PRİON HASTALIKLARI
Yazanlar: İ. Hakan Gürvit, Bedia Samancı
Son güncelleme tarihi: 14.10.2020
Bu bölüm, sayfa sayısı kısıtlaması nedeni ile kitabın basılı halinde kısaltılmıştır. Daha ayrıntılı metin için bölümün uzun versiyonuna başvurulabilir.
Prion hastalıklarının tarihine kısaca bakıldığında; Hans Gerhard Creutzfeldt ilk olguyu 1920 yılında bildirdikten sonra 1921’den itibaren Alfons Maria Jakob art arda beş olgu bildirir. Olgularına “spastik psödoskleroz” adını verir ve literatür tararken Creutzfeldt’in olgusuyla da karşılaşır ve kendi olgularına benzeterek birlikte anar. 1959’da ise Foré yerlilerine özgü Kuru hastalığının ilk otopsi serileri birbiri ardına yayınlanır. Amerikalı veteriner Hadlow 1960’ta scrapie ve kuru arasındaki çarpıcı benzerlikleri fark eder. 1966 yılında kurunun ve Creutzfeldt-Jacob hastalığının (CJH) scrapie gibi deneysel olarak bulaştırılabilirliği gösterilmiştir ve böylelikle “bulaştırılabilir (transmissable) spongiform ensefalopatiler” (TSEler) ismi scrapie, kuru ve CJH’yi içine alan bir insan ve hayvan hastalıkları grubunu kapsayan bir ana sınıf olarak yerleşmiş olmuştur. TSE keşfi Gajdusek’e 1976 Nobel Tıp Ödülü’nü kazandıracaktır. Stanley Prusiner 1982 yılında sorumlu ajanın nükleik asit genomuna sahip olmayan bir “proteinimsi enfeksiyöz partikül” olduğunu ileri sürmüş ve “prion” kavramını ortaya atmıştır ve bu keşfi nedeniyle 1997 yılı Nobel Tıp Ödülü’nü kazanmıştır.
CJH’nin nedeni olarak nörodejenerasyona neden olan diğer proteinopatilerde olduğu gibi, fiziksel konformasyonunu değiştirerek patojen haline dönüşen ve bu haliyle bir şablon oluşturup normal hücresel proteini kendine benzetmeye başlayan, böylelikle çoğalıp biriken bir protein olan prionun (PrP) bulunması alanı kökten değiştirmiştir. Bundan böyle aynı zamanda bulaşabilir bir proteinopati olarak prion hastalıklarından (PrH) veya prionopatilerden söz edilecektir. Oesch 1985 yılında PrP’yi kodlayan geni (PRNP) 20. kromozomun küçük koluna haritalandırmıştır. PRNP biri protein kodlayan olmak üzere iki ekzon ve bir intron şeklinde basit bir genomik yapıdadır. Genin açık okuma çerçevesindeki (ORF) nokta ve insersiyon mutasyonları kalıtsal prion hastalıklarının nedenleridir.
PRİON PROTEİNİ VE PRİON HASTALIKLARI
PrP normal olarak vücut dokularında, bu arada özellikle merkezi sinir sisteminde (MSS), nöron membranında yerleşiktir, astrositlerde de bulunur. Bu normal protein hücresel PrP (PrPc) adını alır ve dış membrana bir glikozilfosfatidilinositol (GPI) çapası ile bağlıdır; buradan endositoz ile geri dönüştürülür. Bir hücre yüzeyi, iki de transmembran şekilleri vardır. Proksimal karboksi ucu (C-terminus) α-sarmallı (üç sarmal) ve iki anti-paralel iç içe geçmiş β-katmanlı yapıda, buna karşılık distal amino ucu (N-terminus) oktapeptid tekrarlı [(PHGGGWGQ) X 4] amino asit yapısındadır. PrH varyantlarında farklı biçimlerde glikozillenen iki tane N-glikozilasyon bölgesi vardır (181. ve 197. rezidülerde).
Kesin işlevi bilinmemekle birlikte bir membran proteini olması nedeniyle sinyal iletim yolaklarında işlev gören bir ligand olduğu ileri sürülmüştür. Glutamaterjik sinapslarda, amiloid ile birlikte reseptör endositozu ve uzun süreli depresyonu tetiklediği ve dolayısıyla AH mekanizmalarına katıldığı ileri sürülmüştür. En kabul gören işlevi hücre içi bakır transportunda oynadığı roldür. PrPc her normal hücresel protein gibi proteolitik enzimlerle (proteazlarla) degrade edilir. Başlangıçta PrP geni nakavt (PRNP-/-) transgenik farelerde (sadece kodlayan eksonun susturulduğu) herhangi bir klinik bulgu gözlenmemesi PrP’nin varkalımsal düzeyde bir işlevi olmadığını düşündürmüştür. Ancak sonrasında genin daha yaygın bir şekilde susturulduğu (kodlayan ekson ve intron) yeni yöntemlerde yaşlı hayvanlarda başlıca serebellar bulgular olmak üzere bazı eksiklik bulgularının ortaya çıktığı kaydedilmiştir. Zaman içinde bunun nedeninin PRNP ile onun komşu geni arasındaki genlerarası düğümlenme ile bu farelerin beyinlerinde komşu genin ektopik ifadesi olduğu bulunmuştur. Bu genin (PRDN) kodladığı proteine PrP ile C-terminalinde %25 homoloji taşıdığı için prion-ilişkili protein Doppel (Dpl) adı verilmiş ve ataksinin nedeni olarak Dpl’nin ektopik ifadesi gösterilmiştir.
Yukarıda söz edildiği gibi dokuda hastalık yapan PrP şeklinin mevcudiyeti bir tohum gibi davranarak PrPc’yi kendine benzetmeye başlar. Dokuda PrPc yoksa, PrPSc de çoğalıp hastalık yapamaz; örneğin, PRNP-/- transgenik farelerde inokülasyon TSE yaratmaz. Dönüşüm PrPc’nin α-sarmallı yapısının proteazlara (proteinaz K, lizozomal enzimler) dirençli β-katmanlı şekle katlanmasıyla gerçekleşir. Bu katlanmayla PrPc’de %3 kadar olan β-katman %43’e ulaşır ve proteaz direnci çok fazla artmış olur. Hastalık yapıcı şekle, yaygın biçimde, koyunlardaki TSE’den (scrapie) alıntıyla PrPSc adı verilir. Proteaz direnci dolayısıyla PrPres, Dünya Sağlık Örgütü tarafından da PrPTSE şeklinde de kullanılmaktadır. PrPTSE başlıca (fakat sadece değil) MSS’de yaygın birikintiler veya amiloid plaklar şeklinde çöker.
Prion hastalığının yayılımındaki farklılıklar, inkübasyon zamanı, PrPTSE topografik dağılım tarzı, nöropatolojik değişikliklerin bölgesel şiddeti gibi kendine özgü fenotipik hastalık özelliklerine neden olan farklı “prion suşları”nın tanımlanmasına yol açmıştır. PrPc proteinaz K (PK) ile çok büyük ölçüde sindirilirken, PrPTSE PK’ye dirençli bir çekirdek kısma sahiptir. PrPTSE birikintilerinin PK ile amino tarafından proteolitik kesimi Parchi ve arkadaşlarının 1996’da Tip 1 ve Tip 2 adlarını verdikleri iki majör suşu üretir. Tip 1 82. rezidüden kesilmiş, 21 kDa moleküler kütleye sahip iken, Tip 2 97. rezidüden kesilmiş, 19 kDa moleküler kütleye sahiptir. İyatrojenik CJH’de 20 kDa bir ara form saptanmıştır. Ayrıca, 154-162. rezidüden kesilmesiyle 12-13 kDa’lık bir üçüncü tip olan PrP-CTF12/13, Tip 1’in proksimalde (karboksi tarafta) GPI çapasının da kesilerek (147-153. rezidüden) daha da kısaltılmasıyla 7-8 kDa bir dördüncü tip olan PrP7-8 oluşur. Yeni tanımlanan “değişken proteaz-duyarlı prionopati (VPSPr) isimli sporadik CJH’deki farklı proteaz-dirençli suşlar aşağıda ilgili başlık altında tartışılacaktır.
RNP kodon 129 M/V polimorfizmi TSE’lerin fenotipik hastalık özelliklerini belirleyen diğer faktördür. Normal popülasyonda genotip sıklıkları MM %37, MV %51, VV %12 iken, sporadik CJH’de MM %71,6, MV %11,7, VV %16,7 bulunmuştur. PRNP D178N mutasyonunda kodon 129’da metionin (M) homozigotluğu ile valin (V) homozigotluğunun birbirlerinden çok farklı iki kalıtsal PrH’ye neden olması çok çarpıcıdır. Benzer şekilde V taşıyıcılığı sporadik ve iyatrojenik PrH’lerde daha fazla plak benzeri patoloji ile daha uzun inkübasyon zamanı ve hastalık süresi ile ilişkilidir.
Suş ve polimorfizm birlikte; sporadik, edinsel ve kalıtsal tüm PrH fenotiplerini belirlerler. Tip 1 ağırlıkla M ile (MM homozigot sporadik CJH’lilerin (sCJH) %95’i Tip 1’e sahip), Tip 2 ağırlıkla V (V taşıyıcı sCJH’lilerin %86’sı Tip 2’ye sahip) ile ilişkilidir. Suş-M/V kombinezonlarına göre sporadik PrH’lerde altı farklı fenotipten oluşan yeni bir sınıflama sistemi önerilmiştir: MM1, MV1, VV1, MM2, MV2 ve VV2. Birkaç istisna sıralanabilir. MM1 ve MV1 tıpatıp bir fenotip sergilediğinden MM(V)1 olarak birleştirilmiştir. MM2 ise birbirine hiç benzemeyen kortikal ve talamik tutulumlu iki ayrı fenotip ile sunulur; bu nedenle MM2C ve MM2T isimlerini alırlar. MV2 olanlardan Kuru benzeri plaklar sergileyenler MV2K olarak sınıflandırılarak MM2C’ye benzer MV2C hastalarından ayırt edilmişler, diğerleri ise MM(V)2C olarak birleştirilmiştir. Zaman içinde suşların her olguda mutlaka saf bir şekilde bulunmadığı her iki suşun aynı beyinde karma olarak görülebileceği de ortaya konmuştur. Karma olgular tüm olguların %35’ine kadar ulaşmakta ve M/V statüsünden etkilenmektedir (MM %43, VV %15). Baskın suş PrPTSE’nin %95 kadarı ve tüm beyinde saptanabilirken, ikincil suş ancak fokal olarak saptanabilmektedir. Dolayısıyla, karma olgular farklı fenotiplere karşılık gelmemekte, fakat baskın suşun fenotipini sergilemektedirler. Böyle olgularda baskın suşun önce yazıldığı bir notasyon kullanılmaktadır (örneğin,, MM1 + 2C veya MM2C + 1).
İnsan PrH’leri (Tablo 1) %85-90 oranında sporadik ortaya çıkarlar (sCJH). Geri kalan %1-2 edinsel (enfeksiyöz), %5-15 de kalıtsal (ailevi veya genetik) olarak ortaya çıkar. İnsan prion hastalıklarının hemen tümü, hasta beyninden hazırlanan ekstrenin primatlar dahil bir deney hayvanına inokülasyonuyla deneysel olarak veya bir prion hastalığı taşıdığı bilinmeyen bir donörden alınmış büyüme hormonu veya kornea, dura mater gibi bir dokunun nakliyle insanlara kazara (iyatrojenik olarak) bulaştırılabilir (iCJH). Sporadik CJH’de PrPC'nin PrPTSE'ye dönüşümünün kendiliğinden (veya muhtemelen PRNP'nin somatik bir mutasyonu yoluyla) meydana geldiği düşünülmektedir. Kalıtsal PrH’lerde, PRNP mutasyonlarının PrPC'yi PrPTSE'ye dönüştüren değişime (yanlış katlanma) daha duyarlı hale getirdiği ileri sürülmüştür.
Tablo 1. Prion hastalıkları.
|
İnsan Hastalıkları |
|
|
Sporadik |
Sporadik CJH (sCJH) Sporadik Fatal İnsomnia (sFI) Değişken Proteaz Duyarlı Prionopati (VPSPr) |
|
Edinsel |
Kuru İyatrojenik CJH (iCJH) Varyant CJH (vCJH) |
|
Kalıtsal |
Ailevi CJH (fCJH) Familyal Fatal İnsomnia (FFI) Gerstmann-Sträussler-Scheinker Hastalığı (GSS) Prion Proteini Serebral Amiloid Anjiopati (PrP-CAA) Huntington Hastalığı Benzeri – 1 (HDL-1) |
|
Hayvan Hastalıkları |
|
|
Koyun |
Scrapie |
|
Sığır |
Bovin Spongiform Ensefalopati (BSE) |
|
Vizon |
Bulaşabilen Vizon Ensefalopatisi (TME) |
|
Geyik |
Kronik Kas Erimesi Hastalığı (CWD) |
|
Kedi |
Felin Spongiform Ensefalopati (FSE) |
Prion Hastalıklarında Laboratuvar
Laboratuvar incelemeleri tanıyı kesinleştirmese de, duyarlılığı arttırarak katkıda bulunur. Ancak, klasik laboratuvar bulguları bazı klinik sunumlar için tipikken (örneğin, MM(V)1) bazılarında hiç görülmeyebilir (örneğin, VV2).
Elektroensefalografi (EEG), hastalık seyrinin bir dönemi için hastalığa özgü periyodik 1 Hz keskin-dalga komplekslerini (PSWC), hastaların üçte ikisinde gösterir (Şekil Pr01).
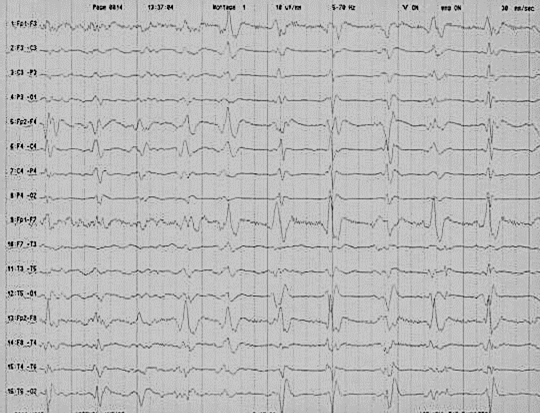
Şekil 1. Tipik CJH EEG’si: Periyodik keskin dalga aktivitesi.
MRG’de difüzyon ağırlıklı görüntülerde (DWI) ortaya çıkan, çoğunlukla ADC karşılığı olan, zaman içinde sırasıyla FLAIR ve T2 ağırlıklı görüntülerde de beliren kortikal (şeritlenme) ve başta bazal ganglia, sonra da talamus olmak üzere izlenen yüksek sinyalin, hızlı seyirli bir demansta CJH için oldukça yüksek duyarlılık ve özgüllük taşıdığı çeşitli çalışmalarda bildirilmiştir (Şekil 2). Bu bulgunun voltaj kapılı potasyum kanalopatili olgularda da izlenebildiği unutulmamalıdır.

Şekil 2. Sporadik CJH’de MRG. İki ayrı sporadik CJH hastası. a. Subkortikal baskın tarz: T2 ağırlıklı aksiyel kesitte (sol üst) daha belirsiz olan kaudat ve putamen hiperintensiteleri, FLAIR koronal kesitte (üst orta) biraz daha belirginleşiyor ve DWI aksiyel kesitte (üst sağ) en belirgin hale geliyor. b. Kortikal şeritlenme ve subkortikal yüksek sinyal ile birlikte olan tarz: FLAIR aksiyel kesitte (alt sol) bazal ganglia ve silik talamik hiperintensite, DWI aksiyel kesitlerde (alt orta ve alt sağ) 2 yanlı medial frontal kortikal şeritlenme. (İstanbul Tıp Fakültesi Nöroloji Anabilim Dalı, Davranış Nörolojisi ve Hareket Bozuklukları Birimi arşivinden).
CJH’de tipik olarak BOS’ta hücre görülmez. Beyin-omurilik sıvısı (BOS) proteini hafif yükselmiş olabilir (<100 mg/dl). BOS’ta nöronal yıkım ürünleri total tau ve 14-3-3 proteinin düzeylerinin belirgin yükselmesinin hastalık için yüksek duyarlılık gösterdiği ileri sürülmektedir. Total tau düzeyinin tanısal doğruluğunun, nöron spesifik enolaz (NSE) ve 14-3-3’e göre daha yüksek olduğu bildirilmiştir, ama buna rağmen DWI-ADC MRG tüm diğer biyoişaretleyicilerden daha doğru tanı koydurucudur.
Son zamanlarda geliştirilen “protein misfolding cyclic amplification assay (PMCA)” ve “real-time quaking-induced conversion (RT-QuIC)” isimli in vitro amplifikasyon teknikleri ile dokularda ve BOS gibi sıvılarda başka yöntemlerle saptanamayacak kadar az miktarda bulunan ve PrPTSE tarafından tetiklenen PrPc’nin hatalı katlanmasının in vitro olarak amplifiye edilerek saptanabilmesi sağlanmaktadır.
PMCA, vCJH dışında, Tip 1 için duyarlı olmadığından daha sınırlı kullanıma sahiptir. Buna karşın RT-QuIC için, majör alttiplerde daha fazla olmak üzere, yüksek ortalama duyarlılık (yaklaşık %77-92), ve özgüllük (%99-100) değerleri bildirilmiştir. İkinci kuşak RT-QuIC ile sonuç alma süresi azaltılmış, duyarlılık daha yükseltilmiştir. iCJH ve kalıtsal CJH’lerde de RT-QuIC çok kullanışlı bir tanı aracıdır.
Yine de kuşkuda kalındığında beyin biyopsisine başvurulmalıdır. CJH kuşkusuyla yapılan bir beyin biyopsisinde 2. kez kullanılmayacak cerrahi aletler seçilir.
PrH Nöropatolojisi
Mikroskopik olarak spongiform değişiklikler, astrositik gliozis ve nöron kaybı şeklindeki triad ile birlikte mikroglial aktivasyon klasik nöropatolojidir. Tip 2 ve V taşıyıcıları ek olarak plak oluşumları sergilerler ve klasik tetrad nispeten geri planda olabilir veya hiç olmayabilir. İmmünhistokimyasal (IHC) boyama ile PrPTSE reaktivitesi ve Western blot (WB) ile proteaza dirençli PrPTSE suşunun gösterilmesi tanı koydurucu olur.
Nöropatolojik imza sayılabilecek olan spongiform (süngersi) değişiklikler, küçük (<10μm) vakuollerle karakterizedir. Spongiform değişiklik korteksin yanısıra bazal ganglia, talamus, serebellum ve beyinsapında görülebilir. Spongiform değişikliğin bölgesel varyasyonları fenotipik varyantlara özgü patolojik topografileri oluşturur.
Nöron kaybı spongiform değişikliklere aynı bölgelerde eşlik eder. Bununla birlikte lokal yatkınlık ve dirençlilik alt bölgeleri ayırt edilir. Kortekste hippokampal CA1-4 alt sektörleri göreli olarak az nöron kaybı sergiler veya hiç nöron kaybı sergilemez. Talamusta ventral-anterior ve medio-dorsal çekirdekler daha ağırlıklı olarak tutulur. Serebellumda Purkinje hücreleri göreli olarak korunurken, internal granüler tabakada ağır nöron kaybı izlenir. Beyinsapı tutulduğunda nöron kaybı ağırlıkla mezensefalik tektum ve substantia nigradadır.
SPORADİK PRİON HASTALIKLARI
Bu başlık en sık görülen klasik CJH, çeşitli varyantları ve sporadik fatal insomni (sFI) ile altı moleküler alttip ve onların fenotipik karşılıklarını içerir. Artık VPSPr de sporadik prion hastalıkları başlığı altında sınıflandırılmaktadır.
Sporadik CJH
Tablo 2’de gösterilen sporadik CJH için tanı kriterleri Dünya Sağlık Örgütü (WHO) tarafından 1998’de yayınlanan kriterlerin 2018 yılında ABD Centers for Disease Control and Prevention (CDC) tarafından güncellenmiş şeklidir. WHO kriterleriyle çok benzer bir algoritma ve terminoloji kullanılmış, sadece laboratuvar yöntemlerine tipik EEG ve BOS 14-3-3 düzeyi yanı sıra tipik MRG ve RT-QuIC bulguları da eklenmiştir.
Tablo 2. Sporadik Creutzfeldt-Jacob Hastalığı İçin Güncellenmiş “ABD Centers for Disease Control and Prevention (CDC)“ Tanı Kriterleri (2018).
|
KESİN • Standard nöropatolojik tekniklerle; ve/veya IHC ile; ve/veya WB ile konfirme PK dirençli PrP; ve/veya scrapie ilişkili filaman (SAF) varlığı |
|
MUHTEMEL • Nöropsikiyatrik bozukluk ile birlikte BOS’ta veya diğer dokularda pozitif RT-QuIC VEYA 1. Hızlı progresif demans; ve aşağıdaki 4 klinik özellikten en az ikisi: • Miyoklonus • Görsel veya serebellar bulgular • Piramidal/ekstrapiramidal bulgular • Akinetik mutizm VE aşağıdaki laboratuvar testlerinden en az birinde pozitif sonuç • Her hangi süreli bir hastalık sırasında tipik bir EEG (periyodik keskin-dalga kompleksi-PSWC) • İki yıldan daha kısa süreli bir hastalık sırasında pozitif BOS 14-3-3 • DWI veya FLAIR-MRI’da kaudat/putamen’de veya en az iki kortikal alanda (temporal, parietal, oksipital) yüksek sinyal VE rutin incelemelerde alternatif bir tanıyı gösteren bulguların olmaması |
|
MÜMKÜN Hızlı progresif demans; ve yukarıdaki 4 klinik özellikten en az ikisi VE yukarıdaki laboratuvar testlerinde bir vakayı “muhtemel” olarak sınıflandıracak pozitif bir sonucun olmaması VE iki yıldan daha kısa hastalık süresi VE alternatif bir tanıyı gösteren rutin incelemelerin yokluğunda |
MM(V)1 – Klasik CJH ve Heidenhain Varyantı
Klasik (miyoklonik) hastalık, Heidenhain varyantı ile birlikte, %70 sıklığıyla en sık görülen sCJH tipidir. Moleküler alttip MM(V)1’in fenotipik sunumudur.
Hastalık coğrafi bölge, etnik köken veya cinsiyet ayırt etmeksizin bütün dünyada görülebilmekte ve yıllık sıklığı yaklaşık 1-1,5/1,000,000 kişi olarak hesaplanmaktadır. Konu üzerine uzmanlaşmış büyük merkezlerde CJH hastalarından oluşan geniş vaka serilerinde ortalama yaşın 70 yıl, ortalama hayatta kalmanın 4 ay kadar olduğu hesaplanmaktadır. Belirtilerin başlamasından sonraki 1 yıl içinde tüm olguların %85-90’ının ölmüş olduğu kaydedilmiştir.
Hastalık olguların yaklaşık %25’inde uykusuzluk, iştahsızlık, kilo kaybı, huzursuzluk, halsizlik gibi spesifik olmayan prodromal belirtilerle başlar. Prodromal belirtiler olsun olmasın, klasik olarak hızlı seyirli bir demans tablosu gösteren bu durumda başlangıç belirtileri yarıdan fazla olguda genel kognitif yıkım, sonra sırasıyla ataksi, afazi, kortikal körlükten agnozilere görsel işleme problemleri, miyoklonus ve hipokinetik ağırlıklı hareket bozukluğu şeklindedir. Bunlar haftalar içinde hızla ağır bir demansa ve sonunda akinetik mutizme evrilir. Hezeyan ve halüsinasyonlar şeklinde psikotik özellikler görülebilir. Genellikle hastalığın orta evrelerinde tabloya katılan, akustik ve hatta taktil uyaranlarla ortaya çıkarılabilen irkilme miyoklonisi tanıya güç katan klinik bulgulardandır.
Klasik laboratuvar bulguları tipik olarak bu varyantta yüksek sıklıklarda saptanır. DWI’da bazal ganglia ve kortekste yüksek sinyal %70’ler düzeyinde görülür. Vurgulanması gereken görüntüleme özellikleri olarak, medial temporal lob (MTL) ve talamik yüksek sinyalin bu varyantta görülmemesidir. MTL yüksek sinyali diğer bir hızlı seyirli demans nedeni otoimmün limbik ensefalitin özelliklerindendir ve bu özellik önemli bir ayırıcı tanı kriteridir. Talamik yüksek sinyal diğer sCJH fenotiplerinde (örneğin, VV2, MV2K ve MV2C ) ve vCJH’de görülebilir. BOS’ta başlangıçtan kısa süre sonra patolojik olarak yükselen 14-3-3 ve total tau ve aylar içinde görülme sıklığı %80’e ulaşan EEG’de PSWC klinik tanıya yardımcıdır.
Dolayısıyla, 60’lı yaşlarda çok hızlı seyirli demans geliştirmiş, irkilme miyoklonileri sergileyen bir hastada DWI’da neokortikal ve bazal ganglia yüksek sinyali, EEG’de PSWC, BOS’ta pozitif 14-3-3 ve total tau tanı koydurucudur. PRNP kodon 129’da M homozigotluğunun veya MV heterozigotluğunun saptanması ve RT-QuIC pozitifliği tanıyı doğrular.
Karma MM(V)+2C alttipi oldukça sıktır ve fenotipin %43’ünü oluşturur. Klinik ve nöropatolojik olarak saf alttipten ayırt edilemez. Sadece Tip 2 yükü arttıkça hastalık süresinin uzadığı ve başlangıçtaki serebellar bulguların seyrekleştiği kaydedilmiştir.
Hastalığın başlangıç evresinde haftalar boyu izole kalabilen görsel semptomlar 1954’te Meyer, Leigh ve Bagg tarafından Heidenhain varyantı olarak adlandırılmıştır. Orijinal olarak Heidenhain tarafından 1929 yılında tarif edilmiş olan bu varyantta anosognozinin eşlik edebileceği kortikal körlüğe (Anton sendromu) kadar varabilen çeşitli yokuş-yukarı görsel işleme bozuklukları (optik distorsiyonlar, astereopsi, akromatopsi, akinetopsi gibi) olabileceği gibi posterior kortikal atrofi (PKA) benzeri yokuş-aşağı görsel işleme bozukluklarının baskın olduğu (simultanagnozi, prosopagnozi gibi) tablolarla da sunulabilir. Klinik sunumdan bekleneceği üzere klasik patoloji oksipital loblarda yoğunlaşmıştır. Bu klinik sunum nüansına rağmen klasik CJH ile MM(V)1 moleküler statü dahil hemen tüm laboratuvar ve nöropatolojik bulgularda ortaklaşır.
VV2 – Brownell-Oppenheimer (Ataksik) Varyantı
Ataksi ile başlangıç ilk kez 1965’te Brownell ve Oppenheimer tarafından bildirilmiştir ve Brownell-Oppenheimer varyantı olarak da adlandırılır.
VV2 moleküler alttipinin klinik sunumu en sık ikinci fenotiptir (sCJH’lerin yaklaşık %16’sı). Ortalama yaş 65 yıl ve ortalama hastalık süresi 6,3 aydır. Ataksi ile sunulur ve demans orta evreden itibaren katılır. Yaklaşık üçte bir olguda miyoklonus hastalık süresince görülmez.
DWI’da yüksek sinyal bazal ganglia ve talamusta izlenir. Kortikal yüksek sinyal genellikle singulat girusa sınırlıdır ve neokortekste izlenmez. PSWC ancak %10 sıklıkta saptanır. BOS biyoişaretleyicileri yüksek sıklıkta pozitiftir.
Dolayısıyla, geç 50’li yaşlarda çok hızlı seyirli ataksi ve sonrasında kognitif yıkım geliştirmiş bir hastada DWI’da bazal ganglia ve talamusta yüksek sinyal ve BOS’ta pozitif 14-3-3 ve total tau tanı koydurucudur. PRNP kodon 129’da V homozigotluğunun saptanması ve RT-QuIC pozitifliği tanıyı doğrular.
MV2K – Uzun Süreli (Kuru Plaklı) Ataktik Varyant
MV2 moleküler alttipinin kuru plaklı varyantı üçüncü sıklıktaki (%10) sCJH fenotipidir. Ortalama yaş 65 yıl ve ortalama hastalık süresi 16 aydır. Klinik sunumda ataksi ve demans baskındır.
PSWC izlenmez. DWI’da yüksek sinyal kortekste frontal loblara ve singulat giruslara sınırlı olarak görülebilir. Tipik olarak bazal ganglia ve talamusta izlenir. Talamik yüksek sinyal bazen pulvinar çekirdeğe sınırlıdır ve bu durumda “pulvinar bulgusu”ndan söz edilir. BOS biyoişaretleyicileri için en düşük duyarlılıklar bu fenotipte bildirilmiştir (%57-89).
Dolayısıyla, 60’lı yaşlarda hızlıca seyreden demans ve ataksisi olan bir hastada DWI’da bazal ganglia ve talamusta yüksek sinyal tanı koydurucudur. PRNP kodon 129’da MV heterozigotluğunun saptanması ve RT-QuIC poztifliği tanıyı doğrular.
MM(V)2C – Uzun Süreli (Kortikal) Varyant
Yukarda değinildiği gibi gerek MM2 ve gerekse de MV2 moleküler alttipleri iki farklı fenotipe ayrılır. Bu fenotip baskın olarak MM2C, ama daha az sayıda da MV2C olgularından oluşur. sCJH içindeki sıklığı %1-2 arasındadır. Ortalama başlangıç yaşı 68 yıl ve ortalama sağkalım 20 aydır. Klinik sunumda jeneralize kognitif yıkım baskındır. Serebellar bileşen yoktur veya mevcutsa bile hafiftir. Miyoklonus genellikle yoktur.
PSWC görülmez. DWI’da kortikal yüksek sinyal özellikle temporal loblarda belirgindir. Talamik yüksek sinyal de eşlik edebilir. Bazal gangliada nadirdir. BOS biyoişaretleyicilerinin pozitivitesi düşüktür (%61-78). Birinci kuşak RT-QuIC duyarlılığı düşükken (%42), ikinci kuşak ile daha iyileştirilmiştir (%78).
Dolayısıyla, 60’lı yaşlarda hızlıca seyreden bir demans geliştiren, fakat EEG’de PSWC ve BOS’ta pozitif biyoişaretleyiciler olmayan bir hastada RT-QuIC negatif dahi olsa DWI’da kortikal şeritlenme bu tanıyı düşündürür. PRNP kodon 129’da homozigot MM veya heterozigot MV saptanması tanıyı destekler. Yine de beyin biyopsisi gerekebilir.
VV1 – Genç Başlangıçlı Varyant
Sıklığı %1 kadardır. Ortalama başlangıç yaşı 39 yıl, ortalama sağkalım süresi 15 aydır. Klinik sunum başlıca davranışsal varyant fronto-temporal demans (dvFTD) tarzındadır. Aylar boyunca bu tablo izole kalabilir. Son evrelere kadar ataksi ve miyoklonus gibi motor bulgular görülmez.
EEG’de PSWC görülmez. DWI’da kortikal yüksek sinyal, diğer fenotipler arasında en belirgin VV1’dedir. Bazal ganglia ve talamusta ise nadiren görülür. BOS biyoişaretleyicilerinin duyarlılığı en yüksek bu fenotiptedir ve %100’e ulaşır.
Dolayısıyla, diğer açılardan her bakımdan klasik dvFTD gibi sunulmuş genç bir hastada DWI’da kortikal şeritlenme ve BOS’ta pozitif 14-3-3 ve total tau tanı koydurucudur. PRNP kodon 129’da V homozigotluğunun saptanması ve RT-QuIC pozitivitesi tanıyı doğrular.
MM2T – Sporadik Fatal Insomnia (Talamik Varyant)
Sıklığı %1 kadardır. Moleküler olarak nadiren MV2T de olabilir. Ortalama başlangıç yaşı 43 yıl, ortalama sağkalım 30 aydır. Klinik ve nöropatolojik olarak kalıtsal form olan familyal fatal insomni (FFI) ile ayırt edilemez. Nörodejeneratif hastalıklar içinde benzersiz biçimde kalıtsal formuna göre çok daha nadir görülür. Şimdiye kadar bildirilmiş kısıtlı sayıda olgunun ilki Prusiner ekibi tarafından 1999 yılında bildirilmiş 44 yaşında bir erkektir.
Klinik tablo uyku bozukluğu, disotonomi ve klasik CJH bulguları triadı şeklindedir. Uyku bozukluğu tipik olarak uykuya dalma zorluğu, sık uyanmalar, ani rüyalar, aşırı gündüz uykululuğu olarak sıralanabilir ve erken dönemden itibaren baskındır. Ağır ve sürekli uykusuzluk sırasında ani rüyalar ve buna eşlik eden motor hiperaktivasyon, stereotipik hareketler (oneirik stupor) ile sempatik aktivasyon agrypnia excitata (agitata) olarak adlandırılır. Oneirik stupor günlük yaşam aktivitelerinin taklidi olan otomatik basit jestlere verilen isimdir. Bu durum, göreli kısa süreli epizotlar şeklinde olan klasik REM uykusu davranış bozukluğuna göre daha uzun süreli, nerdeyse süreklidir. Disotonomi hipertansiyon, taşikardi, hiperhidroz, hiperpireksi, impotans, lakrimasyon ve hipersalivasyon olarak izlenir ve tabloya sonradan da katılabilir. Klasik CJH bulguları kognitif ve motor olarak ayrılır. Kognitif bozukluğun tespiti, söz edilen sürekli rüya durumu nedeniyle güçtür. Test edilebildiğinde, hastalarda diğer sCJH’lerde izlenen demans bileşenlerinden çok, dikkat ve çalışma belleği bozuklukları saptanır. Psikiyatrik bulgular sıktır ve halüsinasyonlar, kişilik değişikliği, depresyon, anksiyete, saldırganlık, disinhibisyon şeklinde olabilir. Motor bulgular, ataksi, dizartri, disfaji, okülomotor bozukluklar (diplopi, yumuşak izleme bozuklukları), piramidal bulgular erken dönemde ayırt edilebilir. Miyoklonus orta evrelerden itibaren eklenir.
EEG’de PSWC ve DWI’da yüksek sinyal görülmez. Klasik BOS biyoişaretleyicileri negatif olabilirken (14-3-3, total tau), nörofilaman hafif zincir proteininin (Nfl) yükseldiği bildirilmiştir. İkinci kuşak RT-QuIC ile duyarlılık %80’e çıkmıştır. İnsan PrH’ları içinde benzersiz olarak, gerek sporadik ve gerekse de familyal FI’da tanı koydurucu laboratuvar incelemeleri polisomnografi (PSG) ve FDG-PET olmaktadır. PSG’de tipik olarak normal uyku mimarisinin (sirkadyen uyku-uyanıklık ve ultradiyen REM-NREM ritimler) giderek bozulması izlenir. Toplam uyku süresinin kısalması ile birlikte uyku iğcikleri ve K kompleks miktarları azalır. Yavaş dalga uykusu (SWS) öncelikle, sonra da REM uykusu bozulur. FDG-PET ile korunmuş kortikal metabolizmaya karşı izole talamik hipometabolizma tipik bulgudur.
Dolayısıyla, uyku bozukluğu sergileyen bir hastada psikiyatrik ve motor bulgular, PSG’de uyku mimarisinin bozulması tanıyı düşündürür. Ayırıcı tanıda diğer agrypnia excitata sunumları olan Morvan sendromu ve delirium tremens ile özellikle NMDAR başta olmak üzere otoimmün ensefalitler göz önüne alınmalıdır. FDG-PET ile talamik hipometabolizma bu tanı olasılıkları ile ayırıcı tanıda en önemli özelliktir. RT-QuIC ile BOS veya olfaktor mukoza pozitivitesi ve PRNP kodon 129 M homozigotluğu tanıyı doğrular.
Değişken Proteaz Duyarlı Prionopati (VPSPr)
VPSPr az sayıda hastada tanımlanmıştır. Tüm genotipler bir arada ortalama başlangıç yaşı 70 yıl (48-87), ortalama hastalık süresi 24 aydır (7-91). Klinik sunumda psikiyatrik bulgular (başlıca disinhibisyon, apati, duygudurum değişiklikleri), afazi (başlıca anomi ve dizartri) ve kognitif bozukluk (başlıca frontal yürütücü bozukluk) bir triad olarak tanımlanmıştır ve bunlara ataksi ve Parkinsonizm şeklinde motor bulgular eşlik edebilir. Klasik CJH laboratuvar bulgularının tanıya yardımcılığının düşük olduğu saptanmıştır. Tanıda en yararlı yöntem ikinci kuşak RT-QuIC olarak öne çıkmaktadır.
Edinsel Prion Hastaliklari
İyatrojenik CJH: Kornea nakli yapılan, prion bulaşmış cerrahi aletlerle nöroşirürjikal operasyon geçiren, enfekte derin elektrodların kullanıldığı, insan hipofiz bezi ekstreleri alan veya dura mater greftlemesi yapılan kişilerde gösterilmiştir. Klinik tablo sporadik hastalığın 129MM(V)1 ve 129VV2 alttiplerine çok benzer. MRG’de sporadik hastalığa benzer bulguların yanı sıra serebellumda difüzyon kısıtlılığı görülebilir.
Bulaşma ile hastalığın ortaya çıkması arasında geçen süre (inkübasyon periyodu), bulaşmanın doğrudan beyne (örneğin cerrahi gereçler, dural greftler), ya da sistemik olarak (örneğin hipofiz ekstresi) olmasına bağlı olarak, 2 ila 40 yıl arasında değişir. Kan ve kan ürünleri ile hastalığın bulaşması riski bir endişe kaynağı olsa da vCJH dışında tam olarak doğrulanmamıştır. Hasta kanının şempanzelere nakli bu hayvanlarda hastalığa yol açmazken, farelerde doğrudan beyne zerkedilmesiyle hastalık ortaya çıkmıştır. Sonradan vCJH geliştiren hastalardan yapılmış kan transfüzyonu öyküleri olan 2 ayrı vCJH olgusu bildirilmiştir.
İnkübasyon süresi değişkendir. Dura mater olgularında kodon PRNP 129MM’nin daha sık ve inkübasyon süresi kısadır. Kinik hızlı seyirli demanstır. Buna karşılık büyüme hormonu olgularında PRNP kodon 129VV daha sık, inkübasyon süresi uzundur ve klinik daha çok progresif ataksidir.
Kuru: Papua Yeni Gine’de Fore yerlileri arasında tarif edilmiş ve hastalığa neden olduğu kanıtlanan bölgeye özgü dinsel ritüelin 1950’lerin sonunda yasaklanıp terkedilmesiyle 2000’li yıllardan itibaren artık görülmez olmuştur. Forelerin geleneklerine göre, aile büyüğünün ölümünden sonra, onun bilgeliğini yeni kuşaklara geçirebilmek için beyni o ailenin kadınları ve her iki cinsiyetten çocukları tarafından yenir, ayrıca beden ve yüze sürülür. Etkenin konjonktivalar, deri ve sindirim sisteminden alınmasını izleyen inkübasyon periyodu sonrasında, hastalık süresi ve klinik özellikler açılarından sCJH’yi andıran bir tablo ortaya çıkar. Hastalık deneysel olarak şempanzelere de geçirilebilmiş ve izleyerek onların da kendi aralarında taşınabilmiştir. Ritüelin yukarda belirtildiği gibi her iki cinsten çocuklara ve erişkin kadınlara sınırlı olması dolayısıyla ilginç bir yaş ve cinsiyet dağılımı izlenir. Olguların %60’ı erişkin kadınlar, %2’si erişkin erkekler ve %38’i her iki cinsiyetten çocuk ve ergenlerdir.
Her üç kodon 129 genotipi ile de Kuru bildirilmişse de heterozigotluğun inkübasyon süresini çok uzattığına hatta hastalıktan koruduğuna dair bildirimler vardır. Elli yılın üzerinde inkübasyon süreleri bildirilmiştir. Başlangıç yaşı 4 ila 60 gibi oldukça geniş bir aralıktadır. MM homozigotlarda hastalık başlangıcı daha erken yaştadır. Hastalık süresi ortalama 12 aydır (3-23). Bazı araştırmalarda, kuru epidemisinden kurtulan Forelerde 127. kodonda koruyucu gibi görünen bir PRNP polimorfizmi (G127V) tanımlanmıştır.
Klinik tablo hemen daima ataksi ile sunulur. Tremor, koreiform ve atetoid hareketler şeklinde hiperkinetik hareket bozuklukları eşlik eder. Disinhibisyon baskın kişilik değişikliği erken dönemde görülür. Uygunsuz öfori ve kompulsif gülmeler ayırıcı özelliktir (hastalık popüler olarak “gülerek ölüm” veya “gülme hastalığı” olarak da adlandırılmıştır). Kognitif yıkım seyirde geç dönem özelliğidir.
Nöropatolojik olarak spongiform değişiklik, astrogliozis ve nöron kaybı triadı tipiktir. Oksipital loblar, hippokampus ve insulanın göreli korunması ile birlikte tüm kortikal alanlar tutulmuştur. Striatum ve serebellum da tutulur. Bununla birlikte nöropatolojik olarak baskın özellik Kuru plaklarıdır. Bu plaklar yuvarlak veya oval, koyu renk bir çekirdeğin çevresinde daha soluk bir halo ile çevrilidir. En sık serebellar granüler tabakada görülürken azalan sıklıkta bazal ganglia, talamus ve serebral kortekste saptanırlar. WB ile PrPTSE Tip 2 özelliktedir.
Yeni Varyant CJH: İlk kez 1995 yılında Birleşik Krallık’ta görülen “yeni varyant CJH” (vCJH) 2000 yılında aynı ülkede 28 vakayla yıllık zirve sayısına ulaşmış ve sonra düşüşe geçmiştir. 2018 yılına kadar 178 vaka bildirilen bu ülkede 2006 yılından itibaren yıllık vaka sayısı beşin altına inmiştir. Birleşik Krallık dışında 27’si Fransa’dan olmak üzere şimdiye kadar 51 vaka bildirilmiştir. Türkiye’den bildirilen vaka yoktur. Hastalığın başlangıç yaşı sporadik hastalığa göre daha erken olup ortalama 27 yıl (12-74 yıl), ortalama hastalık süresi 14,5 aydır. Hastalık duyusal ve psikiyatrik belirti ve bulgularla başlar. Altı ay kadar süren bu başlangıç evresinden sonra önce ataksi, sonrasında da demans, istemsiz hareketler ve inkontinans gibi özellikler tabloya katılırlar. Laboratuvarda, sCJH’nin tipik EEG bulguları görülmez, genellikle spesifik olmayan yavaşlama görülür. BOS’ta 14-3-3 proteini olguların %50’sinde pozitiftir. MRG’de tipik pulvinar bulgusu (“hokey sopası” ismi verilen iki taraflı talamusun pulvinar çekirdeğinde, putamen ve kaudat çekirdeğin başındaki sinyal artışları) %70 sıklıkta görülmüştür. PMCA yöntemi tanı duyarlılığını çok arttırmıştır.
PRNP analizinde hastalığın ilk yirmi yılındaki tüm olguların kodon 129 M homozigot oldukları görülmüştür. Kodon 129’da V sahibi olmak inkübasyon süresini uzattığından bu durum hastalık etmenine aynı zaman çerçevesinde maruz kalan V sahiplerinin henüz klinik belirti geliştirmemiş olabilecekleri, 129V sahipleri dolayısıyla vaka sayılarının yeniden artabileceği endişesi bu kitabın 2011 tarihli ikinci baskısında ifade edilmişti. Nitekim, 2016 yılında 129MV olan ilk vCJH vakası otopsi doğrulamalı olarak bildirildi.
Diğer insan prion hastalıkları ve bu arada sCJH’nin tersine, lenforetiküler sistem de tutulduğundan tonsiller biyopsi ile hastalık yapan prion proteinin (PrPSc) gösterilmesi vCJH için tanı koydurucudur. Azalan konsantrasyonlarda dalak, lenf düğümleri, sürrenal bez, timus ve rektumda da saptanabilir.
Avrupa’da vCJH olguları, ilk deli dana olgusunun (Bovine Spongiform Encephalopathy – BSE) bildiriminden 10 yıl sonra görülmeye başlandı. İzleyerek, vCJH’nin BSE etkeni ile kontamine dana eti yenmesi sonucu gelişebileceğini gösteren çok sayıda delil bulundu. Birleşik Krallık içi ve dışı olguların çok büyük bir bölümünün de yine bu ülke kökenli dana etine maruz kalma sonucu olduğu düşünülmektedir. İnsandan insana kan nakliyle geçiş üç olguda bildirilmiştir.
Nöropatolojik olarak, vCJH’de, sCJH’den farklı olarak “süslü (florid) plak” adı verilen prion plakları ayırıcı özelliktir. Bunlar çok sayıda ve yaygın olarak görülür. Süslü plak merkezi eozinofilik amiloid çekirdeğin önce soluk radyan ışıltılımı fibriller, onun etrafında da vakuoller ve küçük PrPTSE birikinti kümeleri ile çevrelenmiş şekilde görülür. Başlıca neokorteks, talamus, striatum ve beyinsapı tutulur. Spongiform değişiklik tüm kortikal tabakalarda yamamsı bir tarzdadır. Serebellum moleküler tabakasında ve striatumda birleşen özellik gösterir. Posterior talamus spongiform değişiklikten çok baskın olarak gliozis ve ağır nöron kaybı sergiler. Hippokampus ve beyinsapı göreli olarak korunmuştur.
Kalıtsal Prion Hastalıkları
Kalıtsal prion hastalıkları tüm insan PrH’larının %15’ine yakınını oluştururlar ve otozomal dominant bir tarzda, 20. kromozomda kodlanan PRNP geninde oluşan çok sayıda mutasyon sonucu kalıtılırlar. Mutasyonlar hatalı anlamlı (missense), anlamsız (nonsense) veya oktapeptid tekrar insersiyonları (OPRI) veya delesyonları (OPRD) şeklindedir. Bildirilen altmıştan fazla mutasyonun arasında beş hatalı anlamlı nokta mutasyonu (P102L, D178N, V180I, E200K ve V210I) tüm kalıtsal PrH’lerin %85’inin nedenidir. Bunlardan P102L Gerstmann-Sträussler-Scheinker (GSS), diğer dördü fCJH nedenleridir. E200K ve D178N yüksek penetranslı mutasyonlarken (aile öyküsü yüksek ihtimaldir), V180I ve V210I düşük penetranslıdır (aile öyküsü olmayabilir). Ortaya çıkan hastalık klinik, laboratuvar bulguları ve nöropatoloji açılarından sCJH’ye büyük ölçüde benzer. Fenotip ve patolojiyi çok büyük sıklıkla mutasyon değil moleküler alttip (kodon 129 M/V polimorfizmi ve PrPTSE Tip 1 ve Tip 2 suşları) belirler; yani sCJH için her bir moleküler alttip-fenotip ilişkisi fCJH için de geçerlidir. Sadece, stop kodonlu anlamsız mutasyonlar, GSS’ye neden olan biri dışında, hemen daima prion proteini serebral amiloid anjiopatiye (PrP-CAA) neden olurlar. OPRI’lerde dörde kadar olan insersiyonlar seyir açısından üç yıldan kısa sürme (hızlı seyirli), beş ve fazla sayıda olanlar üç yıldan uzun sürme (yavaş seyirli) eğilimindedirler. Tip 1 ve Tip 2 suşlar daima B tarzdadır (diglikozile). Az sayıda istisnai durumda moleküler alttip farklı bir patolojiye yol açabilir. GSS, Huntington hastalığı benzeri-1 (HDL-1) ve PrP-CAA fenotiplerinin sporadik karşılıkları yoktur. Kodon 129 M homozigotluğu genel olarak, V taşıyıcılığına göre hastalık yaşını daha erkene ve hastalık süresini daha kısaya taşır. D178N mutasyonunda, kodon 129 M homozigotluğu FFI’ye, V taşıyıcılığı fCJH’ye neden olur. PrP7-8 GSS’de serebral parankimal, PrP-CAA’da serebrovasküler amiloidoza neden olur.
Ailevi CJH (fCJH): fCJH ile ilintili 21 mutasyon bildirilmişse de bunların kayda değer bir bölümünün patojenitesi sorgulanabilir. E200K ve D178N dünya çapındaki yaygınlıkları ve yüksek penetransları ile başlıca genetik nedenler gibi görünmektedir. Bizim arşivimizdeki iki ailenin incelenebilen üyeleri de D178N-129MM’dirler.
sCJH’ye göre daha erken yaşta başlayıp (30-55 yıl), daha uzun sürebilir (4-60 ay). Klinik tablo klasik sCJH’ye benzer ve ataksi ve diğer motor bulgularla birlikte hızlı seyirli bir demans ve psikiyatrik bozukluklar baskındır. Tipik spontan veya irkilme miyoklonusu seyirde sıklıkla mevcuttur. Ek olarak, görsel bulgular, hipokinetik ve hiperkinetik ekstrapiramidal bulgular, supranükleer paralizi, yabancı el fenomeni ve epileptik nöbet de bildirilen klinik bulgular arasındadır.
Tipik laboratuvar bulguları sCJH’ye göre biraz daha az sıklıkta görülür. EEG’de PSWC göreli olarak düşük sıklıkta saptansa da DWI’da kortikal ve striatal yüksek sinyal ve BOS’ta 14-3-3 ve total tau pozitivitesi yüksek sıklıktadır. RT-QuIC tanıyı doğrular (E200K’da %81, V203I’da %100) ve genetik analizle sorumlu mutasyonun gösterilmesi tanıyı kesinleştirir.
Fatal Familyal İnsomni (FFI): PRNP genindeki D178N-129MM mutasyonu FFI için günümüze kadar bildirilen tek mutasyondur. Yukarıda söz edildiği gibi aynı mutasyon sCJH olarak da sunulabilir. Klinik fenotipi belirleyen kodon 129 polimorfizmidir ve FFI olguları M homozigotlarıdır (daha az sayıda heterozigot FFI bildirilmiştir). Şimdiye kadar 40 ailede 100 olgu bildirilmiştir. Sorumlu PrPTSE, sCJH-MM2T’de olduğu gibi Tip 2’dir (2B). Tipik hasta 50-60 yaşları arasındadır. Hastalık süresi ortalama 15,6 aydır (8-72 ay).
Klinik fenotip ve nöropatolojik olarak sFI’dan farksızdır. sFI’da olduğu gibi klasik laboratuvar bulguları tanıya yardımcı olmaz. Tipik PSG bulguları ve FDG-PET’te talamik hipometabolizma tanı koydurucudur. İkinci kuşak RT-QuIC duyarlılığı FFI’da %83’lere çıkmıştır.
Gerstmann-Sträussler-Scheinker (GSS): İlk kez hastalığa adını veren üç yazar tarafından 1935 yılında Viyanalı bir ailede (“H” Ailesi) tanımlanmış ve 1989 yılında P102L mutasyonunun bulunmasıyla tanımlanan ilk kalıtsal prion hastalığı olmuştur. Orijinal H ailesinin günümüzde 9 kuşağı boyunca 221 üyesi içinde GSS tanısı almış 20 birey bulunmaktadır. Şimdiye kadar 16 mutasyon bildirilmişse de, fCJH için olduğu gibi bunların tümünün patojenitesi kuşkuludur. P102L, P105L, A117V, H187R ve F198S en sık bildirilen mutasyonlardır. Bizim arşivimizdeki tek olgu P105L mutasyonu taşımaktadır.
Başlangıç yaşı 40’lı yaşlar ila 60’lı yaşlar arasında değişir. Beş-altı yıllık hastalık süresiyle (aylar süren çok kısa ve on yıl süren çok uzun biçimleri de nadiren bildirilmiştir) uzun süreli bir PrH’dir.
Klinik olarak başlıca ataksiyle sunulan, seyir içinde parkinsonizm ve piramidal bulguların (başlıca spastik paraparezi) ve geç dönemde demansın katıldığı bir tablodur. Bu fenotip P102L-GSS’nin klasik tablosudur. P102L büyük sıklıkla 129M’dir. P102L-129V olarak bildirilen bir olguda hastalık 33 yaşında nöbetlerle başlamıştır. P105L mutasyonu sahibi beş Japon ailede hasta bireyler spastik paraparezi ile sunulmuş, 5-10 yıllık seyir boyunca demans geç dönemde gelişmiştir. A117V ataksi olmaksızın saf demans ile sunulur ve “telensefalik GSS” olarak da adlandırılır. F198S-129V mutasyonlu Indiana Ailesi piramidal, parkinsonizm ve serebellar motor bulgular, demans, sakkadik göz hareketlerinde bozulma sergilemiştir. Yaş aralığı 40-70 yıldır ve hastalık 1-5 yıl sürmektedir. Nöropatolojik olarak tipik GSS değişiklikleri yanı sıra gri maddede çifte sarmallı filamanlar şeklinde hiperfosforile tau proteini içeren nörofibriler yumaklar (NFYler) görülmüştür. Spongiform değişiklik ender ve sadece plakların çevresindedir. Serebral amiloidoz en belirgin olarak GSS’de görülür. Tipik spongiform değişiklik belirgin değildir.
Klasik laboratuvar bulguları EEG PSWC ve DWI’da yüksek sinyal görülmez. BOS biyoşaretleyicilerinin pozitifliği %20 kadardır. Buna karşılık ikinci kuşak RT-QuIC duyarlılığı %90’a ulaşmıştır.
WB ile ortaya konan bantlar GSS’deki PrPTSE bileşimin oldukça heterojen olduğunu göstermiştir. N-terminus 30, 25, 20 kDa yanı sıra C-terminus 16-17 kDa ve 12-14 kDa PrPTSE suşları saptanmıştır. Ayrıca saptanan PrP PK-duyarlı suşun PK dirençli banttan daha fazla olduğu gösterilmiştir. Ancak, GSS mutasyonlarının doğrudan sonucu olarak N ve C budanmış non-glikolize 7-8 kDa suşun (PrP7-8) tüm GSS fenotiplerinde ortak özellik olduğu anlaşılmaktadır.
Prion Proteini Serebral Amiloid Anjiopati (PrP-CAA): 1996’da genç yaşta yavaş ilerleyici demansı başlayan ve 21 yıl yaşayan bir hastanın 129MM olacak şekilde Y145X anlamsız mutasyonunu taşıdığı, otopsisinde amiloid-β ile değil ama PrP ile immünreaktivite gösteren yaygın serebrovasküler amiloidoz olduğu görüldü. WB ile sorumlu suşun PrP7-8 olduğu görüldü. Bunun ardından Q160X (129MV) ve Y226X (129MV) ve Y163X anlamsız mutasyonları saptandı. Genel olarak hastalık erken yaşta başlamaktadır. Serebral amiloidozis dışında periferik amiloid tutulumu da olabilmekte ve buna bağlı periferik siniri tutulum bulguları izlenebilmektedir.
Huntington Hastalığı Benzeri-1 (HDL-1): Huntington hastalığı (HH) bulguları gösteren ama genetik olarak HH tanısı gösterilemeyen bir ailede PRNP’de kodlayan bölgede 192 nukleotidlik bir heterozigot insersiyonun ekstra sekiz kopyalık bir oktapeptid tekrarlı uzamış bir PrP’ye yol açtığı keşfedildikten sonra prion genindeki mutasyonların HH’nı taklit edebileceği anlaşılmıştır. Aynı mutasyon klasik HH kliniği gösterebilse de, uzun hastalık seyirleri ile nöropsikiyatrik bulgulara yol açıp HH’nın temel bulgusu olan kore yapmadan da seyredebilir.
Tedavi
Prion hastalıklarının henüz bir tedavisi yoktur. Ancak, PrPC’nin PrPSc’ye dönüşümünü engellemek gelecekteki tedavi stratejilerinin temel hedefi gibi durmaktadır. PrPC’ye bağlanarak stabilizasyonunu sağlamak bir yol olabilir. Diğer bir yol da PrPSc’yi destabilize edecek ajanların geliştirilmesi ile olabilir. PrP geni taşımayan (PrP0/0) nakavt transgenik farelerin doğal hayvanlara göre ayırt edilebilir bir farklılık göstermemeleri insanda da PrP genini hedefleyecek tedavileri çekici kılmaktadır. Ancak şu aşamada uygulanabilen tedaviler yalnızca semptomatiktir.
Tetrasiklin grubu antibiyotiklerin spesifik anti-prion etkileri in vitro modeller ve hayvan modellerinde gösterilmiş ve bu nedenle klinik çalışmalarda denenmişlerdir. Doksisiklin göreli geniş sayıda hastanın dahil edildiği bir çift-kör, plasebo kontrollü rastgele faz II çalışmada etkisiz kalmıştır. Bu çalışmanın nispeten geç evre hastalarda yapıldığı düşünülerek gerçekleştirilen bir faz II küçük çalışma ve bir gözlemsel çalışmanın verileri doksiklinin sağkalımı marjinal bir düzeyde uzattığını ortaya koymuştur.
Bu bölüm, sayfa sayısı kısıtlaması nedeni ile kitabın basılı halinde kısaltılmıştır. Daha ayrıntılı metin için online edisyona başvurulabilir (bölümün uzun web versiyonu için tıklayınız).
Kaynaklar
1. Andrew SE, ve ark. Huntington disease without CAG expansion: phenocopies or errors in assignment? Am J Hum Genet 1994; 54 :852-863.
2. Baiardi S ve ark. Revisiting the Heidenhain Variant of Creutzfeldt-Jakob Disease: Evidence for Prion Type Variability Influencing Clinical Course and Laboratory Findings. J Alzheimers Dis 2016; 50 :465-476.
3. Brown K, Mastrianni JA. The prion diseases. J Geriatr Psychiatry Neurol 2010; 23 :277–298.
4. Brownell B, Oppenheimer DR. An Ataxic Form of Subacute Presenile Polioencephalopathy (Creutzfeldt-Jakob Disease). J Neurol Neurosurg Psychiatry 1965; 28 :350-361.
5. Ghetti B ve ark. Prion protein amyloidosis. Brain Pathol 1996; 6, 127-145.
6. Green AJE. RT-QuIC: a new test for sporadic CJD. Pract Neurol 2019; 19 :49-55.
7. Laplanche JLve ark. Prominent psychiatric features and early onset in an inherited prion disease with a new insertional mutation in the prion protein gene. Brain 1999; 122 :2375-2386.
8. Lugaresi E, Provini F. Agrypnia excitata: clinical features and pathophysiological implications, Sleep Med Rev 2001, 5(4): 313-322. doi: 10.1053/smrv.2001.0166.
9. Parchi P ve ark. Phenotypic variability of sporadic human prion disease and its molecular basis: past, present, and future. Acta Neuropathol 2011; 121 :91-112.
10. Rossi M ve ark. Understanding Prion Strains: Evidence from Studies of the Disease Forms Affecting Humans. Viruses 2019;11 :309.