPT mutasyonu PSP ve KBD
fenotipi sergileyen hastalarda da bildirilmiştir.
MAPT mutasyonları
bir dizi farklı tau patolojisine yolaçar. Bunların ilki
başlıca 4R taudan oluşan, AH’ye benzemeyen nörofibriler inklüzyonlardır
ve 4R tau mikrotübül bağlanmasını etkileyen veya 4R/3R
oranını arttıran ekson 10 mutasyonları sonucu görülürler
(en sık olan P301L ilkine neden olur). Bu birikintilerde AH NFY’lerinin
PHF’lerinden morfolojik farklılıklar izlenir. Hem nöronlar hem de
glia etkilenir. Buna karşılık, ekson 9, 11, 12 ve 13
mutasyonları 3R ve 4R tauyu birlikte etkiler ve ya NFY-PHF’lere ya da
PiC’lere benzeyen inklüzyonlar yaratır. Glial etkilenme daha azdır.
Progranulin geni (PGRN)
Yeni yüzyıldan itibaren giderek artan
gözlemler 17q21’e bağlantılandığı halde bütün çabalara
rağmen MAPT mutasyonu bulunamayan ve aynı zamanda da tau
patolojisi sergilemeyip sadece ubikuitin immunoreaktivitesi gösteren (Ub+T-)
ailelerin sayısının FTD-17-tau ailelerini aşmaya
başladığını ortaya koydu. Sonunda 2006 içinde 2
ayrı grup tarafından bu Ub+T- FTD-17 ailelerinden sorumlu genin
progranulin geni (PGRN) olduğunu buldular. Bu genin
kodladığı progranulin proteini bir büyüme faktörüdür ve
gelişim, yara onarımı, inflammasyon ve tümör oluşumu gibi
bir dizi farklı süreçle ilişkilendirilmiştir. Bildirilen
mutasyonların tümü sıfır mutasyonlardır ve muhtemelen
proteinin işlev yitimine neden olarak etki etmektedirler. Kısa süre
önce keşfedilmesine rağmen şimdiye kadar toplam 212 ailede 68
farklı mutasyon bildirilmiştir. Tüm FTLD’lerin %5-10’unu familyal
FTD’lerin ise %15-25’ini oluşturdukları tahmin edilmektedir.
Başlangıç yaşı
aynı aile içinde dahi oldukça değişkendir (38-79 arası).
Klinik olarak FTDdv yanısıra PNFA fenotipi ve KBD fenotipi bildirilmiştir.
Mesulam dil bozuklukları kendi PPA tanımı ile uyumlu 2 ailede
yine PGRN mutasyonu bildirmiştir.
PGRN
mutasyonları nöropatolojik olarak yüzeyel kortikal tabakalarda spongiform
değişikliklerle Ub+T- inklüzyonlara yolaçar. Bunlar genellikle nöronların
sitoplazmalarında olmakla birlikte (NCI’lar), intranükleer de olabilirler
(NII). Distrofik nöritler (DN) de görülür. PGRN’nin bulunmasından
kısa zaman sonra bir biri ardına bildirilen otopsi serilerinde PGRN
mutasyonlu bireylerin beyinlerindeki bilinmeyen ubikuitinize proteinin TDP-43
olduğu ortaya konmuştur. TDP-43 ekson atlama ve transkripsiyon
düzenlemeden sorumlu bir nükleer proteindir ve 1. kromozomda kodlanır (TARDP
geni). İzleyerek sporadik Ub+T- FTD’lerin de (FTLD-U) %90’dan
fazlasının transaktif cevaplı-DNA’ya bağlanan protein 43
(TDP-43) immünoreaktivitesi gösterdiği bulunmuş ve TDP-43
proteinopatileri FTLD’nin de sporadik ve familyal büyük bir bölümünden sorumlu
yeni bir proteinopati olarak nörodejeneratif hastalıklar
sınıflamasına dahil edilmiştir. PGRN mutasyonu
ilintili familyal FTD-TDP, NCI, NII ve DN inklüzyonları birlikte
göstererek FTLD-U subtiplerinden Tip 3 olarak da
sınıflanılır.
Diğer sorumlu genler
Daha 1987 gibi erken bir tarihte
tanımlanmış olan ve baskın olarak davranışsal
değişiklikler ve bir ölçüde dilsel özellikler ve geri planda KBD
benzeri motor özellikler sergileyen geniş bir Danimarka ailesinde
bağlantı analizi kromozom 3’ü işaretlemişti. Geninin
bulunması için bir 10 yıl geçmesi gerekti: kromatin modifiye edici
protein veya diğer ismiyle yüklü multivesiküler protein 2B geni (CHMP2B).
Bu gendeki mutasyonların CHMP2B’nin hücresel lokalizasyonunu bozduğu
gösterilmiştir. Anılan bu Danimarkalı aile
dışında şimdiye kadar 2. bir aile bildirilmemiştir,
dolayısyla FTD-3 kategorisi halen sadece bu orijinal aile ile
sınırlıdır. FTD-3 nöropatolojik olarak bir
FTLD-U’dur. Ancak, TDP-43 immünoreaktivitesi göstermediği gibi bu yıl
geriye kalan %10 kadar TDP-43 negatif FTLD-U’larda mevcudiyeti ortaya konan
”fused in” sarkoma proteini (FUS) immunoreaktivitesi de negatiftir. Böylelikle,
artık FTLD nöropatolojisinin %1’inden azına
sınırlanmış olan FTLD-U kategorisinin tek temsilcisi
burdaki ubikuitinize protein de keşfedilene kadar FTD-3’tür diyebiliriz.
Bu gendeki 2 yeni mutasyon daha sonradan saf ailevi ALS ile
ilişkilendirmiştir.
Inklüzyon cisimciği myoziti (IBM)
ve Paget hastalığı ile birlikte herediter FTD sergileyen aileler
valosin içeren proteini (VCP) kodlayan 9. kromozomda p21.1-p12 lokusuna
haritalanırlar. VCP bir AAA-ATPaz süperfamilyası üyesidir ve hücre
çevrimi kontrolü, membran füzyonu ve ubikuitin-proteasom degradasyon
yolağı gibi bir dizi hücresel aktiviteden sorumludur. Watts ve
arkadaşları toplam 66 hastada buldukları 6 farklı mutasyonu
bildirmişlerdir. Hastalık klinik olarak erişkin
başlangıçlı kavşak tipi myopati, omurga ve kalça
ağrılarıyla karakterize Paget hastalığı ve
kırklı yaşlarda başlayan dilsel varyant
ağırlıklı FTLD ile karakterizedir. Triad bütün
hastaların ancak %12’sinde görülür ve en sık izole sunum %30 kadar
IBM şeklindedir. Dolayısıyla bu hastalara daha
sıklıkla nöromüsküler hastalıklar birimlerinde
rastlanılması beklenir. Nöropatolojik olarak hippokampusun göreli
kurtulduğu başlıca neokortikal NII inklüzyonları (Tip 4)
şeklinde FTD-TDP birikintileri görülür.
Yakınlarda FTD-ALS sergileyen 6
Fransız ailesi kromozom 9p21.3-p13.3’e
bağlantılandırılmıştır. 27 aday genin hiç
birinde mutasyon bulunamamıştır. Hastalar %32 oranında
izole FTD, %29 izole ALS (FALS) ve %39 oranında da her ikisinin
bileşimi şeklinde sunulmuşlardı. TDP-43-pozitif NCI’lar
(Tip 2 FTLD-U) korteks ve medulla spinaliste saptandı. Bulunamayan bu
genin tüm FTD-ALS’nin %60’ından sorumlu olabileceği ileri
sürülmüştür.
Kromozom 1’de kodlana TDP-43 genindeki
mutasyonlar FALS ile ilişkilendirilmiş ve TARDP mutasyonlu
FALS olguları ALS10 olarak da sınıflanmıştı.
Yakın tarihte ALS olmaksızın FTD’ye neden olan 2 ayrı TARDP
mutasyonu bildirildi. Bunlardan ilki FTDdv fenotipi sergileyen bir
kadındı. Diğeri ise FTDdv, PSP özellikleri ve koreye sahipti ve
nöropatolojik olarak başlıca subkortikal çekirdekler ve
beyinsapının TDP-43+ inklüzyonlarla tutulmuş olduğu
görüldü.
Son olarak, 16. kromozomda kodlanan ve
TDP-43-negatif FTLD-U olgularının büyük bölümünde ubkuitinize
edilmiş protein olduğu çok yakınlarda gösterilmiş olan
“fused in” sarkoma proteini (FUS) genindeki mutasyonlar da FALS
olgularıyla ilişkilendirilmiş ve FUS mutasyonlu FALS
olguları ALS6 olarak
sınıflandırılmıştı. Yine yakın
tarihlerde bir FUS mutasyonlu olgunun FTD-ALS sergilediği
bildirildi.
Nöropatoloji
Nöropatolojik olarak makroskopik açıdan FTLD
antitelerinin ayırıcı özelliği daha önce de
değinildiği gibi lober atrofidir. En sık olarak asimetrik
fronto-temporal kombine atrofi görülürken, bunu izole frontal, anterior
temporal ve parietal lob atrofileri izler. Hastalık ilerledikçe
atrofi tarzı simetrikleşme eğilimi gösterir. AH’nin
aksine, tutulum tarzının nasıl ilerleyeceği önceden kestirilemez.
Mikroskopik olarak tau+ inklüzyonlar
(FTLD-tau), Ub+T- inklüzyonlar (FTLD-U) olarak sınıflanırlar.
FTLD-U’nun %90’ına yaklaşan bir kısmında ubikuitinize
proteinin TDP-43 olduğu (FTLD-TDP) bulunmuştur (Neumann et al. ,
2006). Geri kalan %10 kadar FTLD-U’nun da çok yakında ubkuitinize
proteiinlerinin FUS olduğu (FTLD-FUS) bulunmuş ve FTLD-U
sınıfı henüz ubikuitinize proteini bulunamayan az sayıdaki
CHMP2B mutasyonlu familyal FTLD hastasına sınırlı
kalmıştır. Çok az sayıdaki hastada ise geçen
yüzyılın ”ayırtettirici histopatolojisi olmayan demans-DLDH”
veya non-spesifik gliozis tanımını hakedecek şekilde her
hangi bir inklüzyon (FTLD-ni) görülmez. Tümünün de ortak özelliği tutulan
alanlarda saptanan nöron kaybı, gliozis ve yüzeyel lineer
spongiozistir. FTLD-tauda derin kortikal tabakalarda şiş, balonlaşmış
görünümlü Pick hücreleri saptanabilir. PiH’in ayırıcı
özelliği olan Pick cisimcikleri nöronların içinde görülen büyük ve
yuvarlak inklüzyonlardır (bkz. Şekil 19). Tau antikorlarıyla
farklı tarzlarda boyanmaları ve nörofilamanlar, çift sarmallı filamanlar,
kısa düz ve uzun kıvrımlı fibriller gibi farklı eleman
içerikleriyle, oldukça homojen bir yapı olan NFY’lerden farklı olarak
değişkenlik gösterirler. Bunlar PiH’e özgü olarak Bielschowsky
boyası ile boyanırken, Gallyas ile boyanmazlar. Pick cisimciklerine
tutulmamış neokortikal alanlarda rastlanılmıyor olsa da
sayıları atrofiyle korele etmez ve daima atrofik loba kıyasla
hippokampusta daha bol miktarda bulunurlar. Tutularak atrofiye
katılan ve Pick cisimciklerinin görülebildiği subkortikal yapılar
arasında olgudan olguya değişecek şekilde neostriatum,
medial talamus, daha hafif düzeyde olmak üzere de substantia nigra, hipotalamus
ve locus coeruleus bulunur. KBD’de nöronal inklüzyonlar bazen Pick
cisimciklerini andırsa da daha çok halka şeklinde
yumaklardır. Pick cisimciklerinden farklı olarak Bielschowsky
ile gayet zayıf boyanırlarken, bu kez Gallyas ile iyi
boyanırlar. Düz kısa fibrillerden oluşurlar. Nöronal
τ birikimleri Pick cisimciklerine benzer yapılardan çok yaygın
birikimler ve yumaklar tarzındadırlar. Yaygın subkortikal
tutulum içinde substantia nigra nöronlarında kortikobasal cisimcikler de
denilen globoz NFY’ler ayırt edilir. Nöronal
inklüzyonların yanı sıra yaygın astrositik τ
inklüzyonları (GCI’lar) tanı koydurucudur. Bunlara amiloid
içermeyen glial plaklar adı da verilir. Oligodendrogliada da
yaygın biçimde τ ekspresyonu ve arjirofilik inklüzyonlar
görülür. FTLD-TDP inklüzyonları daha önce sözedildiği gibi NCI,
NII ve DN şekillerinde olur ve bunların biri ya da bir
kaçının baskınlığı ve tutulum tarzına göre 4
ayrı alt tipe sınıflandırılmıştır.
FTLD-FUS atipik FTLD-U (aFTLD-U), bazofilik inklüzyon cisimciği
hastalığı (BIBD) ve nöronal orta boy filaman inklüzyonu
hastalığı (NIFID) alt tiplerini kapsar. Kromozom 17
ilintili FTLD ve diğer ailevi FTLD’ler de sporadik proteinopatilerin
nöropatolojisini andırırlar.
Şekil 19. Pick
Cisimcikleri. Bielschowsky gümüş boyama ile arjantofilik intraselüler
inklüzyonlar (Kaynak: http://www.binderlab.northwestern.edu/pickbodies.html)
Tedavi
Nedene yönelik bir tedavinin
doğal olarak FTLD mekanizmasında temel rolü oynayan tau, TDP-43 ve
FUS proteinlerinin patolojik katlanmasını engellemeyi hedeflemelidir.
AH’nin mekanizma temelli tedavi staratejillerinden tau hedefli stratejilerin
ilerde FTLD’lerde de kullanılabileceği düşünülebilir. FTLD’de
mekanizma temelli tedavilerin yerleşebilmesi için farklı proteinopatilerin
de klinik olarak tanınabilmesi tabii ki bir zorunluluk olacaktır.
Bazı bildirimlerde SSRI’ların yararı üzerinde
durulmaktadır. ChEI’lerin ve memantinin yararı
gösterilmemiştir. Çeşitli davranışsal belirtilerin
tedavisi için AH tedavisinde önerilenlere paralel bir yol izlenebilir. Parkinsonizm
büyük ölçüde L-dopa’ya cevapsızsa da erken dönemde alınabilecek olan
sınırlı cevaptan yararlanılabilir.
Tablo 19. Frontotemporal
Lobar Dejenerasyon Klinik Tanı Uzlaşı Kriterleri (Neary et al.
1998)
Liste 1.
FTD’nin klinik tanı özellikleri: Klinik profil
Kişilik değişikliği ve sosyal
davranış bozukluğu, başlangıçta ve
hastalığın seyri boyunca temel özelliklerdir. Algı,
mekansal yetenekler, praksis ve belleğe ilişkin enstrümental işlevler
salim ya da göreli olarak iyi korunmuştur.
I. Temel tanı özellikleri
A.
Sinsi başlangıç, yavaş seyir
B. Sosyal
kişiler arası davranışın erken dönemde bozulması
C. Kişisel
davranışın düzenlenmesinin erken dönemde bozulması
D.
Erken dönemde emosyonel küntlük
E. İç
görünün erken dönemde kaybı
II. Destekleyici tanı özellikleri
A.
Davranışsal bozukluk
1. Kişisel
hijyen ve kendine bakımda azalma
2. Zihinsel
rijidite ve esneklik kaybı
3.
Çelinebilirlik ve sebatsızlık
4. Hiperoralite
ve beslenme tarzında değişiklikler
5. Perseveratif
ve stereotipik davranış
6. Kullanma
davranışı
B.
Konuşma ve dil
1.
Konuşma çıktısında değişiklik
a. Kendiliğindenliğin
kaybı, az konuşma
b.
Basınçlı konuşma
2.
Stereotipik konuşma
3.
Ekolali
4.
Perseverasyon
5.
Mutizm
C.
Fiziksel bulgular
1.
İlkel refleksler
2.
İnkontinans
3.
Akinezi, rijidite, tremor
4.
Düşük ya da labil kan basıncı
D.
İncelemeler
1. Nöropsikoloji:
ağır amnezi, afazi veya algısal-mekansal bozukluk
olmaksızın frontal lob testlerinde anlamlı bozukluk
2. Elektroensefalografi:
klinik olarak aşikar demansa karşılık konvansiyonel EEG’nin
normal olması
3. Beyin
görüntülemesi (yapısal ve/veya işlevsel): ağırlıkla
frontal ve/veya anterior temporal bozukluk
Liste 2.
Progresif tutuk afazinin klinik tanı özellikleri: Klinik profil
Ekspresif dil bozukluğu başlangıçta ve
hastalığın seyri botunca temel özelliktir. Diğer
bilişsel işlevler salimdir veya göreli olarak iyi korunmuşlardır.
I.
Temel tanı özellikleri
A. Sinsi
başlangıç, yavaş seyir
B. Tutuk
spontan konuşma ve şunlardan en az biri: agramatizm, fonemik
parafaziler, anomi
II.
Destekleyici tanı özellikleri
A.
Konuşma ve dil
1.
Kekeleme veya oral apraksi
2.
Tekrarlama bozukluğu
3.
Aleksi, agrafi
4.
Kelime anlamının erken dönemde korunmuş olması
5.
Geç dönemde mutizm
B.
Davranış
1. Sosyal
yeteneklerin erken dönemde korunmuş olması
2. Geç
dönemde FTD’ye benzer davranışsal değişiklikler
C. Fiziksel
bulgular: geç dönemde kontrlateral primitif refleksler, akinezi, rijidite ve
tremor
D. İncelemeler
1. Nöropsikoloji:
ağır amnezi ve algısal-mekansal bozukluk olmaksızın
tutuk afazi
2. Elektroensefalografi:
normal veya hafif asimetrik yavaşlama
3. Beyin
görüntülemesi (yapısal ve/veya işlevsel): ağırlıkla
dominan (genellikle sol) hemisferi tutan asimetrik bozukluk
Liste 3. Semantik
afazi ve asosiyatif agnozinin (SD) klinik tanı özellikleri: Klinik profil
Semantik bozukluk (kelime anlamının ve/veya
nesne kimliğinin anlaşılmasında bozulma)
başlangıçta ve hastalık seyri boyunca temel özelliktir.
Otobiyografik bellek de dahil olmak üzere diğer
bilişsel işlevler salimdir veya göreli olarak iyi korunmuştur.
I. Tanı özellikleri
A. Sinsi
başlangıç, yavaş seyir
B.
Aşağıdakilerle karakterize dil bozukluğu:
1. Progresif,
akıcı, içeriği boş spontan konuşma
2. Adlandırma
ve anlama bozukluğu ile ortaya konan kelime anlamının kaybı
3. Semantik
parafaziler ve/veya
C.
Aşağıdakilerle karakterize algısal bozukluk:
1. Prosopagnozi:
aşina yüzlerin tanınmasında bozulma ve/veya
2. Asosiyatif
agnozi: nesne kimliğinin tanınmasının bozulması
D. Algısal
eşleme ve çizerek kopyalamanın korunmuş olması
E. Tek
kelime tekrarlamanın korunmuş olması
F. Yüksek
sesle okuma ve dikte ile ortografik olarak düzgün kelimeler yazma
yeteneklerinin korunmuş olması
II. Destekleyici tanı özellikleri
A.
Konuşma ve dil
1.
Basınçlı konuşma
2.
İdyosenkratik kelime kullanımı
3.
Fonemik parafazilerin bulunmayışı
4.
Yüzey disleksi ve disgrafisi
5.
Hesabın korunması
B.
Davranış
1.
Sempati ve empatinin kaybı
2.
Kısıtlı takıntılı uğraşılar
3.
Hasislik
C.
Fiziksel bulgular:
1. Primitif
refleksler yoktur veya geç dönemde görülür
2. Akinezi,
rijidite ve tremor
D.
İncelemeler
1.
Nöropsikoloji:
a. Kelime
anlama ve adlandırma ve/veya yüz ve nesne tanıma bozukluğuyla
ortaya konan ağır semantik kayıp
b. Fonoloji,
sentaks ve elemanter algısal süreçler, mekansal yetenekler ile gündelik
belleğin korunmuş olması
2.
Elektroensefalografi: normal
3. Beyin
görüntülemesi (yapısal ve/veya işlevsel): ağırlıkla anterior
temporal bozukluk (simetrik veya asimetrik)
CREUTZFELDT-JACOB VE DIĞER PRION HASTALIKLARI
Hans Gerhard Creutzfeldt ve Alfons Maria Jakob’un
1920 ve 1921’de ardarda bildirdiği olgu serilerinin Spielmeyer
tarafından 1922 yılında Creutzfeldt-Jakob
hastalığı (CJH) olarak adlandırılması
sonrası bunların birbirlerine ve bugün bilinen şekliyle CJH’ye
benzerlikleri çok kuşkuluysa da, artık söz konusu hastalık
tartışmaya yer bırakmayacak biçimde anılan
araştırmacıların isimleri ile anılmaktadır.
Gajdusek 1968 yılında deneysel kuru hastalığı olan
şempanze beyni ve sporadik CJH’sı olan insan beyinlerinden
hazırlanmış materyelleri primatlara inoküle ederek bu
hayvanlarda hastalık geliştirmeyi başarmış ve
bulaştırılabilen spongiform ensefalopatiler (TSE)
kavramını geliştirmiştir. Aynı yıl içinde CJH’yi
subakut sklerozan panansefalit tarzı bir “yavaş virüs
hastalığı” olarak tanımlamıştır. Stanley
Prusiner’in 1982 yılında yavaş virüs kavramını yerle
bir edip, CJH’nin nedeni olarak diğer proteinopatilerde olduğu gibi,
fiziksel konformasyonunu değiştirerek patojen haline dönüşen
(PrPSc) ve bu haliyle bir şablon oluşturup normal hücresel
proteini (PrPc) kendine benzetmeye başlayıp, böylelikle
çoğalıp biriken ve bulaşabilen bir protein olan prionu (PrP)
bulması alanı kökten değiştirmişti. Bundan böyle
aynı zamanda bulaşabilir bir proteinopati olarak prion
hastalıklarından söz edilecektir. Oesch 1985 yılında PrPc’yi
kodlayan geni (PRPN) 20. kromozomda
haritalandırmıştır.
CJH ve yeni varyant CJH’nin de (nvCJH)
aralarında bulunduğu insan prion hastalıkları (bkz. Tablo
20) %85-90 oranında sporadik ortaya çıkarlar. Geri kalan %1-2
enfeksiyöz, yani prion hastalığı gösterilmiş bir kaynaktan
bulaşıcı yolla edinilen ve %5-15’de ailevi, yani 20. kromozomda
kodlanan prion proteini (PrP) geninde bir mutasyon sonucu otozomal dominan
kalıtımla geçen şekillerdir. İnsan prion
hastalıklarının tümü, hasta beyninden hazırlanan ekstrenin
bir primata inokülasyonuyla bu hayvana bulaştırılabilir.
Sporadik Prion Hastalıkları
Sporadik fatal insomni son derece nadir bir
hastalık olduğu için bu başlık sporadik CJH ile pratik
olarak aynı anlama gelir. Hastalık coğrafi bölge, etnik
köken veya cinsiyet ayırd etmeksizin bütün dünyada görülebilmekte ve
yıllık sıklığı yaklaşık 1/1,000,000
kişi olarak hesaplanmaktadır. Bu, ülkemizde kabaca her yıl
65-70 yeni CJH vakasının ortaya çıktığı
anlamına gelmektedir. Başlangıç yaşı ortalama
60’dır. Hastalık çok hızlı seyirlidir. Konu
üzerine uzmanlaşmış büyük merkezlerde CJH hastalarından
oluşan geniş vaka serilerinde ortalama hayatta kalmanın 4 ay
kadar olduğu hesaplanmaktadır. Belirtilerin
başlamasından sonraki 1. yıl içinde tüm olguların
%90’ının ölmüş olduğu kaydedilmiştir.
Tablo 20. Prion
Hastalıkları
|
İnsan Hastalıkları |
|
|
Sporadik |
CJH Fatal Sporadik Insomnia – FSI |
|
İnfeksiyöz |
CJH
(iyatrojenik) vCJH Kuru |
|
Ailevi |
CJH
(ailevi) Fatal Familyal Insomnia - FFI Gerstmann-Sträussler-Scheinker
Hastalığı (GSS) |
|
Hayvan Hastalıkları |
|
|
Koyun |
Scrapie |
|
Sığır |
Bovin
Spongiform Ensefalopati (BSE) |
|
Vizon |
Bulaşabilen
Vizon Ensefalopatisi (TME) |
|
Geyik |
Kronik
Kas Erimesi Hastalığı (CWD) |
|
Kedi |
Felin
Spongiform Ensefalopati (FSE) |
Hastalık olguların yaklaşık
%25’inde uykusuzluk, iştahsızlık, kilo kaybı, huzursuzluk,
halsizlik gibi spesifik olmayan prodromal belirtilerle başlar.
Prodromal belirtiler olsun olmasın, başlangıçtaki kişilik
değişikliği, zihinsel işlevlerde bozulma, ataksi, kortikal
körlükten agnozilere görsel işleme problemleri şeklindeki belirti ve
bulgular, aylar içinde hızla ağır bir demansa ve sonunda
akinetik mutizme yolaçar. Kişilik değişikliği, en
başlarda kişinin daha kolay öfkelenir ya da daha hasis olması
gibi önceden getirdiği kişilik özelliklerinin abartılması
şeklindeyken, giderek FTD özellikleri çeşitli bileşimlerde
sergilenmeye başlar. Hezeyan ve halüsinasyonlar şeklinde
psikotik özellikler görülebilir. Kognitif işlevlerde yıkım
kısa bir süre için progresif afazi veya kortikal körlüğe gidebilen
progresif simultanagnozi benzeri izole kalabilse de zaman içinde genel bir
yıkım tablosu yerleşir. Serebellar ataksi ve
ekstrapiramidal bulgular CJH’nin farklı alt biçimlerinde tabloya
çeşitli dönemlerde katılırlar. Hastalığın
orta evrelerinde akustik ve hatta taktil uyaranlarla ortaya
çıkarılabilen irkilme miyoklonisi tanıya güç katan klinik
bulgulardandır.
Hastalık, yukarda anılan tipik
tarzı yanı sıra farklı isimlerle anılan ve
aşağıda sözedilecek prion ”suşlarının” tipi PRPN
ve kodon 129 polimorfizmine göre (prion proteini geninin 129. kodonundaki
metionin veya valin mevcudiyetine göre) değişen klinik tarzlarda da
görülebilir (Tablo 21). Yeni varyant CJH (vCJH), öteden beri bilinen bu
tarzlara göre “yeni” olduğu için bu şekilde isimlendirilmiştir.
Tablo 21.
Diğer Sporadik CJH Klinik Tarzlarının Özellikleri
Kısaltmalar: AV:
anteroventral çekirdek; BG: basal ganglia; Cer: serebellum; Cx: korteks; M: metionin;
MD: mediodorsal çekirdek; PSW: periyodik yavaş dalga; V: valin
Laboratuar incelemeleri tanıyı
kesinleştirmese de, katkıda bulunur. Elektro-ensefalografi, bir
dönem için hastalığa özgü periyodik 1 Hz keskin-dalga komplekslerini
gösterir (PSW; Şekil 20). PSW ve klinik özellikleri başvuru ve
seyir içindeki sıklıklarını 209 hastalık bir Amerikan
serisinde analiz eden bir 1993 çalışmasında miyoklonus
başlangıçta %5 iken seyir sırasında %88’e, ekstrapiramidal
bulgular %1’den %56’ya, serebellar bulgular %33’ten %70’e, görsel bulgular
%19’dan %41’e, PSW’nin %0’dan %44’e yükseldiği bildirilmiştir. MRG’de
en yüksek duyarlılıkla difüzyon ağırlıklı
görüntülerde (DWI) ortaya çıkan, zaman içinde sırasıyla FLAIR ve
T2 ağırlıklı görüntülerde de beliren kortikal (kortikal
şeritlenme) ve başta bazal ganglia, sonra da talamus olmak üzere
yüksek sinyal, hızlı seyirli bir demansta CJH için yüksek
duyarlılık ve özgüllük taşıdığı çeşitli
çalışmalarda bildirilmiştir (Şekil 21). Bir Japon
çalışmasında 36 olguda DWI-MRG
duyarlılığı %92.3, özgüllüğü %93.8, buna
karşılık PSW duyarlılığı %50, BOS 14-3-3
protein düzeyi %84, BOS NSE protein düzeyi %73.3 bulunmuştur. Bir Amerikan
çalışmasında ise 40 olguda DWI+FLAIR’in birlikte
duyarlılıkları %91, özgüllükleri ise %95 olarak
bildirilmiştir. Bu bulgunun sadece voltaj kapılı potasyum
kanalopatili olgular tarafından taklit edilebileceğine dair bir
bildirim mevcuttur. Manyetik rezonans ya da bilgisayarlı tomografi (BT)
ile kortikal atrofi, bazen de bazal gangliada sinyal değişiklikleri
görülebilir. BOS’da nöronal yıkım ürünleri total tau ve 14-3-3
proteinin düzeylerinin belirgin yükselmesinin hastalık için yüksek
duyarlılık gösterdiği ileri sürülmektedir.
Ayırıcı tanıda diğer hızlı seyirli demans
nedenleri göz önüne alınmalıdır. Yine de kuşkuda
kalındığında beyin biyopsisine
başvurulmalıdır. CJH kuşkusuyla yapılan bir
beyin biyopsisinde 2. kez kullanılmayacak cerrahi aletler seçilir.
Mikroskopik olarak spongiform değişiklikler, immunhistokimyasal
boyama ile PrPSc reaktivitesi ve Western blot ile proteaza dirençli
PrPSc’nin gösterilmesi tanı koydurucu olur. Tablo 22’de
gösterilen tanı kriterleri Dünya Sağlık Örgütü tarafından
1998’de yayınlanmıştır ve MRG ve başta total tau
düzeyi olmak üzere BOS biyoişaretleyicilerini içermediğinden demode
olmuş gibi görünmektedir.
Şekil 20. Tipik
CJH EEG’si: Periyodik keskin dalga (PSW) aktivitesi
Şekil 21. Sporadik
CJH’da MRG.
İki
ayrı sporadik CJH hastası. a. Subkortikal baskın tarz: T2
ağırlıklı aksiyel kesitte (sol üst) daha belirsiz olan
kaudat ve putaminal hiperintensiteleri, FLAIR koronal kesitte (üst orta) biraz
daha belirginleşiyor ve DWI aksiyel kesitte (üst sağ) en belirgin
hale geliyor. b. Kortikal şeritlenme ve subkortikal yüksek sinyal
ile birlikte olan tarz: FLAIR aksiyel kesitte (alt sağ) basal ganglia ve
silik talamik hiperintensite DWI aksiyel kesitlerde (alt orta ve alt sağ)
2 yanlı medyal frontal kortikal şeritlenme. (İstanbul Tıp
Fakültesi Nöroloji Anabilim Dalı, Davranış Nörolojisi ve Hareket
Bozuklukları Birimi arşivinden ).
Tablo 22. Creutzfeldt-Jacob
hastalığı için Dünya Sağlık Örgütü (WHO) Klinik
Tanı Kriterleri
Muhtemel CJH
1.
İlerleyici demans
2.
Tipik EEG
3.
Aşağıdakilerden en az ikisi:
·
Miyoklonus
·
Görsel bozukluk
·
Serebellar bulgular
·
Piramidal veya ekstrapiramidal bulgular
·
Akinetik mutizm
Mümkün CJH
1. İlerleyici
demans
2. Atipik
EEG veya EEG bilgisi mevcut değil
3.
Aşağıdakilerden en az ikisi:
·
Miyoklonus
·
Görsel bozukluk
·
Serebellar bulgular
·
Piramidal veya ekstrapiramidal bulgular
·
Akinetik mutizm
4. Demans
süresi iki yıldan daha az
Kesin CJH
1. Karakteristik
nöropatoloji
2. Beyin
dokusunda Western blot analizi ile proteaza dirençli prion proteininin
saptanması
3.
Nöropatolojik incelemede scrapie ile ilintili fibrillerin saptanması
4.
Hastalıklı beyin dokusu inokülatı ile deneysel
bulaşmanın gösterilmesi
İnfeksiyöz Prion
Hastalıkları
İyatrojenik CJH, kornea nakli
yapılan, prion bulaşmış cerrahi aletlerle
nöroşirürjikal operasyon geçiren, infekte derin elektrodların
kullanıldığı, insan hipofiz bezi ekstreleri alan veya dura
mater greftlemesi yapılan kişilerde gösterilmiştir. Klinik
tablo sporadik hastalığa çok benzer. Bulaşma ile
hastalığın ortaya çıkması arasında geçen süre
(inkübasyon periyodu), bulaşmanın doğrudan beyne (örneğin
cerrahi gereçler, dural greftler), ya da sistemik olarak (örneğin hipofiz
ekstresi) olmasına bağlı olarak, 2 ila 40 yıl arasında
değişir. Kan ve kan ürünleri ile hastalığın
bulaşması riski bir endişe kaynağı olsa da, tam
doğrulanmamıştır. Hasta kanının şempanzelere
nakli bu hayvanlarda hastalığa yol açmazken, farelerde doğrudan
beyne zerkedilmesiyle hastalık ortaya
çıkmıştır. Sonradan vCJH geliştiren hastalardan
yapılmış kan transfüzyonu öyküleri olan 2 ayrı vCJH olgusu
bildirilmiştir. Bir çalışmada, nöroşirürjikal olsun veya
olmasın, ameliyat sayısı arttıkça hastalık riskinin de
arttığı gösterilmiştir. Brown ve arkadaşları
1974-2000 arası bildirilmiş olan 267 iyatrojenik CJH olgusunun analizini
bildirdiler. İnkübasyon sürelerinin 30 yıla kadar
çıkabildiği bu olguların 139’u kadavra kökenli büyüme hormonu
(74 Fransız, %0.3-4.4) kullanmış, 114’üne ise dura mater grefti
(67 Japon, %0.02-0.05) yapılmıştı. Dura mater olguları
kodon PRNP129MM sık, inkübasyon süresi kısa ve klinik olarak
hızlı seyirli demansa sahiplerdi. Buna karşılık büyüme
hormonu olgularında PRNPkodon129VV sık, inkübasyon süresi uzun
ve klinik olarak progresif ataksi sergilenmişti.
Kuru, Yeni Gine’de Fore yerlileri
arasında tarif edilmiş ve hastalığa neden olduğu
kanıtlanan bölgeye özgü dinsel ritüelin terkedilmesiyle de artık
görülmez olmuştur. Foreliler’in geleneklerine göre, aile
büyüğünün ölümünden sonra, onun bilgeliğini yeni kuşaklara
geçirebilmek için beyni o ailenin kadınları ve her iki cinsiyetten
çocukları tarafından yenir, ayrıca beden ve yüze sürülür.
Etkenin konjonktivalar, deri ve sindirim sisteminden alınmasını
izleyen inkübasyon periyodu sonrasında, hastalık süresi ve klinik
özellikler açılarından sporadik CJH’yi andıran bir tablo ortaya
çıkar. Hastalık deneysel olarak şempanzelere de
geçirilebilmiş ve izleyerek onların da kendi aralarında
taşınabilmiştir.
vCJH, Britanya, Fransa ve
İrlanda’da yaklaşık 50 genç hastada 1990’ların
ortalarından itibaren bildirilmeye başlanmıştır.
Henüz ABD’den ve ülkemizden bildirilen olgu yoktur. Birleşik
Krallık’ta bildirilen 35 olgunun ortalama başlangıç
yaşı 29’dur (aralık: 18-53). Aynı seride ölüme kadar
geçen süre ortalaması 16 aydır. Hastalık duysal ve
psikiyatrik belirti ve bulgularla başlar. Altı ay kadar süren
bu başlangıç evresinden sonra önce ataksi, sonrasında da demans,
istemsiz hareketler ve inkontinans gibi özellikler tabloya
katılırlar. Laboratuarda, sporadik CJH’nin tipik EEG
bulguları görülmez. BOS’ta 14-3-3 proteini olguların %50’sinde
pozitiftir. MRG’de tipik pulvinar bulgusu %70
sıklığında görülmüştür. PRPN analizinde tüm
olguların kodon 129 metionin homozigot oldukları görülmüştür.
Kodon 129’da valin sahibi olmak inkübasyon süresini uzattığından
bu durum hastalık etmenine aynı zaman çerçevesinde maruz kalan valin
sahiplerinin henüz klinik belirti geliştirmemiş olabilecekleri
endişesini doğurmuştur. Diğer insan prion
hastalıkları ve bu arada sporadik CJH’nin tersine, tonsiller biyopsi
ile hastalık yapan prion proteinin (PrPSc) gösterilmesi vCJH
için tanı koydurucudur. vCJH, beyin dokusunda da, sporadik CJH’den
farklı olarak spongiform değişikliklerden çok prion
plaklarına neden olur.
Avrupa’da vCJH olguları, ilk deli
dana olgusunun (Bovine Spongiform Encephalopathy – BSE) bildiriminden 10
yıl sonra görülmeye başlandı. İzleyerek, vCJH’nin BSE
etkeni ile kontamine dana eti yenmesi sonucu gelişebileceğini
gösteren çok sayıda delil bulundu. BSE primatlara doğrudan
beyne inokülasyon yoluyla bulaştırılabildi ve beyin dokusundaki
patolojik değişikliklerin, vCJH’de görülenin benzerleri olduğu
izlendi. Sonrasında, BSE ağız yoluyla da maymunlara
bulaştırılabildi. Sığır PrP’si eksprese
eden transjenik farelerde hem BSE, hem de vCJH beyin ekstreleri ile inokülasyon
sonucu ortaya çıkan hastalıkların inkübasyon zamanları,
nöropatolojik değişiklikler ve PrP’nin glikoform profilleri
açılarından birbirinin tıpkısı olduğu
saptandı.
Ailevi prion
hastalıkları
Ailevi
prion hastalıkları (Tablo 23), otozomal dominant bir tarzda,
20. kromozomda kodlanan PrP geninde (PRPN) oluşan çok sayıda
mutasyon sonucu kalıtılır. Bildirilen 50’den fazla
mutasyonun %95’i 4 nokta mutasyonu (kodon 102, 178, 200 ve 210) ve 2 oktapeptid
(5 ve 6) insersiyonu şeklindedir. Biz de PRPN kodon 178 Asp-Asn
mutasyonu taşıyan aileden 10 yıl içinde 2 olgu izledik. Ortaya
çıkan hastalık klinik ve laboratuar bulguları
açılarından sporadik CJH’ye çok benzer (ailevi CJH - fCJH) veya daha
nadiren klinik fenotip çok farklı olabilir (GSS ve FFI). Birbirinin
tıpkısı mutasyonlar bile aynı genin farklı bir bölgesindeki
(kodon 129) nükleik asid yapı taşının biri ya da
diğeri olmasına göre farklı klinik tiplere yolaçabilirler (kodon
178 nokta mutasyonunda, kodon 129’da metionin homozigotluğu FFI’ye, valin
homozigotluğu CJH’ye neden olur). Farklı klinik fenotiplerin
özellikleri için Tablo 23’e bakınız.
Tablo 23. Ailevi
Prion Hastalıklarının Klinik Özellikleri
|
fCJH |
Sporadik
CJH’ye göre daha erken başlayıp (geç 40 – erken 50’ler), daha uzun
sürebilir (22-24 ay). |
|
GSS |
40’lı
yaşların ortalarında başlar ve 5 yıl kadar
sürer. Serebellar ataksi ve spastik paraparezi erken dönemde
görülürken, demans ileri evrede katılır. |
|
FFI |
İlerleyici
uykusuzluk, disotonomi, dizartri, ataksi, miyoklonus, piramidal bulgular
erken bulgulardır. Demans ileri evrede görülür. |
Nöropatoloji
TSE’lerin temel nöropatolojik
özellikleri nöron kaybı, gliozis ve status spongiozistir.
Spongiozis, parenkim hücreleri ve nöronal proçeslerin vakuolizasyonuna
karşılık gelir. TSE lezyonları inflamatuar değildir.
Lezyonların dağılımı spesifik prion
hastalığının anatomik yatkınlığı ve
klinik fenotipini de yansıtır. Kuru’da serebellar gri maddeye
sınırlıdır. CJH’de o olgudaki semptomatolojiyi
yansıtır tarzda beyinde gri ve beyaz maddede değişkenlik
gösterir. Ultrastrüktürel olarak, prion çomakları da denilen
fibriller mevcuttur. En fazla Kuru, GSS ve vCJH’de, en seyrek olarak da
sporadik CJH’de olmak üzere amiloid boyaları ile boyanan plaklar
görülür. Ancak, bu plakların cevherleri AH’de olduğu gibi Aβ değil
PrPSc ile immun reaksiyon verir. vCJH’de görülen eozinofilik
merkez ve soluk kenarlı plaklar süslü (“florid”) adını
alır. Farklı prion hastalıklarının patolojik
özellikleri için Tablo 24’ e bakınız.
Tablo
24 İnsan
Prion Hastalıklarının Nöropatolojisi
|
Hastalık |
Nöropatoloji |
|
Kuru |
Kuru
plakları, spongiozis |
|
CJH |
Nöron
kaybı, spongiozis, gliozis |
|
fCJH |
Nöron
kaybı, spongiozis, gliozis |
|
nvCJH |
Süslü
plaklar ve yaygın spongiozis |
|
FFI |
Talamik
gliozis ve nöron kaybı |
|
GSS |
Plaklar,
gliozis, +/- spongiozis |
Şekil
22. Prion
nöropatolojisinde mikroskopi. solda spongiozis izlenirken sağda PrP (+)
amiloid plaklar izlenmekte. (Kaynak: http://www.path.sunysb.edu/faculty/woz/NPERESS/webclass2p2.htm)
Prion Kavramı
Prionlar genetik materyel
taşımayan infeksiyöz patojenlerdir. CJH’nin SSPE gibi bir
“yavaş virüs infeksiyonu” olduğu düşünülen bundan 20 yıl
kadar önce yukardaki cümleyi söylediğinde alaya alınan Stanley
Prusiner’in, çoğalma için genetik kodun şart olduğunu
düşünen genel geçer biyoloji paradigmasına bu gerçekten de taban
tabana zıt gibi görünen iddiası, artık tartışma
götürmez bir kesinlik kazanmış, hatta kendisi de bu nedenle 1997’de
Nobel Tıp Ödülü ile ödüllendirilmiştir. PrP, daha önce sözü
edildiği gibi 20. kromozom tarafından kodlanan, normal işlevi
aydınlatılamamış olan bir hücresel proteindir (PrPc).
PrPc, infeksiyöz PrP’ye (PrPSc) α-sarmallı
fiziksel konformasyonel yapısını kaybedip β
tabakalarına katlanarak yeni bir konformasyonel yapı kazanarak
dönüşür. Yani, PrPSc, normal proteinle tamamen aynı
kimyasal yapıya, amino asid dizisine sahipken, infeksiyöz özellik bu yeni
fiziksel yapıdan kaynaklanmaktadır. Suşa spesifik
özelliklerini genlerde kodlayan nükleik asid genomlu patojenlerin tersine,
prionlar suşa spesifik özelliklerini tamamen PrPSc’nin tersiyer
yapısında kodlamaktadırlar. PrPSc proteaza
dirençli, çözünemeyen ve ekstraselüler olarak biriken bir özellik
gösterir. PrPSc birikimi çevre dokuda nöron kaybı,
astrositik gliozis ve vakuolizasyona neden olur. Bu vakuoller giderek
birleşerek süngersi (spongiform) bir görünüm alırlar.
Transgenetik çalışmalarla da kanıtlandığı gibi,
PrPSc PrPC için bir şablon oluşturmakta ve PrPC
molekülü yeni bir PrPSc oluşturacak şekilde
katlanmaktadır. Başka bir deyişle, tek bir PrpSc molekülü
dahi PrP geni tarafından normal olarak üretilmekte olan PrPC’den
zaman içinde (inkübasyon süresi) hastalık belirtilerini göstermeye yetecek
miktarda birikmiş PrPSc üretebilmektedir.
Dolayısıyla da, PrPSc üretimi, ister ailevi olgularda
olduğu gibi PrP genindeki farklı mutasyonlarla, ister
infeksiyöz olgularda olduğu gibi infeksiyöz materyelin ortama
sunulmasıyla tetiklensin, çoğalma mekanizmasının yukarda
tanımlanan şekilde olduğu anlaşılmaktadır.
Bu durum da bir kalıtsal hastalığın nasıl aynı
zamanda infektif olabileceğini açıklamaktadır. Sporadik
olgulardaki etyoloji henüz açığa
çıkarılamamıştır. Ancak, bunun bir somatik
mutasyon sonucu veya PrPC’nin PrPSc’ye kendiliğinden
dönüşümü sonucu olduğu ileri sürülmektedir. Birbirinden bu
kadar farklı anatomik yatkınlık tarzları ve klinik
fenotipik özellikler gösteren prion hastalıklarının altında
farklı konformasyonel yapılara sahip, farklı PrPSc
suşlarının yattığı düşünülmektedir.
Tedavi
Prion hastalıklarının
henüz bir tedavisi yoktur. Ancak, PrPC’nin PrPSc’ye
dönüşümünü engellemek gelecekteki tedavi stratejilerinin temel hedefi gibi
durmaktadır. PrPC’ye bağlanarak
stabilizasyonunu sağlamak bir yol olabilir. Diğer bir yol da
PrPSc’yi destabilize edecek ajanların geliştirilmesi ile
olabilir. PrP geni taşımayan (PrP0/0) nakavt transjenik
farelerin doğal hayvanlara göre ayırdedilebilir bir
farklılık göstermemeleri insanda da PrP genini hedefleyecek
tedavileri çekici
kılmaktadır.
HAREKET BOZUKLUĞU VE
DEMANS
Huntington hastalığı,
PSP, multi sistem atrofi (MSA) gibi bir grup hastalıkta
hiperkinetik, akinetik-rijid, serebellar ya da bunların içiçe
geçmesinden oluşan motor bulgular, klinik tabloda zamansal ve
ağırlık olarak “subkortikal” tarzda bir zihinsel bozukluğun
önünde olurlar. Bu antiteler ve tedavileri “Hareket Bozuklukları”
bölümünde daha ayrıntılı biçimde
tartışılmıştır.
Huntington hastalığı
(HH), otozomal dominant bir nörodejenerasyondur. Koreye zaman içinde
demans eşlik etmeye başlar. PSP ile birlikte “subkortikal”
demansların prototipi sayılan bu tabloda bradifreni, zihinsel
esneklikte kayıp, soyutlama, planlama bozuklukları şeklinde bir
frontal yürütücü bozukluğa, tanımanın korunduğu sekonder
yakın bellek bozukluğu eşlik eder; diğer kognitif
işlevler göreli korunmuştur. Patolojik olarak Huntingtin
genindeki artan trinükleotid (C-A-G) tekrarları sonucu oluşan
eksitotoksik hasarın, tipik klinik sendroma yol açan anatomik
yatkınlığına bağlıdır. Mikroskopisinde
başlıca neostriatumda nükleer huntingtin birikimleri görülür.
Progresif supranükleer paralizi
(PSP)’de aksiyel baskın bir akinetik-rijid bozukluk ve özellikle
aşağıya bakış felci şeklinde bir vertikal
bakış paralizisi mevcuttur. Diğer oküler hareket
bozuklukları da bulunur. Özellikle elektro-okülogram (EOG) ile de
saptanabilen sakkadik hareketlerde yavaşlama hemen bütün olgularda mevcut
olan tipik özelliktir. Provoke olmayan sık düşmeler öykünün
başlıca özelliklerindendir. Demans HH’dekine benzer. Göz
bulguları yanı sıra otonom tutulumun beklenmemesi MSA, LCD ve
idyopatik Parkinson hastalığı gibi α-sinükleopatili
diğer parkinsonizm sendromlarından ayırmaya yardım
eder. MRG’de alışık bir göz özellikle sagital T1
kesitlerde mezensefalon atrofisini ayırt edebilir. Volümetrik olarak
saptanan globus pallidus atrofisinin normal kontrollerin yanı sıra
PH’lılar ve LCD’lilerden de ayırabildiğine ilişkin bir
bildirim bulunmaktadır. SPECT ve PET gibi fonksiyonel görüntüleme
araçları ile genellikle simetrik frontal hipoperfüzyon/metabolizma saptanır.
Patolojik olarak bir taupati olarak sınıflanabilecek bir
nörodejenerasyondur. Taupati kendini intranöronal globoz NFY’ler, nöropil
iğcikler şeklinde ortaya koyar. Nöron kaybı ve gliozis
eşlik eder. Bu patoloji, beyinsapında superior colliculi dahil
peri-akuaduktal gri madde, pre-tektal alan yanı sıra, substantia
nigra pars reticulata, subtalamik çekirdek ve globus pallidusta
saptanır. Kortikal tutulum daha seyrek ve hafiftir.
Başlıca τ birikimleri ve daha da seyrek olarak nöron kaybı,
gliozis ve mikrovakuolizasyon görülür. KBD ve PSP’nin tipik klinik
sunumları dahil birbirlerini taklit edebildikleri yapılan otopsi
doğrulamalı çalışmalardan bilinmektedir.
KBD’ye yukarda “Progresi Apraksi” ve
“Fronto-temporal Demans” bölümleri içinde değinilmişti.
Multi-sistem Atrofi (MSA)
tabloları, içiçe geçmiş, ancak birinin ön planda olduğu
“Striato-nigral Dejenerasyon” (SND), “Olivo-ponto-serebellar Atrofi” (OPCA) ve
“Shy-Drager Sendromu” (SDS) tablolarından oluşur. SND’de
simetrik ve tremorsuz bir akinetik-rijid sendrom, OPCA’da ataksi, SDS’de otonom
disfonksiyon temel özelliktir. Yeni nomenklatürde SDS’nin
bağımsız bir antite olarak varlığına son
verilmiş ve OPCA şekli için MSA-C (serebellar MSA), SND şekli
için MSA-P (parkinsonyen MSA) kullanılmaktadır. Disotonomi her
iki bozukluğa da olgudan olguya değişen zamanlama ve
şiddetlerle katılmaktadır. Hafif düzeyde ve demans
şiddetine genellikle ulaşmayan bir yürütücü bozukluk şeklinde
kognitif disfonksiyon eşlik eder. Patolojik olarak bir α-sinükleopatidir. Dolayısıyla,
diğer α-sinükleopatilerde olduğu gibi MSA’larda da REM-uykusu
davranış bozukluğu sıktır. Glial fibriler
α-sinüklein inklüzyonları MSA’ların ayırıcı
patolojik özelliğidir.
Wilson hastalığı,
Hallervorden-Spatz hastalığı, Kufs hastalığı
metabolik bozukluklara bağlı yirmili yaşların demans
hastalıkları arasındadır. Genç
başlangıçlı demansların ayırıcı
tanısına girerler. İlk iki hastalık tipik olarak hareket
bozuklukları ön planda olan sendromlara sahiptir. Genç erişkin
demansları arasında olduğu için burada anılan Kufs’a
demansın yanısıra epilepsi eşlik eder. Whipple
hastalığı da, bazen PSP’yi taklit eden diğer nadir hareket
bozukluğu – demans sendromları arasındadır.
SEKONDER DEMANSLAR
Vasküler Demans
Bundan 30 yıl önce “serebroskleroz”
adıyla en sık, 20 yıl önce “Multi-infarkt demans” (MID)
adıyla AH’den sonra 2. sıklıktaki demans olduğu
düşünülen VaD, son yıllardaki kliniko-patolojik korelasyon
çalışmalarıyla sıralamadaki yerini iyiden iyiye kaybetmiş
durumdadır. Bu çalışmalarda klinik olarak VaD
tanısı almış olguların büyük bölümünün nekropsilerinde
gerçekte AH oldukları veya hem vasküler, hem de AH patolojilerini birlikte
gösteren “karma (mixed) demans” (MIX) oldukları görülmüştür.
İnme için risk faktörlerinin büyük bölümünün AH için de risk faktörleri olduğu
gösterilmiştir.
VaD için çok sayıda
yayınlanmış tanı kriterleri mevcuttur. Bunlar
arasında duyarlılık ve özgüllük açılarından tek
başına optimuma yaklaşan olmadığı gibi, içerme ve
dışlama kriterleri nedeniyle birbirleri arasında da ciddi
uzlaşmazlıklar bulunmaktadır. Tablo 25’de sunulan
NINDS-AIREN kriterleri, “Muhtemel Vasküler Demans” (VaD)
tanısı içindir. İçleme ve dışlama kriterleri
açısından oldukça katı, bu nedenle
duyarlılığı düşük, ancak özgüllüğü oldukça yüksek
bir kriterler dizisidir. “Hachinski İskemik Skoruı” (HIS),
VaD’ı DAT’dan ve MIX’den ayırmak amaçlı olarak BT öncesi dönemde
geliştirilmiş, BT sonrasında bir dizi
modifikasyonları önerilmiş, klinik uygulamada hala bir ölçüde yerini
koruyan bir ölçektir (Tablo 26). Duyarlılık ve
özgüllük çalışmaları, 18 puanlık versiyonda VaD için 7 ve
üstü, DAT için 4 ve altı puanları kabul edilebilir bulmuştur.
Tablo 25.
Muhtemel Vasküler Demans Tanısı İçin NINDS-AIREN Kriterleri
1.
Aşağıdaki dışlama kriterleri bulunmaksızın
demansın mevcudiyeti:
Bilinç bozukluğu, delirium, psikoz, nöropsikolojik
muayeneyi engelleyecek nitelikte ağır afazi veya sensoryal-motor
değişiklikler, kognitif değişiklikleri açıklayabilecek
nitelikte sistemik veya beyin (Alzheimer hastalığı gibi)
hastalıkları.
2.
Serebrovasküler Hastalık.
3.
1 ve 2 arasında aşağıdaki tarzda bir
ilişki:
a)
Saptanabilen bir inmeyi izleyen 3 ay içinde başlayan demans
b)
Kognitif işlevlerin ani bozulması veya kognitif bozuklukların
dalgalanan tarzda basamaksı ilerleyişi.
Muhtemel vasküler demans tanısıyla uyumlu
olacak klinik özellikler şunlardır:
1.
Erken dönemde yürüyüş bozukluğu.
2.
Dengesizlik veya kendiliğinden düşmeler öyküsü.
3.
Erken dönemde, ürolojik nedenlerle açıklanamayan idrara sık
çıkma, acele etme veya diğer üriner belirtiler.
4.
Psödobulber paralizi.
5.
Kişilik veya duygudurum değişiklikleri, abuli, depresyon,
emosyonel inkontinans veya psikomotor retardasyon ya da yürütücü
işlevlerde bozuklukları da içerecek şekilde diğer
subkortikal bozukluklar.
Vasküler demans tanısı ihtimalini azaltan
özellikler şunlardır:
1.
Beyin görüntülemesinde karşılık gelecek lezyonlar
olmaksızın, bellek bozukluğu ve kognitif işlevlerde
ilerleyici bozulmanın erken dönemde mevcudiyeti.
2.
Kognitif bozukluk dışında diğer fokal nörolojik
bulguların bulunmaması.
3.
BT veya MRG’de serebrovasküler lezyonların bulunmaması.
Tablo 26.
Hachinski İskemik Skoru (Orijinal versiyon ve bir modifikasyonu)
|
Klinik Özellik |
Hachinski (1975) |
Loeb-Gandolfo (1983) |
|
Ani
başlangıç |
2 |
2 |
|
Basamaksı
kötüleşme |
1 |
|
|
Dalgalanan
seyir |
2 |
|
|
Noktürnal
konfüzyon |
1 |
|
|
Kişiliğin
göreli korunması |
1 |
|
|
Depresyon |
1 |
|
|
Somatik
yakınmalar |
1 |
|
|
Emosyonal
inkontinans |
1 |
|
|
Hipertansiyon
öyküsü |
1 |
|
|
İnme
öyküsü |
2 |
1 |
|
Jeneralize
ateroskleroz |
1 |
|
|
Fokal
nörolojik belirtiler |
2 |
2 |
|
Fokal
nörolojik bulgular |
2 |
2 |
|
Bilgisayarlı Tomografi |
|
|
|
Hipodens
alan – izole |
|
2 |
|
Hipodens
alan – multipl |
|
3 |
|
Maksimum skor |
18 |
10 |
|
Vasküler
sınır |
7-18 |
5-10 |
|
Karma
sınır |
5-6 |
3-4 |
|
Dejeneratif
sınır |
0-4 |
0-2 |
MID tanımı kardiyo-embolik veya
atero-trombotik inmelerin aynı hastada tekrarlaması sonucu bu tür bir
inmeye eşlik etmesi beklenen sekelin yanı sıra, tabloya demans
şiddetinde bir zihinsel bozukluğun da katılmasını
öngörür. Oysa ki çeşitli VaD serilerinde bu tür tablolar bütün
VaD’lıların düşük bir oranını
oluşturmaktadır. Bu düşük temsile kardiyo/serebrovasküler
hastalığa ilişkin sorunların
ağırlığının (hemipleji, yatağa
bağımlılık, vb.) bu tür hastaları demansla
uğraşan uzmanların önüne daha az getirmesi yanısıra,
afazi gibi optimal mental durum değerlendirmesini engelleyecek sorunlar
nedeniyle bu tür hastalarda demans tanısının daha az konuyor
olması olabilir. Sıklıkla hipertansiyona bağlı,
daha seyrek olarak amiloid anjiyopati ve genetik nedenler de dahil diğer
küçük damar hastalığı seyrinde görülen VaD serilerin
yarıdan çoğunu oluşturmaktadır. Bu VaD tabloları
akut iskemiye bağlı olarak derin gri madde yapıları ve
frontal lobların beyaz maddesinde multipl laküner infarktlar (laküner
durum) veya stratejik lokalizasyonlu tek bir derin infarkt sonucu
olabileceği gibi, Binswanger hastalığının
mekanizmasında ileri sürüldüğü gibi peri ve paraventriküler beyaz
maddenin kronik hipoperfüzyonu sonucu yaygın beyaz madde gliozisi ile de
ortaya çıkabilir.
Tipik bir VaD için, inme gibi bir vasküler olay
çevresinde akut başlangıç, yine ayırdedilebilir bir vasküler
olaya kadar bir ölçüde düzelmeyle birlikte durağan bir plato dönemi ve
yeni olay sonrasında basamaksı kötüleşme ile seyreden kognitif
yıkım öyküsü esastır. Klinik tablo VaD alt tiplerine özgü
değişiklikler gösterir. MID, serebral hemisferleri
besleyen ana arterlerin proksimal ve/veya distal tıkanmalarına
bağlı olarak, bu alanlarda oluşan infarktüslerin devreden
çıkardığı işlevleri yansıtır. Bu
durumda beklenen tablo, sol hemisfer inmelerinin afazik bozukluklarıyla,
sağ hemisfer inmelerinin ihmal sendromu ve vizyo-spasyal
bozukluklarının karışımı şeklindedir.
Yakın bellek de nadiren primer bir bozukluk söz konusudur. Demansa
hemi- veya tetraparezi, yüzeyel duyu bozuklukları ve görme alanı
kusurları sıklıkla eşlik etmektedir.
Laküner durum, “subkortikal demans” olarak
tanımlanan tablonun protipidir. Seyir yukarda tanımlanan
şekilde akut başlangıç ve basamaksı ilerleme
tarzındadır. Demans profili bellekte tanımanın
korunduğu sekonder bozukluk, frontal yürütücü bozukluk
şeklindedir. Davranışsal özellikler arasında
bradifreniden apatiye giden psikomotor yavaşlama ve emosyonel labilite ön
plandadır. Somatik nörolojik bulgular ağırlıkla
hemiparezi sekelleri, piramidal bulgular, alt beden yarısı (veya
vasküler) parkinsonizm şeklinde yürüyüş bozuklukları psödobulber
paralizi-dizartri tarzındadır. Üriner inkontinans erken dönemde
tablonun esaslı parçalarından birini oluşturmaktadır.
Afazi ve hemianopsi gibi “kortikal” kognitif ve algısal bulguların
görülmesi beklenmez. Beyin görüntülemesinde tabloyla
ilişkilendirilebilecek derin gri madde yapıları ve frontal beyaz
madde içindeki laküner lezyonların varlığı tanı
koydurucu olacaktır. Ancak tablo her zaman bu kadar tipik
olamayabilir. Binswanger hastalığı (BH) seyrinde
peri-ventriküler beyaz maddenin kronik hipoperfüzyona bağlı
ilerleyici gliozisinde doku kaybı sıklıkla sessiz infarktlar
şeklinde olabilir. Bu nedenle öykü içinde akut başlangıç
veya basamaksı ilerleme bazen hiç ayırt edilemeyebilir veya birkaç
gün sürüp düzelen hafif bir konuşma/yutma bozukluğu, taraf zaafı
şeklinde bir minör inmeyle tablonun biraz daha bozulduğu
kaydedilir. Klinik tablo laküner duruma çok benzer. Subkortikal
beyaz maddede anılan bozukluk kendisini BT’de beyaz madde
yoğunluğunda yaygın azalma (lökoareozis), MRG’de T2 ve proton
ağırlıklı incelemede peri ve paraventriküler bölgelerde
birleşme eğilimi (konfluans) gösteren yüksek sinyal odakları
şeklinde ortaya koyar (Şekil 23).
Medyal talamus, kaudat çekirdek başı,
kapsula interna genusunun ventral bölümünü içeren stratejik lokalizasyonlu
infarktlar bazen tek başlarına demans tablosunu ortaya
çıkarabilirler. Demans profili tutulan stratejik alana göre
değişir. Örneğin, medyal talamusta dorsomedyal çekirdekle
birlikte mamillo-talamik traktusun infarktı frontal yürütücü ve amnestik
bozuklukların bir karmasını oluştururken, kaudat çekirdek
başı infarktlarında saf bir frontal yürütücü bozukluk görülür.
Bu durumda, yeni bir vasküler olay olmazsa akut demans tablosu zaman içinde
kendiliğinden düzelebileceği gibi, nörolojik defisiter bulgular da
mevcut olmayabilir (Tablo 27).
Notch proteini genindeki mutasyonlar klinik tablo
ve görüntüleme açılarından BH’ya çok benzeyen “Cerebral Autosomal
Dominant Arteriyopati Subkortikal İnfarktlar Lökoensefalopati”
(CADASIL) isimli ailevi hastalığa neden olur. Bu
ailelerde migren öyküsü ile birlikte BH benzeri demans, otozomal dominant
kalıtım tarzıyla birlikte bulunur.
VaD tanısı pratik olarak iskemik
vasküler lezyonlara bağlı demanslara karşılık olarak
kullanılsa da, büyük bir hemorajik inmeyi izleyerek (intraserebral kanama,
subaraknoid kanama) hasta sağ kalırsa, sekel tablo demans ile uyumlu
statik bir ensefalopati olabilir.
VaD tedavisi serebrovasküler hastalık
tedavisi ilkelerini izler. Başta hipertansiyon ve diabet olmak üzere
vasküler risk faktörlerinin kontrolü, asetil salisilik asit ve benzeri
anti-agregan uygulama, kardiyak veya arterden artere emboli riskinde
anti-koagülasyon, kritik ekstra-kranyal karotis interna daralmalarında
uygun olgularda karotis endarterektomisi VaD’ın önlenmesi ve denetiminde
de etkili olacaktır. Bir NMDA reseptör antagonisti memantin ve AH’de
kullanılan kolinesteraz inhibitörleri VaD’da da etkin olduklarına
dair çok güçlü olmasa da deliller mevcuttur. Depresyon ve diğer duygudurum
bozukluklarının uygun tedavisine
çalışılmalıdır. Vasküler parkinsonizm ve
yürüyüş bozuklukları L-dopaya sıklıkla
cevapsızdır, ancak bu ajanlar uygun dozlara çıkılıp
belli bir cevap alındığı takdirde
kullanılabilir.
Şekil 23. BT ve MRG’de
İskemik Beyaz Madde Hasarının Görünüşü.
BT’de lökoareozis; lateral ventriküller düzeyinden geçen
kesitte paraventriküler beyaz madde
dansitesi azalmış. B. MRG’de aynı
olgu; T2 ağırlıklı kesitte beyaz maddede yüksek sinyal.
Tablo 27.
Vasküler demans alt tipleri
|
|
Multi-infarkt
Demans |
Laküner
Durum |
Stratejik
İnfarkt |
Binswanger
Sendromu |
|
Etyoloji |
Kardiyo-emboli,
karotis hastalığı, intra-kranyal in-situ tromboz |
Arteriolo-skleroz, amiloid anjiyopati,
diğer küçük damar hastalığı |
Arteriolo-skleroz, amiloid anjiyopati,
diğer küçük damar hastalığı, intra-kranyal in-situ
tromboz |
Arteriolo-skleroz, amiloid anjiyopati,
diğer küçük damar hastalığı, CADASIL |
|
Damar
alanı |
Tek
arter |
Tek
arter, küçük arteriyol |
Tek
arter |
Sınır
sulama alanı, küçük arteriyoller |
|
Beyin
hasarı mekanizması |
Akut
iskemi |
Akut
iskemi |
Akut
iskemi |
Kronik
hipoperfüzyon |
|
Patoloji |
Birden
fazla bölgesel infarkt |
Multipl
lakünler |
Stratejik
lokalizasyonlu tek infarkt |
Demyelinizasyon
ve aksonal kayıp |
|
Görüntüleme |
Kortiko-subkortikal
infarkt alanları |
Derin
gri madde ve frontal beyaz maddede laküner infarktlar |
Medyal
talamus, kaudat çekirdek, kapsula interna genusunda laküner infarkt |
Peri,
paraventriküler beyaz maddede lökoareozis / hiperintensite |
|
Lokalizasyon |
Geniş
boyutlu nöro-kognitif şebekeler |
Kortiko-striato-talamo-kortikal
devreler |
Kortiko-striato-talamo-kortikal
devreler |
Kortiko-kortikal
bağlantılar |
|
Klinik |
Afazi,
vizyo-spasyal bozukluk, hemiparezi, hemianopsi |
Yürütücü
bozukluk, psödobulber paralizi, vasküler parkinsonizm, üriner inkontinans |
Yürütücü
bozukluk, amnezi |
Yürütücü
bozukluk, yürüme bozukluğu, üriner inkontinans, sıklıkla
yavaş seyir |
Chui’den modifiye edilmiştir.
Normal Basınçlı
Hidrosefali
Normal basınçlı hidrosefali
(NPH), BH’ya benzer bir triada sahiptir. İlk kez Adams ve Hakim
tarafından tanımlanmıştır ve dolayısıyla
Adams-Hakim sendromu olarak da anılır. Demans genellikle hafif
şiddettedir ve zamanlama olarak yürüyüş bozukluğundan sonra
başlar. Yürüyüş bozukluğu genellikle ilk, bazen de tek
başvuru yakınmasıdır. Triadın diğer özellikleri
ancak sorgulamayla (daha sık idrara çıkma, idrar için acele etme, GYA’ları
etkilemeyen unutkanlık) ve ayrıntılı bir mental durum
muayenesi ile ortaya konur. Yürüyüş bozukluğu
başlangıçta hafif bir serebellar ataksiyi andırırken
giderek parkinsonyen özellikler kazanır. Yürüyüşü
başlatmak güçtür, yürüyüş kısa adımlarladır ve
adımlama sırasında ayakları yerden kaldırmak güç
gibidir (manyetik yürüyüş). Postüral dengesizlik, düşmeler ve
donma fenomeni görülebilir. İleri evrelerde giderek ayakta durma,
hatta oturma güçleşir. Sfinkter kusuru başlangıçta idrar
için acele etme (“urgency”) tarzındayken ileri evrelerde tam bir
inkontinans yerleşir. Kognitif bozukluk aslında
sıklıkla demans şiddetine ulaşmaz. Bazen depresyonla
karıştırılabilecek bir apati eğilimi eşlik
eder. Nöropsikolojik muayenede, limbik tarzda olmayan tanımanın
korunduğu bir bellek bozukluğu ve yürütücü bozuklukla giden bir
demans profiliyle birlikte, görüntülemede kortikal atrofi ya da subkortikal
iskemik değişiklikler olmaksızın ventriküler dilatasyon NPH
özellikleridir. Fizyopatolojisinde BOS
akışkanlığında bir bozulmanın
yarattığı BOS basınç artışının
ventrikül sistemini genişletmesinden sonra konveksite ve ventriküler
basınçlar arasındaki dengesizliğin zaman içinde kaybolması
ileri sürülmektedir. BOS dolaşım bozukluğunu tetikleyen
olay bir subaraknoid kanama gibi öyküde ayırt edilebilen bir olay
olabileceği gibi (sekonder NPH) böyle bir öykü mevcut olmayabilir
(idyopatik NPH). BOS boşaltıcı LP’ler ile klinik düzelme
(yaklaşık 20-50 cc ile özellikle yürüyüşte düzelme) tanı
koydurucudur. Bu düzelme sıklıkla geçici ama bazen uzun süreli
de olabilir. Ancak negatif bir LP testi NPH’yı
dışlamaz. Sürekli basınç monitorizasyonu ve bazı
hidrodinamik testlerin de tanı koydurucu olduğu ileri sürülmektedir.
Tedavi ventrikülo-peritoneal “shunt” şeklindedir. Cerrahi
sonrası iyi prognoz öngörücüleri olarak yürüyüş bozukluğunun
zamanlamada ilk belirti olması, kognitif bozukluğun görece kısa
süreli olması, NPH’nın sekonder olması ve LP testiyle pozitif
sonuç, kötü prognoz öngörücüleri olarak da zamanlama ve şiddet açısından
ön planda kognitif bozukluk, MRG’de aşikar kortikal atrofi ve yaygın
beyaz madde bozukluğunun varlığı ileri sürülmüştür.
Diğer Sekonder Demanslar
Hızlı seyirli
demansların ayırıcı tanısına “paraneoplastik
limbik ensefalit” de katılır. Sistemik kanserler içinde
en sıklıkla akciğer kanseri ile ilintilidir. Sistemik
kanserin saptanmasından bir kaç yıl önce ortaya çıkabilir.
Depresyon ve letarji gibi prodromal belirtilerle başlangıç sıktır.
Araya giren nöbetler tabloyu alevlendirebilir. Tablo
yerleştiğinde kognitif olarak bir izole amnestik durumdur. Buna
diurnal ritim bozuklukları, disinhibisyon gibi kişilik
değişiklikleri eşlik eder. Görüntüleme tümüyle normal
olabileceği gibi, Herpes simpleks ensefaliti tutulum tarzına benzer
bir şekilde başlıca mezyal temporal alanlarda yüksek sinyal
saptanabilir. BOS’ta sıklıkla protein
artışı ve lenforaji görülür. Yine, BOS’ta anti-Hu antikoru
pozitifliği tanıya yardım eder. Son zamanlarda başta NMDA
ve AMPA reseptörleri ve voltaj kapılı potasyum kanalı (VGKC)
olmak üzere tedaviye göreli olarak iyi cevaplı kanalopatilerle limbik
ensefalit olguları bildirileri artmaktadır.
İnfeksiyöz (tuberküloz,
nörosifilis, kriptokok, Lyme, HIV) inflamatuar (sarkoidoz, özellikle Hashimoto
tiroiditi olmak üzere vaskülitik ve non-vaskülitik otoimmun
meningo-ensefalitler) ve neoplastik subakut-kronik menengo-ensefalitler
hızlı seyirli demanslar ayırıcı tanısında
değerlendirilmesi gereken durumlardır. Özellikle
inorganik cıva olmak üzere toksinler de hızlı seyirli demansları
taklit edebilir. MRG (kontrast madde ile leptomeningeal boyanma,
granülomatöz parenkim lezyonları), BOS incelemeleri (spesifik patojenin
üretilmesi, spesifik seroloji, sitoloji, hücre ve protein
artışı), serebral anjiyografi ve toksik tarama anılan
farklı durumların ayırıcı tanısında
kullanılan laboratuar yöntemleridir.
Wernicke-Korsakoff
Hastalığı, gerçekte bir izole amnestik durum
olsa da, eşlik eden psikotik bozukluklar ve kişilik
değişikliğiyle hastanın
bağımsızlığını bozarak sıklıkla
bir demans gibi davranır. B1 vitamini (thiamin) eksikliğine
bağlı olarak limbik (corpus mamillare, dorsomedyal ve pulvinar
talamik çekirdekler) ve beyinsapı (peri-akuaduktal gri madde)
yapılarının hasarlanması sonucu gelişir.
Konfüzyon, oftalmoparezi, ataksi ve nistagmustan oluşan ve haftalar süren
bir akut dönem sonrası, bu bulgular çeşitli derecelerde düzelirken
kronik tablo yerleşir. En sıklıkla, kronik alkolizm
zemininde ortaya çıksa da, ülkemizde açlık grevleri de belli
başlı nedenleri arasındadır. Bu durumda, tabloyu
ortaya çıkaran etkenin açlığın kendisi değil de,
genellikle B1 içermeyen şekerli parenteral sıvılarla
sonlandırılmasına bağlı iyatrojenik bir etken
olduğu görülmüştür.
Nöro-Behçet ve multipl skleroz
hastalıklarının kronik evrelerinde kognitif bozukluk
şiddeti demans düzeyine ulaşabilir.
KAYNAKLAR ve OKUMA ÖNERILERI
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
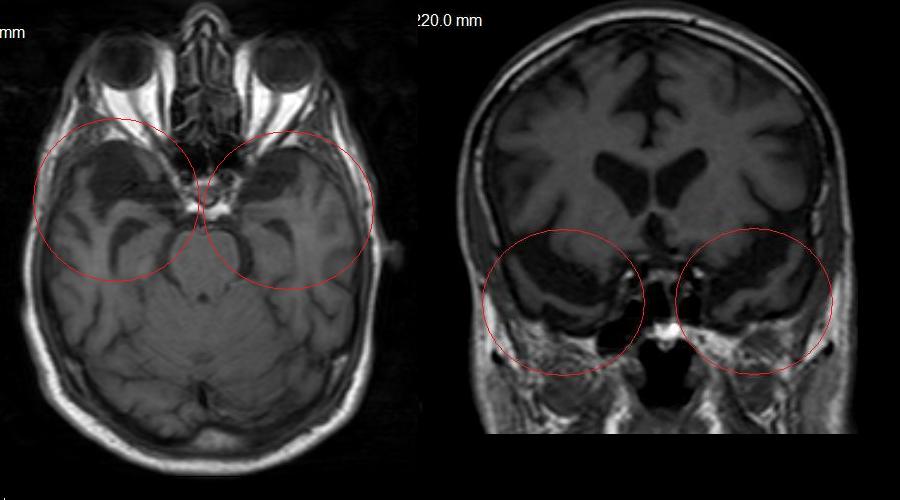{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
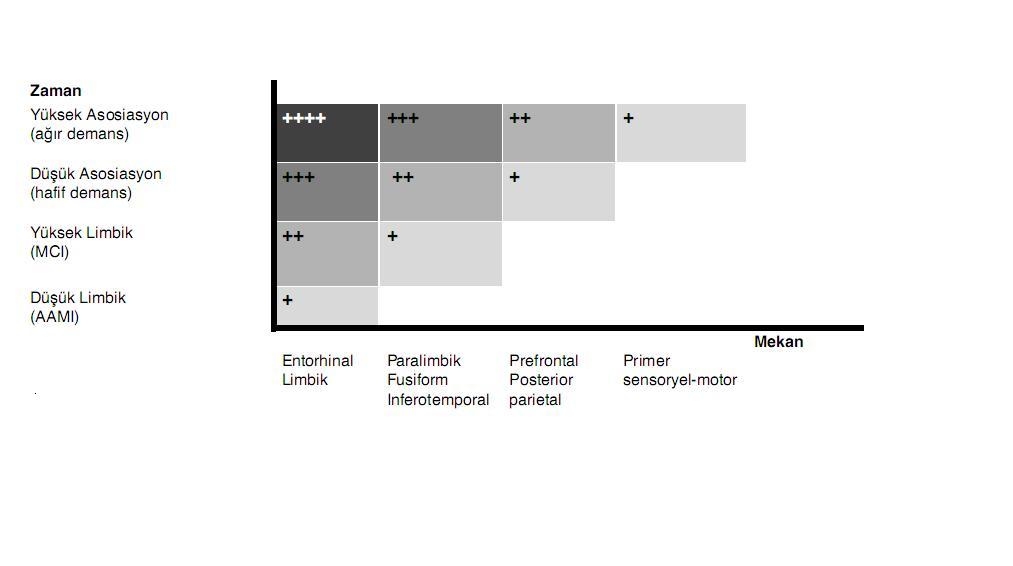{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
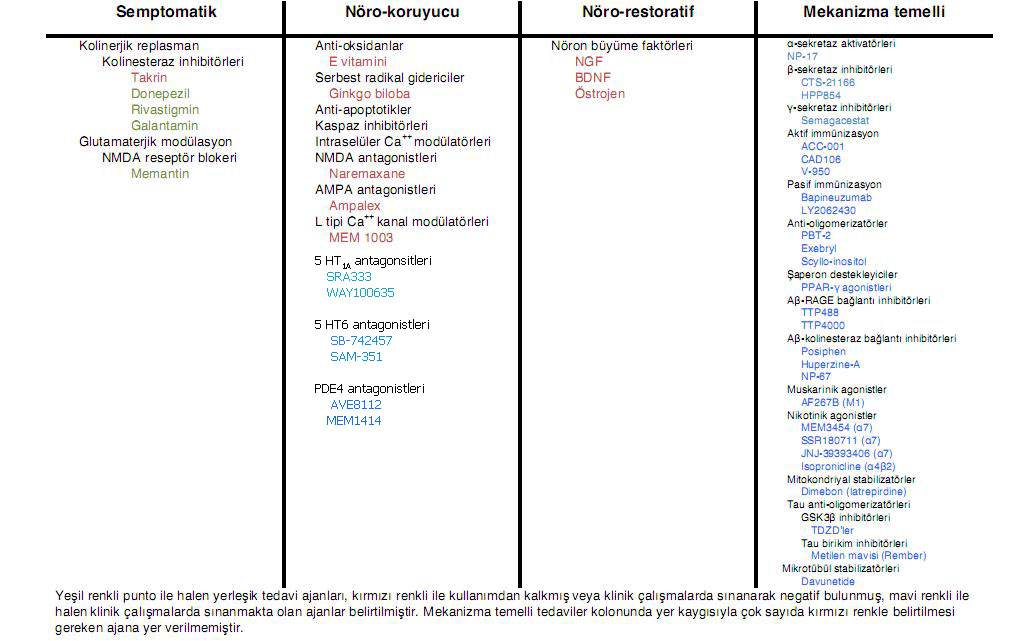{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}